

Abstract
Cancer‐associated fibroblasts (CAFs) are a major component of the tumor microenvironment that mediate resistance of cancer cells to anticancer drugs. Tranilast is an antiallergic drug that suppresses the release of cytokines from various inflammatory cells. In this study, we investigated the inhibitory effect of tranilast on the interactions between non–small cell lung cancer (NSCLC) cells and the CAFs in the tumor microenvironment. Three EGFR‐mutant NSCLC cell lines, two KRAS‐mutant cell lines, and three CAFs derived from NSCLC patients were used. To mimic the tumor microenvironment, the NSCLC cells were cocultured with the CAFs in vitro, and the molecular profiles and sensitivity to molecular targeted therapy were assessed. Crosstalk between NSCLC cells and CAFs induced multiple biological effects on the NSCLC cells both in vivo and in vitro, including activation of the STAT3 signaling pathway, promotion of xenograft tumor growth, induction of epithelial‐mesenchymal transition (EMT), and acquisition of resistance to molecular‐targeted therapy, including EGFR‐mutant NSCLC cells to osimertinib and of KRAS‐mutant NSCLC cells to selumetinib. Treatment with tranilast led to inhibition of IL‐6 secretion from the CAFs, which, in turn, resulted in inhibition of CAF‐induced phospho‐STAT3 upregulation. Tranilast also inhibited CAF‐induced EMT in the NSCLC cells. Finally, combined administration of tranilast with molecular‐targeted therapy reversed the CAF‐mediated resistance of the NSCLC cells to the molecular‐targeted drugs, both in vitro and in vivo. Our results showed that combined administration of tranilast with molecular‐targeted therapy is a possible new treatment strategy to overcome drug resistance caused by cancer‐CAF interaction.
Keywords: cancer‐associated fibroblast, drug resistance, tranilast
Crosstalk between NSCLC cells and CAFs induced multiple biological effects on the NSCLC cells including activation of the STAT3 signaling pathway, promotion of xenograft tumor growth, induction of EMT, and acquisition of resistance to molecular‐targeted therapy. Combined administration of tranilast with molecular‐targeted therapy reversed the CAF‐mediated resistance of the NSCLC cells to the molecular‐targeted drugs.

Abbreviations
- CAF
cancer‐associated fibroblast
- DAPI
4′,6‐diamidino‐2‐phenylindole
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transition
- FFPE
formalin‐fixed and paraffin‐embedded
- IL
interleukin
- JAK
Janus kinase
- NSCLC
non–small cell lung cancer
- RAS
rat sarcoma
- STAT3
signal transducer and activator of transcription 3
- TERT
telomerase reverse transcriptase
- TGF‐β
transforming growth factor‐beta
- TME
tumor microenvironment
- TUNEL
TdT‐mediated dUTP nick end labeling
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Lung cancer is the leading cause of cancer death worldwide. 1 Significant advances have been made in the treatment of non–small cell lung cancer (NSCLC) with the development of drugs targeting oncogenic driver mutations or immune checkpoint molecules, 2 which have enabled precision medicine. However, while most molecular‐targeted therapies initially show significant effect, acquired resistance to the drugs eventually develops. Efforts are being made to develop complementary therapies to maximize the effects of the targeted therapies or overcome the acquired drug resistance. A variety of resistance mechanisms to anticancer drugs, especially to molecular‐targeted drugs, have been elucidated, including second‐site mutations in the kinase domain, bypass signaling pathway activation, copy number changes, and histologic transformation, 3 , 4 although many questions still remain.
Cancer‐related fibroblasts (CAFs) are one of the major components of the tumor stroma and contribute significantly to the tumor microenvironment (TME). It has been shown that CAFs release several cytokines, such as interleukin‐6 (IL‐6), TGF‐β, and epidermal growth factor (EGF), which activate multiple signaling pathways in cancer cells to promote tumor cell proliferation, invasion, metastasis and chemoresistance. 5 , 6 , 7 , 8 It has been reported that activation of the IL‐6/STAT3 signaling pathway in NSCLC cells by substances secreted by CAFs is a critical contributor to CAF‐mediated drug resistance. 5 Therefore, CAF‐targeted therapy is gaining attention as one of the complementary treatment strategies for cancer.
