

Abstract
Overexpression of ubiquitin‐specific protease 28 (USP28) is found in hepatic carcinoma. It is unclear whether the deubiquitinase plays a role in hepatocarcinogenesis. Deregulation of the Wnt signaling pathway is frequently associated with liver cancer. Transcription factor 7‐like 2 (TCF7L2) is an important downstream transcription factor of the Wnt/β‐catenin signaling pathway, but the mechanisms by which TCF7L2 itself is regulated have not yet been revealed. Here, we report that USP28 promotes the activity of the Wnt signaling pathway through maintaining the stability of TCF7L2. We further show that FBXW7 is the E3 ubiquitin ligase for TCF7L2. By regulating the levels of TCF7L2, USP28 modulates the Wnt/β‐catenin signaling in liver cancer and USP28 depletion or inhibition by a small molecule inhibitor leads to a halt of growth in liver cancer cells. These results suggest that USP28 could be a potential therapeutic target for liver cancer.
Keywords: deubiquitination, liver cancer, TCF7L2, USP28, Wnt signaling
we reported that USP28 promotes the activity of the Wnt signaling pathway through maintaining the stability of TCF7L2. We further showed that FBXW7 is the E3 ubiquitinatin ligase for TCF7L2. By regulating the levels of TCF7L2, USP28 modulates the Wnt/β‐catenin signaling pathway in liver cancer. As a result, liver cancer cells are sensitive to USP28 depletion or inhibition by a small molecule inhibitor. Our results suggest that USP28 could be a potential therapeutic target for liver cancer.

Abbreviations
- APC
adenomatous polyposis coli
- CPD
Cdc4 phosphodegron
- DUB
deubiquitinase
- DVL
Disheveled
- FBXW7
F‐box/WD repeat‐containing protein 7
- GSK3β
glycogen synthase kinase‐3β
- HCC
hepatocellular carcinoma
- HIF1α
hypoxia‐inducible factor 1‐alpha
- LEF1
lymphoid enhancer‐binding factor 1
- LSD1
lysine‐specific demethylase 1
- Notch1
neurogenic locus notch homolog protein 1
- qPCR
quantitative PCR
- SCC
squamous cell carcinoma
- SIM
SUMO‐interacting motif
- SUMO
small ubiquitin‐like modifier
- TCF
transcription factor
- TCF7L1
transcription factor 7‐like 1
- TCF7L2
transcription factor 7‐like 2
- UBA
ubiquitin‐associated domain
- UIM
ubiquitin‐interacting motif
- USP
ubiquitin‐specific protease
1. INTRODUCTION
Liver cancer is a devastating disease currently without effective targeted therapies. Hepatocellular carcinoma is the primary form of liver cancer that arises from mutations in key growth regulatory genes, among which is β‐catenin 1 , 2 , 3 .β‐Catenin is the mediator of Wnt signaling. In the resting state, β‐catenin is ubiquitinated by SCFTrCP after phosphorylation by GSK3β present in a multisubunit protein complex containing DVL, AXIN1, and APC, and subsequently degraded by the proteasome. 4 , 5 , 6 , 7 When Wnt engages with its receptor Frizzled, the DVL/APC/GSK3β complex is recruited to the cell membrane, leaving β‐catenin unphosphorylated and thus stabilized. β‐Catenin can then enter the nucleus to activate TCF/LEF family transcription factors. In HCC, mutations in CTNNB1 occur in 28%–40% of patients and AXIN1 in approximately 11% of patients. 1 These mutations stabilize β‐catenin, causing the Wnt signaling to stay constantly on. TCF7L2/TCF4 is a member of the TCF/LEF family of transcription factors. It maintains stemness and promotes proliferation in embryonic tissues and adult stem cells, as well as in a wide variety of tumors including liver cancer. 3 , 8 , 9 , 10
The ubiquitination of proteins is a highly dynamic process catalyzed by a three‐enzyme (E1, E2, and E3) cascade that adds ubiquitin moiety to protein substrates to form ubiquitin chains of different linkages, and by a battery of DUBs that act to remove ubiquitin modifications. The USP family comprises the largest group among the DUBs. 11 USP28 is a member of this family and acts on a diverse array of substrates, including c‐MYC, 12 , 13 c‐Jun, 14 NICD, 14 HIF1α, and LSD1. 15 Through regulating these substrates, USP28 is involved in DNA damage repair, 16 apoptosis, epigenetic regulation, and cell cycle control. Thus, it could potentially contribute to the development of a variety of tumors. 17
Here we report that USP28 plays a role in liver cancer. Its expression is elevated in the tumor cells and is correlated with Wnt signaling. We further show that TCF7L2, like β‐catenin, is regulated by ubiquitination‐mediated proteasomal degradation and USP28 is a deubiquitinase that maintains the stability of TCF7L2. By regulating the levels of TCF7L2, USP28 modulates the Wnt/β‐catenin signaling in liver cancer and USP28 depletion or inhibition by a small molecule inhibitor leads to a halt of growth in liver cancer cells. Our results suggest that USP28 could be a potential therapeutic target for liver cancer.
2. MATERIALS AND METHODS
2.1. Cell culture
HEK293T, HepG2‐C3A, and HuH6 were purchased from ATCC and cultured in DMEM (C11995500BT; Gibco) supplemented with 1% penicillin/streptomycin (BL505A; Biosharp) and 10% FBS (HB‐FBS‐500; HAKATA) at 37°C with 5% CO2. Cycloheximide (C7698; Sigma‐Aldrich), MG132 (S2619; Selleck), and AZ1 (S8904; Selleck) were used at the specified conditions.
2.2. Plasmids
We made use of the gateway cloning strategy to overexpress USP28, FBXW7, and TCF7L2. The corresponding cDNA was first cloned into a pENTRY vector and then transferred into pHAGE 18 through LR recombination. Other plasmids including shRNAs in pLKO.1 were constructed using conventional cloning.
2.3. Virus production and infection
Lentivirus packaging and the infection of cells were carried out as described. 29 The shRNAs sequence used are described in Table S1.
2.4. Western blot analysis and coimmunoprecipitation
The analysis of protein expression levels by western blotting and the detection of protein–protein interaction through coimmunoprecipitation were carried out as described. 29 The Abs used are described in Table S2.
2.5. Ubiquitination assay
The cells (transfected with various plasmids) were first treated with 10 μM MG132 (S2619; Selleck) for 8 h, harvested, and lysed in denaturing buffer (20 ml pH 8.0 Tris–HCl, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P‐40, 1% sodium deoxycholate, 1% SDS, plus protease inhibitors). The lysates were heated at 100°C for 15 min followed by dilution with NETN buffer to reduce SDS content to 0.1%. The diluted lysates were then incubated with appropriate Abs (bound to agarose beads). The agarose beads were washed, boiled in loading buffer, and loaded onto SDS‐PAGE gel for electrophoresis.
