

Abstract
Trastuzumab‐induced cardiotoxicity interferes with continued treatment in approximately 10% of patients with ErbB2‐positive breast cancer, but its mechanism has not been fully elucidated. In this study, we recruited trastuzumab‐treated patients with ≥30% reduction in left ventricular ejection fraction (SP) and noncardiotoxic patients (NP). From each of these patients, we established three cases of induced pluripotent stem cell‐derived cardiomyocytes (pt‐iPSC‐CMs). Reduced contraction and relaxation velocities following trastuzumab treatment were more evident in SP pt‐iPSC‐CMs than NP pt‐iPSC‐CMs, indicating the cardiotoxicity phenotype could be replicated. Differences in ATP production, reactive oxygen species, and autophagy activity were observed between the two groups. Analysis of transcripts revealed enhanced kallikrein5 expression and pro‐inflammatory signaling pathways, such as interleukin‐1β, in SP pt‐iPSC‐CMs after trastuzumab treatment. The kallilkrein5‐protease‐activated receptor 2 (PAR2)‐MAPK signaling pathway was more activated in SP pt‐iPSC‐CMs, and treatment with a PAR2‐antagonist suppressed interleukin‐1β expression. Our data indicate enhanced pro‐inflammatory responses through kallikrein5‐PAR2 signaling and vulnerability to external stresses appear to be the cause of trastuzumab‐induced cardiotoxicity in SP.
Keywords: cardiotoxicity, inflammation, kallikrein, patient‐derived iPS cell, trastuzumab
Using the induced pluripotent stem cell‐derived cardiomyocytes model generated from patients with severe trastuzumab‐induced cardiotoxicity, we newly found that the toxicity may be attributed to increased pro‐inflammatory responses mediated through an enhanced kallikrein5‐PAR2 signaling pathway.
Abbreviations
- cTnT
cardiac troponin‐T
- CTRCD
cancer therapy‐related cardiac dysfunction
- CV
contraction velocity
- DOX
doxorubicin
- HER2/ErbB2
human epidermal growth factor receptor 2
- I‐191
4‐(8‐(tert‐butyl)‐6‐(4‐fluorophenyl)imidazo[1,2‐b]pyridazine‐2‐carbonyl)‐3,3‐dimethylpiperazin‐2‐one
- IL1β
interleukin‐1β
- iNOS
inducible nitric oxide synthase
- iPSC
induced pluripotent stem cell
- KLK
kallikrein
- LVEF
left ventricular ejection fraction
- NP
noncardiotoxic patient group
- NRG1
neuregulin‐1
- OCR
oxygen consumption rate
- PAR2
protease‐activated receptor 2
- pt‐iPSC‐CM
patient‐specific iPSC‐derived cardiomyocyte
- qRT‐PCR
quantitative real‐time RT‐PCR
- rKLK5
recombinant KLK5
- RNA‐Seq
RNA sequencing
- ROS
reactive oxygen species
- RV
relaxation velocity
- SNP
single nucleotide polymorphism
- SP
severely cardiotoxic patient group
- Tmab
trastuzumab
- UA
ursolic acid (3β‐hydroxy‐12‐ursen‐28‐ic acid)
1. INTRODUCTION
Human epidermal growth factor receptor 2 is overexpressed in approximately 20% of breast cancer and is associated with poor prognosis. Trastuzumab, an anti‐ErbB2 humanized mAb released in 1998, dramatically reduces cancer recurrence and mortality. 1 , 2 However, unpredictable cardiotoxicity occurs in approximately 5%–15% of patients. 3 , 4 , 5 Trastuzumab‐induced cardiotoxicity is unpredictable as it is not cumulative, and the main phenotype shows a diffuse decrease in left ventricular wall motion and a decrease in myocardial wall thickness, with morphology resembling dilated cardiomyopathy. 6 Currently, there is no alternate drug with comparable efficacy to Tmab; thus, there is a great concern that interruption or discontinuation of treatment due to the cardiotoxicity could lead to disease progression and diminished effectiveness in preventing recurrence of breast cancer. 7 However, the precise details of the mechanism of cardiotoxicity have not been fully elucidated, and early prediction and prevention of the onset of toxicity are still not feasible.
In the heart, ErbB2 promotes downstream signaling following heterodimerization between ErbB4 and NRG1, secreted from cardiac and vascular endothelial cells. The NRG1‐ErbB2/4 axis plays an important role as protective and prosurvival pathways in response to cardiac stress. 8 , 9 , 10 , 11 Trastuzumab binds to the extracellular domain of ErbB2, impairing its activation of downstream signaling through dimerization. 12 Previous studies have shown that ErbB2 inhibition leads to mitochondrial dysfunction and abnormal intracellular metabolism, which could weaken the myocardium and make it vulnerable to myocardial injury, leading to cardiac dysfunction. 13 , 14 , 15 , 16 , 17 Nevertheless, despite the negative impact of Tmab, cardiotoxicity does not occur in all treated patients. So we hypothesized that individual differences in molecular mechanisms might influence cardiotoxicity.
The known risks of developing cardiotoxicity include age, a history of anthracycline treatment, and cardiovascular events. 18 , 19 However, unpredictable cases of cardiotoxicity also occur in clinical practice, in patients with none or few risk factors. Some genome‐wide association studies have explored the hypothesis that Tmab‐induced cardiotoxicity might occur in patients with a particular genetic background in addition to the aforementioned acquired risk factors, and several SNPs have been identified. 20 However, functional analysis at the cardiomyocyte‐cellular level has not been performed thus far, and it is unclear whether these SNPs are mechanistically associated with Tmab toxicity. Analysis of cardiomyocytes from patients would be the ideal validation approach, but myocardial biopsy is highly invasive and cannot be routinely carried out. As an alternative, disease‐specific iPSC‐derived organ models with the genetic background of the target disease have been developed. These disease models have been widely used as tools to elucidate the causes and mechanisms of disease development and search for optimal drugs. 21
In the current study we aimed to elucidate the mechanisms and biological characteristics underlying Tmab‐induced cardiotoxicity. To achieve this, we generated pt‐iPSC‐CMs from breast cancer patients who received Tmab‐based treatments and evaluated differences in cellular function and gene expression between groups with and without Tmab‐induced cardiotoxicity.
2. MATERIALS AND METHODS
2.1. Generation of iPSCs and cardiac differentiation of pt‐iPSC‐CMs from peripheral blood of patients with breast cancer
Induced pluripotent stem cells for the differentiation of cardiomyocytes were generated from PBMCs obtained from six Tmab‐treated patients with breast cancer who had or did not have cardiotoxicity (detailed information is listed in Table 1). In brief, activated T cells (1 × 106) were reprogrammed to iPSCs using a CytoTune‐iPS 2.0 Sendai Reprogramming kit (ID Pharma Co. Ltd). The iPSC lines were differentiated into pt‐iPSC‐CMs using a small molecule‐based monolayer differentiation protocol and were maintained as previously reported. 22 , 23 Cardiomyocyte purity was assessed using cardiomyocyte detector miRNA switches (aceRNA Technologies). 24 Detailed methods are described in Data S1 (Figure S1A,B, materials and methods).
TABLE 1.