Drug repositioning (or repurposing/reprofiling) is the process of finding new therapeutic indications for existing drugs. It is an attractive approach toward rapid drug discovery and development at a relatively low cost and high efficiency because clinical and pharmacokinetic data of existing drugs are already established. 9 One of the most notable examples of a repositioned drug is thalidomide, which was initially developed for morning sickness and was subsequently successfully repositioned as an anticancer drug for multiple myeloma. 9
Here, we examined the usefulness of tranilast (N‐[3,4‐dimethoxycinnamoyl]‐anthranilic acid) as a possible therapeutic agent for targeting CAFs. Tranilast is an antiallergic drug that is used clinically for bronchial asthma and hypertrophic scars. 10 The drug is known to suppress collagen production and release of TGF‐β from fibroblasts 11 and of IL‐11 from macrophages. 12 In this study, we investigated the microenvironmental interactions between the CAFs and NSCLC cells in coculture models in vitro and in vivo. We then assessed the inhibitory effect of tranilast on their crosstalk and its potential to overcome CAF‐mediated acquisition of drug resistance in cancer treatment.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
Three EGFR‐mutant NSCLC cell lines (HCC827 [EGFR exon19del E746‐A750], HCC4011 [EGFR L858R], and PC9 [EGFR exon19del E746‐A750]) and two KRAS‐mutant NSCLC cell lines (H23 [KRAS G12C] H2009 [KRAS G12A]) were used in this study. All the NSCLC cell lines were purchased from the American Type Culture Collection. Three CAFs named CAF1, CAF2, and CAF3 were obtained from surgically resected three NSCLC specimens derived from each three patient, immortalized by retroviral transduction of hTERT to stabilize the cell phenotype and avoid cellular senescence, and cultured in Roswell Park Memorial Institute (RPMI)‐1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified incubator under 5% CO2. Studies using clinical specimens were approved by the Okayama Medical School and Hospital's Research Ethics Committee (approval number: #1906‐033), and informed consent was obtained from individual patients for the use of their materials. Osimertinib, an EGFR‐tyrosine kinase inhibitor, was purchased from ChemScene; selumetinib, an MEK inhibitor, was purchased from Selleckchem; and tranilast was purchased from Tokyo Chemical Industry.
2.2. Coculture of NSCLC cells with fibroblasts
Cancer cells were placed in the bottom compartment (2 × 104 cells per chamber) and CAFs (2 × 105) in the top compartment of transwell polycarbonate membrane cell culture inserts placed in a six‐well plate; the bottom and top compartments were separated by a filter with a pore size of 0.4 mm (Sarstedt).
2.3. Cell viability assay
The sensitivity to each drug was determined by the MTS assay. Cells were plated at a density of 3000 cells per well in 96‐well plates and treated with a preset concentration of each drug. Cell viability was assayed after 72 hours of treatment using the CellTiter 96 aqueous bromide one solution reagent (Promega). For measurement of the IC50 value, eight replicate determinations were obtained in three independent experiments. Data were analyzed by nonlinear regression, using Graphpad Prism Ver. 6.0.3 (GraphPad Software).
2.4. Western blotting
The total cell lysate was extracted with a lysis buffer, a mixture of RIPA buffer, phosphate inhibitor cocktails 2 and 3 (Sigma‐Aldrich) and complete Mini Protease Inhibitor Cocktail (Roche). Western blot analysis was performed using the following primary antibodies: STAT3, p‐STAT3 (Tyr705), E‐cadherin, vimentin, IL‐6 (D3K2N), and GAPDH (Cell Signaling Technology). The secondary antibody used was HRP‐conjugated anti‐rabbit IgG (Santa Cruz Biotechnology). We evaluated the substances secreted from the CAFs into the culture medium by the centrifugal filter device Amicon Ultra 10 K (Pall). To detect specific signals, the membrane was examined using the ECL Prime Western Blotting Detection System (GE Healthcare) and LAS‐3000 (Fujifilm). The relative band intensities were quantified using ImageJ software (National Institute of Health).
2.5. Gene expression assay
Total RNA from cultured cells was isolated using the RNeasy Mini Kit (Qiagen) and reverse‐transcribed with the High‐Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative RT‐PCR (qRT‐PCR) was performed on the StepOnePlus Real‐Time PCR system (Thermo Fisher Scientific) using TaqMan gene expression assays (Thermo Fisher Scientific). The gene expression levels were calculated using the delta‐delta CT method. GAPDH was used as the endogenous control. The assays were repeated three times. Data are expressed as the means ± SE.
2.6. Colony formation assay
Cancer cells (2 × 104 cells per chamber) were cultured in a six‐well plate with or without CAFs (2 × 105) placed in the top compartment of a transwell polycarbonate membrane cell culture insert and treated with the molecular‐targeted drugs and tranilast for 7 days. After fixation in 4% formaldehyde, the cells were stained using 0.2% crystal violet. Colony counts were measured using the ImageJ software (National Institutes of Health).