2.6. RNA extraction and quantitative RT‐PCR
Total RNA was extracted from the lysates according to the manufacturer of TRIzol (T9424; Sigma‐Aldrich). cDNA was produced using a reverse transcription kit (PrimeScript RT Master Mix, RR036B; Takara). The mRNA expression of related genes was detected using the qPCR kit (SYBR Advantage qPCR Premix [2x] SDS, S4748; Takara). The primers used are described in Table S3.
2.7. Cell proliferation assay
The cells were treated with the corresponding virus. After 3 days, 1000 cells per well were placed in 96‐well plates for each group. When the cells adhered to the wall, 100 μl mixture was added to each well at a specific time in the ratio of MTS reagent (G5421; Promega) : complete medium = 1:5. After incubation at 37°C for 2 h, the cells were detected at 490 nm wavelength.
2.8. Colony formation assay
After the cells were infected with the respective viruses and selected with puromycin treatment, they were placed in 6‐well plates with 4000 cells per well. After 5–7 days, the colonies were fixed with 4% paraformaldehyde for 20 min, stained with crystal violet (C0121; Beyotime) for 20 min, rinsed with water, and photographed.
2.9. Animals
The Usp28 KO mice were obtained with CRISPR/Cas9 gene editing technique (Viewsolid Biotech). To generate HepG2 and HuH6 xenografts, 1 × 107 cells were inoculated subcutaneously under the armpit of BALB/c nude mice and allowed to grow to a certain size. The tumors were removed and divided into 2 × 2 × 2 mm tumor cubes, and the cubes were inoculated subcutaneously under the armpit of nude mice. After the tumors grew to approximately 100 mm3, the mice were randomly divided into two groups and dosed every other day by oral gavage at 200 mg/kg of AZ1 or the vehicle. The tumor sizes were measured every 3 days with a Vernier caliper. After 15 days of growth, the tumors were removed for analyses. The BALB/c nude mice were purchased at 6–8 weeks of age from GemPharmatech. All animals were housed in a specific pathogen‐free facility and provided with food and water. All animal experiments were carried out according to the guidelines approved by the Animal Care and Use Committee of the First Affiliated Hospital of Zhejiang University.
2.10. Statistical analysis
Data processing and plotting were carried out with GraphPad Prism 8.0. The difference between two groups was analyzed with Student's t‐test. A p value of less than 0.05 was considered statistically significant.
3. RESULTS
3.1. USP28 is important for Wnt signaling in HCC cells
To determine whether USP28 contributes to HCC, we extracted USP28 expression data from The Caner Genome Atlas database and found that its expression was significantly higher in tumor tissues than in the surrounding normal tissues (Figure 1A), and later stage tumors expressed higher levels of USP28 than earlier stage tumors (Figure 1B). In addition, the expression levels of USP28 inversely correlated with patient survival (Figure 1C). These data implicate USP28 in the development of HCC. To explore how USP28 might be involved in HCC development, we undertook a Gene Set Enrichment Analysis and found a significant positive correlation between USP28 expression and the expression of Wnt signaling pathway genes (Figure 1D), suggesting that USP28 might contribute to Wnt signaling. To determine if that is the case, we depleted USP28 expression in HepG2 and HuH6 cells, two HCC cell lines carrying β‐catenin‐stabilizing mutations, and assessed the Wnt signaling strength in these cells through a reporter assay (TOPFlash). As shown in Figure 1E, the depletion of USP28 dramatically suppressed the reporter luciferase activities. In addition to Wnt signaling, the expression of USP28 in HCC is also correlated with other cancer‐related signaling pathways such as mTOR, insulin signlaing, etc. (Figure S1).
FIGURE 1.
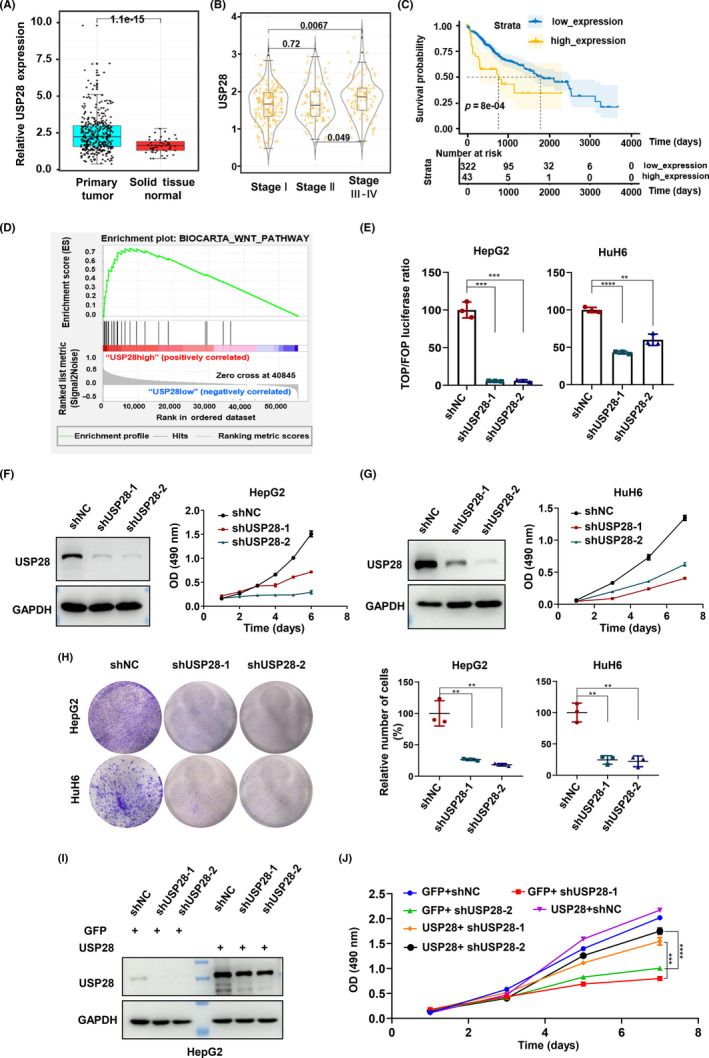
Ubiquitin‐specific protease 28 (USP28) is important for Wnt signaling in hepatocellular carcinoma cells. (A) Expression of USP28 in liver cancer samples and normal tissues (paracancer). Three hundred and fifty‐one of the samples were cancerous and 61 were paracancer. (B) Distribution of USP28 expression in different pathological stages of liver cancer. WILCOX test was used to calculate the differences among the three groups and p values were directly calculated. (C) Relationship between USP28 expression and survival in patients with liver cancer. Software X‐tile was used to obtain the value of cut‐off. Cut‐off (USP28) = 1.8068. All data are from The Cancer Genome Atlas database with 351 data in (A–C). (D) Correlation between USP28 and Wnt signaling pathway was examined using Gene Set Enrichment Analysis. (E) TOPFlash reporter assay in HepG2 and HuH6 cells depleted of USP28 by two independent shRNAs. Results are expressed as relative (TOP/Renilla)/(FOP/Renilla) luciferase activity. (F, G) Western blot analysis of USP28 and the cell viability detection in HepG2 (F) and HuH6 (G) cells with USP28 depleted by shRNA‐mediated knockdown. Cell viability was determined using MTS assay and the absorbance was 490 nm. (H) Ability for plate clone formation in HepG2 and HuH6 cells depleted of USP28 by two independent shRNAs. (I) Western blot analysis of USP28 in HepG2 cells depleted of USP28 by two independent shRNAs followed the exogenous overexpression of USP28. (J) Cell viability of HepG2 depleted of USP28 by two independent shRNAs followed the exogenous overexpression of USP28 determined using MTS assay. All data are representative of three independent experiments (mean ± SEM). Two‐tailed Student's t‐tests were applied to examine statistical significance. **p < 0.01; ***p < 0.001; ****p < 0.0001.