Classification definitions and patient characteristics of induced pluripotent stem cell lines derived from patients with breast cancer
| Line code | Age (years) | Sex | LVEF (%) | Evaluation of risk factors | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Minimum | Decline range | Age | High BMI | AF | HT | DM | Smoker | PMH | Cumulative dose anthracycline | Post RTx | BNP ≥100 pg/ml | |||
| >65 years | >30 kg/mg2 | ||||||||||||||
| SP1 | 58 | F | 69 | 30 | 39 | − | − | − | − | − | − | − | 360 | − | 8.5 |
| SP2 | 63 | F | 75 | 33 | 42 | − | − | − | − | − | − | − | 389.2 | − | 30 |
| SP3 | 65 | F | 66 | 14 | 52 | − | − | − | − | − | − | − | 400 | − | 41 |
| NP1 | 57 | F | 70 | 60 | − | − | − | − | − | − | − | − | 300 | − | 44 |
| NP2 | 70 | F | 65 | 63 | − | + | − | − | − | + | − | − | 450 | − | 57 |
| NP3 | 50 | F | 68 | 64 | − | − | − | − | − | − | − | − | 350 | − | 23 |
Abbreviations: AF, atrial fibrillation; BMI, body mass index; BNP, brain natriuretic peptide; DM, diabetes mellitus; F, female; HT, hypertension; LVEF, left ventricular ejection fraction; M, male; NP, noncardiotoxic patient group; PMH, past cardiovascular‐related disease history; RTx, radiotherapy; SP, severely cardiotoxic patient group.
2.2. Immunofluorescence staining and confocal microscopy
Patient‐specific iPSC‐derived cardiomyocytes maintained on iMatrix‐coated 35‐mm glass bottom dishes (Matsunami Glass Ind.) were stained with the following primary Abs: rabbit anti‐cTnT (ab45932; Abcam) and mouse anti‐α‐actinin (ab18061; Abcam). Alexa Fluor 488 goat anti‐rabbit or Alexa Fluor 594 goat anti‐mouse Abs (Thermo Fisher Scientific) were used as secondary Abs.
2.3. Calcium imaging
Ca2+ signals of pt‐iPSC‐CMs seeded on iMatrix‐coated 35‐mm glass bottom dishes were monitored with Cal520‐AM (AAT Bioquest). Detailed protocols are available in Data S1. For inhibitor experiments, pt‐iPSC‐CMs were seeded into iMatrix‐coated black clear bottom 96‐well plates and assays were carried out using a FlexStation 3 microplate reader (Molecular Devices).
2.4. Action potential signal
Patient‐specific iPSC‐derived cardiomyocytes seeded on 35‐mm glass bottom dishes were stained with a FluoVolt Membrane Potential Kit (Thermo Fisher Scientific). FluoVolt images were captured using the same device and the same setting used for Ca2+ imaging.
2.5. Field potential recordings
The field potential recordings were undertaken using a multielectrode array system (MEA 2100; Multi Channel Systems) as described previously. 25 Prior to recordings, pt‐iPSC‐CMs were plated onto a 60‐electrode standard multielectrode array. Data acquisition was carried out using MC_Rack program software.
2.6. Drug treatment
Patient‐specific iPSC‐derived cardiomyocytes were treated with Tmab (anti‐HER2 humanized mAb; BioVision, Inc.) or DOX (Sigma‐Aldrich) for 7 days. Human IgG (Fujifilm Wako Chemicals) or DMSO (Sigma‐Aldrich) was used as a negative control treatment unless noted. The medium containing fresh drug was replaced every 2–3 days. The KLK5 antagonist, UA (Sigma‐Aldrich), 26 the PAR2 antagonist (small molecules), I‐191 (AOBIOUS), 27 the PAR2 antagonist (peptides), FSLLRY‐NH2 (Tocris Bioscience), 28 , 29 and Recombinant Human Kallikrein 5 (R&D Systems) were cotreated for inhibition experiments.
2.7. Contractility analysis
Spontaneous beating activity of monolayer cardiomyocytes was recorded with an all‐in‐one KEYENCE BZ‐X800 fluorescence microscope. During data collection, cells were maintained under controlled conditions at 37°C with 5% CO2 and 95% air in a stage‐top microscope incubator (Tokai Hit). Motion vector analysis was carried out by high‐resolution motion capture tracking using the SI8000 Cell Motion Imaging System (Sony Corporation). Detailed protocols are described in Figure S2.
2.8. Quantitative plate‐based assays
Patient‐specific iPSC‐derived cardiomyocytes were seeded on 96‐well plates subjected to plate‐based assays after drug treatment using SpectraMax 340PC384, SpectraMax L, and FlexStation 3 microplate readers. CYTO‐ID autophagy detection kits 2.0 (Enzo Life Sciences) were used for autophagy assays, ROS‐Glo H2O2 assays (Promega) were used for ROS detection, JC‐1 assays (Enzo Life Sciences) were used for mitochondrial membrane potential measurement, and Cell Titer Glo 2.0 assays (Promega) were used for ATP content, in accordance with the manufacturers' instructions.
2.9. Seahorse extracellular metabolic flux assay
Mitochondrial respiration in pt‐iPSC‐CMs was measured using the Seahorse Bioscience XF24 Flux Analyzer with XF Cell Mito Stress Test Kit (Agilent Technologies) following the manufacturer's instructions. Briefly, cells seeded on XF24 microplates treated with Tmab or DOX. Oxygen consumption rate (pmol/min) was normalized to baseline OCR and a total cell count of each well was carried out using the CyQuant Cell Proliferation assay (Thermo Fisher Scientific).
2.10. Immunoassays
Human KLK8, KLK5, cTnI, and NRG1 levels were measured in pt‐iPSC‐CMs cultured supernatants using the Human KLK8/Kallikrein‐8 ELISA Kit PicoKine (BOSTER Bio), Human KLK5/Kallikrein‐5 ELISA Kit PicoKine (BOSTER Bio), Human Cardiac Troponin I ELISA kit (RayBiotech), and Human NRG1 DuoSet ELISA kit (R&D Systems), respectively, in accordance with the manufacturers' instructions.
2.11. Western blot analysis
All pt‐iPSC‐CMs were lysed in RIPA buffer supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Nacalai Tesque). The protein bands were detected by enhanced chemiluminescence. Table S2 lists all Abs used in this study. MB231, a HER2‐negative breast cancer cell line, was used as a negative control, and SK‐BR‐3, a HER2‐positive breast cancer cell, was used as a positive control for ErbB2.
2.12. RNA sequencing and data analysis
Total RNAs prepared from pt‐iPSC‐CMs in the presence or absence of 1 μM Tmab were subjected to RNA sequencing analysis. The obtained Fastq data were processed using various pipelines to obtain the quantifying counts. Differentially expressed genes were identified using a threshold of false discovery rate of less than 0.05 and minimal fold change greater than 3. Detailed methods and pipelines used for the analysis are described in Data S1.
2.13. Quantitative RT‐PCR
All qRT‐PCRs were carried out using the gene‐specific primers and universal probe library probes (Roche). Relative gene expression levels were calculated using the housekeeping gene GAPDH as an internal control per the 2−ΔΔCt method. Detailed methods are described in Data S1, and the primers and probes are listed in Table S3.
2.14. Study approval
All patients provided written informed consent prior to undergoing any procedures. Human research was reviewed and approved by the Juntendo University Hospital Institutional Review Board (Approval Number: 2019160).
2.15. Statistical analysis
Statistical analyses were undertaken using GraphPad Prism 8. All data are expressed as mean ± SEM. Box and whisker plots show the 25th to 75th percentiles and minimum to maximum values, with the median indicated. Statistical comparisons were made using one‐way ANOVA followed by Tukey's multiple comparison test, or two‐tailed unpaired Student's t test, or nested t‐test 30 , 31 between two groups. Values of p < 0.05 were considered to indicate statistical significance.
Detailed methods are available in the Data S1.
