

SUMMARY
Although mutations in mitochondrial-associated genes are linked to inflammation and susceptibility to infection, their mechanistic contributions to immune outcomes remain ill-defined. We discovered the disease-associated gain-of-function allele Lrrk2G2019S (leucine-rich repeat kinase 2) perturbs mitochondrial homeostasis and reprograms cell death pathways in macrophages. When the inflammasome is activated in Lrrk2G2019S macrophages, elevated mitochondrial ROS (mtROS) directs association of the pore-forming protein gasdermin D (GSDMD) to mitochondrial membranes. Mitochondrial GSDMD pore formation then releases mtROS, promoting a switch to RIPK1/RIPK3/MLKL-dependent necroptosis. Consistent with enhanced necroptosis, infection of Lrrk2G2019S mice with Mycobacterium tuberculosis elicits hyperinflammation and severe immunopathology. Our findings suggest a pivotal role for GSDMD as an executer of multiple cell death pathways and demonstrate that mitochondrial dysfunction can direct immune outcomes via cell death modality switching. This work provides insights into how LRRK2 mutations manifest or exacerbate human diseases and identifies GSDMD-dependent necroptosis as a potential target to limit Lrrk2G2019S-mediated immunopathology.
In Brief:
Disease-associated mutations in the kinase Lrrk2 promote association of gasdermin D with mitochondrial membranes, which induces release of reactive oxygen species, a switch toward necroptotic cell death, and hyperinflammatory pathology.
INTRODUCTION
Despite a growing appreciation for mitochondria as critical regulators of antimicrobial defenses and cell death (Riley and Tait, 2019; West and Shadel, 2017), we still know very little about how mitochondrial perturbations drive protective or pathogenic immune responses in the context of actual human disease. Mutations in mitochondrial-related genes involved in processes ranging from mitophagy (PARK2, PARL, PINK1) to fission and fusion of the mitochondrial network (OPA, MFN2) have been repeatedly linked to chronic inflammation and susceptibility to infection with pathogens like Mycobacterium leprae and Mycobacterium tuberculosis (Mtb), which cause leprosy and tuberculosis, respectively (Patrick and Watson, 2021). One notable mitochondrial-associated gene with poorly understood connections to inflammation and immunity is leucine-rich repeat kinase 2 (LRRK2). LRRK2 is a multiple domain-containing protein that functions both as a GTPase and a kinase (Anand and Braithwaite, 2009; Bae and Lee, 2015). Because mutations in LRRK2 constitute the greatest known genetic component of familial Parkinson’s disease (PD) (Goldwurm et al., 2005; Gosal et al., 2005; Khan et al., 2005), much of what is known about LRRK2 comes from studies of central nervous system (CNS) cells, where it functions in a number of cellular pathways (Cookson, 2012; Wallings et al., 2015). In non-CNS cells, LRRK2 has been linked to intracellular membrane trafficking through RAB phosphorylation (Steger et al., 2016), endolysosomal dynamics (Hartlova et al., 2018; Herbst et al., 2020), and mitochondrial homeostasis (Eberhardt et al., 2020; Hsieh et al., 2016; Ludtmann et al., 2019).
We do not fully understand why mutations in LRRK2 upset the immune milieu to trigger inflammatory disorders like Crohn’s disease (Derkinderen and Neunlist, 2018; Hui et al., 2018) or confer susceptibility to mycobacterial infection (Fava et al., 2016; Marcinek et al., 2013; Zhang et al., 2009). Our recent study of LRRK2 knockout macrophages suggested that LRRK2 controls innate immunity via maintenance of mitochondrial homeostasis (Weindel et al., 2020). Motivated by these findings, we looked to the gain-of-function LRRK2G2019S allele as a model to elucidate how mitochondrial mutations impact innate immune and infection outcomes. The LRRK2G2019S mutation is surprisingly prevalent in humans. In Ashkenazi Jews and North Africans, the allele accounts for a significant proportion of PD (Khan et al., 2005; Ozelius et al., 2006). LRRK2G2019S is also associated with overall increased risk of certain cancers (Agalliu et al., 2015) and there is growing interest in how this allele influences infection and inflammation (Herbst and Gutierrez, 2019; Shutinoski et al., 2019). Here, in studies of mice and flies, we describe an ancient connection between LRRK2’s role in mitochondrial homeostasis and immunity and report that mitochondrial ROS (mtROS) renders Lrrk2G2019S mitochondria susceptible to gasdermin D (GSDMD) pore formation that leads to necroptotic cell death.
RESULTS
Mitochondria in Lrrk2G2019S cells are fragmented and prone to depolarization in response to cellular stress.
Loss of LRRK2 triggers mitochondrial depolarization and network fragmentation (Ho et al., 2018; Ho et al., 2019; Weindel et al., 2020). To determine how the human disease-associated constitutively active LRRK2G2019S allele (Athanasopoulos et al., 2018; Berger et al., 2010; Lobbestael et al., 2012; Luzon-Toro et al., 2007) alters mitochondrial dynamics, we began with a qualitative assessment of the mitochondrial network in embryonic fibroblasts from wild-type (WT) and Lrrk2G2019S mice. Immunofluorescence (IF) confocal microscopy revealed increased mitochondrial fragmentation in Lrrk2G2019S cells compared to WT (Fig. 1A). Consistent with Lrrk2G2019S promoting mitochondrial fission by interacting with and phosphorylating the mitochondrial fission protein DRP1 (Ho et al., 2018; Perez Carrion et al., 2018; Su and Qi, 2013; Wang et al., 2012), p616 DRP1 levels were higher in Lrrk2G2019S bone marrow derived macrophages (BMDMs) by immunoblot (Fig. 1B).
Figure 1. Lrrk2G2019S promotes mitochondrial dysfunction marked by network fragmentation and susceptibility to depolarization upon cellular stress.

A. Mitochondria (anti-TOM20; green) in WT and Lrrk2G2019S MEFs. Nuclei (DAPI; blue). B. Immunoblot and quantification (n=2) of pDRP1 (pSer616 = activation; pSer637 = inhibition) in WT and Lrrk2G2019S BMDMs relative to total DRP1 with ACTIN loading control. C. FSC-A (FACS plots and histograms) of isolated mitochondria from WT and Lrrk2G2019S BMDMs relative to bead standards +/− 10 μM Mdivi-1 for 16h. D. Mitochondrial size distribution in WT and Lrrk2G2019S BMDMs based on size standard polystyrene beads +/− 10 μM Mdivi-1. E. Mitochondrial membrane potential measured by flow cytometry of TMRE (585/15) in WT and Lrrk2G2019S BMDMs. Histograms display (585/15) x-axis. Quantitation on right. F. Mitochondrial membrane potential measured by flow cytometry of JC-1. JC-1 aggregation = normal mitochondrial membrane potential (610/20); JC-1 monomers = low membrane potential (530/30) x-axis. Histograms display (610/20) y-axis. Quantification on right. G. JC-1 in WT and Lrrk2G2019S BMDMs treated with rotenone (2.5 μM for 3h) followed by ATP (5 mM for 5 and 30 min). Right histograms display (610/20) x-axis. H. Drosophila melanogaster lipid droplets (Nile red; red), mitochondria (ATP5A; green), and nuclei (DAPI; blue) in WT (CT(w1118)), hLRRK2, hLRRK2-G2019S, and hLRRK2-G2019S-K1906M-expressing flies. Expression driven by a fat body-specific promoter (CGGal4>). Statistical analysis: Data are mean of three or more biological replicates (unless otherwise noted). Error bars depict SEM. Statistical significance determined using a two-tailed Student’s T test (B, E, F), or one-way ANOVA with Sidak’s post-test (C): * = p<0.05, ** = p<0.01, *** = p<0.001.
To quantitatively measure mitochondrial fragmentation in WT and Lrrk2G2019S BMDMs, we developed a strategy to measure mitochondrial size differences down to the sub-micron level via flow cytometry. Polystyrene beads of 1, 2, and 4 μm were used to create a gating strategy whereby the size of MitoTracker green-stained mitochondria could be compared to the size of bead standards using the forward (FSC) optical detector (MacDonald et al., 2019; Schneider et al., 2019) (Fig. 1C and S1A–B). Consistent with DRP1 hyperactivation, we measured more “small” mitochondria (<1 μm) and fewer “large” mitochondria (>4 μm) in Lrrk2G2019S BMDMs compared to WT (Fig. 1D). Importantly, treatment with low concentrations of the DRP1 inhibitor Mdivi-1 (10μM) significantly reduced mitochondrial fragmentation in Lrrk2G2019S macrophages (Fig. 1C–D), without impacting total mean fluorescence intensity (Fig. S1C), supporting a role for pDRP1 in promoting mitochondrial hyper-fission in these cells.
Because mitochondrial fragmentation can alter membrane potential (Liesa and Shirihai, 2013), we measured mitochondrial membrane depolarization in Lrrk2G2019S macrophages using two cell-permeant dyes: Tetramethylrhodamine (TMRE), which is sequestered in healthy, negatively charged mitochondria, and JC-1, which exists as red aggregates in healthy mitochondria and green cytosolic monomers that increase as mitochondrial membrane potential is lost. TMRE fluorescence intensity and JC-1 aggregates were both decreased in Lrrk2G2019S BMDMs, indicating mitochondrial membrane depolarization (Fig. 1E–F). JC-1 aggregates were further reduced in Lrrk2G2019S macrophages following treatment with the complex I inhibitor, rotenone + ATP (Fig. 1G).
The mouse genotype used for the majority of our studies is B6.Cg-Tg(Lrrk2*G2019S)2Yue/J, which overexpresses Lrrk2G2019S on a BAC (Li et al., 2007). As an important control, we measured no differences in TMRE in BMDMs isolated from B6.Cg-Tg(Lrrk2)6Yue/J mice, which overexpress WT LRRK2 on a BAC, and matched B6 controls (Fig. S1D). We also measured mitochondrial membrane potential in BMDMs isolated from B6.Cg-Lrrk2tm1.1Hlme/J mice (a.k.a. Lrrk2G>S/+), in which the G2019S mutation has been engineered into the LRRK2 genomic locus. We detected a subtle but statistically significant decrease in TMRE in Lrrk2G>S/+ BMDMs (Fig. S1E–F). These data argue that the G2019S mutation and not LRRK2 protein abundance is the main driver of mitochondrial depolarization in Lrrk2G2019S macrophages.
Several labs have successfully used Lrrk2G2019S-expressing flies as a model to study LRRK2 function (Cording et al., 2017; Liu et al., 2008; Ng et al., 2009). To test whether expression of hLRRK2 influences mitochondrial homeostasis in an evolutionarily conserved fashion, we generated transgenic flies that express hLRRK2 alleles in the fat body, a tissue that controls energy metabolism and plays a major role in innate immunity in insects (Anand et al., 2012; Bosch et al., 2020). Visualizing lipid droplets (NileRed) and mitochondria (anti-ATP5A) in flies expressing hLRRK2, hLRRK2-G2019S, and hLRRK2-G2019S-K1906M (a kinase dead allele (Lin et al., 2010)), we discovered that mitochondrial network density was decreased by the hLRRK2 and hLRRK2-G2019S alleles, but unaltered in the fat body of flies expressing the kinase dead allele (Fig. 1H; green). Lipid droplets, which store triacylglycerols and serve as a fuel source for mitochondrial OXPHOS, were larger in hLRRK2 and hLRRK2-G2019S flies compared to those expressing hLRRK2-G2019S-K1906M or controls (Fig. 1H; red). Flies expressing hLRRK2 alleles via a ubiquitous promoter displayed similar phenotypes (Fig. S3I, mock). These findings suggest that Lrrk2G2019S alters mitochondrial homeostasis and energetics in macrophages ex vivo and at an organismal level, in evolutionarily distant animals.
Lrrk2G2019S macrophages do not exhibit altered transcriptional responses following innate stimuli.
Based on our previous work (Weindel et al., 2020), we hypothesized that fragmented, depolarized mitochondria could impact basal levels of inflammatory mediators in Lrrk2G2019S macrophages (Fig. 2A). However, we measured no differences in expression of Ifnb1 transcripts, IFN-β protein, or interferon stimulated gene (ISG) transcripts in WT vs. Lrrk2G2019S BMDMs at rest (Fig. 2B–C). Protein levels of key IFN signaling molecules were also comparable between the two genotypes (Fig. S2A–B), as were levels of Ifnb1 and ISG expression when cells were treated with a panel of innate agonists (Fig. S2C–D) or infected with Mycobacterium tuberculosis (Mtb), an intracellular bacterial pathogen that activates IRF3 via cGAS-dependent cytosolic DNA sensing (4h) (Fig. S2E) (Collins et al., 2015; Wassermann et al., 2015; Watson et al., 2015). Other pro-inflammatory cytokines were similarly expressed in WT and Lrrk2G2019S macrophages at 4h post-Mtb infection (Fig. S2F). We concluded that Lrrk2G2019S does not promote chronic engagement of cytosolic nucleic acid sensors, nor does it significantly alter the ability of BMDMs to activate inflammatory gene expression following innate immune stimuli.