Suppressing the Wnt signaling is predicted to result in impairments of proliferation. Indeed, the depletion of USP28 led to much slowed growth of HepG2 (Figures 1F and S2A,B) and HuH6 (Figures 1G and S2C,D) cells. The colony formation assay also showed similar results (Figure 1H). More importantly, the growth defects in USP28‐depleted HepG2 cells could be rescued by re‐expression of the deubiquitinase (Figure 1I,J), eliminating potential off‐target effects of the two shRNA constructs used to silencing USP28 expression.
Given the TOPFlash results, we reasoned that USP28 as a deubiquitinase might function to maintain the stability of the TCF/LEF family of transcription factors, much like what it does to c‐MYC. To determine whether that is the case, we knocked down USP28 expression and examined the protein levels of LEF1, TCF7, TCF7L1, and TCF7L2. Except TCF7, the other three transcription factors were all downregulated in USP28‐depelted cells (Figure 2A). We chose TCF7L2 for further studies as it is the most intensively studied TCF/LEF family member in terms of tumor development. First, we wanted to know whether the overexpression of TCF7L2 could rescue the TOPFlash activities suppressed by USP28 depletion (Figure 1E). As shown in Figure 2B,C, exogenous TCF7L2 could indeed restore TOPFlash activities in USP28‐depleted cells, although the rescue was not 100%, likely because the other TCF/LEF members were still downregulated. More importantly, the exogenous TCF7L2 not only rescued the TOPFlash activities but also the growth defects of the USP28‐depleted cells (Figure 2D). Furthermore, we assessed the expression of Wnt target genes in USP28‐depleted cells. As expected, the expression of CCND1 and AXIN2 decreased in both HepG2 and HuH6 cells (Figure 2E,F). To extend these in vitro observations, we examined the levels of TCF7L2 in the liver of WT and Usp28 KO mice (Figure S3A‐C). As shown in Figure S3D, the absence of Usp28 resulted in the decreases of TCF7L2 levels as well as cyclin D1 levels. Taken together, these data strongly suggest that USP28 contributes to Wnt signaling through maintaining the stability of TCF/LEF transcription factors.
FIGURE 2.
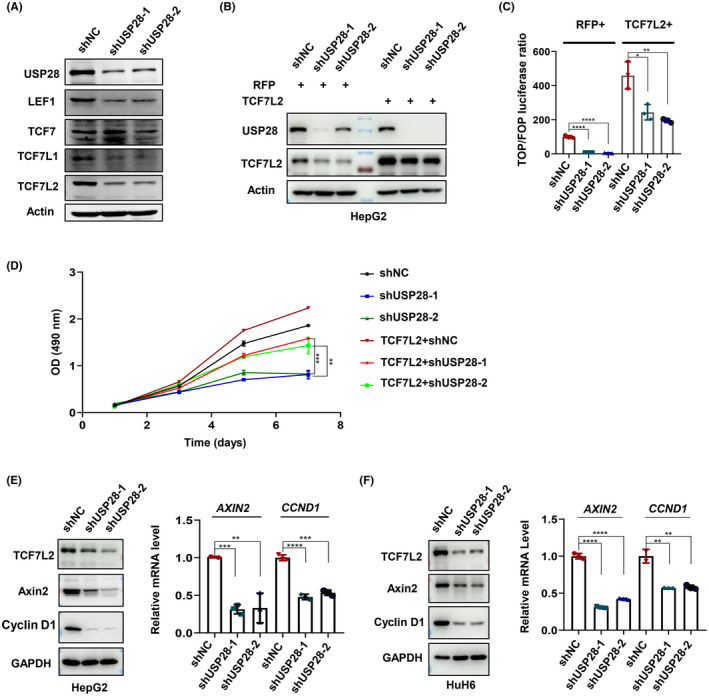
Ubiquitin‐specific protease 28 (USP28) positively regulates Wnt signaling through transcription factor 7‐like 2 (TCF7L2). (A) Western blot analysis of TCF/LEF family transcription factors in HepG2 cells. (B) Western blot analysis of TCF7L2 in HepG2 cells depleted of USP28 by shRNA‐mediated knockdown followed the overexpression of TCF7L2. (C) TOPFlash reporter assay in HepG2 cells depleted of USP28 by two independent shRNAs followed the overexpression of TCF7L2. All data were normalized to shNC in the RFP control group. (D) Cell viability of HepG2 cells depleted of USP28 by two independent shRNAs followed the overexpression of TCF7L2 determined using MTS assay. (E, F) Western blot and quantitative RT‐PCR analyses of TCF7L2, AXIN2, and CCND1 expression in HepG2 (E) and HuH6 (F) cells depleted of USP28 by two independent shRNAs. All data are representative of three independent experiments (mean ± SEM). Two‐tailed Student's t‐tests were used to examine statistical significance. **p < 0.01; ***p < 0.001; ****p < 0.0001
3.2. USP28 interacts with and modulates the ubiquitination of TCF7L2
To confirm that USP28 maintains the stability of TCF7L2 as a deubiquitinase, we first asked whether MG132, a proteasome inhibitor, could prevent the downregulation of TCF7L2 in USP28‐depleted cells. Indeed, it could (Figure 3A). Next, we measured the half‐life of TCF7L2. As shown in Figures 3B and S4A,B, the depletion of USP28 shortened the half‐life, whereas the overexpression of USP28 extended it (Figure S3C). In addition, the overexpression of USP28 could only increase the protein levels of TCF7L2, not its mRNA levels (Figure S3D). These data indicate that USP28 maintains the stability of TCF7L2.
FIGURE 3.