3. RESULTS
3.1. Characteristics of patients selected for pt‐iPSC‐CM establishment
Twenty‐four of 468 patients with breast cancer (5.1%) who received Tmab at Juntendo University Hospital between 2010 and 2019 were diagnosed with CTRCD. 32 Cardiac function‐related data were collected in the Onco‐Cardiology division. Patients in CTRCD group, who developed severe cardiotoxicity with a decline in LVEF of ≥30% from baseline, was defined as SP. Patients that did not develop cardiotoxicity for at least 1 year after completion of treatment were defined as NP. Three patients with matched backgrounds (low clinical risk score) 33 in both groups were selected and pt‐iPSC‐CMs were generated (Figure 1A). The patient characteristics and LVEF changes of both groups are shown in Table 1. Patients in SP discontinued Tmab according to guidelines, 34 and even after 3–5 years of cardioprotective therapy, LVEF did not recover to baseline (Figure 1B). There was no clinically significant difference in parameters indicating cardiac function before treatment in the two groups (Table S1).
FIGURE 1.
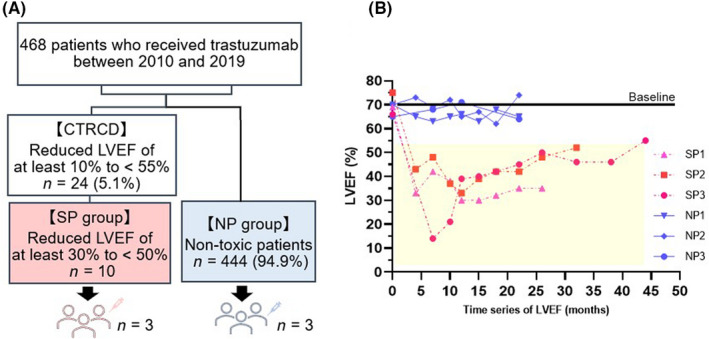
Characteristics of selected patients with breast cancer for generation of patient‐specific induced pluripotent stem cell‐derived cardiomyocytes. (A) Flow diagram of patient selection. Twenty‐four of 468 patients with breast cancer (5.1%) who received trastuzumab (Tmab)‐based treatment were diagnosed with cancer therapy‐related cardiac dysfunction (CTRCD). Three patients in each group were selected from the severely cardiotoxic patient group (SP), patients with a reduced left ventricular ejection fraction (LVEF) of at least 30% from baseline. (B) Time series of LVEF in the six patients. Cardiac‐function values within the yellow background indicate that Tmab could not be administered. NP, patients without cardiotoxicity
3.2. Establishment of pt‐iPSC‐CMs and evaluation of cell function
Patient‐specific iPSC‐derived cardiomyocytes were generated from the six selected patients (SP, n = 3; NP, n = 3). The karyotype was confirmed prior to the start of induction (Figure 2A). Next, structural and electrophysiological functional assessments were undertaken to ensure that the established pt‐iPSC‐CMs, with reference to previously published data, could be used to accurately assess cell function. 25 , 35 , 36 On day 8 postdifferentiation, beating myocardium was observed (Figure 2B, Video S1). The purity of pt‐iPSC‐CMs was assessed using CM detector RNA Switch, confirming that metabolic purification was sufficient (Figure 2C). The expression of sarcomere structures was confirmed by fluorescent immunostaining with cTnT and α‐actinin staining (Figure 2D). The expression of cTnT and α‐actinin, and the target receptors of Tmab, ErbB2, and ErbB4, was confirmed by western blotting (Figure 2E). The expression of ErbB2 and 4 receptors, and the extracellular secretion of NRG1 protein level detected in the culture supernatants was also confirmed in all six lines of pt‐iPSC‐CMs (Figure S1C,D). Furthermore, we confirmed Ca2+ transients and electrophysiological properties characteristic of cardiomyocytes (Figure 2F–H).
FIGURE 2.
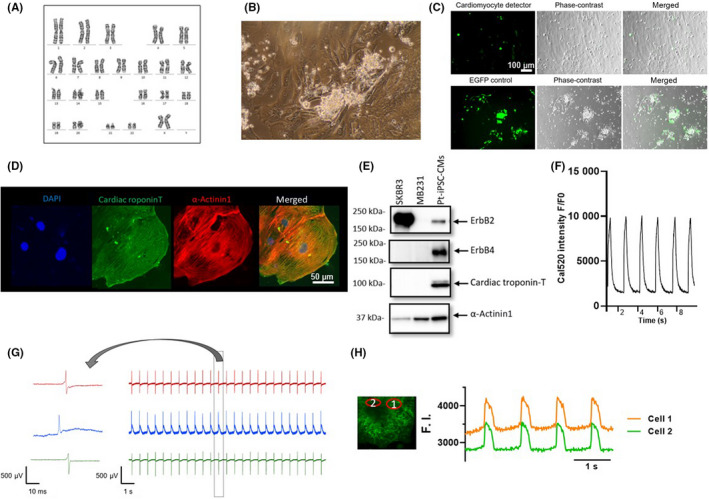
Generated patient‐specific induced pluripotent stem cell‐derived cardiomyocytes (pt‐iPSC‐CMs) show a functional cardiomyocyte phenotype. (A) Representative image of normal karyotype of iPSC lines by G‐band analysis. (B) Representative phase‐contrast image. (C) Confirmation of sufficient purification after metabolic purification using CM detector RNA switch. EGFP is expressed in cells other than cardiomyocytes (upper row). (D) Images of immunostaining with DAPI (blue), cardiac troponin‐T (cTnT; green) and α‐actinin (red). (E) Expression of western blot of ErbB2, ErbB4, cTnT, and α‐actinin receptors. MB231 was used as a negative control and SK‐BR‐3 as a positive control for ErbB2. (F) Recording of Ca2+ transients observed with the Ca2+ indicator, Cal520. (G) Electrophysiological activities of spontaneously beating pt‐iPSC‐CMs and enlarged single beat profile using the multielectrode array. (H) Membrane potential recording showing cardiomyocyte‐specific action potentials recorded with the voltage sensitive dye, FluoVolt. F.I., fluorescence intensity
3.3. Trastuzumab‐induced reduction in contractility more evident in SP
First, we evaluated the contractility of drug‐treated pt‐iPSC‐CMs using four parameters to confirm the recapitulation of the patient's phenotype (Figure 3A). The myocardial contractility was decreased by Tmab treatment in all the lines tested (Figure 3B). The rate of contractility change was more evident on day 7 than on days 2–5 (Figure S2B). Doxorubicin was also simultaneously examined as a positive control (Figure S3). Following Tmab treatment, a significant decrease in CV and RV was observed in all lines of pt‐iPSC‐CMs. Moreover, there was a greater reduction in CV and RV in SP than in NP (Figure 3B,C). The contraction–relaxation duration and spontaneous beats show no significant difference (Figure 3D,E). These data indicate that our model replicates the clinical phenotype of patients receiving Tmab treatment.
FIGURE 3.