Figure 2. Lrrk2G2019S promotes cell death during intracellular bacterial infection and inflammasome activation.
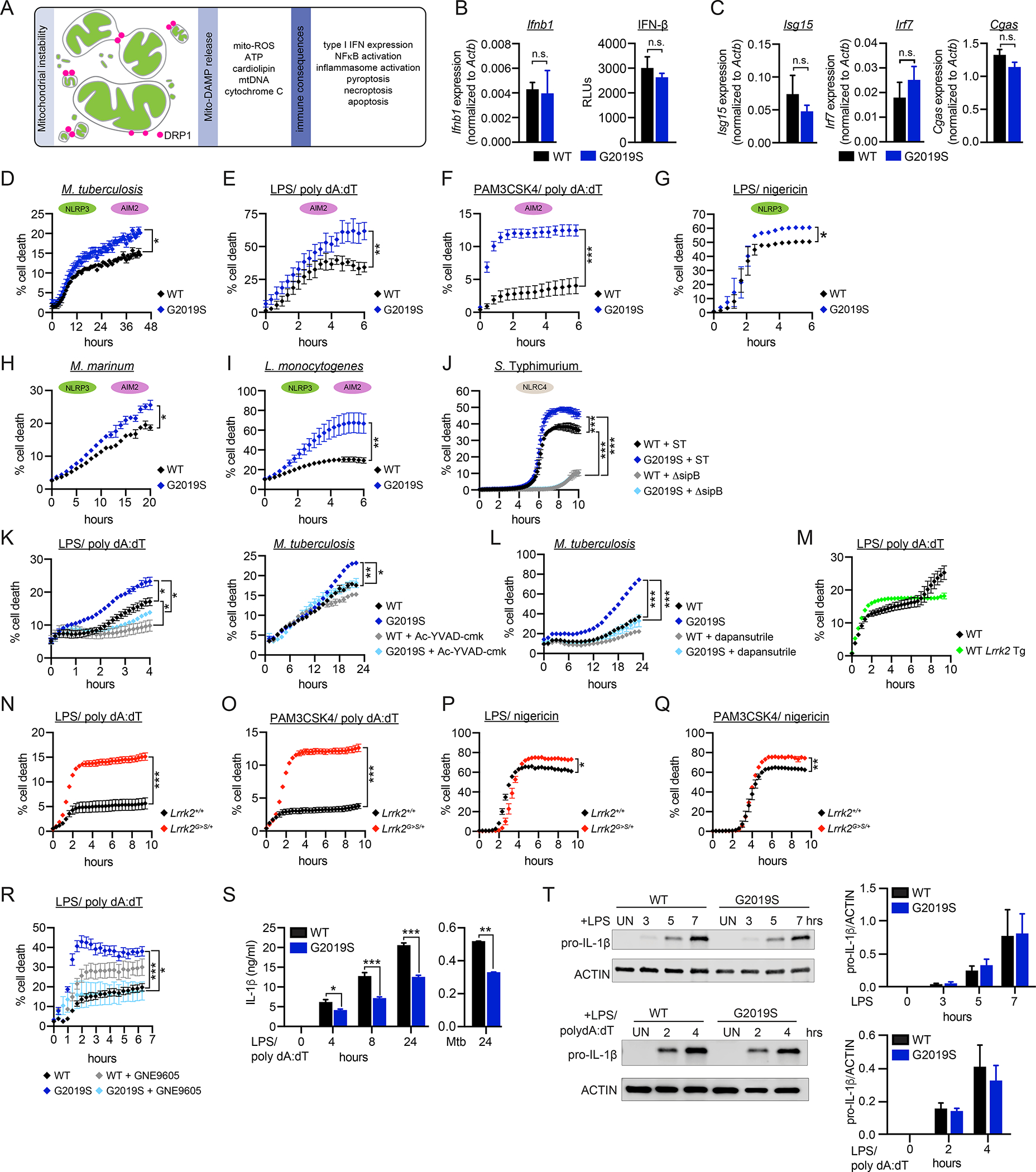
A. Innate immune consequences of mitochondrial network instability. B. Basal Ifnb1 transcript (left; measured by RT-qPCR) and protein levels (right; relative light units (RLUs) measured by ISRE reporter cells) in WT and Lrrk2G2019S BMDMs. C. Basal ISG transcript levels in WT and Lrrk2G2019S BMDMs measured by RT-qPCR. D. % cell death in WT and Lrrk2G2019S BMDMs over a time course of infection with Mtb (MOI 5). Cell death measured by propidium iodide incorporation (% = PI+ cells/total cells*100). E. % cell death in WT and Lrrk2G2019S BMDMs over a time course of AIM2 stimulation. All AIM2 stimulations were performed in BMDMs by 10 ng/mL LPS priming for 3h followed by 1 μg/mL poly dA:dT. Time courses begin with the introduction of poly dA:dT. F. As in E but with Pam3CSK4 priming (10 ng/ml). G. % cell death in WT and Lrrk2G2019S BMDMs over a time-course of NLRP3 stimulation (10 ng/ml LPS priming for 3h followed by 25 μM nigericin). H. As in D, but with Mycobacterium marinum (MOI 5). I. As in D, but with Listeria monocytogenes (MOI 2). J. As in D but with Salmonella enterica (serovar Typhimurium); WT SL1334 and ΔsipB (MOI 0.5). K. (left) AIM2 stimulation as in E or Mtb infection as in D (right) but with caspase-1/11 inhibitor Ac-YVAD-cmk (100 μM). L. As in D but with NLRP3 inhibitor dapansutrile added upon infection (20 μM). M. AIM2 stimulation as in E but with WT and WT Lrrk2 Tg BMDMs. N. AIM2 stimulation as in E but with Lrrk2+/+ and Lrrk2G>S/+ (LRRK2 G2019S KI) BMDMs. O. As in N but with 10 ng/ml Pam3CSK4 priming. P. As in N but with NLRP3 stimulation with LPS/nigericin. Q. As in N but with Pam3CSK4/nigericin. R. As in E but with LRRK2 inhibitor GNE9605 (1 μM). S. Extracellular IL-1β protein levels measured by ELISA over a time course of AIM2 stimulation (as in E) or at 24h after infection with Mtb (MOI 10) in WT and Lrrk2G2019S BMDMs. T. Protein levels of pro-IL-1β after LPS treatment (top) and LPS/poly dA:dT treatment (bottom) in WT and Lrrk2G2019S BMDMs. Quantification on right. n=2. Statistical analysis: n=3 or more unless otherwise noted. Statistical significance determined via two-tailed Student’s T test (B, C, T), a one-way ANOVA with Sidak’s post-test (S, T), or two-way ANOVA with Tukey’s post-test (D-R).
Lrrk2G2019S macrophages are prone to caspase-1-mediated cell death in response to intracellular bacterial infection and inflammasome activation.
Mitochondrial instability is also a well-known trigger of several regulated cell death pathways (Fig. 2A) (Tait and Green, 2013). To determine if Lrrk2G2019S BMDMs are prone to regulated cell death, we infected WT and Lrrk2G2019S BMDMs with Mtb (MOI = 5) and measured propidium iodide (PI) incorporation over time. Over a 48h Mtb infection, a higher percentage of Lrrk2G2019S BMDMs were PI+ compared to WT controls (Fig. 2D). This cell death was MOI-dependent (Fig. S2H) and concomitant with uptake of Mtb bacilli (Fig. S2I–J). Since we did not observe significant differences in Mtb CFUs in WT and Lrrk2G2019S BMDMs (Fig. S2G), we do not believe enhanced cell death is due to differences in bacterial replication.
Several cell death modalities are simultaneously engaged during Mtb infection of macrophages, including apoptosis, pyroptosis (via the NLRP3 (Dorhoi et al., 2012) and to a lesser extent AIM2 (Saiga et al., 2012; Wassermann et al., 2015) inflammasomes), and necroptosis (Roca and Ramakrishnan, 2013; Roca et al., 2019). To pinpoint the type of cell death that Lrrk2G2019S BMDMs are prone to, we treated cells with agonists to stimulate specific cell death modalities. Apoptosis triggers like etoposide and ABT737 did not elicit more cell death in Lrrk2G2019S cells; if anything, fewer Lrrk2G2019S cells were PI+ after these treatments (Fig. S2K–L). However, cell death was notably enhanced in Lrrk2G2019S BMDMs post-stimulation of the AIM2 inflammasome via either LPS or Pam3CSK4 priming (3h), followed by transfection with the synthetic dsDNA sequence poly dA:dT (Fig. 2E–F and S2M). Direct stimulation of the NLRP3 inflammasome recapitulated this phenotype (Fig. 2G and S2N). In response to nigericin, maximal cell death occurred rapidly, creating a narrow window in which to capture Lrrk2G2019S-dependent cell death enhancement. To stimulate the NLRP3 inflammasome in a more biologically relevant fashion, we infected WT and Lrrk2G2019S BMDMs with the intracellular bacterial pathogens Mycobacterium marinum (MOI 5) and Listeria monocytogenes (MOI 2) and again observed higher % cell death in Lrrk2G2019S macrophages (Fig. 2H–I). Engagement of the NLRC4 inflammasome during Salmonella enterica serovar Typhimurium infection also caused significantly higher cell death in Lrrk2G2019S BMDMs in a SPI-1-dependent fashion (Fig. 2J). Cell death in Lrrk2G2019S BMDMs triggered by LPS/polydA:dT or Mtb infection was caspase-dependent (Fig. 2K) and, in the case of Mtb infection, could be rescued by treating cells with the NLRP3 inhibitor dapansutrile (Fig. 2L). We measured no significant increase in inflammasome-mediated cell death in BMDMs overexpressing WT LRRK2 (Fig. 2M and S2O), but Lrrk2G>S/+ knock-in BMDMs were exquisitely sensitive to multiple cell death triggers (Fig. 2N–Q and S2P). Importantly, inhibition of LRRK2 kinase activity with the highly potent and selective inhibitor GNE-9605 rescued cell death in the Lrrk2G2019S Tg BMDMs (Fig. 2R). These data argue that activation of the AIM2, NLRP3, or NLRC4 inflammasomes is necessary and sufficient to elicit enhanced cell death of Lrrk2G2019S BMDMs and strongly implicate Lrrk2G2019S kinase activity in this cell death.
As another canonical readout of AIM2 inflammasome activation, we measured IL-1β release in WT and Lrrk2G2019S BMDMs. Despite exhibiting enhanced caspase-1-dependent cell death following inflammasome activation (Fig. 2K), Lrrk2G2019S BMDMs release markedly less IL-1β post-AIM2 stimulation and at 24h post-Mtb infection (Fig. 2S). To begin to explain the reduced IL-1β release in Lrrk2G2019S BMDMs, we tested if upregulation of inflammasome components was reduced. We measured no significant differences in expression of pyroptotic cell death mediators (Fig. S2Q) or inflammasome associated cytokines (Il18 and Il1b (Fig. S2R)) in resting WT and Lrrk2G2019S BMDMs. Pro-IL-1β protein levels and ASC speck formation were also comparable (Fig. 2T and S2S). Therefore, while inflammasome activation triggers enhanced cell death in Lrrk2G2019S BMDMs, this death is not concomitant with enhanced release of pyroptotic inflammatory mediators, suggesting that Lrrk2G2019S BMDMs die via an alternative cell death modality.
Inflammasome activation triggers additional mitochondrial stresses in Lrrk2G2019S macrophages.
We hypothesized that underlying mitochondrial dysfunction in Lrrk2G2019S BMDMs (Fig. 1) reprograms cell death signaling downstream of inflammasome activation to cause non-pyroptotic cell death. To define how mitochondrial homeostasis is altered by inflammasome activation in Lrrk2G2019S BMDMs, we measured mitochondrial membrane potential at 2h post-LPS/poly dA:dT treatment by flow cytometry. Resting Lrrk2G2019S and Lrrk2G>S/+ BMDMs showed reduced TMRE fluorescence intensity that was further reduced by LPS/poly dA:dT treatment (Fig. 3A and S3A), indicative of AIM2-dependent mitochondrial depolarization. AIM2-mediated depolarization was accompanied by accumulation of mtDNA in the cytosol of Lrrk2G2019S Tg macrophages (Fig. 3B–C). Consistent with enhanced cytosolic mtDNA release, levels of type I IFN transcripts were higher in Lrrk2G2019S macrophages at 4–5h post-AIM2 activation (Fig. 3D and S3B) and at 24h post-Mtb infection (Fig. 3E). Another mtDAMP (damage associated molecular pattern) with connections to both inflammasome activation and cell death is mitochondrial reactive oxygen species (mtROS). To define the repertoire of free radicals in Lrrk2G2019S during AIM2 inflammasome activation, we used the fluorogenic probe cellROX and the mitochondrial-targeted superoxide indicator mitoSOX. Signal from both probes was significantly higher in Lrrk2G2019S macrophages at 2h post-LPS/poly dA:dT treatment (Fig. 3F–G). Because genes in the oxidative stress-responsive NRF2 regulon were not differentially expressed in Lrrk2G2019S BMDMs (Li and Kong, 2009) (Fig. S3C), we hypothesized that mitochondrial defects were responsible for increased mtROS production in Lrrk2G2019S BMDMs.