2.7. Xenograft mouse model
Four‐week‐old BALB/c‐nu/nu female mice were purchased from Charles River Laboratories. All mice were provided with sterilized food and water and housed in a barrier facility under a 12:12‐hour light‐dark cycle. PC9 cells (1 × 106), CAF1 cells (1 × 106), or PC9 cells (5 × 105) mixed with CAF1 cells (5 × 105) (1:1 ratio) were suspended in 100 μL of RPMI‐1640 with Matrigel Basement Membrane Matrix (Corning) mixture (1:1 ratio) and injected subcutaneously into the backs of the mice. The tumor xenografts were measured using digital calipers, and the tumor volumes were calculated using the formula, volume = 1/2 × ([shortest diameter]2 × [longest diameter]). When the tumor volumes exceeded approximately 150 mm3, the mice were randomly allocated to one of four groups: the control group, the osimertinib (5 mg/kg/days) group, the tranilast (200 mg/kg/days) group, and the osimertinib (5 mg/kg/days) plus tranilast (200 mg/kg/days) group (n = 7 per group). The drugs were suspended in 0.5 w/v (%) methylcellulose and administered by oral gavage five times a week. The tumor volumes were measured twice a week. For analysis of apoptosis, formalin‐fixed and paraffin‐embedded (FFPE) tissue samples from the xenograft tumors were stained using the DeadEnd Fluorometric TUNEL System (PROMEGA). The cells were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI). The number of positively stained cells as a percentage of the total number of cells was counted under a fluorescence microscope in five random fields and averaged. Data are expressed as the mean ± SE. The protocol for the animal experiments was approved by the Animal Care and Use Committee, Okayama University (permit number: OKU‐2020564).
3. RESULTS
3.1. Cancer‐associated fibroblasts facilitate the malignant behaviors of NSCLC cells
To assess the microenvironmental interactions between the CAFs and NSCLC cells, we performed coculture of NSCLC cell lines with CAFs using the Boyden chamber system and evaluated the changes in the phosphorylation status of STAT3 in the NSCLC cells by Western blotting. As expected, coculture of NSCLC cells with CAFs increased phospho‐STAT3 in PC9 cells (Figure 1A). Next, the role of CAFs within a tumor on the tumor growth was evaluated in vivo. PC9 cells alone or PC9 cells plus CAF1 cells were subcutaneously implanted into the flanks of nude mice. As shown in Figure 1B, the group that received subcutaneous implantation of PC9 cells plus CAFs showed more aggressive growth of the tumors; no mass formation was observed in the group that received subcutaneous implantation of CAFs alone. Next, we examined the effect of CAFs in facilitating EMT in the tumor cells. As shown in Figure 1C, HCC4011 cells cultured with CAFs showed morphological changes to a spindle shape and elongated formation as compared with control cells cultured without CAFs, while the differences were not clear in other cell lines (Figure S1). Furthermore, HCC4011, H2009, and H23 cells showed upregulation of the mesenchymal marker vimentin, but on the other hand, PC9 and HCC827 cells did not show significant changes (Figure 1D and Figure S1). This suggests that CAF induces EMT in the NSCLC cells, while the tendency to undergo EMT differs depending on the cell line. We also investigated the acquisition of drug resistance mediated by CAFs in the in vivo model. Mice bearing tumor xenografts of PC9 cells alone or of PC9 cells plus CAF1 cells were treated with osimertinib (5 mg/kg) by oral gavage five times a week. Osimertinib treatment was less effective at slowing the growth of tumor xenografts of PC9 cells plus CAFs than of tumor xenografts of PC9 cells alone (Figure 1E).
FIGURE 1.
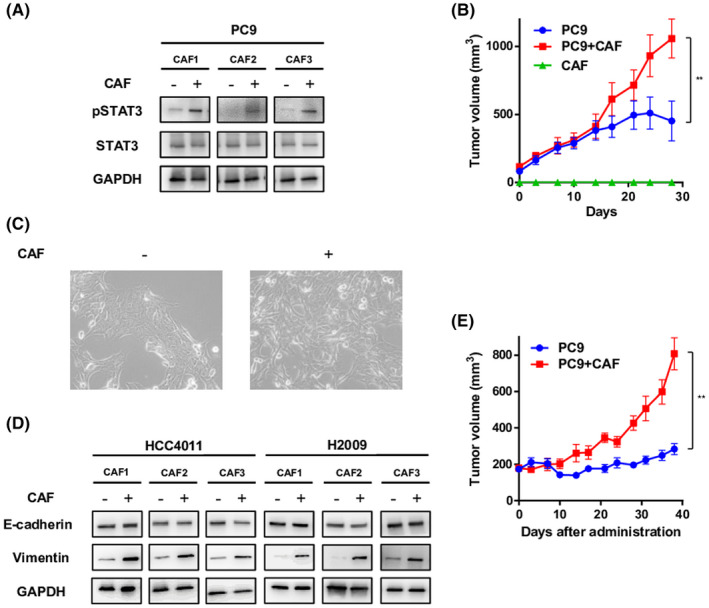