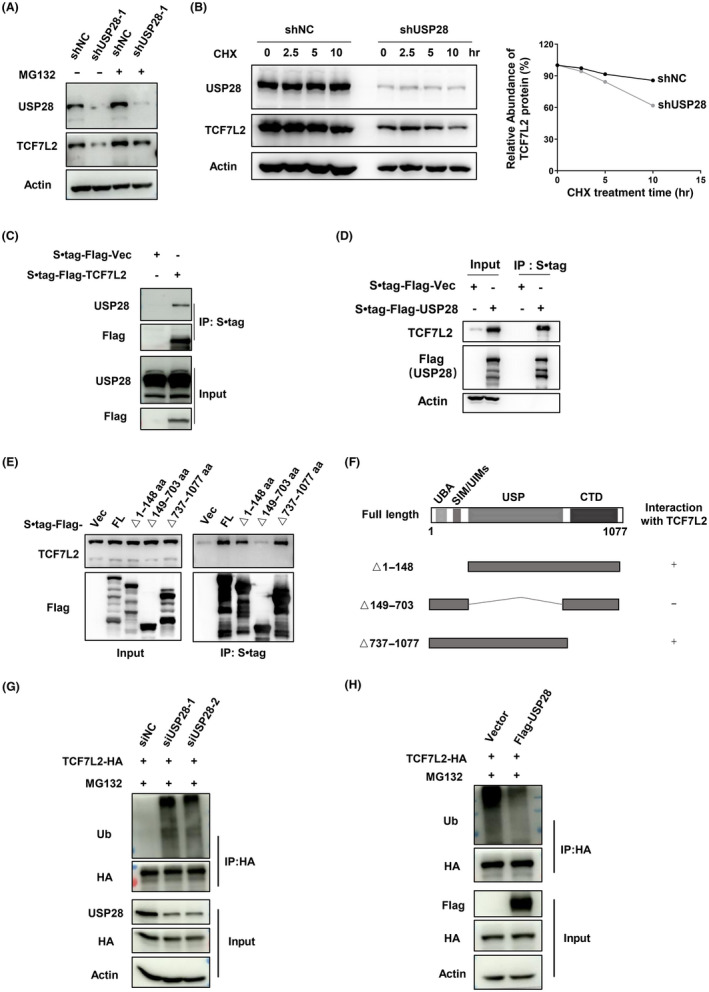
Ubiquitin‐specific protease 28 (USP28) interacts with and modulates ubiquitination of transcription factor 7‐like 2 (TCF7L2). (A) Western blot analysis of TCF7L2 in HEK293T cells treated with MG132 (20 μM) for 3 h followed USP28 depletion by shRNA‐mediated knockdown. (B) Half‐life analysis of TCF7L2 protein in HEK293T cells with USP28 depletion. Cells were treated with cycloheximide (CHX, 100 μg/ml) for the indicated time periods and harvested for analysis. Protein band intensities were measured with Image J software and plotted. (C) Immunoprecipitation of S·tag‐Flag‐TCF7L2 expressed in HEK293T cells followed by western blot analysis. (D) Immunoprecipitation of S·tag‐Flag‐USP28 expressed in HEK293T cells followed by western blot analysis. (E) Immunoprecipitation of S•tag‐Flag‐USP28 (full length and truncation mutants, see F) in HEK293T cells to examine the interaction between USP28 and TCF7L2. (F) Schematic diagram of the deletion mutants of USP28. (G, H) Ubiquitination assay of TCF7L2 in HEK293T cells with USP28 depleted (G) or overexpressed (H). Cells were treated with MG132 (10 μM) for 8 h before sample collection. IP, immunoprecipitant
To obtain more direct evidence that USP28 is a deubiquitinase for the transcription factor, we next examined the interaction between USP28 and TCF7L2. Immunoprecipitation of ectopically expressed Flag‐tagged TCF7L2 could bring down endogenous USP28 (Figure 3C), and vice versa (Figure 3D), indicating an interaction between the two. USP28 is composed of 1077 amino acid residues with a UBA, a UIM, and a SIM in the N‐terminus. Both UBA and UIM are mainly responsible for the recognition and recruitment of ubiquitinated substrates to USP28, while SIM attracts SUMO1/2 to enable SUMOylation of USP28, a modification that inhibits the DUB activity of USP28. 19 To map the region of USP28 responsible for the interaction, we expressed various deletion or truncation mutants of USP28 and examined their ability to interact with TCF7L2. As shown in Figure 3E, the USP domain of USP28 is indispensable for interacting with TCF7L2, while the UIM/UAB and other regions of USP28 are not required for the interaction (Figure 3F). Furthermore, we expressed TCF7L2‐HA, immunoprecipitated the flagged TCF7L2, and examined ubiquitin modification on TCF7L2 through immunoblotting for ubiquitin. As shown in Figure 3G, the amount of ubiquitin detectable in TCF7L2 immunoprecipitates significantly increased with the knockdown of USP28. In contrast, the overexpression of USP28 resulted in decreased ubiquitination of TCF7L2 (Figure 3H). Taken together, these results indicate that USP28 regulates the stability of TCF7L2 through interacting with and deubiquitinating the transcription factor.
Interestingly, although USP28 seems to regulate the abundance of TCF7L1 and LEF1 (Figure 2A), no interaction between USP28 and TCF7L1/LEF1 could be detected (Figure S4E). The knockdown of USP28 did result in an increase in the ubiquitination of LEF1 (Figure S3F) but did not increase that of TCF7L1 (Figure S3G). These data suggest that USP28 is a DUB for LEF1 and USP28 most likely regulates TCF7L1 indirectly.
3.3. FBXW7 mediates the ubiquitination of TCF7L2
Multiple pieces of evidence suggest that USP28 antagonizes the ubiquitination process carried out by the SCFFBXW7 complex. 12 , 14 , 20 , 21 To determine whether FBXW7 is the E3 for TCF7L2, we silenced its expression first. As shown in Figures 4A,B and S5A,B, the depletion of FBXW7 resulted in the stabilization of TCF7L2 and extended its half‐life. In contrast, the overexpression of FBXW7 led to a reduction in TCF7L2 levels (Figure 4C) and shortened its half‐life (Figure S4C). These data strongly suggest that the SCFFBXW7 complex is an E3 ubiquitin ligase for TCF7L2.
FIGURE 4.