Figure 3. Inflammasome activation triggers additional mitochondrial stresses that alter metabolism and promote cell death in Lrrk2G2019S macrophages.
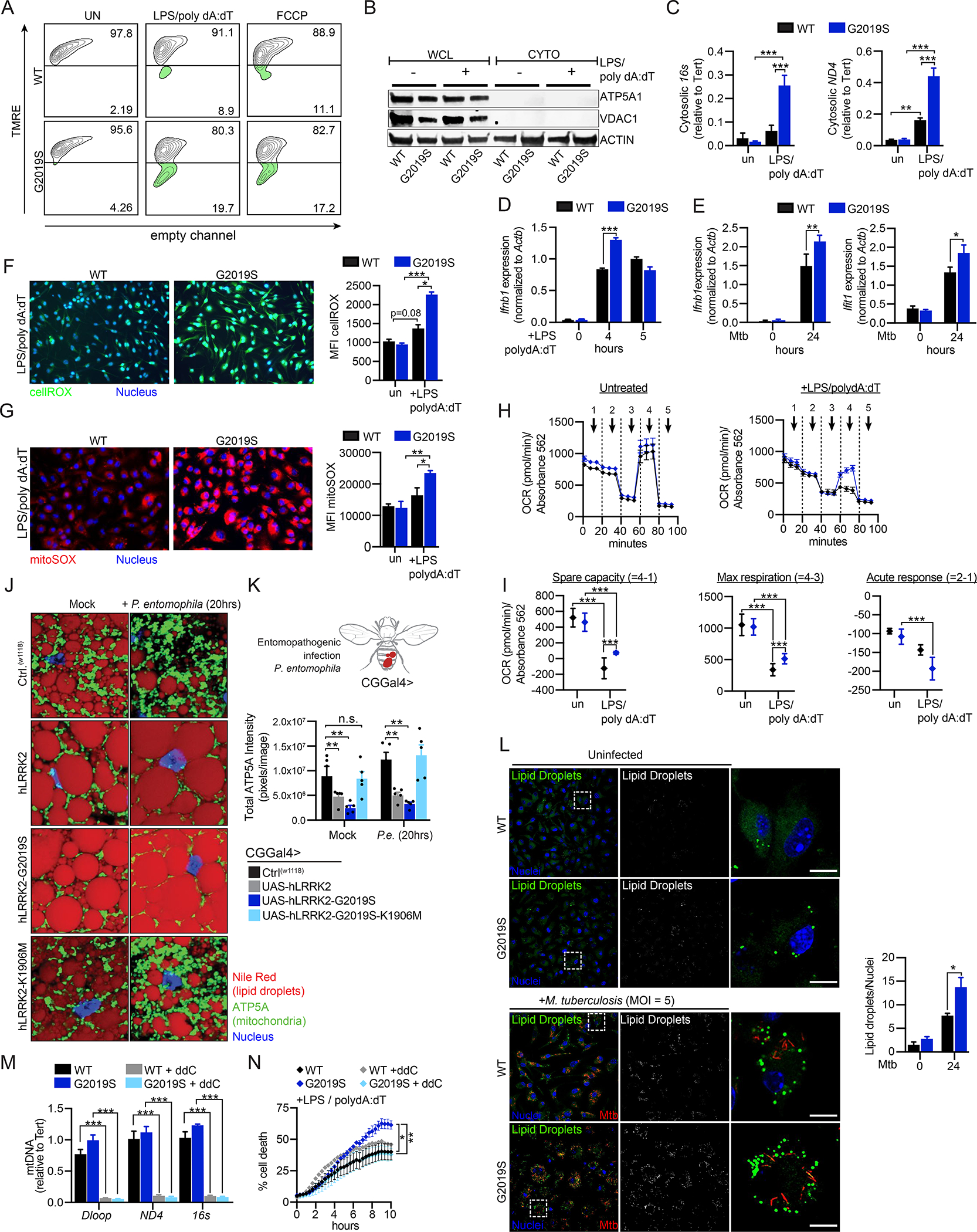
A. TMRE staining of WT and Lrrk2G2019S BMDMs in unstimulated cells and 2h post-AIM2 stimulation. 50 μM FCCP (30 min) used as a positive control. B. Total and cytosolic fractions from WT and Lrrk2G2019S BMDMs 3h post-AIM2 stimulation. Mitochondrial proteins ATP5A1 and VDAC1 show purity of the cytosolic fraction with ACTIN loading control. C. qPCR of mtDNA (16s and ND4) from cytosolic fractions in B., quantified relative to total nuclear DNA (Tert) in unstimulated and AIM2 stimulated (3h) WT and Lrrk2G2019S BMDMs D. Ifnb1 transcript levels by RT-qPCR in WT and Lrrk2G2019S BMDMs at 0, 4, and 5h post-AIM2 stimulation. E. Ifnb1 and Ifit1 transcript levels by RT-qPCR in WT and Lrrk2G2019S BMDMs at 0 and 24h post-Mtb infection (MOI 5). F. WT and Lrrk2G2019S BMDMs stained with cellROX (green) and live cell nuclear stain NucBlue (blue) 2h post-AIM2 activation. (right) Mean fluorescence intensity (MFI) measured using GEN5 software (Biotek) expanding a primary mask created around each nucleus by 10 μm. G. As in F but with the mitochondrial targeted superoxide dye mitoSOX. (right) MFI. H. Oxygen consumption rate (OCR) measured by Agilent Seahorse Metabolic Analyzer in WT and Lrrk2G2019S BMDMs: untreated (left) and AIM2 stimulated (+10 ng/mL LPS 3h, 1 μg/mL poly dA:dT 1h)(right). I. Spare respiratory capacity, maximal respiration, and acute response measured in untreated and AIM2-stimulated WT and Lrrk2G2019S BMDMs. J. Lipid droplets (Nile Red; red), mitochondria (ATP5A; green), and nuclei (DAPI; blue) in WT (CT(w1118)), hLRRK2, hLRRK2-G2019S, and hLRRK2-G2019S-K1906M-expressing Drosophila melanogaster (CGGal4> promoter) treated with sucrose (mock) or infected with Pseudomonas entomophila (P.e.) for 20h. K. Quantification of mitochondrial network intensity in 3–5 whole fat body images taken in mock and P.e.-infected hLRRK2-expressing flies. L. Lipid droplet staining (LipidTox; 1x; 30min) in un- and Mtb-infected (MOI 5) WT and Lrrk2G2019S BMDMs 24h post-infection. Lipid droplets/nucleus calculated with ImageJ (153 WT cells counted; 101 Lrrk2G2019S). M. qPCR of total Dloop, ND4 and 16s in WT and Lrrk2G2019S BMDMs +/− 4 days of 10 μM ddC treatment. N. % cell death over a time course of AIM2 activation +/− ddC treatment. Statistical analysis: n=3 or more unless otherwise noted. Statistical significance determined via two-tailed Student’s T test (L), two-way ANOVA with Tukey’s post-test (N), or a one-way ANOVA with Sidak’s post-test (C-G, I, K, M).
Lrrk2G2019S macrophages remain reliant on oxidative phosphorylation following engagement of pattern recognition receptors.
To test whether altered mitochondrial respiration could contribute to mtROS accumulation in Lrrk2G2019S BMDMs, we assayed their oxygen consumption (OCR) and extracellular acidification rates (ECAR). While basal metabolic capacity was similar between WT and Lrrk2G2019S BMDMs, spare capacity and maximal respiration were elevated in Lrrk2G2019S BMDMs during macrophage activation, both in the context of LPS treatment alone (Fig. S3D–E) and following LPS/polydA:dT treatment (Fig. 3H–I). This suggests that Lrrk2G2019S BMDMs fail to undergo the well-characterized “Warburg shift” (Kelly and O’Neill, 2015), and instead continue to rely on OXPHOS following macrophage activation. OCR defects were evident only in the initial macrophage respiratory burst; WT and Lrrk2G2019S BMDMs treated with LPS for 24h underwent the OXPHOS-to-glycolysis transition normally (Fig. S3F). No differences in glycolytic capacity (ECAR) between WT and Lrrk2G2019S BMDMs were observed in response to any stimulus (Fig. S3G–H). We propose that the failure of Lrrk2G2019S mitochondria to metabolically reprogram from OXPHOS to glycolysis places additional demands on the electron transport chains of fragmented mitochondria and contributes to elevated ROS levels (Fig. 3F–G).
We next asked whether mitochondria similarly fail to respond to immune stimuli in hLRRK2-G2019S expressing flies. Flies were orally infected with the bacterial entomopathogen strain Pseudomonas entomophila (P.e.) alongside mock-infected controls. After 20h of infection, we visualized mitochondria and lipid droplets in the fat body as in Fig. 1H. An expected increase in mitochondrial density, demonstrative of the increased demand for energy output upon infection (Zhao and Karpac, 2021), was evident in WT and hLRRK2-G2019S-K1906M-expressing Drosophila, but little to no change was observed in the density of the mitochondrial network in hLRRK2 and hLRRK2-G2019S-expressing flies (Fig. 3J–K and S3I–J). Likewise, lipid droplet size and number qualitatively decreased in response to P.e. infection in WT and hLRRK2-G2019S-K1906M flies but remained unchanged in flies expressing hLRRK2 and hLRRK2-G2019S. We also measured significantly higher numbers of lipid droplets in Lrrk2G2019S macrophages during Mtb infection (Fig. 3L), suggesting that LRRK2’s role in regulating cellular energy homeostasis during bacterial infection is evolutionarily conserved.
To link defects in mitochondrial homeostasis with Lrrk2G2019S cell death, we measured PI incorporation after AIM2 activation in cells treated with the nucleoside analog 2′,3′-dideoxycytidine (ddC) for 5 days to deplete mitochondria/mtDNA (Fig. 3M). While mtDNA depletion was sufficient to rescue inflammasome-mediated cell death in Lrrk2G2019S BMDMs, it did not significantly impact PI incorporation in WT cells (Fig. 3N). This argues that mitochondrial metabolism and/or mtDNA release uniquely contributes to inflammasome-triggered cell death in Lrrk2G2019S macrophages.
Cell death, mitochondrial depolarization, and mitochondrial ROS accumulation in Lrrk2G2019S macrophages is GSDMD-dependent.
We next sought to elucidate the molecular mechanisms responsible for uncoupling caspase-1-dependent inflammasome-triggered cell death and IL-1β release in Lrrk2G2019S macrophages. In addition to promoting cleavage of pro-IL-1β upon inflammasome activation, caspase-1 also cleaves gasdermin-D (GSDMD) to form N-GSDMD (He et al., 2015; Shi et al., 2015), which oligomerizes to form pores that translocate IL-1β out of the cell and cause pyroptosis (Ding et al., 2016; Mulvihill et al., 2018; Orning et al., 2019). To test if GSDMD contributes to Lrrk2G2019S-dependent cell death, we measured PI incorporation after AIM2 stimulation in the presence of two inhibitors of GSDMD pore formation: disulfiram and necrosulfamide. Both were sufficient to return PI incorporation in Lrrk2G2019S macrophages to WT levels (Fig. 4A–B), as was siRNA knockdown of GSDMD (Fig. 4C and S4A). A simple explanation for GSDMD driving Lrrk2G2019S -dependent cell death is that N-GSDMD forms more pores in the plasma membrane of Lrrk2G2019S BMDMs, leading to more pyroptosis. However, if this were the case, one would expect more IL-1β release, which we did not observe (Fig. 2S). Therefore, we hypothesized that GSDMD contributes to Lrrk2G2019S-dependent cell death via an alternative mechanism. Recent studies demonstrate that gasdermins can traffic to the mitochondria during NLRP3 inflammasome activation to enhance inflammasome activity via GSDMD-mediated mitochondrial stress and mtDAMP release (Rogers et al., 2019). To test if GSDMD’s interaction with the mitochondrial network is altered in Lrrk2G2019S BMDMs, we used IF microscopy to assess GSDMD colocalization (red, anti-GSDMD) with mitochondria (green, anti-TOM20) after AIM2 activation. Significant accumulation of GSDMD at the mitochondria following AIM2 stimulation was seen in both WT and Lrrk2G2019S BMDMs, with higher levels on Lrrk2G2019S mitochondria via microscopy (Fig. 4D and S4B) and biochemical cellular fractionation (Fig. 4E and S4C). Having linked lower mitochondrial membrane potential and excessive mtROS production to cell death in Lrrk2G2019S BMDMs (Figs. 3A and 3G), we asked if GSDMD contributed to these defects by treating cells with the GSDMD inhibitor disulfiram during AIM2 activation. Remarkably, disulfiram treatment was sufficient to return both Lrrk2G2019S BMDM mitochondrial membrane potential (Fig. 4F–G) and cellular ROS (Fig. 4H) to WT levels. These data indicate that AIM2 stimulation triggers N-GSDMD mitochondrial association, leading to membrane depolarization and ROS accumulation, and that this occurs preferentially in Lrrk2G2019S macrophages.