Effect of cancer‐associated fibroblasts (CAFs) on the progression and acquisition of drug resistance of non–small cell lung cancer (NSCLC) through activation of STAT3 signaling and induction of epithelial‐mesenchymal transition (EMT). A, PC9 cells were cocultured with CAFs for 7 d, and the expression level of pSTAT3 was analyzed by Western blotting. B, PC9 cells alone, CAF1 cells alone, or PC9 cells plus CAF1 cells were subcutaneously implanted into the flanks of nude mice, and the tumor sizes were monitored. Each group consisted of five mice. Data shown are the means ± SE; **p < 0.01. C, HCC4011 cells were cocultured with CAFs, and morphological changes were observed. D, HCC4011 and H2009 cells were cocultured with CAFs for 72 h, and the expression levels of EMT markers were analyzed by Western blotting. E, PC9 cells alone or PC9 cells plus CAF1 cells were subcutaneously implanted into the flanks of nude mice. When the tumors reached a diameter of approximately 150 mm3, the mice were treated with 5 mg/kg of osimertinib five times a week. The tumor volumes were then determined on the indicated days after the start of treatment. Each group consisted of seven mice. Data shown are the means ± SE; **p < 0.01
3.2. Tranilast suppresses STAT3 activation via decrease of CAF‐derived IL‐6 and inhibits EMT
We examined the inhibitory effect of tranilast on CAF‐induced STAT3 activation and EMT in the NSCLC cells. Treatment with tranilast alone showed little effect on the cell growth of any of the five lung cancer cell lines or three CAFs, with IC50 values of 150‐250 μM (Figure S2). Next, cancer cells were cultured with or without CAF1 cells for one week in the presence or absence of 100 μM tranilast, and the protein expressions of STAT3 and EMT markers in the tumor cells were analyzed. Coculture with CAFs increased the expression level of pSTAT3 in the tumor cells, and tranilast inhibited CAF‐mediated pSTAT3 activation (Figure 2A). Identical results were confirmed using the other sublines of CAFs (Figure S3). To further explore the mechanism of STAT3 suppression by tranilast, we examined the expression of IL‐6, a cytokine that activates the JAK1/STAT3 signaling pathway. Treatment with tranilast for 48 hours decreased the mRNA expression of IL6 in CAFs (Figure 2B) and of IL‐6 secretion from CAFs into the culture medium (Figure 2C). In terms of the inhibitory effect on EMT, tranilast decreased the expression of vimentin in the tumor cells, suggesting a partial inhibitory effect on CAF‐induced EMT (Figure 2D). This finding was also confirmed with the other CAF sublines (Figure S4). Next, HCC4011 and H2009 cells were treated with IL‐6 (1 ng/mL) for 72 hours, and the EMT status was analyzed by Western blotting to assess whether the EMT inhibitory effect by tranilast was due to the suppressive effect on IL‐6 secretion from CAFs. However, IL‐6 treatment did not induce EMT in this model (data not shown). Furthermore, a TGF‐β–induced EMT model was used to evaluate the direct effect of tranilast on the cancer cells. As shown in Figure 2E,F, following TGF‐β (3 ng/mL) stimulation, HCC4011 cells showed morphological changes to a spindle shape and elongated formation, and HCC4011 and H2009 cells showed upregulation of vimentin, suggesting the occurrence of EMT in the cells; however, addition of tranilast showed a partial inhibitory effect on TGF‐β–induced EMT (Figure 2G). These results suggest that tranilast directly prevents EMT of cancer cells, although the detailed mechanisms still remain elusive.
FIGURE 2.
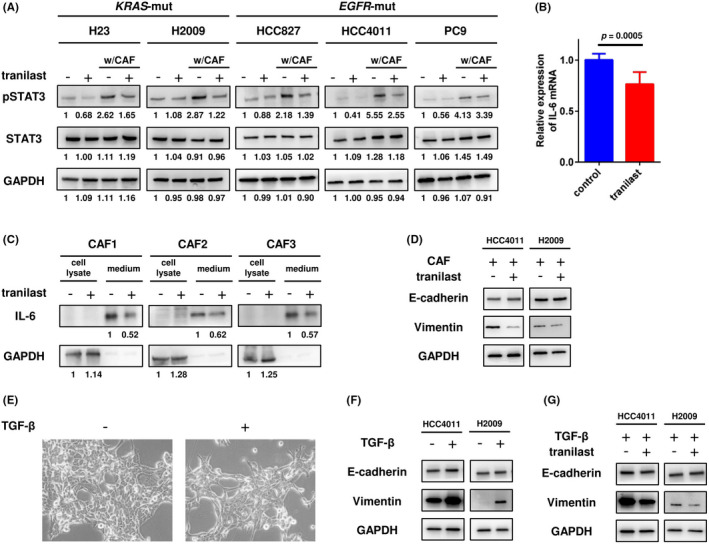