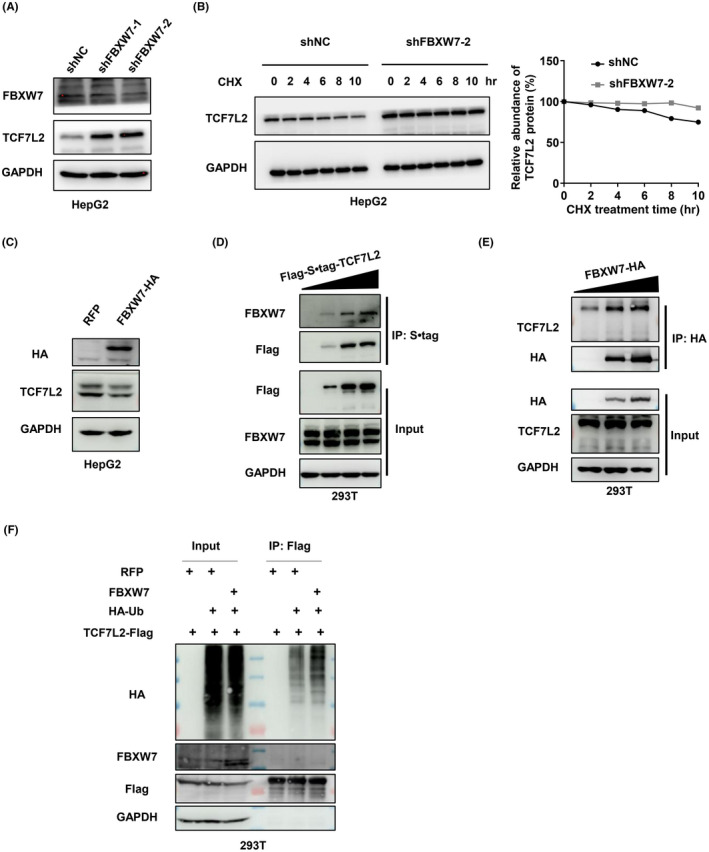
FBXW7 mediates ubiquitination of transcription factor 7‐like 2 (TCF7L2). (A) Western blot analysis of TCF7L2 in HepG2 cells depleted of FBXW7 by two independent shRNAs. (B) Half‐life analysis of TCF7L2 protein in HepG2 cells with FBXW7 depleted. The cells were treated with cycloheximide (CHX,100 μg/ml) for the indicated time periods and harvested for analysis. Protein band intensities were measured with ImageJ software and plotted. (C) Western blot analysis of TCF7L2 in HepG2 cells with exogenous overexpression of FBXW7. (D, E) FBXW7 interacts with TCF7L2. Immunoprecipitation of S•tag‐Flag‐TCF7L2 (D) or FBXW7‐HA (E) in HEK293T cells transfected with increasing amounts of corresponding plasmids. (F) Ubiquitination assay of TCF7L2 in HEK293T cells with or without FBXW7 overexpression. Flag‐tagged TCF7L2 was immunoprecipitated and blotted for HA‐ubiquitin. Cells in (F) were treated with MG132 (10 μM) for 8 h before sample collection
Next, we wanted to determine whether FBXW7, as the adaptor protein responsible for recruiting substrates for ubiquitination, could interact with TCF7L2. We therefore carried out an immunoprecipitation experiment. As shown in Figure 4D,E, Flag‐tagged TCF7L2 could bring down FBXW7 in a dose‐dependent manner, and similarly, HA‐tagged FBXW7 could bring down TCF7L2. As expected, the overexpression of FBXW7 increased the ubiquitination of TCF7L2 (Figures 4F and S5D). These results indicate that FBWX7 can interact with and ubiquitinate TCF7L2. In addition, we examined whether FBXW7 could regulate TCF7L1 and LEF1, another two TCF/LEF family members regulated by USP28 (Figure 2A). The overexpression of FBXW7 could reduce the protein levels of TCF7L1 and LEF1 (Figure S4E), but no interaction could be detected between FBXW7 and the two transcription factors (Figure S4E), and the knockdown of FBXW7 did not alter the ubiquitination levels of TCF7L1 or LEF1 (Figure S4G,H). These results suggest that FBXW7 is not an E3 for these two TCF/LEF family members and likely regulates their protein levels indirectly.
The substrates of FBXW7 usually contain a conserved CPD motif (T/SPXXS/T), which needs to be phosphorylated at the first and last Thr or Ser residues in order to be recognized by FBXW7 (Figure 5A). 22 , 23 There are four potential CPD motifs in TCF7L2 (Figure 5B). To determine which one of these four is responsible for interacting with FBXW7, we mutated each of them by replacing the Thr or Ser residues with Ala and expressed the mutants together with tagged FBXW7. Immunoprecipitation showed that the third potential CPD motif in TCF7L2 (residues 509–513, SPPPS) was responsible for interaction with FBXW7, as TCF7L2‐APPPA could no longer be brought down by FBXW7 (Figure 5C). Furthermore, unlike TCF7L2 with the other three potential CPD motifs mutated, TCF7L2‐APPPA was stable in the presence of overexpressed FBXW7 with a much‐extended half‐life (Figures 5D,E and S5A–D). Consistently, exogenously expressed FBXW7 could not increase the levels of ubiquitination of TCF7L2‐APPPA as it could over WT TCF7L2 (Figure 5F). Furthermore, both of the Ser residues in SPPPS are critical as mutating either one of them resulted in the resistance to exogenously expressed FBXW7 (Figure S5E). However, mutating the Ser residues to Asp did not mimic their phosphorylation as TCF7L2‐DPPPD did not become unstable as expected (Figure S5F).
FIGURE 5.
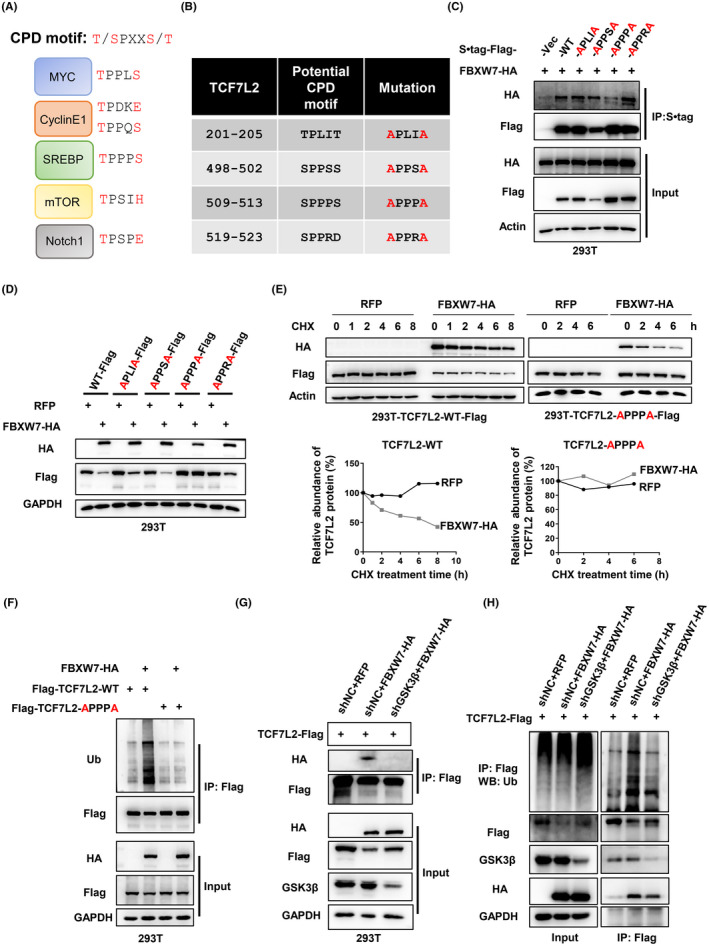
Identification of the Cdc4 phosphodegron (CPD) motif in transcription factor 7‐like 2 (TCF7L2). (A) CPD motifs in known FBXW7 substrates. (B) Potential CPD motifs in TCF7L2 and intended mutations. (C) Immunoprecipitation of S•tag‐Flag‐TCF7L2 (full length and various CPD mutants) expressed in HEK293T cells followed by western blot analysis. (D) Western blot analysis to examine the expression levels of TCF7L2 with potential CPD motifs mutated under FBXW7 overexpression. (E) Overexpression of FBXW7 could shorten the half‐life of WT TCF7L2, but not the half‐life of TCF7L2‐APPPA. The cells were treated with cycloheximide (CHX, 100 μg/ml) for different times. Protein band intensities were measured and plotted. (F) Ubiquitination assay. Flag‐tagged TCF7L2 or TCF7L2‐APPPA was coexpressed with FBXW7‐HA, immunoprecipitated, and blotted for ubiquitin. The coexpressions in HEK293T cells were realized through lentiviral infection and the cells were treated with MG132 (10 μM) for 8 h before sample collection. (G) Glycogen synthase kinase‐3β (GSK3β) was required for the interaction between FBXW7 and TCF7L2. TCF7L2‐Flag and FBXW7‐HA were coexpressed in HEK293T cells depleted of GSK3β and their interaction was examined through immunoprecipitation and western blot analysis. (H) Ubiquitination of TCF7L2 by FBXW7 depends on GSK3β function. The ubiquitination assay was carried out as in (F)
The CPD motif is conserved in TCF7L2 from other species (Figure S6G), indicating its evolutionary importance. However, other members of TCF/LEF family do not contain this CPD motif.