Figure 4. GSDMD mediates mitochondrial dysfunction and cell death during inflammasome activation in Lrrk2G2019S BMDMs.
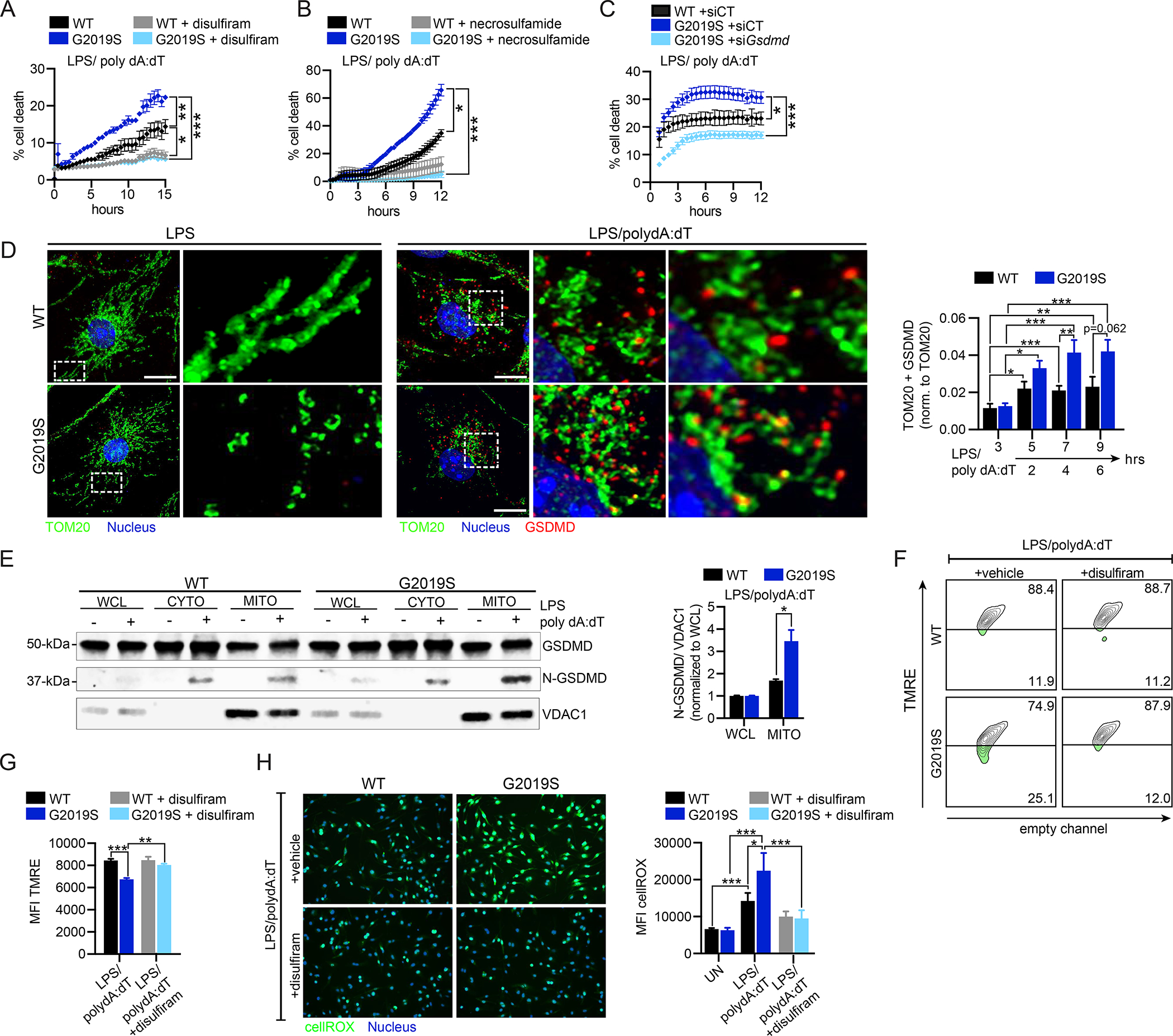
A. % cell death following AIM2 activation in WT and Lrrk2G2019S BMDMs +/− 1 μM disulfiram added 1h pre-AIM2 activation. B. As in A but with 20 μM necrosulfamide treatment. C. % cell death following AIM2 activation in WT and Lrrk2G2019S BMDMs transfected with siGsdmd or an untargeted negative control siRNA (siCT). D. Mitochondria (anti-TOM20; green) and GSDMD (anti-GSDMD; red) in WT and Lrrk2G2019S BMDMs 3h post-LPS treatment (top) or 4h post-AIM2 activation. Nuclei visualized by DAPI (blue). (right) Fiji-based analysis of TOM20+ GSDMD aggregates normalized to total TOM20 over a time course of AIM2 stimulation in WT and Lrrk2G2019S BMDMs. E. N-GSDMD mitochondrial association in WT and Lrrk2G2019S BMDMs via biochemical fractionation and immunoblot, with VDAC1 to control for mitochondrial membrane enrichment. (right) N-GSDMD relative to VDAC1 normalized to whole cell lysate. n= 2. F. TMRE staining of WT and Lrrk2G2019S BMDMs +1 μM disulfiram or DMSO (vehicle), followed by 2h AIM2 activation, by flow cytometry. G. as in F but TMRE MFI. H. CellROX (green) staining in WT and Lrrk2G2019S BMDMs +1 μM disulfiram or DMSO (vehicle) at 2h post-AIM2 stimulation (live cell nuclei staining with NucBlue). (right) cellROX MFI. Statistical analysis: n=3 or more unless otherwise noted. Statistical significance determined via a two-tailed Student’s T test (F), a two-way ANOVA with Tukey’s post-test (A-C), or a one-way ANOVA with Sidak’s post-test (E, H, J).
N-GSDMD forms pores in the macrophage mitochondrial network during AIM2 inflammasome activation.
We next investigated the extent to which inflammasome-triggered N-GSDMD mitochondrial association occurs in WT macrophages. Using flow cytometry, we measured GSDMD mitochondrial association by co-staining isolated mitochondria with MitoTracker and anti-GSDMD antibodies after AIM2 activation. We detected an increase in GSDMD association with mitochondria 2h after AIM2 activation, which was dependent on GSDMD’s ability to form pores (Fig. 5A). We also observed GSDMD-dependent mitochondrial membrane depolarization in WT BMDMs following AIM2 stimulation (Fig. 5B), although notably, with slower kinetics than previously observed in Lrrk2G2019S cells (4h vs. 2h). To directly address the capacity of N-GSDMD to damage mitochondria, we created a minimal in vitro system. Briefly, N-GSDMD was generated by combining recombinantly-expressed full length GSDMD with the catalytically active mouse CASP11 (residues 100–373, wtCASP11) or a catalytically dead form of the same protein (C254A; mutCASP11) (Fig. 5C and S5A). Only wtCASP11 was capable of cleaving GSDMD, and it cleaved similar amounts of GSDMD in wild type and Lrrk2G2019S mitochondrial preparations (Figs. 5D and S5B). Incubation of mitochondria isolated from WT BMDMs with GSDMD/wtCASP11 depolarized 81.6% of mitochondria in 1h (Fig. 5E). Comparing WT and Lrrk2G2019S depolarization under similar conditions (30 min incubation), we measured 48.3% depolarization in mitochondrial isolated from Lrrk2G2019S samples vs. 30.8% in WT mitochondria (Fig. 5F). These data demonstrate that N-GSDMD is necessary and sufficient to depolarize mitochondrial membranes upon inflammasome activation. To begin to define what makes Lrrk2G2019S mitochondria susceptible to GSDMD association and pore formation, we treated WT BMDMs with menadione, which generates mitochondrial ROS via redox cycling (Loor et al., 2010; Tao et al., 2020). Treatment of WT macrophages with menadione (25 μM) was sufficient to generate ROS (Fig. S5C) and promote N-GSDMD association with mitochondria via biochemical fractionation (Fig. 5G, S5D) and flow cytometric analysis of GSDMD+ isolated mitochondria (Fig. 5H). These findings suggest that mtROS is the main driver of GSDMD mitochondrial association in macrophages after inflammasome activation.
Figure 5. N-GSDMD directly mediates depolarization of macrophage mitochondrial membranes following AIM2 activation.

A. GSDMD association (anti-GSDMD; PE, x-axis) with the mitochondrial network measured by flow cytometry of isolated mitochondria 4h after AIM2 activation +1 μM disulfiram or DMSO (vehicle) B. TMRE staining of WT BMDMs treated with 1 μM disulfiram or DMSO (vehicle), followed by AIM2 activation for 4h C. Schematic of recombinant proteins used in in vitro experiments D. GSDMD cleavage in vitro via recombinant wt or mutCASP11 in the presence of mitochondrial extracts from WT and Lrrk2G2019S BMDMs (n=2) E. FACS plot of TMRE staining of mitochondria isolated from WT BMDMs in the presence of full length GSDMD and wtCASP11 or mutCASP11. 1h incubation. Quantitation at lower right. F. As in E. but with mitochondria isolated from WT and Lrrk2G2019S BMDMs. 30 min incubation G. N-GSDMD mitochondrial association in WT and Lrrk2G2019S BMDMs via biochemical fractionation and immunoblot during AIM2 activation +/− 25 μM menadione. VDAC1; control for mitochondrial membrane enrichment. (right) N-GSDMD relative to VDAC1 normalized to WCL H. As in A but 2h after AIM2 activation, WT BMDMs +/− 25 μM menadione Statistical analysis: n=3 or more unless otherwise noted. Statistical significance determined via a one-way ANOVA with Sidak’s post-test (A, B, E, F, H).
MtROS drives necroptosis in Lrrk2G2019S macrophages
Collectively, our data suggest that Lrrk2G2019S macrophages die via activation of a non-pyroptotic cell death pathway driven by mtROS-dependent N-GSDMD pore formation in the mitochondrial network. MtROS has been previously implicated in necroptosis, a form of inflammatory programmed cell death in which RHIM domain-containing proteins RIPK1/RIPK3 activate the pore-forming protein MLKL downstream of cell intrinsic and extrinsic stimuli (Fig. 6A) (Fulda, 2016; Shindo et al., 2013; Zhang et al., 2020). Thus, we set out to determine whether Lrrk2G2019S BMDMs die via necroptosis following inflammasome activation. pMLKL accumulated in Lrrk2G2019S macrophages after AIM2 stimulation (Fig. 6B) and during M. tuberculosis infection (Fig. 6C) and MLKL, RIPK1, and RIPK3 were each necessary to enhance cell death in Lrrk2G2019S macrophages following AIM2 stimulation (Figs. 6D–F; S6A–B). This was not due to differences in necroptotic protein expression between WT and Lrrk2G2019S cells (Fig. S6C–D). Lrrk2G2019S cell death was independent of caspase-8 and did not exhibit hallmarks of apoptosis (Fig. S6E–F), arguing against involvement of the newly described PANoptosome (Samir et al., 2020). While siRNA knockdown of the necroptosis adapter ZBP1 significantly enhanced cell death in Lrrk2G2019S macrophages (Fig. 6G), this death could be rescued by RIPK3 inhibition (Fig. S6G–H). Knocking down the TLR-4 adapter TRIF (Meylan et al., 2004) resulted in a similar phenotype (Fig. 6H and S6I). These data suggest that adapters like ZBP1 and TRIF do not directly promote Lrrk2G2019S cell death but do likely regulate the pools of available necroptosis mediators like RIPK3.
Figure 6. GSDMD-dependent alteration of mitochondrial homeostasis triggers RIPK1/RIPK3/MLKL-dependent necroptotic cell death in Lrrk2G2019S BMDMs.