Inhibitory effect of tranilast on cancer‐associated fibroblast (CAF)‐induced pSTAT3 upregulation and epithelial‐mesenchymal transition (EMT) in non–small cell lung cancer (NSCLC) cells. A, NSCLC cell lines were cultured in the presence of tranilast for 7 d, and the expression level of pSTAT3 was analyzed by Western blotting. B, After tranilast treatment for 48 h, the mRNA expression levels of IL‐6 in the CAFs were measured by real‐time PCR. Each measurement was conducted in triplicate, and the data are representative of three independent experiments (mean ± SE) conducted using different CAF sublines (CAF1, CAF2, and CAF3). C, Secretion of IL‐6 in the culture medium of the CAFs was assessed by Western blotting. D, NSCLC cell lines and CAFs were cocultured in the presence of tranilast for 7 d, and the EMT status was analyzed by Western blotting. E, HCC4011 cells were treated with TGF‐β, and morphological changes were observed. F, After TGF‐β treatment for 72 h, the EMT status in the HCC4011 and H2009 cells was analyzed. G, HCC4011 and H2009 cells were treated with TGF‐β in the presence or absence of tranilast for 72 h, and the EMT status was analyzed by Western blotting
3.3. Combined treatment with tranilast plus molecular‐targeted drugs suppressed the growth of NSCLC cells in an in vitro coculture model of NSCLC cells and CAFs
In the section above, we described the inhibitory effects of tranilast on STAT3 activation and EMT in cancer cells, which suggests that combined administration of tranilast with molecular‐targeted agents may be an effective therapeutic strategy to counter CAF‐mediated acquisition of drug resistance. Therefore, we assessed the efficacy of combined tranilast plus osimertinib or selumetinib treatment in an in vitro coculture model of NCSLC cells and CAFs. Tranilast treatment alone showed a minimal inhibitory effect on the growth of the cancer cells, while osimertinib/selumetinib treatment alone showed a strong growth‐inhibitory effect in the EGFR‐mutant/KRAS‐mutant lung cancer cells. When the cancer cells were cocultured with CAFs, they were less sensitive to the aforementioned treatments; however, combined tranilast plus osimertinib/selumetinib treatment resulted in improved cancer cell sensitivity to osimertinib/selumetinib (Figure 3A). Similar results were obtained with the other CAF sublines (Figures S5A and S5B). Quantification of the results using the ImageJ software revealed significant differences (Figure 3B).
FIGURE 3.
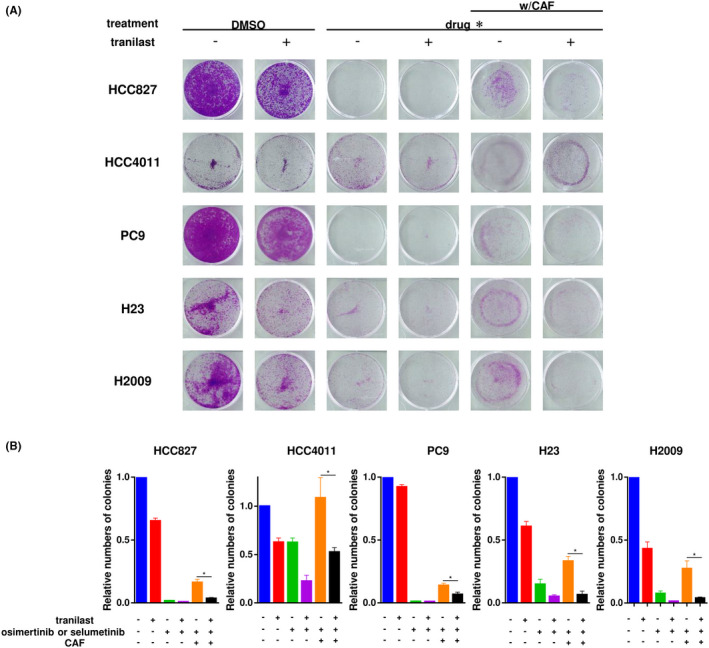
Effect of tranilast on cancer‐associated fibroblast (CAF)‐induced drug resistance in non–small cell lung cancer (NSCLC) cells. A, NSCLC cell lines were cultured with or without CAFs in the presence or absence of tranilast for 7 d. *: osimertinib for HCC827, HCC4011, and PC9 cells, and selumetinib for H23 and H2009 cells. B, The number of colonies was quantified using the ImageJ software. Each condition was assayed in duplicate, and the data are representative of three independent experiments. Data shown are the means ± SE; *p < 0.05, **p < 0.01
3.4. Combined tranilast plus osimertinib treatment inhibited tumor growth in a mouse xenograft model of EGFR ‐mutant NSCLC cells plus CAFs
To validate the aforementioned in vitro findings, we tested the efficacy of combined osimertinib plus tranilast treatment in a mouse mixed‐cell (PC9 cells plus CAF1 cells) xenograft model. Mice bearing mixed‐cell xenografts were treated with osimertinib alone, tranilast alone, or a combination of the two drugs. Although tranilast alone was found to exert a mild inhibitory effect in the colony formation assay in vitro, tranilast monotherapy had minimal effect on the tumor growth in vivo. Osimertinib monotherapy caused only partial shrinkage of the tumor, whereas combined osimertinib plus tranilast treatment yielded an enhanced antitumor effect. The drug combination was well‐tolerated throughout the 38‐day treatment period (Figure 4A). TUNEL staining showed the greatest number of apoptotic cells in the combined‐treatment group (Figure 4B,C).