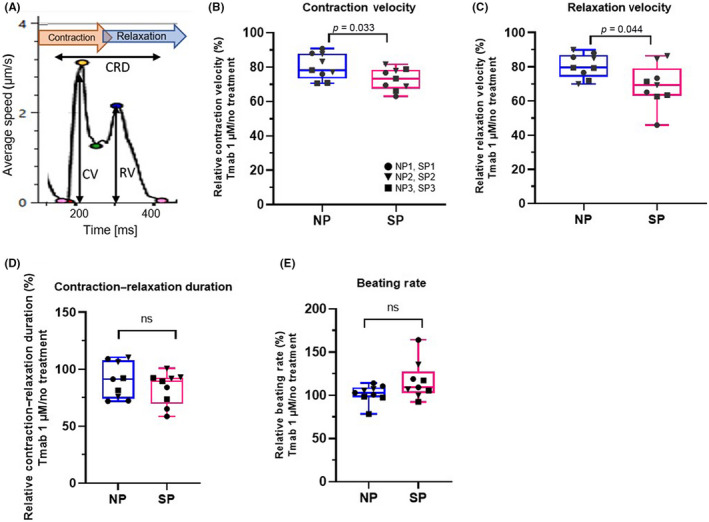
Contractility analysis of patient‐specific induced pluripotent stem cell‐derived cardiomyocytes (pt‐iPSC‐CMs) following trastuzumab (Tmab) treatment with motion vector analysis. (A) Illustration of a representative motion waveform with associated parameters. (B–E) Relative change in parameters for contractility following Tmab treatment: (B) contraction velocity (CV), (C) relaxation velocity (RV), (D) contraction–relaxation deformation (CRD) distance, and (E) beating rate. Data were obtained using all the lines of pt‐iPSC‐CMs and the assay was repeated three times. NP, noncardiotoxic patient group; SP, severely cardiotoxic patient group
3.4. Differences in intracellular metabolic function and vulnerability between SP and NP
Next, we evaluated the cardiotoxic effects of Tmab on pt‐iPSC‐CMs, using plate‐based assays. The vulnerability of cardiomyocytes to DOX was also evaluated. A decrease in cell viability was indicated by a decrease in ATP content. Higher Tmab concentrations resulted in a greater decrease in cell viability in SP compared with NP. Doxorubicin resulted in an approximate halving in the number of cells, with no difference between the two groups (Figure 4A). The generation of ROS was significantly higher in SP than NP following Tmab treatment, and even higher after DOX treatment, suggesting that SP might generally be more susceptible to cardiac remodeling (Figure 4B). The autophagy activity level decreased following Tmab treatment, but there was no significant difference between SP and NP at the 1 μM dose. In contrast, a significant increase was observed in SP with both the higher dose of Tmab and DOX (Figure 4C,D). The release of cTnI, a marker of myocardial damage, did not increase with Tmab treatment, but did with DOX treatment, with higher cTnI release detected in SP than NP, indicating SP vulnerability (Figure 4E). Intracellular Ca2+ transients mediating myocardial contraction was measured using a fluorescent Ca2+ indicator. Both Tmab and DOX decreased the amplitude of Ca2+ transients, but there was no significant difference between the two groups (Figure 4F). Taken together, these results suggest that intercellular metabolism and vulnerability differ between SP and NP.
FIGURE 4.
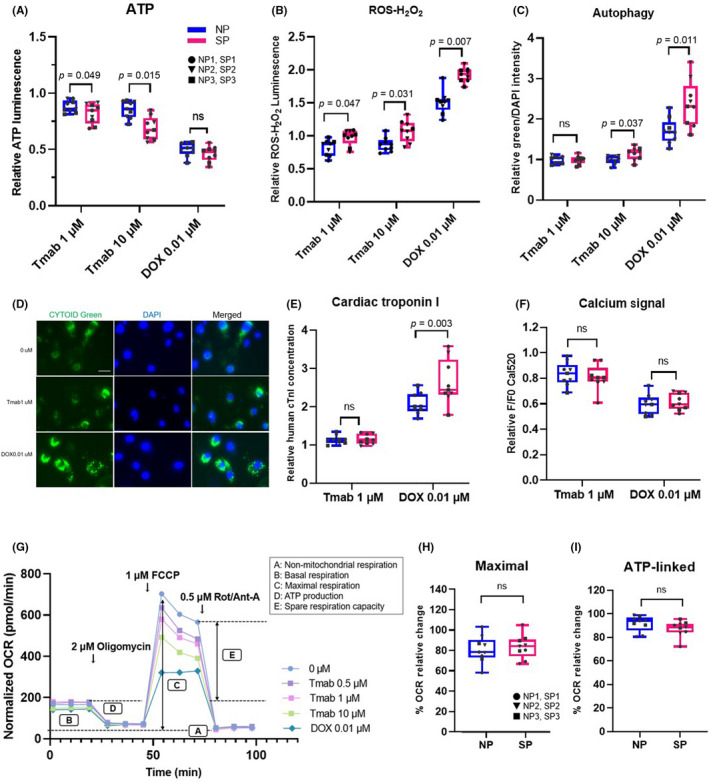
Quantitative plate‐based assays and mitochondrial stress test of patient‐specific induced pluripotent stem cell‐derived cardiomyocytes (pt‐iPSC‐CMs) following trastuzumab (Tmab) or doxorubicin (DOX) treatment. (A–C) Relative change in each assay with drug treatment. (A) Quantification of ATP content measured using Cell Titer‐Glo assay. (B) Quantification of reative oxygen species (ROS) generation measured using ROS‐Glo H2O2 assay. (C) Autophagy level measured using CYTO‐ID staining. (D) Representative images of immunostaining of CYTO‐ID green. (E) Quantification of cardiac troponin I in the culture medium with ELISA. (F) Amplitudes of peak Ca2+ transients. (G, H) Oxygen consumption rate (OCR) was measured using the Seahorse XF24 extracellular‐flux analyzer. (G) Representative plot of OCR measured following drug treatment. Each evaluation parameter and sequential addition (oligomycin, FCCP, and Rot/Ant‐A) is shown in the graph. (H) Maximal respiration shown with relative OCR rate. (I) Mitochondrial membrane potential was measured using JC‐1 dye. Data were obtained using all lines of pt‐iPSC‐CMs and the assay was repeated three times. NP, noncardiotoxic patient group; ns, not significant; SP, severely cardiotoxic patient group; FCCP, carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone; Rot/Ant‐A, rotenone/antimycin A
3.5. Mitochondrial dysfunction due to ErbB2 inhibition
We next undertook mitochondrial stress tests. Both pt‐iPSC‐CMs showed a decrease in OCR following Tmab and DOX treatment (Figure 4G). Maximal respiratory volume decreased in a dose‐dependent manner with Tmab, indicating that ErbB2 inhibition attenuates mitochondrial function, but there was no obvious difference between the two groups (Figure 4H). Other end‐points listed in Figure 4G showed similar results (Figure S4). Mitochondrial membrane potential change was reduced with DOX but not Tmab (Figure 4I). Collectively, these data show that in our model of pt‐iPSC‐CMs, ErbB2 inhibition by Tmab causes mitochondrial dysfunction. However, as this effect did not differ between the two groups, it suggests there are other significant effectors on intracellular energy metabolism involved in the mechanism of reduced contractility.
3.6. Characteristic gene expression in SP pt‐iPSC‐CMs
To elucidate the molecular mechanisms of the dysfunctional differences between SP and NP pt‐iPSC‐CMs, we performed RNA‐Seq using both Tmab‐treated (1 μM) and untreated pt‐iPSC‐CMs from both groups (Figure 5A). Differentially expressed genes between SP versus NP and Tmab‐treated versus untreated groups were found (false discovery rate <0.05 and fold change >3; Tables S4–S7). Among these, we searched for genes of interest that have previously been associated with cardiotoxicity. We paid special attention to KLK5, a member of the KLK‐related peptidase family, and KLK8, a downstream member of KLK5, which were specifically expressed in all SP pt‐iPSC‐CMs. In addition, several genes involved in inflammation regulation, including IL1β, TNFRSF8, and PTGS1, were more highly expressed in SP than NP during Tmab treatment. To confirm the data obtained by RNA‐Seq, we undertook qRT‐PCR of the selected genes, as shown in Table S3. Results of these genes are shown in Figure 5B–F. Moreover, KLK5 and KLK8 were measured in immortalized lymphoblasts from SP and NP patients, but no significant expression was observed in these cells, suggesting that their expression was cardiac‐specific.