A. Cell-intrinsic and cell-extrinsic signaling cascades that trigger necroptosis. Red = steps in the pathway tested for involvement in Lrrk2G2019S cell death B. Phospho-MLKL aggregation at 6h post-AIM2 stimulation in WT and Lrrk2G2019S BMDMs. (right) pMLKL aggregates/nuclei quantified using Fiji. C. As in B but at 24h post-Mtb infection (MOI 2) D. % cell death following AIM2 activation in WT and Lrrk2G2019S BMDMs transfected with an siRNA against Mlkl (siMlkl) or a non-targeting control siRNA (siCT) E. As in D, but +/− the RIPK1 inhibitor GSK2982772 (10 μM) F. As in D, but +/− RIPK3 inhibitor GSK872 (1 μM) G. As in D, but after transfection with siZbp1 H. As in G but with siTrif I. CellRox (green) and NucBlue (blue) staining in WT and Lrrk2G2019S BMDMs 2h post-AIM2 activation +/− RIPK3 inhibitor GSK872 (1 μM) or DMSO (vehicle). MFI on right. J. % cell death following AIM2 activation in WT and Lrrk2G2019S BMDMs +/− the mitochondrial ROS scavenger Necrox-5 (25 μM) K. % cell death following menadione treatment (25 μM) in WT BMDMs after AIM2 activation +/− GSK872 (1 μM) L. Extracellular IL-1β protein levels as measured by ELISA at 6 h post AIM2 stimulation in WT and Lrrk2G2019S BMDMs +/− RIPK3 inhibitor GSK872 (1 μM) Statistical analysis: n=3 or more unless otherwise noted. Statistical significance determined using a two-tailed Student’s T test (B, C), a two-way ANOVA with Tukey’s post-test (D-H, J, K), or a one-way ANOVA with Sidak’s post-test (I, L).
Previous studies have shown that activated RIPK3 promotes aerobic respiration leading to ROS production through pyruvate dehydrogenase activation (Yang et al., 2018). To test how RIPK3 activation contributes to mtROS following inflammasome activation, we measured cellROX after AIM2 stimulation in WT and Lrrk2G2019S cells with and without GSK872 treatment. While inhibiting RIPK3 was sufficient to return ROS levels in Lrrk2G2019S cells to those of WT (Fig. 6I), this rescue was incomplete, likely due to GSDMD-mediated damage sustained by the mitochondria following AIM2 stimulation. Importantly, treatment with the mitochondrial ROS scavenger/necroptosis inhibitor Necrox-5 completely rescued Lrrk2G2019S cell death following AIM2 stimulation (Fig. 6J) and treatment of WT BMDMs with menadione induced cell death following AIM2 stimulation in a RIPK3-dependent fashion (Fig. 6K). Lastly, RIPK3 inhibition restored IL-1β release in Lrrk2G2019S BMDMs to that of WT cells following AIM2 activation (Fig. 6L). This suggests that IL-1β release is disrupted in Lrrk2G2019S BMDMs via competition between necroptotic cell death (pMLKL pore formation = no IL-1β release) and pyroptotic cell death (GSDMD pore formation = IL-1β release). Together, our data argue that mtROS is the main driver of a pyroptosis-to-necroptosis shift in Lrrk2G2019S macrophages and provide evidence for a RIPK3-mediated feed-forward loop that enhances mtROS production and cell death.
Bacterial infection promotes hyperinflammation and pathogenesis in hLRRK2-G2019S flies.
Having measured a defect in the ability of hLRRK2-G2019S-expressing D. melanogaster to upregulate mitochondrial dynamics during infection with P. entomophila (Fig. 3J–K), we asked whether flies exhibit increased susceptibility to pathogen challenge. Briefly, flies were orally infected with P. entomophila for 20h and survival was monitored for 10–11 days (Fig. 7A). Fat body expression of both hLRRK2 and hLRRK2-G2019S (CGGal4>) or ubiquitous expression of hLRRK2-G2019S (DaGal4>) correlated with decreased fly survival, whereas hLRRK2-K1906M expression provided protection (Fig. 7B). These phenotypes correlated with bacterial burdens at 20h post-infection (Fig. S7A). Additionally, inflammatory gene expression (Drs, Atta, and Dipt), which we have linked to P.e. susceptibility (Zhao and Karpac, 2021), was higher in hLRRK2-G2019S flies (Fig. 7C and S7B), suggesting that LRRK2 expression and kinase activity correlate with hyperinflammation and hyper-susceptibility to bacterial infection in Drosophila.
Figure 7. Lrrk2G2019S plays an evolutionarily conserved role in promoting hyperinflammation and susceptibility to bacterial pathogens.

A. P.e. infection timeline B. Survival curves of WT (W118), hLRRK2- (UAS-hLRRK2), hLRRK2-G2019S- (UAS-hLRRK2 G2019S), and hLRRK2 G2019S-K1906M- (UAS-hLRRK2 G2019S-K1906M) expressing flies (fat body tissue-specific expression (CGGal4>) and ubiquitous expression (DaGal4>)) over a 10-day period after a 20h P.e. infection C. Innate immune response gene expression after 20h P.e. infection, relative to Act5c, by RT-qPCR D. Intracellular TNF-α protein levels in PBMCs 4h after stimulation with 1 μg/mL LPS. Fold change of MFI in resting vs. stimulated cells E. Mtb infection timeline with number of mice sacrificed at each time point F. Mtb colony forming units (CFUs) recovered from the lung and spleen of WT and Lrrk2G2019S infected mice at day 21 and 77 post-infection. total n = 30. G. As in F but from Lrrk2+/+ and Lrrk2G>S/+ mice, n = 18 H. Inflammatory nodules in the lungs of WT and Lrrk2G2019S Mtb-infected mice at day 21 and 77 post-infection. White arrows indicate lesions I. H&E stain of inflammatory nodules in the lungs of WT and Lrrk2G2019S Mtb-infected mice at day 21 and 77. % inflammation (right). Black arrows indicate regions of inflammation J. H&E stain of neutrophils within an inflammatory nodule in the lung of WT and Lrrk2G2019S mice at day 21 and 77 post-Mtb infection. Arrows indicate degenerate neutrophils (white), lymphocytes (teal), and macrophages (yellow) K. (left) Semiquantitative score of pulmonary inflammation based on granulomatous nodules in none (0), up to 25% (1), 26–50% (2), 51–75% (3) or 76–100% (4) of fields. (right) Pathology scoring of PMNs in the lungs of WT and Lrrk2G2019S Mtb-infected mice at day 21 and 77 L. PMNs in the lungs of WT and Lrrk2G2019S mice at day 21 by flow cytometry M. Inflammatory cytokines transcripts from lung homogenates by RT-qPCR. N. RIPK1, RIPK3, IFNβ, and IL1β protein levels in lung homogenates (50 μg total protein/lane) at day 21 post-Mtb infection; (right) Quantification O. Serum levels of IL-6, IL-17a, IL-1α, and LIX, in WT and Lrrk2G2019S mice via cytokine array at day 21 post-Mtb infection Statistical analysis: n as indicated. Statistical significance determined using either a one-way ANOVA with Sidak’s post-test (C, D), or a Mann-Whitney U test (F, G, I, K-O).
Lrrk2G2019S promotes hyperinflammation and immunopathology during Mycobacterium tuberculosis infection.
To further translate our findings to models of human disease, we asked whether Lrrk2G2019S mice have altered circulating immune cell populations. Building on earlier work (Li et al., 2007; Moehle et al., 2015), we measured populations of immune cells in blood isolated from WT and Lrrk2G2019S mice by flow cytometry. We found that while Lrrk2G2019S and WT mice have similar numbers of circulating immune cells (Figs. S7C, E, F), certain Lrrk2G2019S myeloid populations express modestly higher levels of inflammatory mediators like TNF-α in response to TLR stimulation (Fig. 7D–S7D).
Cell death modality usage is a key determinant of Mtb infection outcomes, with apoptotic macrophage cell death generally being anti-bacterial and necrosis being pro-bacterial (Srinivasan et al., 2014). To test our prediction that Lrrk2G2019S mice would exhibit exacerbated pathogenesis in response to Mtb infection, we used a low dose aerosol infection model to administer approximately 100 Mtb bacilli per mouse to a cohort of littermate age-matched male and female WT and Lrrk2G2019S mice, sacrificing mice at days 21 and 77 (Fig. 7E). Bacterial burdens were dramatically higher in the lungs and spleens of Lrrk2G2019S mice at day 77, with a moderate but significant increase seen in Lrrk2G2019S lungs as early as day 21 (Fig. 7F). Lrrk2G>S/+ (KI) mice were similarly permissive to Mtb replication, with higher CFUs in the lung at day 77 post-infection (Fig. 7G). Gross examination revealed extensive inflammatory nodules within the lungs of Lrrk2G2019S Tg mice compared to WT controls at day 21 and 77 (Fig. 7H). Lungs from Lrrk2G2019S mice trended towards greater inflammatory infiltrates at day 21, and by day 77 Lrrk2G2019S lungs were almost completely obscured by coalescing inflammatory cell infiltrates (Fig. 7I). Qualitatively, the lungs of Lrrk2G>S/+ KI mice also exhibited signs of exacerbated inflammation (Fig. S7G). Increased granulomatous nodules and PMNs were seen in Lrrk2G2019S lungs by H&E staining (Fig. 7J–K), and enhanced neutrophil recruitment (CD45+ B220− CD11b+ Ly6G+) was measured in Lrrk2G2019S lungs at day 21 post-infection by flow cytometry (Fig. 7L). Consistent with higher neutrophil recruitment, pro-inflammatory cytokine transcripts were higher in the lungs of Lrrk2G2019S and Lrrk2G>S/+ Mtb-infected mice at early and late infection time points (Fig. 7M, S7I, and S7K). ISG transcripts were also higher in the lungs of Lrrk2G2019S and Lrrk2G>S/+ mice during Mtb infection (Fig. S7J–K), paralleling Lrrk2G2019S BMDMs following AIM2 activation ex vivo (Fig. S3B). Previous studies have shown that accumulation of necroptosome proteins is indicative of necroptosis (Lim et al., 2019; Liu et al., 2018); we measured significantly higher levels of RIPK1 and RIPK3 by immunoblot in lung homogenates from Mtb-infected Lrrk2G2019S mice at day 21 (Fig. 7N). Although circulating levels of many soluble immune mediators were similar in WT and Lrrk2G2019S Mtb-infected mice (Fig. S7L), Lrrk2G2019S Mtb-infected mice had high IL-6 and low IL-1α serum levels (Fig. 7O), consistent with cells favoring necroptotic over pyroptotic cell death (Di Paolo and Shayakhmetov, 2016). Additionally, release of circulating cell-free mtDNA was enhanced in Lrrk2G2019S mice at days 21 and 63 post-Mtb infection, indicating higher levels of mitochondrial stress (Fig. S7M–N). These findings suggest that Mtb infection triggers more necroptosis in the lungs of Lrrk2G2019S mice, leading to local hyperinflammation, immunopathology and poor infection outcomes.
DISCUSSION
Cellular surveillance of mitochondrial integrity is a key checkpoint in the control of programmed cell death and release of inflammatory mediators. Here, we provide evidence that inflammatory phenotypes attributed to the human disease-associated allele Lrrk2G2019S are due in large part to cell death modality switching driven by altered mitochondrial homeostasis. Specifically, our data support a model whereby mtROS licenses N-GSDMD to switch between a plasma membrane-associated executioner of pyroptosis to a mitochondrial-associated initiator of necroptosis. We report that upon bacterial infection of Lrrk2G2019S macrophages, inflammasome activation proceeds normally, with caspase-1 cleaving GSDMD and pro-IL-1β. However, instead of forming pores in the plasma membrane to elicit IL-1β release and pyroptotic cell death, GSDMD is recruited to the mitochondria by excess ROS generated by overuse of the electron transport chain. At the mitochondria, N-GSDMD pores cause additional damage, leading to release of mtDAMPs and activation of RIPK1/RIPK3. RIPK3, likely through activation of pyruvate dehydrogenase (Yang et al., 2018), amplifies mtROS production, further enhancing RIPK and MLKL activation, and triggering necroptosis.
These data lend additional support to an emerging model whereby flexible usage of cell death proteins evolved as a host adaptation against intracellular pathogens that suppress or subvert cell death pathways (Fritsch et al., 2019; Man et al., 2013; Mascarenhas et al., 2017; Pierini et al., 2012; Tsuchiya et al., 2019; Van Opdenbosch et al., 2017). It is tempting to speculate that although cells evolved flexible cell death pathways as a response to intracellular pathogens, these switches are also tripped by mitochondrial damage and mtDAMP release due to the ancient bacterial origins of the mitochondrion. While balancing the distribution of GSDMD between mitochondrial and plasma membranes seems like a good way to modulate cell death outcomes, we still do not why N-GSDMD evolved to associate with mitochondria in the first place. It is possible that GSDMD forms pores in mitochondria to amplify cell death signals via release of cytochrome C (Rogers et al., 2019) or to limit transcriptional upregulation of genes that promote cellular proliferation by releasing mtDNA and stimulating cGAS/STING (Huang et al., 2021). It is also possible that GSDMD association with mitochondria is a carry-over from the Rickettsial origins of mitochondria, as N-GSDMD can bind to and kill bacteria in in vitro cultures (Ding et al., 2016).