FIGURE 4.
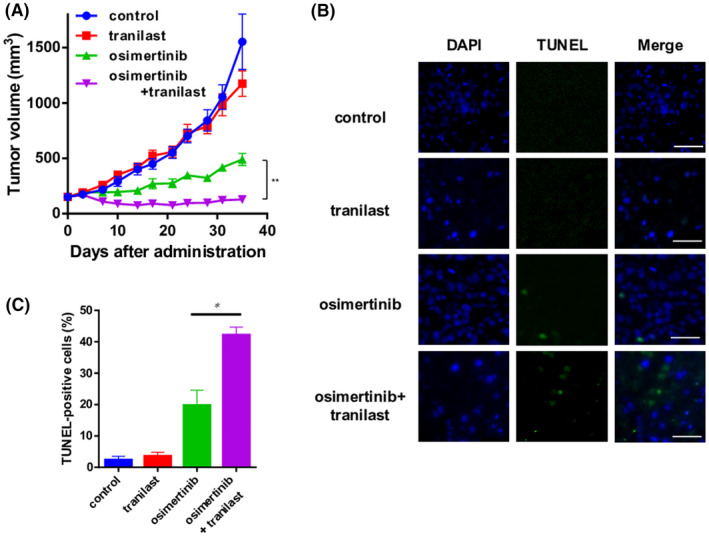
Enhancement of the tumor‐inhibitory effect of tranilast in a mouse xenograft model of non–small cell lung cancer (NSCLC) cells plus cancer‐associated fibroblasts (CAFs). A, A mixture of PC9 cells and CAFs was subcutaneously implanted into the flanks of nude mice. The mice were treated with tranilast (200 mg/kg) alone, osimertinib (5 mg/kg) alone, or a combination of osimertinib plus tranilast by oral gavage five times a week. The tumor volumes were determined on the indicated days after the start of the treatments. Each group consisted of seven mice. Data shown are the means ± SE; **p < 0.01. B, TUNEL staining (green) and DAPI (blue) staining showing the apoptotic neuronal nuclei and all the cell nuclei under microscopy. Scale bars: 50 μm. C, The population of TUNEL‐positive cells is shown as a percentage of the total number of cells. Data represent the means ± SE of five random fields. *p < 0.05. DAPI, 4′,6‐diamidino‐2‐phenylindole; TUNEL, TdT‐mediated dUTP nick end labeling
4. DISCUSSION
Accumulating evidence indicates that the crosstalk between cancer cells and the TME plays critical roles in the processes involved in tumor progression, including cancer initiation, proliferation, invasion, and metastasis. 13 CAFs are the major cellular components of the TME that contribute significantly to tumor aggressiveness and drug resistance by producing various types of cytokines, chemokines, growth factors, and matrix‐degrading enzymes. 14 Therefore, disruption of the TME, especially targeting the CAFs, is attracting attention as a novel and critical complemental therapeutic strategy to molecular‐targeted treatment. In this study, we clarified the inhibitory effects of tranilast on CAF‐induced enhancement of an aggressive tumor phenotype.