It is known that GSK3β is the kinase that phosphorylates the CPD motif to enable FBXW7 to recognize its substrates. 24 , 25 Thus, we went ahead to determine whether GSK3β was also important in regulating TCF7L2. To that end, we found that GSK3β was indeed required for FBXW7 to function as TCF7L2’s E3. First, an interaction could be detected between TCF7L2 and GSK3β (Figure S5H). Second, the depletion of GSK3β expression could increase the levels of TCF7L2 (Figure S5I), and such a depletion could also overcome the action of exogenously expressed FBXW7 (Figure S5J). Moreover, the interaction between TCF7L2 and FBXW7 and the ubiquitination of TCF7L2 by FBXW7 were all dependent on GSK3β (Figure 5G,H).
3.4. Pharmacological inhibition of USP28 regulates TCF7L2 stability and exerts antiproliferative effects against liver cancer cells
Having established the regulation of TCF7L2 by USP28 through genetic manipulations, we next wanted to test the pharmacological inhibition of USP28. To that end, we used a reported inhibitor AZ1. 26 HepG2 and HuH6 cells were treated with different concentrations of AZ1 for 48 h, and AZ1 prominently decreased the levels of USP28 itself and that of TCF7L2, as well as the target genes of TCF7L2, AXIN2, and Cyclin D1 (Figure 6A). As a result, AZ1 treatment also suppressed the Wnt reporter activities (TOPFlash; Figure 6B). Furthermore, AZ1 could inhibit the growth of HepG2 cells with an EC50 value of 12.65 μM and HuH6 cells with an EC50 value of 5.852 μM (Figure 6C). It also inhibited the colony formation ability of both HepG2 and HuH6 cells (Figure 6D). However, neither the exogenous expression of WT TCF7L2 nor the CPD mutant (APPPA) version could resist the growth suppression imposed by AZ1 (Figure S6), indicating that the compound affects more than just TCF7L2 to exert its antiproliferation effects.
FIGURE 6.
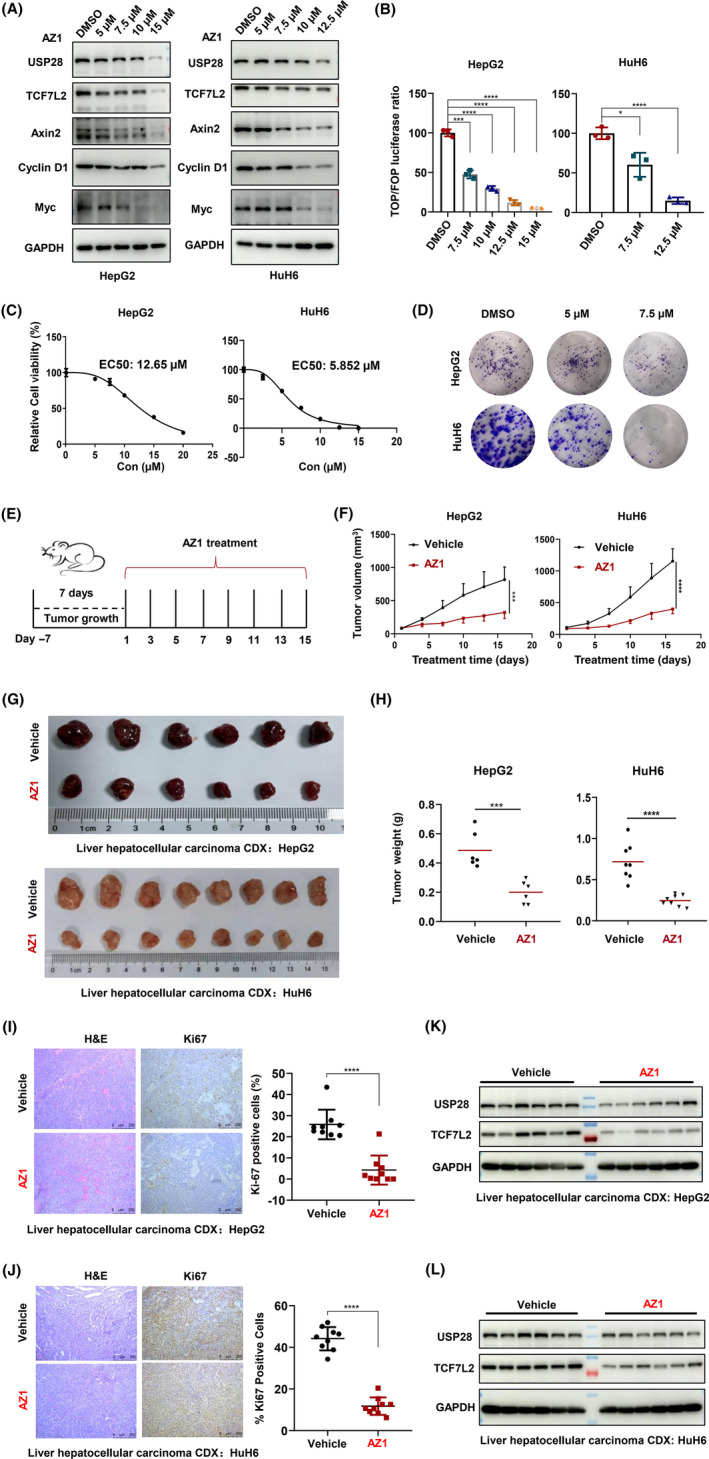
Pharmacological inhibition of ubiquitin‐specific protease 28 (USP28) regulates transcription factor 7‐like 2 (TCF7L2) stability and exerts antiproliferative effect against liver cancer cells. (A) Western blot analysis of USP28, TCF7L2, Axin2, Cyclin D1, and Myc in HepG2 and HuH6 cells treated with different concentrations of AZ1 for 48 h. (B) TOPFlash reporter assay in HepG2 and HuH6 cells treated with AZ1 in different concentrations for 48 h. Results are expressed as relative (TOP/Renilla)/(FOP/Renilla) luciferase activity. All data are representative of three independent experiments (mean ± SEM). (C) Cell viability of HepG2 and HuH6 cells treated with AZ1 in different concentrations for 72 h determined using MTS assay. Absorbance was 490 nm. (D) Ability for plate clone formation of HepG2 and HuH6 cells treated with AZ1 in different concentrations. The above data are representative of three independent experiments (mean ± SEM). (E) Schematic diagram of the animal experimental design. (F) Tumor volume was measured every 2 days beginning on the first day of treatment. CDX from HepG2 cells, n (vehicle) = 6, n (AZ1) = 6. CDX from HuH6 cells, n (vehicle) = 8, n (AZ1) = 8. (G) Representative figure of tumors formed. (H) Tumor weight of the vehicle and AZ1 groups. CDX from HepG2 cells, n (vehicle) = 6, n (AZ1) = 6. CDX from HuH6 cells, n (vehicle) = 8, n (AZ1) = 8. (I, J) H&E and immunohistochemical staining of Ki‐67 after treatment with vehicle or AZ1 for tumor tissues of CDX from HepG2 (I) and HuH6 cells (J) (bar = 50 μm). n (vehicle) = 9, n (AZ1) = 9. Three independent tumor samples were taken from each group, and three independent visual fields were taken from each tumor sample. (K, L) Western blot analysis of USP28 and TCF7L2 in CDX from HepG2 (K) and HuH6 (L) cells treated with vehicle or AZ1. Two‐tailed Student's t‐tests were applied to examine statistical significance. **p < 0.01; ***p < 0.001; ****p < 0.0001