Despite having connected LRRK2-dependent mtROS production to GSDMD-dependent necroptosis, the precise mechanistic contribution LRRK2 kinase activity to Lrrk2G2019S cell death is still not entirely clear. One intriguing possibility is that LRRK2, sometimes annotated as RIPK7, promotes necroptosis by acting as a RIP (receptor interacting protein) kinase family member itself (Rideout and Re, 2017). Such activity could help explain how hLRRK2-G2019S controls hyperinflammation and susceptibility to bacterial infection in Drosophila, where GSDMD and pyroptosis are not conserved. Across insect genomes, one can identify innate immune receptors, adaptors, and transcription factors that encode RHIM-like motifs but lack kinase domains (e.g. PGRP-LE/LC, IMD, and Relish) (Chan et al., 2015). It is conceivable that LRRK2 acts as an evolutionarily conserved kinase bridge, modulating phosphorylation and activity of RHIM-containing innate immune proteins in insects as well as in mammals.
As mutations in LRRK2 are notoriously associated with both inherited and sporadic PD, it will be important to translate our findings to cells in the CNS. A growing literature supports a key role for necroptosome components in promoting inflammatory cell death in the CNS (Iannielli et al., 2018; Onate et al., 2020; Yuan et al., 2019). We propose that cell death modality reprogramming is a major way in which mutations in genes like LRRK2 trigger and/or exacerbate multiple human diseases.
Limitations of the Study
Our study reports that Lrrk2G2019S macrophages are prone to cell death following inflammasome activation via GSDMD-mediated necroptosis. While our findings are consistent with mtROS directing GSDMD to form pores on mitochondrial membranes, it is possible that other functions of LRRK2 related to mitophagy, endolysosomal trafficking, lysosome acidification, etc., also contribute to cell death. To date, we have only studied cell death phenotypes in BMDMs. Other immune cells may respond differently to infection and/or inflammasome stimulation. We already have some evidence of this, as circulating myeloid cells generate higher levels of inflammatory mediators like TNF-α following LPS treatment but Lrrk2G2019S BMDMs do not. Because necroptosis does not carry a clear immune signature, it is difficult to definitively connect the cell death phenotypes we observe in Lrrk2G2019S macrophages with hyper-susceptibility and hyperinflammation during Mtb infection in vivo, especially given that Lrrk2G2019S mice harbor increased bacterial loads. Generation of mouse lines that express Lrrk2G2019S in a Mlkl−/−, Ripk1−/−, or Ripk3−/− genetic background will help clarify the mechanisms through which Lrrk2G2019S-mediated necroptosis alters infection outcomes in vivo.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Robert O. Watson (robert.watson@tamu.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mouse husbandry and strains
Lrrk2G2019S mice (B6.Cg-Tg(Lrrk2*G2019S)2Yue/J, also known as FLAG-LRRK2-G2019S, stock # 012467 were purchased from Jackson Laboratories (Bar Harbor, ME). Femurs from Lrrk2 Tg WT mice (B6.Cg-Tg(Lrrk2)6Yue/J) were kindly provided by Dr. Malu Tansey at the University of Florida. Lrrk2G2019S KI mice B6.Cg-Lrrk2tm1.1Hlme/J also known as LRRK2 G2019S knock-in, stock # 030961 were purchased from the Jackson Laboratories (Bar Harbor, ME). Both Lrrk2G2019S and G2019S KI mice are maintained with filial breeding on a C57BL6/J background and fed 4% standard chow. All mice used in experiments were compared to age- and sex- matched controls. Littermates were used for experiments. Mice used to generate BMDMs were males between 10–16 weeks old. For in vivo infection male and female mice were infected with Mtb at 10–12 weeks. Embryos used to make primary MEFs were 14.5 days post-coitum. All animals were housed, bred, and studied at Texas A&M Health Science Center under approved Institutional Care and Use Committee guidelines.
M. tuberculosis
The Erdman strain was used for all M. tuberculosis infections. Low passage lab stocks were thawed for each experiment to ensure virulence was preserved. M. tuberculosis was cultured in roller bottles at 37 °C in Middlebrook 7H9 broth (BD Biosciences) supplemented with 10% OADC (BD Biosciences), 0.5% glycerol (Fisher), and 0.1% Tween-80 (Fisher) or on 7H10 plates. All work with M. tuberculosis was performed under Biosafety level 3 containment using procedures approved by the Texas A&M University Institutional Biosafety Committee.
M. marinum
Low passage glycerol lab stocks of M. marinum were grown in 7H9 broth (BD Biosciences) supplemented with 10% OADC (BD Biosciences), 0.5% glycerol (Fisher), and 0.1% Tween-80 (Fisher). Cultures were grown at 30 °C shaking in the dark.
L. monocytogenes
Low passage glycerol lab stocks of L. monocytogenes strain 10304s were streaked onto BHI agar plates and incubated at 37 °C overnight.
S. enterica (ser. Typhimirium)
Salmonella enterica serovar Typhimurium (SL1344) was obtained from Dr. Denise Monack, Stanford. S. T. stocks were streaked out on LB agar plates and incubated at 37 °C overnight.
Fly husbandry and strains
w1118 and DaGal4 were obtained from Bloomington Drosophila Stock Center. CGGal4 was provided by C. Thummel. UAS-hLRRK2, UAS-hLRRK2-G2019S and UAS-hLRRK2-G2019S-K1906M were provided by C. Elliott (University of York). All flies were reared on standard yeast and cornmeal-based diet at 25 °C and 65% humidity on a 12 hr light/dark cycle, unless otherwise indicated. The standard lab diet (cornmeal-based) was made with the following protocol: 14 g Agar/165.4 g Malt Extract/ 41.4 g Dry yeast/ 78.2 g Cornmeal/ 4.7 mL propionic acid/ 3 g Methyl 4-Hydroxybenzoate/ 1.5 L water. In order to standardize metabolic results, 50 virgins were crossed to 10 males and kept in bottles for 2–3 days to lay eggs. Wet folded filters (GE healthcare, CAT No.10311843) were inserted in bottles after parental flies were removed. Progeny of crosses were collected for 3–4 days after initial eclosion. Collected progeny were then transferred to new bottles and allowed to mate for 2 days (representing unique populations). The following progeny, approximately 20 female flies, were then separated into each vial (before mock or oral infection treatment) for 10 days at 25 °C and 65% humidity on a 12 hr light/dark cycle. Post-mated female flies were used for all experiments due to sensitivity to Pseudomonas entomophila (P.e.) infection. The UAS-hLRRK2, UAS-hLRRK2-G2019S, UAS-hLRRK2-G2019S-K1906M, CGGal4, and DaGal4 transgenic lines were backcrossed 10 times into the control strain w1118 background, continued backcrossing was performed every 6–8 months to maintain isogeneity. Details on the original generation of the hLRRK2, hLRRK2-G2019S, and hLRRK2-G2019S-K1906M transgenic flies, and the protein expression levels of each allele, can be found in (Cording et al., 2017; Liu et al., 2008; and Lin et al., 2010).
Primary cell culture
Bone marrow derived macrophages (BMDMs) were differentiated from BM cells isolated by washing mouse femurs with 10 mL DMEM 1 mM sodium pyruvate. Cells were then centrifuged for 5 min at 400 rcf and resuspended in BMDM media (DMEM, 20% FBS (Millipore), 1 mM sodium pyruvate (Lonza), 10% MCSF conditioned media (Watson lab)). BM cells were counted and plated at 5×106 cells per 15 cm non-TC treated dishes in 30 mL complete BMDM media. Cells were fed with an additional 15 mL of BMDM media on day 3. Cells were harvested on day 7 with 1X PBS EDTA (Lonza). For ddC depletion of mitochondrial DNA BMDMs were treated with 10 μM ddC on day 3 and 5 of differentiation. Cells were harvested on day 7 of culture. Mouse embryonic fibroblasts (MEFs) were isolated from embryos. Briefly, embryos were dissected from yolk sacs, washed 2 times with cold 1X PBS, decapitated, and peritoneal contents were removed. Headless embryos were disaggregated in cold 0.05% trypsin-EDTA (Lonza) and incubated on ice for 20 min., followed by incubation at 37 °C for an additional 20 min. Cells were then DNase treated with 4 mL disaggregation media (DMEM, 10% FBS, 100 μg/mL DNASE I (Worthington)) for 20 min at 37 °C. Supernatants were isolated and spun at 1000 rpm for 5 min. Cells were resuspended in complete MEF media (DMEM, 10% FBS, 1 mM sodium pyruvate), and plated in 15 cm TC-treated dishes 1 dish per embryo in 25 mL of media. MEFs were allowed to expand for 2–3 days before harvest with trypsin 0.05% EDTA.
METHOD DETAILS
Bacterial infections
Multiplicity of infection (MOI) for each bacterial infection is listed by experiment in the figure legends. For M. tuberculosis infection, the inoculum was prepared by growing bacteria to log phase (OD600 0.6–0.8). Bacterial cultures were spun at low speed (500 rpm) for 5 min to remove clumps. Bacteria were then pelleted with a spin at 3000 rpm 5 min and washed with 1X PBS this step was repeated twice. Resuspended bacteria were briefly sonicated and spun at low speed once again to further remove clumps. The bacteria were diluted in DMEM (Hyclone) + 10% horse serum (Gibco) in vitro infections, or 1X PBS in vivo infections. For in vitro infections plates containing bacteria and cells were spun for 10 min at 1000 rpm to synchronize infection. Following the spin fresh media was added to cells along with inhibitors of interest See “cell stimulation with innate immune agonist” for concentrations.
For M. marinum infection, the inoculum was prepared by growing bacteria to log phase (OD600 0.6–0.8). Bacterial cultures were spun at 2700 rcf for 10 min and washed in 1X PBS. Bacteria were pulled through a syringe with a 26 gauge needle 3 times to create single cell suspension. Bacteria were then centrifuged twice for 2 min at 2000 rcf to remove clumps. The bacteria were diluted in DMEM (Hyclone) + 10% horse serum (Gibco) and added to cells. Bacterial/cell plates were then spun at 1000 rpm for 10 min to synchronize infections after which fresh cell culture media was added. For cell death infection assays with M. marinum cells were incubated at 32°C.
For L. monocytogenes infection, the inoculum was prepared by growing bacteria in BHI overnight at 37 °C. The following morning, cultures were diluted 1:5 in fresh BHI, grown to log phase (OD600 0.5–1.0) at 37 °C 2–3 hrs. Upon reaching mid-log phase, 1 mL of bacteria were pelleted at 5000 rpm for 3 min and washed twice with 1X PBS. Bacteria were diluted in DMEM (Hyclone) and added to cells. spun at 1000 rpm for 5 min then incubated for 10 min at 37°C prior to adding fresh media and proceeding with downstream assays.
For S. Typhimurium infection overnight cultures of bacteria were grown in LB broth containing 0.3 M NaCl and grown at 37 °C until they reached an OD600 of 0.9. On the day of infection cultures were diluted 1:20. Once cultures had reached mid-log phase (OD600 0.6–0.8) 2–3 hrs, 1 mL of bacteria were pelleted at 5000 rpm for 3 min and washed twice with 1X PBS. Bacteria were diluted in DMEM (Hyclone) and added to cells. Bacteria/cells were spun at 1000 rpm for 5 min then incubated for 10 min at 37°C prior to adding fresh media and proceeding with downstream assays.
siRNA knockdown in BMDMs
On day 5 of differentiation BMDMs were reseeded at 0.35×106 cells/ well in triplicate in 12-well non-tissue culture treated plates. The following day media was replaced with 500 μL fresh BMDM media 30 min prior to transfection. Cells were transfected using Fugene SI reagent and 10 μM of siRNA stock against either Gsdmd (s87492), Zbp1 (s233871), Trif (s98708), or Mlkl (s92952). For a negative control, Silencer® Select Negative Control #1 (4390843) was used. Cells were incubated for 24 hrs in transfection media, then allowed to rest for 48h at 37 °C prior to downstream survival experiments.
Cell stimulation with innate immune agonists
BMDMs were plated in 96-well half area plates at 2.5×104 cells/well, 12-well plates at 5×105 cells/well, 6-well plates at 1×106 cells/well. To analyze upregulation of ISGs and NFkB associated genes, cells were stimulated for 4 hrs. with 1 μM CL097 (InvivoGen), 100 ng/mL LPS (InvivoGen), 100 ng/mL Pam3CSK4 (InvivoGen). Cells were transfected for 4 hrs. with 1 μg/mL ISD (Watson lab), 1 μg/mL poly I:C (InvivoGen), 1 μg/mL cGAMP (InvivoGen) using lipofectamine reagent (Thermo Fisher). Cells were transfected for 4 hrs with 1 μM CpG 2395 (IDT) using Gene Juice (EMD Millipore). To analyze AIM2 inflammasome activation, cells were stimulated with either 10 ng/mL LPS (InvivoGen) or Pam3CSK4 (InvivoGen), for 3 hrs followed by 1 μg/mL poly dA:dT (InvivoGen) transfection with lipofectamine (Thermo Fisher) at a 3:1 ratio. To analyze NLRP3 inflammasome activation, cells were stimulated with either 10 ng/mL LPS (InvivoGen) or Pam3CSK4 (InvivoGen), for 3 hrs followed by 25 μM nigericin (ThermoFisher). For rescue assays cells were treated at the time of LPS priming with 100 μM AC-YVAD-CMK (InvivoGen), 25 μM dapansutrile (Cayman Chemicals), 1 μM GNE9605 (Cayman Chemicals), 50 μM Z-IETD-FMK (BD biosciences), 10 μM necrostatin-1 (Calbiochem), 10 μM GSK2982772 (Cayman Chemicals), 1 μM GSK872 (Cayman Chemicals), 25 μM Necrox-5 (Cayman chemicals), 25 μM menadione (Sigma), or 1 hr before AIM2 activation 1 μM disulfiram (Sigma), 20 μM necrosulfamide (EMD millipore).