FIGURE 5.
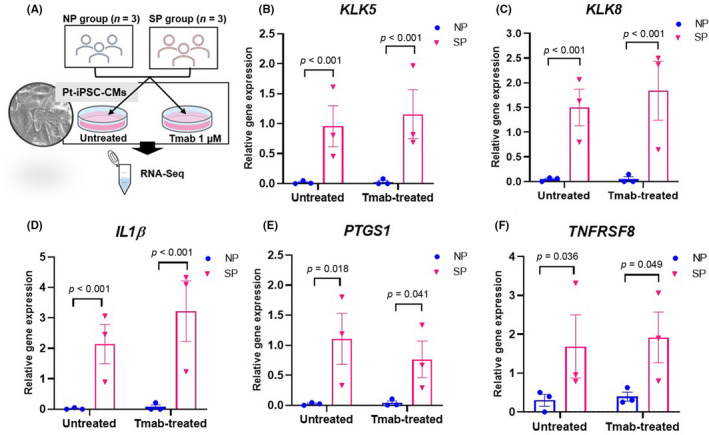
RNA sequencing (RNA‐Seq) of patient‐specific induced pluripotent stem cell‐derived cardiomyocytes (pt‐iPSC‐CMs) and quantitative real‐time RT‐PCR (qRT‐PCR) analysis of genes associated with cardiotoxicity. (A) Schematic illustrating the experimental workflow. RNA‐Seq was undertaken on iPSC‐CMs that were treated for 7 days with or without 1 μM trastuzumab (Tmab). (B–F) Comparisons of various gene expression levels in untreated and Tmab‐treated pt‐iPSC‐CMs using qRT‐PCR analysis (false discovery rate < 0.05 and fold change > 3) showed distinct patterns of gene expression between the severely cardiotoxic patient group (SP; n = 3) versus noncardiotoxic patient group (NP; n = 3) and Tmab‐treated versus untreated. Relative gene expressions of KLK5, KLK8, IL1B, PTGS1, and TNFRSF8 in pt‐iPSC‐CMs are shown. All data are expressed as mean ± SEM
3.7. Differences in the KLK‐PAR2‐MAPK pro‐inflammatory pathway underlie development of cardiotoxicity
Based on mRNA expression characteristics of SP pt‐iPSC‐CMs, we next investigated protein expression in pt‐iPSC‐CMs (Figure 6A). Extracellular secretion of KLK5 and KLK8, as measured by KLK5 and KLK8 protein levels in the culture supernatants of pt‐iPSC‐CMs, was significantly higher in SP than in NP, with the latter having almost undetectable levels (Figure 6B,C). In The Human Protein Atlas, a tissue‐based map of the human proteome, expression levels of KLK5 and KLK8 in the general myocardium are minimal, suggesting that the enhanced expression of these proteins in SP pt‐iPSC‐CMs is characteristic of SP. We next observed that PAR2, a reported substrate for KLK5/KLK8, was expressed in all pt‐iPSC‐CMs lines in western blot analyses (Figure 6D), and its expression levels were significantly higher in SP than NP (Figure 6E). In addition, PAR2 activation levels were assessed in response to treatment with the PAR2 agonist trypsin using intracellular Ca2+ as the indicator. Patients with the SP group responded to trypsin, even at the lower concentration of 1 nM, and showed significantly higher activity than NP with trypsin concentrations of 10 and 100 nM (Figure 6F). Previous reports have indicated that KLK activation of PAR2 triggers pro‐inflammatory signals through downstream signaling of ERK1/2 and JNK. 37 , 38 The phosphorylation levels of ERK1/2 and JNK were significantly higher in SP than NP (Figure 6G–J). Collectively, these findings suggest that enhancement of inflammation‐related pathways could be a biological feature of SP.
FIGURE 6.
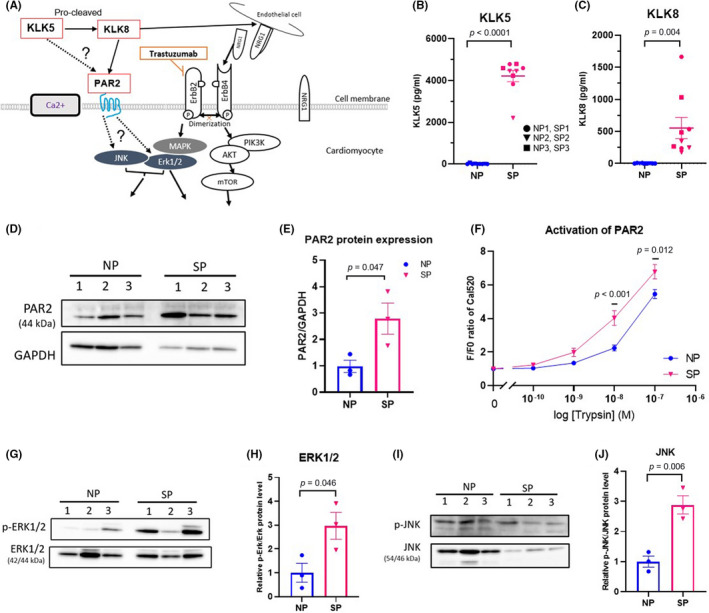
Kallikrein (KLK) drives inflammation status in the severely cardiotoxic patient group (SP) through activated protease‐activated receptor 2 (PAR2). (A) Proposed KLK‐PAR2‐MAPK and ErbB signaling pathways in the patient‐specific induced pluripotent stem cell‐derived cardiomyocyte (pt‐iPSC‐CM) model. Pathways validated in this study are shown as dotted lines. (B, C) KLK5 and KLK8 levels detected in culture supernatant of pt‐iPSC‐CMs, as measured by ELISA. (D) Representative western blot analysis of PAR2 protein expression. (E) Quantification of PAR2 normalized to GAPDH. (F) PAR2 signaling assessed by Ca2+ transients induced by trypsin, a PAR2 agonist. (G, H) Representative western blot and quantification of p‐ERK1/2 and ERK1/2. (I, J) Representative western blot and quantification of p‐JNK and JNK protein expression. Data were obtained using pt‐iPSC‐CMs from six individuals, assays were repeated three to five times for ELISA and Ca2+ transients and twice for western blots. All data are expressed as mean ± SEM. NRG‐1, neuregulin‐1; SP, severely cardiotoxic patient group
3.8. Antipro‐inflammatory effects of KLK‐PAR2 inhibition in pt‐iPSC‐CMs
We hypothesized that the enhanced expression of KLK5 activates PAR2 to transduce its cardiotoxic signal, as evidenced by the increase of IL1β in SP. We then examined whether inhibition of the KLK5‐PAR2‐MAPK pathway could attenuate the expression of inflammation‐related genes. First, we tested the effect of inhibitors on PAR2 activity. The dose and treatment time of the KLK5 antagonist, UA, and the PAR2 antagonists, I‐191 and FSLLRY‐NH2, were determined in preliminary experiments based on the IC50 values of previous reports. 26 , 27 , 29 Compared with the untreated group, PAR2 activity in SP was significantly inhibited by 10 μM of UA, suggesting that KLK5 inhibition can suppress the PAR2 pathway (Figure 7A–C). In addition, treatment with 2 μM I‐191 inhibited the intracellular Ca2+ response to trypsin in both groups, with the inhibition tending to be greater in SP compared with NP (Figure 7D–F). The addition of 100 μM FSLLRY‐NH2 also reduced PAR2 activity, as shown with Ca2+ imaging (Figure S5). Furthermore, downstream IL1β mRNA expression levels, which increased after 1 week of Tmab treatment, were suppressed by I‐191 and FSLLRY‐NH2 (Figure 7G). The contractility of SP pt‐iPSC‐CMs treated with Tmab for 7 days tended to improve with further supplementation of I‐191 or FSLLRY‐NH2 (Figure 7H).