To examine the effect of AZ1 in vivo, we inoculated HepG2 and HuH6 cells in nude mice to generate xenograft tumors. When the tumors reached approximately 100 mm3 in volume, the tumor‐bearing mice were given AZ1 at 200 mg/kg body weight every other day through an oral gavage for 15 days (Figure 6E). AZ1 clearly suppressed the growth of the tumors as shown by tumor volume and weight (Figure 6F–H) as well as by Ki‐67 staining (Figures 6I–J and S8). The AZ1 treatment did not change the histology of the tumors (Figure 6I–J). Consistent with the observations in vitro, the protein levels of USP28 and TCF7L2 in AZ1‐treated tumor tissues were significantly lower than those in the control group (Figure 6K–L).
4. DISCUSSION
As a deubiquitinase, USP28 has been shown to be necessary for the maintenance of the protein levels of c‐MYC, 12 LSD1, 15 HIF1α, 27 Notch1, 14 53BP1, 28 and CLASPIN. 16 Almost all of these substrates, especially c‐MYC, have been implicated in tumor initiation and/or progression. It was shown that the intestinal tumorigenesis induced by the inactivation of Apc was hindered in Usp28‐deficient mice, 14 triple‐ negative breast cancer cells depend on USP28 for proliferation and survival due to its role in maintaining the stability of RecQ family helicases, 29 and SCC cells require USP28 for proliferation due to its function in maintaining the stability of ΔNp63. 30 , 31 In addition, the sensitivity of SCC to chemotherapy can be improved by inhibiting the USP28‐DNp63‐DDR axis. 32 Here we show that this deubiquitinase is also important in liver cancer cells with constitutively active Wnt signaling. Our results show that USP28 contributes to the activity of the Wnt signaling pathway through stabilizing TCF7L2 (Figure 7), at least partially, as it also regulates other members of the TCF/LEF family transcription factors (Figure 2A) as well as Forkhead box protein M1 (FOXM1), another transcription factor involved in Wnt signaling. 33
FIGURE 7.
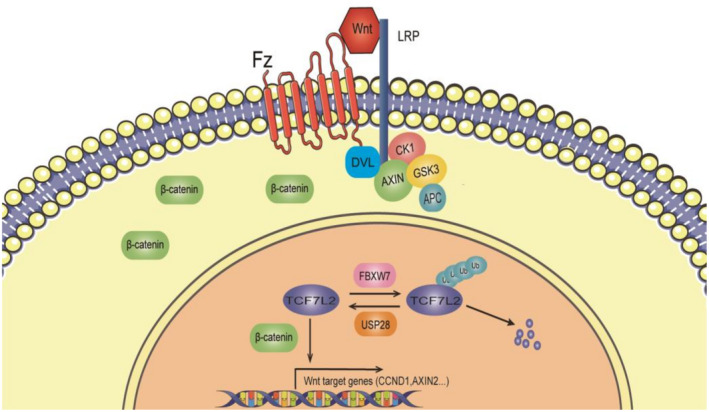
Model of regulation of transcription factor 7‐like 2 (TCF7L2) function through ubiquitination. APC, adenomatous polyposis coli; DVL, Disheveled; Fz, frizzled; GSK3, glycogen synthase kinase‐3; LRP, lipoprotein receptor‐related; USP28, ubiquitin‐specific protease 28
As Usp28‐deficient mice develop normally, the role of Usp28 in the normal Wnt signaling during embryonic development must be minimal or its absence could be mitigated by unspecified redundant mechanisms, for example, suppressing GSK3β activity so that TCF7L2 is not ubiquitinated in the first place. Interestingly, GSK3β is also required for the ubiquitination of β‐catenin, suggesting a concerted effort by the cells in regulating Wnt signaling.
USP28 has not been shown to be an oncogene, and yet it is required by a number of different types of cancer cells (including the liver cancer cells reported here) for proliferation and/or survival, making it an addicted nononcogene. The reason for its addiction in these diverse types of cancer cells is likely that there are a quite few oncodrivers (and the number is increasing) regulated by this deubiquitinase. It is difficult to pinpoint exactly which oncodriver's destabilization leads to the growth‐stopping effects in a particular type of cancer cell after blocking USP28. Rather than the destabilization of a specific oncodriver, we believe the growth‐stopping effect resulted from the destabilization of a combination of oncodrivers, although the destabilization of c‐MYC might contribute the most. Nonetheless, the body of evidence is becoming larger and larger that USP28 is a potentially excellent therapeutic target in a number of malignancies. Hopefully clinical applications will soon emerge.
DISCLOSURE
The authors have no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: N/A.
Informed consent: N/A.
Registry and registration no. of the study/trial: N/A.