Plate based assays for cell death and reactive oxygen species (ROS)
BMDMs were plated in 96-well half area clear bottom plates (Corning) at 2.5×104 cells/well in 50 μL of media using a multichannel pipette. After adherence to plate (approximately 1–2 hrs), an additional 25 μL of media was added to each well containing cells. The following day media was removed and 10 ng/mL LPS was added along with respective inhibitor if applicable (see paragraph above for inhibitor details). When performing cell death assays, just prior to 3 hrs post LPS stimulation 5 μg/ml propidium iodide (PI) (ThermoFisher) was added to each well and exposure/focus was calibrated at 4X magnification on either a Lionheart plate reader or Cytation 5 (BioTek). total cell numbers were determined using NucBlue (Thermo Fisher) (2 drops per mL) in 1X PBS with a subset of the plated cells. For ROS assays, to detect cellular oxidation cellROX green (ThermoFisher) was used. Cells were stained for 30 min in 5 μM cellROX green in culture media at 37 °C. To detect mitochondrial superoxide mitoSOX (ThermoFisher) was used. Cells were stained for 10 min in 5 μM mitoSOX in 1X PBS 2% FBS at 37 °C. For image analysis Gen 3.5 software (BioTek) was used. Dead cells were counted by PI staining of nucleus. Mean fluorescence intensity was calculated from images taken at 10X magnification by creating a mask around NucBlue staining nuclei. A secondary mask was extended for the signal of interest (cellROX (FITC-channel) or mitoSOX (RFP-channel)) 10 μM around each nucleus.
Gene expression analysis by RT-qPCR
For mammalian tissue and cells, RNA was isolated using Direct-zol RNAeasy kits (Zymogen). cDNA was synthesized with Bio-Rad iScript Direct Synthesis kits (BioRad) per manufacturer’s protocol. RT-qPCR was performed in triplicate wells using PowerUp SYBR Green Master Mix. Data was analyzed on a QuantStudio 6 Real-Time PCR System (Applied Biosystems), and quantification of gene expression was performed using a standard curve and normalized to Actb expression levels. For insect tissues, RNA from intact fly carcass (containing mostly fat body) was extracted using Trizol as per manufacturer’s protocol. cDNA was synthesized using Superscript III (Invitrogen). RT-qPCR was performed using SYBR Green, the Applied Biosystems StepOnePlus Real-Time PCR systems. Quantification of gene expression levels were calculated using the ΔCt method and normalized to Act5C expression levels.
Seahorse metabolic assays
Seahorse XF mito stress test kits and cartridges were prepared per Agilent’s protocols and analyzed on an Agilent Seahorse XF 96-well analyzer using WAVE software post analysis. The day before the assay BMDMs were seeded at 5×104 cells/well overnight. For inflammasome activation on the day of the assay cells were treated with 10 ng/mL LPS (Invivogen), followed by 1 μg/mL poly dA:dT (Invivogen) 3 hrs later. After 1 hr of AIM2 activation cells were processed per manufacturer’s directions and analyzed using the Agilent Seahorse Mito Stress Test kit (Agilent). For overnight stimulation with LPS, cells were treated 2 hrs after plating with 10 ng/mL LPS and incubated overnight at 37 °C.
ELISA
Cytokine levels of IL1β, were determined using DuoSet ELISA Development Systems (R&D Systems) per manufacturer’s protocol. Cell culture supernatants from 4 biological replicates (cells plated in 12-well plates 1×106 cells/well) were diluted 1:5 for early time points and 1:10 for later (24h) time points. Mtb infected cells were diluted 1:2. Plates were read at 450 and 570 nm with Gen5 software using a Cytation 5 (Biotek).
Flow cytometry
For submicron analysis of mitochondria by flow cytometry, cells were lifted off non-tissue culture treated plates using 1X PBS EDTA and added to a 96-well V bottom plate. Cells were pelleted by centrifugation at 400 rcf for 3 min. Cells were resuspended in 1X PBS 2% FBS 200 nM MitoTracker green (Invitrogen) and allowed to stain for 15 min at 37 °C. Cells were then washed once with PBS 2% FBS and were resuspend in ice cold mitoFLOW buffer containing 300 mM sucrose, 10 mM Tris (pH 7.4), 0.5 mM EDTA, and 1X Halt Protease Inhibitor Cocktail. Lysis was performed by vortexing cells for 3 min followed by removal of debris by centrifugation at 400 rcf for 5 min at 4 °C. For antibody labeling samples were centrifuged at 12,000 rcf for 10 min at 4 °C and resuspend in 50 μL blocking buffer (5% BSA in mitoFLOW buffer),and incubated on ice for 15 min. Blocking was followed by an additional 20 min incubation on ice with antibodies of interest (GSDMD-PE 1:500, Abcam). Mitochondria were washed 2 times in mitoFLOW buffer and were analyzed on an LSR Fortessa X20 (BD Biosciences). Flow-Jo software was used for post-acquisition analysis MitoTracker green was used to gate on mitochondria.
For JC-1 assays to assess mitochondrial membrane potential, cells were lifted from culture plates with 1X PBS + EDTA. Single cell suspensions were made in 1X PBS 4% FBS. JC-1 dye was sonicated for 5 min with 30 second intervals. Cells were stained for 30 min at 37 °C in 1 μM JC-1 dye, washed twice in PBS 4% FBS and analyzed on an LSR Fortessa X20 (BD Biosciences). Flow-Jo software was used for post-acquisition analysis. Aggregates were measured under Texas Red (610/20 600LP) and monomers under FITC (525/50 505LP). To assess mitochondrial membrane potential under stress, cells were treated for 3 hrs with 2.5 μM rotenone prior to being lifted off the culture plates. 5 μM ATP was then added for 5, or 30 min. As a positive control 50 μM FCCP was added for 15 min.
For TMRE assays to assess mitochondrial membrane potential, cells were lifted from culture plates with 1X PBS EDTA. Single cell suspensions were made in 1X PBS 4% FBS. Cells were stained for 20 min at 37 °C in 25 nM TMRE dye, washed 1X in PBS 4% FBS and analyzed on an LSR Fortessa X20 (BD Biosciences). Flow-Jo software was used for post-acquisition analysis. Fluorescence was measured under PE (585/15). To assess mitochondrial membrane potential during inflammasome activation cells were stimulated with 10 ng/mL LPS for 3 hrs followed by poly dA:dT for an additional 2 hrs prior to being lifted off the plates.
For cell death and apoptosis assays cells, were stimulated with 10 ng/mL LPS for 3 hrs followed by 1 μg/mL poly dA:dT, or 50 μM etoposide (Fisher), or 10 μM ABT737 (ChemCruz), for 6 hrs prior to being lifted off the plates. Single cell suspensions were made in 1X PBS 4% FBS. Cells were stained for 5 min at RT in 5 μg/mL propidium iodide, and 25 nM annexin-V (APC, eBioscience) and were then immediately analyzed on an LSR Fortessa X20 (BD Biosciences).
Ex vivo stimulation and analysis of PBMCs by flow cytometry was performed essentially as described in: Lei et al.(Lei et al., 2021). Briefly, mice were deeply anesthetized and whole blood was collected in sodium heparin tubes. Red blood cells were subjected to 2 rounds of lysis with 1X ACK lysis buffer. Leukocytes were then stimulated in RPMI+10% FBS containing LPS (1 μg/mL) in the presence of protein transport inhibitors brefeldin A and monensin for 4 hrs. Ghost Dye 710 (Tonbo) was used as a live dead stain. Fc receptors were blocked with anti-mouse CD16/CD32 Fc shield (2.4G2, 70–0161, Tonbo), and cells were stained with antibodies against surface proteins CD3 (APC-fire, BioLegend), CD11b (APC Cy7, Tonbo), CD19 (BV605, BioLegend), Ly6C (BV650, BioLegend), MHCII (BV711, BioLegend), Ly6G (PE, Tonbo ), F4/80 (PerCP- C5.5, Tonbo). Permeabilization of cells was achieved with Foxp3/Transcription Factor Staining Buffer Kit (TNB-0607-KIT, Tonbo), and cells were stained with antibodies against intracellular TNF-α (PEC7, BioLegend). Flow cytometry was performed on a 5-laser Cytek Aurora, and Flow-Jo software was used for post-acquisition analysis.
For ex vivo analysis of lung cell populations on day 21 post Mtb infection, the inferior lobe was isolated and washed in 1X PBS followed by mincing and digestion for 1 hr in Dispase (5 U/ml) at 37 °C. Following digestion tissue was passed through 70 μm filters to achieve single cell suspensions. Live dead staining was performed using Ghost dye 510 (Tonbo). Fc receptors were blocked using CD16/CD32 monoclonal antibody (eBiosciences). Cells were stained with antibodies against surface proteins CD11b (BV421, BD Biosciences), CD11c (BV605, BioLegend), CD45 (BV785, BioLegend), CD170 (eFluor-488, eBiosciences), MHCII (PE, BD Biosciences), Ly6G (PerCP-Cy5.5, eBiosciences), Ly6C (APC, eBiosciences), CD206 (APCeFLuor-700 eBiosciences), B220 (APCeFluor-780, eBiosciences). Cells were washed 2 times before fixing in 4% PFA for 15 min at RT. Following fixation cells were washed twice with 1X PBS and incubated overnight at 4 °C. Cell counts were based on live single cells (Ghostlow/−) in 200 μL or 1/3 lung lobe. Flow cytometry was performed on the LSR Fortessa X20, and Flow-Jo software was used for post-acquisition analysis.
Cytoplasmic DNA enrichment
BMDMs were plated in 10 cm dishes at 1×107. The next day, plates were treated with LPS and poly dA:dT as indicated. To harvest, cells were lifted with 1X PBS EDTA. Cells were washed and resuspended in 5 mL 1X PBS. Total DNA was isolated from 2% of resuspended cells and treated with 25 mM NaOH. The samples were boiled for 30 min then neutralized with 50 mM TRIS pH 8.0. The remainder of the cell suspension was pelleted at 3000 rcf for 5 min. Cells were resuspended in 500 μL cytosolic lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 50 μg/mL digitonin, 10 mM EDTA) and incubated on ice for 15 min. Cells were spun down at 1000 rcf to pellet intact cells and nuclei that were then used for obtaining the membrane fraction. The supernatant was transferred to a fresh tube and spun down at 15000 rcf to remove additional organelle fragments and transferred to a fresh tube. Cytosolic protein was obtained by transferring 10% of supernatant to a fresh tube with 4X sample buffer + DTT and boiled for 5 min. Cytosolic DNA was isolated from the remaining supernatant by mixing an equal volume of 25:24:1 phenol: chloroform: isoamyl alcohol, vigorously shaken and centrifuged for 10 min at ~21130 rcf (max speed). The aqueous phase was transferred to a fresh tube and DNA was precipitated by mixing with 300 mM sodium acetate 10 mM MgCl2 and 1 μL glycogen and 3 volumes of 100% ethanol. The DNA was pelleted by centrifugation at max speed for 20 min at 4 °C. The pellet is washed with 1 mL of cold 70% ethanol and centrifuged for 5 min at max speed. The supernatant was removed, and the pellet was air dried for approximately 10 min. The pellet is resuspended with 200 μL EB. For the mitochondrial membrane fraction, the pellet of intact cells previously collected was resuspended in 500 μL membrane lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1% NP-40), vortexed then centrifuged for 3 min at 7000 rcf. 50 μL of the cleared lysate was transferred to a fresh tube and received 4X sample buffer + DTT. Western blot analysis was used to check for contaminating mitochondrial proteins in the cytosolic fraction compared to the membrane fraction. qPCR was performed using total DNA diluted 1:100 and cytosolic DNA diluted 1:2 and measured Tert, 16s, ND4 and DLoop. The total and cytosolic reactions were normalized to Tert to control for variation in cell numbers.