FIGURE 7.
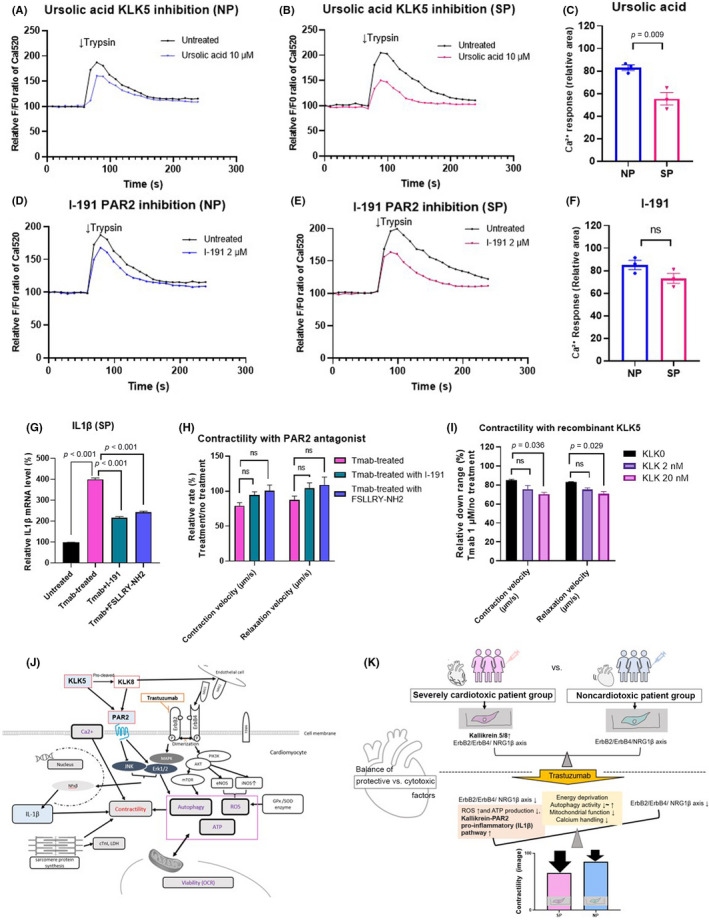
Inhibition of the kallikrein5 (KLK5)–protease‐activated receptor 2 (PAR2) pathway suppresses inflammation and recombinant KLK5‐mediated contractility toxicity in patient‐specific induced pluripotent stem cell‐derived cardiomyocytes (pt‐iPSC‐CM). (A–F) Trypsin (100 nM) induced Ca2+ release in pt‐iPSC‐CMs. (A, B) Representative Ca2+ signaling change with or without the KLK5 antagonist, ursolic acid (UA). (D, E) Representative Ca2+ signaling change with or without the PAR2 antagonist, I‐191. (C, F) Comparison of the relative area of change in Ca2+ response to trypsin under UA and I‐191 treatment. The curved graph area above the F0 baseline was measured using ImageJ. (G) Interleukin‐1β (IL1β) mRNA expression in the severely cardiotoxic patient group (SP) treated with trastuzumab (Tmab) for 7 days ± I‐191 and FSLLRY‐NH2. (H) Contractility in SP treated with Tmab for 7 days ± I‐191 and FSLLRY‐NH2. (I) Contraction and relaxation velocities in noncardiotoxic patient group (NP) treated with Tmab ± recombinant KLK5 (2 nM, 20 nM). (J) Proposed relationship between KLK5/KLK8‐PAR2‐MAPK and ErbB signaling pathways in the pt‐iPSC‐CM model. (K) Schematic showing the multifactorial molecular differences in contractility in SP and NP pt‐iPSC‐CMs and alterations in the balance between protective and cytotoxic effects after Tmab administration. All data are expressed as mean ± SEM. eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; ns, not significant; OCR, oxygen consumption rate; ROS, reactive oxygen species
3.9. Enhancement of cardiotoxicity by KLK5 in NP pt‐iPSC‐CMs
To confirm the involvement of the KLK5‐PAR2 signaling pathway in the development of severe cardiotoxicity, we examined the effects of KLK5 supplementation on NP pt‐iPSC‐CMs. Supplementation of rKLK5 protein in the medium in a dose‐dependent manner resulted in a significant reduction of CV and RV following Tmab treatment (Figure 7I). In addition, supplementation of NP pt‐iPSC‐CM with 20 nM rKLK5 was shown to have toxic effects on intracellular metabolic function, ATP, and ROS production that approximates SP (Figure S6). These observations suggest that enhanced expression of KLK5 in SP and the resultant activation of pro‐inflammatory signaling are involved in the development of Tmab‐induced severe cardiotoxicity (Figure 7J).
4. DISCUSSION
In this report, we selected only patients with severe cardiotoxicity due to Tmab and excluded the clinical risks of cardiotoxicity as much as possible, in order to delineate the difference between NP and SP. As a result, we were able to successfully demonstrate Tmab toxicity in our cell model. There was a significant difference in the extent of toxicity between pt‐iPSC‐CMs from SP and NP, with cardiomyocytes showing altered energy metabolism in response to Tmab. Furthermore, we found enhanced expression of inflammation‐associated genes in SP pt‐iPSC‐CMs. Analysis of protein expression and functional assays confirmed the enhancement of the inflammation‐associated KLK5‐PAR2‐MAPK pathway in SP pt‐iPSC‐CMs, providing evidence supporting the idea that this pathway is an important determinant of Tmab‐induced severe cardiotoxicity.
Inhibition of ErbB2 has been reported to suppress autophagy and evoke mitochondrial dysfunction and accumulation of ROS. 17 , 39 We observed mitochondrial dysfunction by Tmab in a dose‐dependent manner but without significant difference between SP and NP. In contrast, ATP production, ROS levels, and autophagy dysregulation were much more prominent in SP pt‐iPSC‐CMs as compared with NP. We speculated that the shift from suppressed to increased autophagy might be caused, at least in part, by the higher production of ROS, an upstream regulator of autophagy, in SP pt‐iPSC‐CMs. 40 Our data demonstrate the possible contribution of the energy metabolism pathway in the development of cardiotoxicity.
Kallikreins are a family of 15 secreted serine proteases encoded by the largest protease gene cluster in the human genome. Kallikreins are involved in a variety of biological processes and are associated with several diseases, including inflammation and cancer. 41 Gene expressions of KLK5 and KLK8 were higher in SP, with both proteins also detected in the culture supernatant of SP pt‐iPSC‐CMs. One substrate of the KLKs is PAR, from a seven‐transmembrane G protein‐coupled receptor superfamily that includes PAR2, which is activated by KLK5/KLK8. 42 , 43 As subfamilies of the downstream MAPK signaling pathway, ERK1/2, JNK, and p38 are known to have roles in cardiac remodeling such as myocardial fibrosis and inflammation. 42 , 44 , 45 , 46 In the present study, we found enhanced expression of inflammation‐related genes is mediated by the KLK5‐PAR2‐MAPK pathway in SP but not NP pt‐iPSC‐CMs. This led us to speculate that inhibition of this pathway could provide a clue for the therapeutic strategies against Tmab‐induced cardiotoxicity. As shown in Figure 7J, ErbB2 and PAR2 can both activate the MAPK signaling pathway, but they have opposing cardioprotective and pro‐inflammatory functions, respectively. We speculate that the reason for this could be due to an imbalance in the workload of the two signaling pathways: the intracellular ErbB signaling pathway mainly signals through AKT rather than MAPK, whereas the PAR2 signaling pathway branches out to JNK and ERK/MAPK.