In addition to its use as an antiallergic drug, tranilast is also widely used for the treatment to hypertrophic scars, and its safety for clinical use has already been demonstrated. Tranilast inhibits the release of biologically active mediators from mast cells, as well as VEGF‐induced angiogenesis. 10 , 15 , 16 , 17 In addition, tranilast has been reported to have direct anticancer effects by suppressing cancer cell proliferation, migration, and invasion and inhibiting tumor angiogenesis in diverse cancer types, such as breast cancer, gastric cancer, and esophageal cancer. Previous reports indicated that tranilast is generally used in high concentrations. 18 , 19 , 20 , 21 , 22 The major mechanism underlying the efficacy of tranilast is thought to be suppression of the cellular TGF‐β signaling pathway by the drug, although other drug actions could have roles as well. 23 On the basis of these preclinical findings, a phase I/II study of neoadjuvant combined chemotherapy plus tranilast for esophageal cancer was initiated and is currently under way. 24
In this study, we demonstrated, both in vitro, using coculture models of NSCLC cells with CAFs, and in vivo, that tranilast prevented the acquisition of resistance to molecular‐targeted drugs by suppressing the IL‐6/JAK/STAT3 signaling pathway and inhibition of EMT in cancer cells. IL‐6 is one of the major cytokines released into the TME, which activates the cellular IL‐6/JAK/STAT3 signaling pathway, promotes tumor cell proliferation and invasiveness, and inhibits cellular apoptosis. 25 In addition, activation of the IL‐6/STAT3 axis, both in an autocrine manner in oncogene‐addicted cancer cells and in a paracrine manner from CAFs, has been shown to contribute to the acquisition of treatment resistance. 26 , 27 EMT refers to phenotypic conversion of cells from an epithelial type into a mesenchymal type and confers the tumor metastatic behaviors of cancer cell invasion and migration on the tumor cells; it is also known to be another critical mechanism underlying the acquisition of CAF‐mediated drug resistance. 28 TGF‐β is considered to be a key mediator of CAF‐induced EMT. We demonstrated the inhibitory effect of tranilast on CAF‐induced EMT, although the secretion of TGF‐β from CAFs was not suppressed, unlike the case of IL‐6 secretion from these cells (data not shown). Furthermore, TGF‐β–induced EMT was also demonstrated to be suppressed by tranilast treatment. Considering these findings, tranilast appeared to have a direct effect of inhibiting EMT in the cancer cells. Kang et al. showed that tranilast inhibited EMT by inhibiting the TGF‐β/Smad pathway in peritoneal mesothelial cells 29 ; however, we could not confirm any changes in the Smad activity in our NSCLC cell lines (data not shown), suggesting that other signaling pathway(s) than the TGF‐β/Smad may also be involved in the suppression of EMT by tranilast. Further studies are needed for a more precise elucidation of this intriguing biological mechanism.
In summary, we showed that tranilast inhibited CAF‐mediated acquisition of resistance of NSCLC cells to molecular‐targeted therapy by inhibiting the IL‐6/STAT3 axis via its actions on the CAFs and prevented EMT via its direct actions on the cancer cells. Based on these findings, we strongly suggest that combined administration of tranilast with molecular‐targeted therapy is an attractive complementary therapeutic strategy to maximize the effect of molecular‐targeted therapy.
DISCLOSURE
No potential conflicts of interest were disclosed.
ETHICAL APPROVAL
Studies using clinical specimens were approved by the Okayama Medical School and Hospital's Research Ethics Committee (approval number: # 1906–033), and informed consent was obtained from individual patients for the use of their materials. The animal protocol was approved by the Animal Care and Use Committee, Okayama University (permit number: OKU‐2020564).
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5A
Figure S5B
ACKNOWLEDGMENTS
We thank Ms. Fumiko Isobe (Department of Thoracic, Breast, and Endocrinological Surgery, Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, Okayama, Japan) for her technical assistance.
Ochi K, Suzawa K, Thu YM, et al. Drug repositioning of tranilast to sensitize a cancer therapy by targeting cancer‐associated fibroblast. Cancer Sci. 2022;113:3428‐3436. doi: 10.1111/cas.15502
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. doi: 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2. Pao W, Girard N. New driver mutations in non‐small‐cell lung cancer. Lancet Oncol. 2011;12(2):175‐180. doi: 10.1016/s1470-2045(10)70087-5 [DOI] [PubMed] [Google Scholar]