Animal studies: All animal experiments were performed according to the guidelines approved by the Animal Care and Use Committee of the First Affiliated Hospital of Zhejiang University and the National Research Council.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank the core facility in Zhejiang Provincial Key Laboratory of Pancreatic Disease for experimental support. The authors are in debt to Mr. Shenghui Hong for excellent animal support. This work was supported by grants from the National Natural Science Foundation of China (81773032), National Key R&D Program of China (2018YFA0507500), and Innovation Capacity Support Plan of Shaanxi Province (2018TD‐002).
Sun X, Cai M, Wu L, et al. Ubiquitin‐specific protease 28 deubiquitinates TCF7L2 to govern the action of the Wnt signaling pathway in hepatic carcinoma. Cancer Sci. 2022;113:3463‐3475. doi: 10.1111/cas.15509
Xiao Sun and Mengjiao Cai contributed equally to this work.
Contributor Information
Jin Peng, Email: pengjin0000@126.com.
Suxia Han, Email: shan87@mail.xjtu.edu.cn.
Pumin Zhang, Email: pzhangbcm@zju.edu.cn.
REFERENCES
- 1. Bugter JM, Fenderico N, Maurice MM. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer. 2021;21(1):5‐21. [DOI] [PubMed] [Google Scholar]
- 2. Calderaro J, Ziol M, Paradis V, Zucman‐Rossi J. Molecular and histological correlations in liver cancer. J Hepatol. 2019;71(3):616‐630. [DOI] [PubMed] [Google Scholar]
- 3. He S, Tang S. WNT/beta‐catenin signaling in the development of liver cancers. Biomed Pharmacother. 2020;132:110851. [DOI] [PubMed] [Google Scholar]
- 4. Behrens J, Jerchow BA, Wurtele M, et al. Functional interaction of an axin homolog, conductin, with beta‐catenin, APC, and GSK3beta. Science. 1998;280(5363):596‐599. [DOI] [PubMed] [Google Scholar]
- 5. Hart MJ, de los Santos R, Albert IN, et al. Downregulation of beta‐catenin by human Axin and its association with the APC tumor suppressor, beta‐catenin and GSK3 beta. Curr Biol. 1998;8(10):573‐581. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura T, Hamada F, Ishidate T, et al. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta‐catenin, GSK‐3beta and APC and reduces the beta‐catenin level. Genes Cells. 1998;3(6):395‐403. [DOI] [PubMed] [Google Scholar]
- 7. Su Y, Fu C, Ishikawa S, et al. APC is essential for targeting phosphorylated beta‐catenin to the SCFbeta‐TrCP ubiquitin ligase. Mol Cell. 2008;32(5):652‐661. [DOI] [PubMed] [Google Scholar]
- 8. Schepers A, Clevers H. Wnt signaling, stem cells, and cancer of the gastrointestinal tract. Cold Spring Harb Perspect Biol. 2012;4(4):a007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wenzel J, Rose K, Haghighi EB, et al. Loss of the nuclear Wnt pathway effector TCF7L2 promotes migration and invasion of human colorectal cancer cells. Oncogene. 2020;39(19):3893‐3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550‐563. [DOI] [PubMed] [Google Scholar]
- 12. Popov N, Wanzel M, Madiredjo M, et al. The ubiquitin‐specific protease USP28 is required for MYC stability. Nat Cell Biol. 2007;9(7):765‐774. [DOI] [PubMed] [Google Scholar]
- 13. Chakravorty D, Ghosh A, Saha S. Computational approach to target USP28 for regulating Myc. Comput Biol Chem. 2020;85:107208. [DOI] [PubMed] [Google Scholar]
- 14. Diefenbacher ME, Popov N, Blake SM, et al. The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J Clin Invest. 2014;124(8):3407‐3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Y, Wang Y, Yang XH, et al. The deubiquitinase USP28 stabilizes LSD1 and confers stem‐cell‐like traits to breast cancer cells. Cell Rep. 2013;5(1):224‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA‐damage response. Cell. 2006;126(3):529‐542. [DOI] [PubMed] [Google Scholar]
- 17. Wang X, Liu Z, Zhang L, et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018;9(2):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emanuele MJ, Elia AE, Xu Q, et al. Global identification of modular cullin‐RING ligase substrates. Cell. 2011;147(2):459‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wen Y, Shi L, Ding Y, et al. The N‐terminal ubiquitin‐binding region of ubiquitin‐specific protease 28 modulates its deubiquitination function: NMR structural and mechanistic insights. Biochem J. 2015;471(2):155‐165. [DOI] [PubMed] [Google Scholar]
- 20. Diefenbacher ME, Chakraborty A, Blake SM, et al. Usp28 counteracts Fbw7 in intestinal homeostasis and cancer. Cancer Res. 2015;75(7):1181‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schulein‐Volk C, Wolf E, Zhu J, et al. Dual regulation of Fbw7 function and oncogenic transformation by Usp28. Cell Rep. 2014;9(3):1099‐1109. [DOI] [PubMed] [Google Scholar]
- 22. Sailo BL, Banik K, Girisa S, et al. FBXW7 in cancer: what has been unraveled thus far? Cancers (Basel). 2019;11(2):246‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr. The v‐Jun point mutation allows c‐Jun to escape GSK3‐dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8(1):25‐33. [DOI] [PubMed] [Google Scholar]
- 25. Perez‐Benavente B, Garcia JL, Rodriguez MS, et al. GSK3‐SCF(FBXW7) targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene. 2013;32(17):2189‐2199. [DOI] [PubMed] [Google Scholar]
- 26. Wrigley JD, Gavory G, Simpson I, et al. Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem Biol. 2017;12(12):3113‐3125. [DOI] [PubMed] [Google Scholar]
- 27. Flugel D, Gorlach A, Kietzmann T. GSK‐3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28‐dependent degradation of HIF‐1alpha. Blood. 2012;119(5):1292‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knobel PA, Belotserkovskaya R, Galanty Y, Schmidt CK, Jackson SP, Stracker TH. USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double‐strand break metabolism. Mol Cell Biol. 2014;34(11):2062‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J, Dong Y, Ma H, et al. The Deubiquitinase USP28 stabilizes expression of RecQ family helicases and maintains the viability of triple negative breast cancer cells. J Biol Chem. 2021;101443:101443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prieto‐Garcia C, Hartmann O, Reissland M, et al. Maintaining protein stability of Np63 via USP28 is required by squamous cancer cells. EMBO mol Med. 2020;12(4):e11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruiz EJ, Pinto‐Fernandez A, Turnbull AP, et al. USP28 deletion and small‐molecule inhibition destabilizes c‐MYC and elicits regression of squamous cell lung carcinoma. Elife. 2021;10:e71596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prieto‐Garcia C, Hartmann O, Reissland M, et al. Inhibition of USP28 overcomes cisplatin‐resistance of squamous tumors by suppression of the Fanconi anemia pathway. Cell Death Differ. 2022;29(3):568‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen L, Xu Z, Li Q, et al. USP28 facilitates pancreatic cancer progression through activation of Wnt/beta‐catenin pathway via stabilising FOXM1. Cell Death Dis. 2021;12(10):887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1