Interleukin‐1β, which was highly expressed in SP following Tmab treatment, is one of the major pro‐inflammatory cytokines that can cause cardiac dysfunction. 47 , 48 Interleukin‐1β is known to be a potent inducer of iNOS, which contributes to increased ROS production. As such, inhibition of iNOS is reported as cardioprotective when combined with anti‐HER2 and anthracyclines. 49 Moreover, rivaroxaban, an inhibitor of factor Xa currently used as an anticoagulant in clinical practice, causes decreased expression of PAR2. Rivaroxaban suppresses inflammation‐induced genes, including IL1β, in a mouse model of myocardial infarction and shows cardioprotective effects such as reduction of heart failure and inhibition of fibrosis. 50 In this study, we found that I‐191 and FSLLRY‐NH2, negative allosteric modulators and antagonists for PAR2, suppressed the enhancement of IL1β mRNA expression caused by Tmab treatment in SP pt‐iPSC‐CMs. Therefore, patients with Tmab‐induced cardiotoxicity who respond poorly to the general cardioprotectants could show improvement in cardiac function with inhibitors of the KLK5‐PAR‐MAPK‐IL1β pathway. 51 , 52 Furthermore, cohorts such as SP, where KLK‐mediated inflammatory pathways are likely to be enhanced, could find a common denominator in terms of susceptibility to cardiotoxicity. Recently, the involvement of inflammation, as assessed by increased MMP‐2 and GDF‐15, was reported in the MANTICORE clinical trial by diversified evaluation of the clinical characteristics of Tmab‐induced cardiotoxicity using blood tests and imaging. 53
Our study had some limitations. First, the number of patients was small and thus our findings might not be generalizable. Confirmation of our findings in more samples is required to generalize our findings. Second, we have not identified the genetic factors driving enhanced expression levels of KLK5/8. Thus, the mechanisms by which activation of KLK5/8 is enhanced in patients with severe Tmab‐induced cardiotoxicity still needs to be elucidated. Third, the maturation of pt‐iPSC‐CMs might not be completely uniform, which could influence our results by altering the expression levels of factors, such as receptors. Another limitation is that the myocardial environment might not be fully reproduced in vitro, and new responses could be observed in vivo with the involvement of surrounding stromal cells and cells responsible for immunity. This is particularly important in studies on inflammation and therefore, our observation need to be confirmed.
We determined that pt‐iPSC‐CMs recapitulate the individual differences in contractile and intracellular metabolic dysfunction caused by Tmab and show them to be a useful model for studying mechanisms and treatments in drug‐induced cardiotoxicity. The mechanism of cardiotoxicity is multifactorial, including dysfunction of cellular energy metabolism and impairment of the cardioprotection of the ErbB/NRG1 pathway. Our study shows inflammatory factors could also contribute to the cardiotoxicity phenotype (Figure 7K), where suppression of PAR2 signaling in vitro could control the appearance of severe cardiotoxicity. Thus, our study could provide clues for avoiding interruption of Tmab therapy due to cardiotoxicity and developing a new treatment strategy.
FUNDING INFORMATION
Grant‐in‐Aid for Special Research in Subsidies for ordinary expenses of private schools.
DISCLOSURE
The authors declare no competing interests.
Supporting information
Figure S1‐S6
Data S1
Video S1
Table S1‐S7
ACKNOWLEDGMENTS
This work was supported in part by a Grant‐in‐Aid for Special Research in Subsidies for ordinary expenses of private schools. We thank the Laboratory of Molecular and Biochemical Research, Juntendo University Graduate School of Medicine, for technical assistance. The authors sincerely appreciate Clear Science Pty Ltd for language editing.
Sasaki R, Kurebayashi N, Eguchi H, et al. Involvement of kallikrein‐PAR2‐proinflammatory pathway in severe trastuzumab‐induced cardiotoxicity. Cancer Sci. 2022;113:3449‐3462. doi: 10.1111/cas.15508
REFERENCES
- 1. Goldhirsch A, Gelber RD, Piccart‐Gebhart MJ, et al. 2 years versus 1 year of adjuvant trastuzumab for HER2‐positive breast cancer (HERA): an open‐label, randomised controlled trial. Lancet. 2013;382:1021‐1028. [DOI] [PubMed] [Google Scholar]
- 2. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2‐positive breast cancer: planned joint analysis of overall survival from NSABP B‐31 and NCCTG N9831. J Clin Oncol. 2014;32:3744‐3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harbeck N, Ewer MS, De Laurentiis M, Suter TM, Ewer SM. Cardiovascular complications of conventional and targeted adjuvant breast cancer therapy. Ann Oncol. 2011;22:1250‐1258. [DOI] [PubMed] [Google Scholar]
- 4. Eiger D, Franzoi MA, Pondé N, et al. Cardiotoxicity of trastuzumab given for 12 months compared to shorter treatment periods: a systematic review and meta‐analysis of six clinical trials. ESMO Open. 2020;5:e000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nemeth BT, Varga ZV, Wu WJ, Pacher P. Trastuzumab cardiotoxicity: from clinical trials to experimental studies. Br J Pharmacol. 2017;174:3727‐3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ewer MS, Lippman SM. Type II chemotherapy‐related cardiac dysfunction: time to recognize a new entity. J Clin Oncol. 2005;23:2900‐2902. [DOI] [PubMed] [Google Scholar]
- 7. Pivot X, Romieu G, Debled M, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2‐positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol. 2013;14:741‐748. [DOI] [PubMed] [Google Scholar]
- 8. Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257‐270. [DOI] [PubMed] [Google Scholar]
- 9. Zhao YY, Sawyer DR, Baliga RR, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. 1998;273:10261‐10269. [DOI] [PubMed] [Google Scholar]
- 10. Pentassuglia L, Sawyer DB. The role of neuregulin‐1beta/ErbB signaling in the heart. Exp Cell Res. 2009;315:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crone SA, Zhao YY, Fan L, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459‐465. [DOI] [PubMed] [Google Scholar]
- 12. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39‐51. [DOI] [PubMed] [Google Scholar]
- 13. Leemasawat K, Phrommintikul A, Chattipakorn SC, Chattipakorn N. Mechanisms and potential interventions associated with the cardiotoxicity of ErbB2‐targeted drugs: insights from in vitro, in vivo, and clinical studies in breast cancer patients. Cell Mol Life Sci. 2019;77:1571‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fedele C, Riccio G, Malara AE, D'Alessio G, De Lorenzo C. Mechanisms of cardiotoxicity associated with ErbB2 inhibitors. Breast Cancer Res Treat. 2012;134:595‐602. [DOI] [PubMed] [Google Scholar]
- 16. Grazette LP, Boecker W, Matsui T, et al. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin‐induced cardiomyopathy. J Am Coll Cardiol. 2004;44:2231‐2238. [DOI] [PubMed] [Google Scholar]
- 17. Kitani T, Ong SG, Lam CK, et al. Human induced pluripotent stem cell model of trastuzumab‐induced cardiac dysfunction in breast cancer patients. Circulation. 2019;139:2451‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jawa Z, Perez RM, Garlie L, et al. Risk factors of trastuzumab‐induced cardiotoxicity in breast cancer: a meta‐analysis. Medicine (Baltimore). 2016;95:e5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Battisti NML, Andres MS, Lee KA, et al. Incidence of cardiotoxicity and validation of the heart failure association‐international cardio‐oncology society risk stratification tool in patients treated with trastuzumab for HER2‐positive early breast cancer. Breast Cancer Res Treat. 2021;188:149‐163. [DOI] [PubMed] [Google Scholar]