- 3. Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR‐TKI therapy in 155 patients with EGFR‐mutant lung cancers. Clin Cancer Res. 2013;19(8):2240‐2247. doi: 10.1158/1078-0432.CCR-12-2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu HA, Suzawa K, Jordan E, et al. Concurrent alterations in EGFR‐mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of mtor as a mediator of resistance. Clin Cancer Res. 2018;24(13):3108‐3118. doi: 10.1158/1078-0432.CCR-17-2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shien K, Papadimitrakopoulou VA, Ruder D, et al. JAK1/STAT3 activation through a proinflammatory cytokine pathway leads to resistance to molecularly targeted therapy in non‐small cell lung cancer. Mol Cancer Ther. 2017;16(10):2234‐2245. doi: 10.1158/1535-7163.MCT-17-0148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer‐associated fibroblasts induce epithelial‐mesenchymal transition of breast cancer cells through paracrine TGF‐beta signalling. Br J Cancer. 2014;110(3):724‐732. doi: 10.1038/bjc.2013.768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao Q, Yang Z, Xu S, et al. Heterotypic CAF‐tumor spheroids promote early peritoneal metastatis of ovarian cancer. J Exp Med. 2019;216(3):688‐703. doi: 10.1084/jem.20180765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer‐associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res. 2012;10(11):1403‐1418. doi: 10.1158/1541-7786.MCR-12-0307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masuda T, Tsuruda Y, Matsumoto Y, Uchida H, Nakayama KI, Mimori K. Drug repositioning in cancer: The current situation in Japan. Cancer Sci. 2020;111(4):1039‐1046. doi: 10.1111/cas.14318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Komtsu H, Kojima M, Tsutsumi N, et al. Study of the mechanism of inhibitory action of tranilast on chemical mediator release. Jpn J Pharmacol. 1988;46:43‐51. [DOI] [PubMed] [Google Scholar]
- 11. Suzawa H, Kikuchi S, Arai N, Koda A. The mechanism involved in the inhibitory action of tranilast on collagen biosynthesis of keloid fibroblasts. Japan J Pharmacol. 1992;60:91‐96. [DOI] [PubMed] [Google Scholar]
- 12. Hiraide S, Yanagawa Y, Iizuka K. Tranilast inhibits interleukin‐33 production by macrophages. Eur J Pharmacol. 2018;818:235‐240. doi: 10.1016/j.ejphar.2017.10.057 [DOI] [PubMed] [Google Scholar]
- 13. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557‐4566. doi: 10.1158/0008-5472.CAN-18-3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer‐associated fibroblasts. Nat Rev Cancer. 2020;20(3):174‐186. doi: 10.1038/s41568-019-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Azuma H, Banno K, Yoshimura T. Pharmacological properties of N‐(3′,4′‐dimethoxycinnamoyl) anthranilic acid (N‐5′), a new anti‐atopic agent. Br J Pharmacol. 1976;58:483‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Isaji M, Nakajoh M, Naito J. Selective inhibition of collagen accumulation by N‐(3, 4‐dimethoxycinnamoyl)anthranilic acid (N‐5′) in granulation tissue. Biochem Pharmacol. 1987;36:469‐474. [DOI] [PubMed] [Google Scholar]
- 17. Isaji M, Miyata H, Ajisawa Y, Takehara Y, Yoshimura N. Tranilast inhibits the proliferation, chemotaxis and tube formation of human microvascular endothelial cells in vitro and angiogenesis in vivo. Br J Pharmacol. 1997;122:1061‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Darakhshan S, Ghanbari A. Tranilast enhances the anti‐tumor effects of tamoxifen on human breast cancer cells in vitro. J Biomed Sci. 2013;20(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prud'homme GJ, Glinka Y, Toulina A, Ace O, Subramaniam V, Jothy S. Breast cancer stem‐like cells are inhibited by a non‐toxic aryl hydrocarbon receptor agonist. PLoS One. 2010;5(11):e13831. doi: 10.1371/journal.pone.0013831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saito H, Fushida S, Harada S, et al. Importance of human peritoneal mesothelial cells in the progression, fibrosis, and control of gastric cancer: inhibition of growth and fibrosis by tranilast. Gastric Cancer. 2018;21(1):55‐67. doi: 10.1007/s10120-017-0726-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hiroi M, Onda M, Uchida E, Aimoto T. Anti‐tumor effect of N‐[3,4‐dimethoxycinnamoyl]‐anthranilic acid (tranilast) on experimental pancreatic cancer. J Nippon Med Sch. 2002;69:224‐234. [DOI] [PubMed] [Google Scholar]
- 22. Shiozaki A, Kudou M, Ichikawa D, et al. Esophageal cancer stem cells are suppressed by tranilast, a TRPV2 channel inhibitor. J Gastroenterol. 2018;53(2):197‐207. doi: 10.1007/s00535-017-1338-x [DOI] [PubMed] [Google Scholar]
- 23. Darakhshan S, BidmeshkiPour A. Tranilast: A review of its therapeutic applications. Pharmacol Res. 2014;91:15‐28. [DOI] [PubMed] [Google Scholar]
- 24. Shiozaki A, Kudou M, Fujiwara H, et al. Clinical safety and efficacy of neoadjuvant combination chemotherapy of tranilast in advanced esophageal squamous cell carcinoma: Phase I/II study (TNAC). Medicine. 2020;99(50):e23633. doi: 10.1097/MD.0000000000023633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lederle W, Depner S, Schnur S, et al. IL‐6 promotes malignant growth of skin SCCs by regulating a network of autocrine and paracrine cytokines. Int J Cancer. 2011;128(12):2803‐2814. doi: 10.1002/ijc.25621 [DOI] [PubMed] [Google Scholar]
- 26. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene‐addicted cancer cells. Cancer Cell. 2014;26(2):207‐221. doi: 10.1016/j.ccr.2014.05.019 [DOI] [PubMed] [Google Scholar]
- 27. Karakasheva TA, Lin EW, Tang Q, et al. IL‐6 mediates cross‐talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 2018;78(17):4957‐4970. doi: 10.1158/0008-5472.CAN-17-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178‐196. doi: 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang SH, Kim SW, Kim KJ, et al. Effects of tranilast on the epithelial‐to‐mesenchymal transition in peritoneal mesothelial cells. Kidney Res Clin Pract. 2019;38:472‐480. doi: 10.23876/j.krcp.19.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5A
Figure S5B