- 20. Udagawa C, Zembutsu H. Pharmacogenetics for severe adverse drug reactions induced by molecular‐targeted therapy. Cancer Sci. 2020;111:3445‐3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamaguchi A, Ishikawa KI, Inoshita T, et al. Identifying therapeutic agents for amelioration of mitochondrial clearance disorder in neurons of familial Parkinson disease. Stem Cell Rep. 2020;14:1060‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burridge PW, Matsa E, Shukla P, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tohyama S, Fujita J, Hishiki T, et al. Glutamine oxidation is indispensable for survival of human pluripotent stem cells. Cell Metab. 2016;23:663‐674. [DOI] [PubMed] [Google Scholar]
- 24. Miki K, Endo K, Takahashi S, et al. Efficient detection and purification of cell populations using synthetic MicroRNA switches. Cell Stem Cell. 2015;16:699‐711. [DOI] [PubMed] [Google Scholar]
- 25. Kanda Y, Yamazaki D, Kurokawa J, Inutsuka T, Sekino Y. Points to consider for a validation study of iPS cell‐derived cardiomyocytes using a multi‐electrode array system. J Pharmacol Toxicol Methods. 2016;81:196‐200. [DOI] [PubMed] [Google Scholar]
- 26. Matsubara Y, Matsumoto T, Koseki J, Kaneko A, Aiba S, Yamasaki K. Inhibition of human kallikrein 5 protease by triterpenoids from natural sources. Molecules. 2017;22:1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang Y, Yau MK, Lim J, et al. A potent antagonist of protease‐activated receptor 2 that inhibits multiple signaling functions in human cancer cells. J Pharmacol Exp Ther. 2018;364:246‐257. [DOI] [PubMed] [Google Scholar]
- 28. McLarty JL, Meléndez GC, Brower GL, Janicki JS, Levick SP. Tryptase/protease‐activated receptor 2 interactions induce selective mitogen‐activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension. 2011;58:264‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeng X, Zhang S, Xu L, Yang H, He S. Activation of protease‐activated receptor 2‐mediated signaling by mast cell tryptase modulates cytokine production in primary cultured astrocytes. Mediators Inflamm. 2013;2013:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eisner DA. Pseudoreplication in physiology: more means less. J Gen Physiol. 2021;153:e202012826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sikkel MB, Francis DP, Howard J, et al. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113:1743‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zamorano JL, Lancellotti P, Rodriguez Muñoz D, et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for practice guidelines: the task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. 2016;37:2768‐2801. [DOI] [PubMed] [Google Scholar]
- 33. Ezaz G, Long JB, Gross CP, Chen J. Risk prediction model for heart failure and cardiomyopathy after adjuvant trastuzumab therapy for breast cancer. J Am Heart Assoc. 2014;3:e000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Curigliano G, Lenihan D, Fradley M, et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann Oncol. 2020;31:171‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takeda M, Miyagawa S, Fukushima S, et al. Development of in vitro drug‐induced cardiotoxicity assay by using three‐dimensional cardiac tissues derived from human induced pluripotent stem cells. Tissue Eng Part C Methods. 2018;24:56‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eldridge S, Guo L, Mussio J, Furniss M, Hamre J 3rd, Davis M. Examining the protective role of ErbB2 modulation in human‐induced pluripotent stem cell‐derived cardiomyocytes. Toxicol Sci. 2014;141:547‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zuo P, Zhu Y, Li Y, et al. Protease‐activated receptor 2 deficiency in hematopoietic lineage protects against myocardial infarction through attenuated inflammatory response and fibrosis. Biochem Biophys Res Commun. 2020;526:253‐260. [DOI] [PubMed] [Google Scholar]
- 38. Ghorpade DS, Ozcan L, Zheng Z, et al. Hepatocyte‐secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature. 2018;555:673‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mohan N, Shen Y, Endo Y, ElZarrad MK, Wu WJ. Trastuzumab, but not pertuzumab, dysregulates HER2 signaling to mediate inhibition of autophagy and increase in reactive oxygen species production in human cardiomyocytes. Mol Cancer Ther. 2016;15:1321‐1331. [DOI] [PubMed] [Google Scholar]
- 40. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Diamandis EP, Yousef GM. Human tissue kallikrein gene family: a rich source of novel disease biomarkers. Expert Rev Mol Diagn. 2001;1:182‐190. [DOI] [PubMed] [Google Scholar]
- 42. Cao B, Yu Q, Zhao W, et al. Kallikrein‐related peptidase 8 is expressed in myocardium and induces cardiac hypertrophy. Sci Rep. 2016;6:25038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prassas I, Eissa A, Poda G, Diamandis EP. Unleashing the therapeutic potential of human kallikrein‐related serine proteases. Nat Rev Drug Discovery. 2015;14:183‐202. [DOI] [PubMed] [Google Scholar]
- 44. Du JK, Yu Q, Liu YJ, et al. A novel role of kallikrein‐related peptidase 8 in the pathogenesis of diabetic cardiac fibrosis. Theranostics. 2021;11:4207‐4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang M, Du J, Wang Y, et al. Tissue kallikrein‐related peptidase8 protects rat heart against acute ischemia reperfusion injury. Int J Biol Macromol. 2019;140:1126‐1133. [DOI] [PubMed] [Google Scholar]
- 46. Wang M, Ma Y, Zhang T, Gao L, Zhang S, Chen Q. Proteinase‐activated receptor 2 deficiency is a protective factor against cardiomyocyte apoptosis during myocardial ischemia/reperfusion injury. Mol Med Rep. 2019;20:3764‐3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oyama J, Shimokawa H, Momii H, et al. Role of nitric oxide and peroxynitrite in the cytokine‐induced sustained myocardial dysfunction in dogs in vivo. J Clin Invest. 1998;101:2207‐2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsujino M, Hirata Y, Imai T, et al. Induction of nitric oxide synthase gene by interleukin‐1 beta in cultured rat cardiocytes. Circulation. 1994;90:375‐383. [DOI] [PubMed] [Google Scholar]
- 49. Hsu WT, Huang CY, Yen CYT, Cheng AL, Hsieh PCH. The HER2 inhibitor lapatinib potentiates doxorubicin‐induced cardiotoxicity through iNOS signaling. Theranostics. 2018;8:3176‐3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu J, Nishida M, Inui H, et al. Rivaroxaban suppresses the progression of ischemic cardiomyopathy in a murine model of diet‐induced myocardial infarction. J Atheroscler Thromb. 2019;26:915‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Friebel J, Weithauser A, Witkowski M, et al. Protease‐activated receptor 2 deficiency mediates cardiac fibrosis and diastolic dysfunction. Eur Heart J. 2019;40:3318‐3332. [DOI] [PubMed] [Google Scholar]
- 52. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119‐1131. [DOI] [PubMed] [Google Scholar]
- 53. Kirkham AA, Pituskin E, Thompson RB, et al. Cardiac and cardiometabolic phenotyping of trastuzumab‐mediated cardiotoxicity: a secondary analysis of the MANTICORE trial. Eur Heart J Cardiovasc Pharmacother. 2021;8:130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1‐S6
Data S1
Video S1
Table S1‐S7