Immunofluorescence (IF) microscopy
MEFs were seeded at 1×105 cells/well on glass coverslips in 24-well dishes. BMDMs were seeded at 3×105 cells/well on glass coverslips in 24-well dishes. Cells were fixed in 4% PFA for 10 min at RT and then washed three times with PBS. Coverslips were incubated in primary antibody diluted in PBS + 5% non-fat milk + 0.1% Triton-X (PBS-MT) for 3 hrs. Primary antibodies used in this study were phospho-MLKL (Ser358) (D6H3V) (Cell Signaling Technology, #91689; 1:200), GSDMD [EPR20859] (Abcam, ab219800, 1:100), and TOM20 (clone 2F8.1; Millipore Sigma, MABT166, 1:100).Cells were then washed three times in PBS and incubated in secondary antibodies (goat anti-rabbit Alexa Fluor 488; Invitrogen, 1:500, or goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 594; Invitrogen, 1:1000) and DAPI (1:10,000) diluted in PBS-MT for 1 hr. Coverslips were washed twice with 1X PBS and twice with deionized water and mounted on glass slides using Prolong Gold Antifade Reagent (Invitrogen). Z-stack images were obtained using an Olympus IX83 inverted confocal microscope equipped with 60X oil immersion objective. Quantifications were performed using Fiji ImageJ. Images were opened as separate channels and z-stacks were converted to maximum intensity projections. Each channel was thresholded such that the region of interest was masked, and then particles (nuclei) counted and the region of interest (gasdermin D) measured. For LipidTOX assays fixed coverslips were washed gently three times with 1X PBS and incubated in DAPI (1:10,000) for five minutes. Coverslips were washed again gently three times with 1X PBS. LipidTOX neutral stain was diluted 1:1000 in 1X PBS. Cells were incubated with LipidTOX for 30 min in the dark at RT and mounted on microscope slides for imaging. Images were taken on the Olympus FV3000 confocal microscope at 60X. Quantification of lipid droplets was conducted manually. For annexin-V staining for cell death, cells on coverslips were infected with mCherry M. tuberculosis (Erdman) at MOI 2 for 24 and 48 hrs. Cells were washed 1X in cold PBS and incubated with 1.25 μL FITC annexin V-FITC (Biolegend Cat.640906) in cold annexin binding buffer (Biolegend Cat. 422201) for 15 min at RT in the dark. Cells were washed 1X with RT annexin binding buffer and fixed in cold 4% PFA in annexin binding buffer for 10 min. Cells were washed 2X with 1X PBS at RT, washed with DAPI-PBS, and mounted onto slides. Images were taken on Olympus FV3000 confocal microscope at 60X. Quantification of Mtb and Annexin V positive cells was conducted manually.
Mitochondrial isolation
Prior to downstream analysis, mitochondria were isolated using a Mitochondria/Cytosol Fractionation Kit (ab65320). Briefly, 5×106 BMDMs were lifted on 10 cm plates using 1X PBS-EDTA and pelleted at 600 rcf for 5 min at 4 °C. The cells were resuspended in 500 μL 1X Cytosolic Extraction Buffer Mix provided by the kit without DTT or Protease Inhibitors and incubated on ice for 10 min. Cells were homogenized on ice with 100 passes using the tight-fitting pestle B to lyse the cells followed by differential centrifugation. Homogenate was centrifuged at 700 rcf for 10 min at 4 °C to clear un-lysed cells and nuclei. The supernatant was collected and placed into a fresh tube and centrifuged at 10,000 rcf for 30 min at 4 °C. The supernatant was collected into a fresh tube and 4X sample buffer with 5 mM DTT was added. The pellet was either resuspended in 1X PBS for flow cytometry or lysed in sample buffer with 5 mM DTT for western analysis.
Protein quantification by immunoblot
Cells were washed with PBS and lysed in 1X RIPA buffer with protease and phosphatase inhibitors, with the addition of 1 U/mL Benzonase to degrade genomic DNA. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked for 1 hr at RT in LiCOR Odyssey blocking buffer or TBS with 5% BSA. Blots were incubated overnight at RT with the following antibodies: ACTB (Abcam, 1:2000), DRP1 (Cell Signaling, 1:1000); pDRP1 Ser616 (Cell Signaling, 1:1000), pDRP1 Ser637 (Cell Signaling, 1:1000),TFAM (Millipore, 1:1000), VDAC1 (Protein Tech, 1:1000). Membranes were incubated with appropriate secondary antibodies for 2 hrs at RT prior to imaging on a LiCOR Odyssey Fc Dual-Mode Imaging System. For protein isolated from lung, lung tissues were homogenized using Fisherbrand™ Disposable Tissue Grinders with 200 μl 1X PBS containing 20X proteinase and phosphatase inhibitors (Pierce). Samples were transferred to microcentrifuge tubes and spun for 1 hr at 10,000 rcf at 4 °C. Following spin, 200 μl of supernatants were transferred to tubes containing 20X SDS. Samples were boiled for 10 min. Protein concentrations were measured using a bicinchoninic acid assay (BCA) (Thermo Fisher Scientific). After the BCA, 50 μg of protein was run on Mini-PROTEAN TGX Stain-Free precast gels and transferred onto a 0.45 μm nitrocellulose membrane. Following blocking for 1 hr in 5% BSA/TBS, primary antibodies were diluted 1:1000 in 5% BSA/TBST and probed overnight at 4 °C. The membranes were then washed three times with TBST and incubated with the secondary antibody diluted 1:5000 for 2 hrs prior to imaging on a LI-COR Odyssey FC Imaging System. Primary antibodies used were α-RIP1 (Cell Signaling Technology #3493), α-RIP3 (Cell Signaling Technology #95702), and α-IL-1β (Cell Signaling Technology #12507) α-IFNβ (Santa Cruz Biotechnology # sc-57201)
GSDMD/CASP11 protein expression and purification
The cDNAs encoding human and mouse GSDMD were inserted into a modified pET28(a) vector with an N-terminal Avi-His6-SUMO-tag. The proteins were expressed in E. coli BL21 in regular LB medium (BD). When OD600 reached 1.0, the cells were induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and cultured overnight at 16 °C. The E. coli cells were harvested by centrifugation at 5000 rpm for 10 min. Then, the cells were lysed in a lysis buffer containing 50 mM Tris, 300 mM NaCl, pH 8.0 by sonication on ice. After centrifugation at 4 °C, 16000 rpm for 30 min, the supernatants containing the target proteins were loaded onto a nickel-NTA column equilibrated with the lysis buffer. After washing the column with 200 mL washing buffer (20 mM Tris, 500 mM NaCl, 25 mM Imidazole, pH 7.5), the target proteins were eluted with the elution buffer (20 mM Tris, 150 mM NaCl, 250 mM Imidazole, pH 7.5). The Avi-His6-SUMO tag was cleaved using SUMO protease at 4 °C overnight and removed using a nickel-NTA column. Human and mouse GSDMD were further purified by gel filtration chromatography using a Superdex200 (16/60 GL) column (GE Healthcare) in a running buffer containing 20 mM Tris, 150 mM NaCl, at pH 7.5.
To express inactive mouse caspase-11 catalytic domain (residue 100 to 373), it was cloned into a modified pET28(a) vector with an N-terminal His6-SUMO-tag. Cys254 in the active site of caspase-11 was mutated to alanine. Asp285 was replaced with a thrombin cleavage site with the sequence LVPRGS. The inactive mouse caspase-11 catalytic domain was expressed and purified using the same protocol used for GSDMD. After purification on a Superdex200 column, the inactive mouse caspase-11 catalytic domain was incubated with thrombin at 4 °C overnight and further purified on a Superdex200 column in the same running buffer as for GSDMD. To generate active mouse caspase-11 catalytic domain (residue 100 to 373), it was cloned into a modified pET28(a) vector with an N-terminal His6-SUMO-tag and a C-terminal His6-tag. There is a thrombin recognition site LVPRGS between His6- and SUMO-tag. Asp285 was replaced with a thrombin cleavage site. The His6-SUMO-mouse caspase-11 catalytic domain-His6 protein was expressed in E.coli BL21 and purified by nickel-NTA column using the same protocol used for GSDMD. The eluted protein from nickel-NTA column was incubated with SUMO protease and thrombin at 4 °C overnight. The active mouse caspase-11 catalytic domain-His6 protein was purified by a nickel-NTA column and SUMO-tag was removed. Then, it was further purified on a Superdex200 column in the same running buffer as for GSDMD.
All the proteins were stored in a buffer containing 20 mM Tris, 150 mM NaCl, 5 mM DTT, at pH 7.5. To test if the active mouse caspase-11 catalytic domain protein can cleave GSDMD or not, GSDMD was diluted into the reaction buffer containing 50 mM HEPES, 3 mM EDTA, 150 mM NaCl, 0.005% (v/v) Tween-20, 10 mM DTT, pH 7.5. The final concentration of GSDMD was 0.5 mg/ml and the ratio of GSDMD and caspase-11 was 10:1 (w/w). After incubation at 37 °C for 1 h, the samples were analyzed by SDS-PAGE.
In vivo Mtb infections
All infections were performed using procedures approved by Texas A&M University Institutional Care and Use Committee. The Mtb inoculum was prepared as described above. Age- and sex-matched mice were infected via inhalation exposure using a Madison chamber (Glas-Col) calibrated to introduce 100–200 CFUs per mouse. For each infection, approximately 5 mice were euthanized immediately, and their lungs were homogenized and plated to verify an accurate inoculum. Infected mice were housed under BSL3 containment and monitored daily by lab members and veterinary staff. At the indicated time points, mice were euthanized, and tissue samples were collected. Blood was collected in serum collection tubes, allowed to clot for 1–2 hrs at RT, and spun at 1000 rpm for 10 min to separate serum. Serum cytokine analysis was performed by Eve Technologies. Organs were divided to maximize infection readouts (CFUs: left lobe lung and ½ spleen; histology: 2 right lung lobes and ¼ spleen; RNA: 1 right lung lobe and ¼ spleen). For histological analysis organs were fixed for 24 hrs in neutral buffered formalin and moved to ethanol (lung, spleen). Organs were further processed as described below. For cytokine transcript analysis, organs were homogenized in Trizol, and RNA was isolated as described below. For CFU enumeration, organs were homogenized in 5 ml PBS + 0.1% Tween-80, and serial dilutions were plated on 7H10 plates. Colonies were counted after plates were incubated at 37 °C for 3 weeks.
Histopathology
Lungs and spleens fixed with 10% neutral buffered formalin followed by 70% ethanol were subjected to routine processing, embedded in paraffin, and 5 μm sections were cut and stained with hematoxylin and eosin (H&E) or acid-fast stain (Diagnostic BioSystems). A board certified veterinary pathologist performed a masked evaluation of lung sections for inflammation using a scoring system: score 0, none; score 1, up to 25% of fields; score 2, 26–50% of fields; score 3, 51–75% of fields; score 4, 76–100% of fields. To quantify the percentage of lung fields occupied by inflammatory nodules, scanned images of at least 2 sections of each lung were analyzed using Fiji ImageJ (NIH) to determine the total cross-sectional area of inflammatory nodules per total lung cross sectional area(Schindelin et al., 2012).
Immuno-multiplex assay
Sera was analyzed by Eve Technologies: Mouse Cytokine Array/Chemokine Array 13-plex Secondary Panel (MD13). Briefly, sera was isolated following decapitation in Microtainer serum separator tubes (BD Biosciences) followed by sterile filtration 2 times with Ultrafree-MC sterile filters, 10 min at 10,000 rpm (Millipore Sigma). For analysis sera was prediluted 1:1 to a final volume of 100 μL in 1x PBS pH 7.4 and assayed/analyzed in duplicate.
mtDNA from serum
Serum samples from untreated, day 21 or day 63 infected mice were boiled for 10 min and DNA was extracted using a DNeasy Blood & Tissue Kit (Cat # 69504), following the protocol for nonnucleated blood. 100 uL of boiled serum was used for all samples. A standard curve was generated by pooling all samples and serial diluting by a factor of 5 while the sample DNA was diluted 1 to 10. mtDNA was measured by qPCR amplifying DLoop.
Oral Pseudomonas entomophila (P.e.) fly infection
Bacterial entomopathogen strain P.e. was grown in LB medium at 29°C, shaking at 200 rpm overnight. Fresh bacterial cultures were generated daily. The liquid cultures were poured into a sterile centrifuge flask and centrifuged at 4000 rcf at 4 °C for 15 min. The liquid LB medium was removed from the centrifuge flask, and the bacterial pellet was resuspended in a small amount of LB medium. The final bacteria concentration (of the resuspended pellet, OD600=50–60 for P.e.) was adjusted by diluting with additional LB medium. Bacterial cultures were routinely genotyped for accuracy and reproducibility of experiments.
To prepare the inoculum, 2.5% and 5% sucrose (in sterile water) were prepared fresh. The 5% sucrose solution was mixed with an equal volume of bacteria solution (at OD600=50–60 for P.e.) to create the solution used for oral infection (feeding). NOTE: This OD were used for all experiments except for survival analysis. 10 day-old mated female flies (20 per vial) were transferred to a fly food vial containing filter paper completely covering the food. Filter paper was soaked with a solution consisting of either 185 μL bacterial oral infection mix (for infections) or 2.5% sucrose solution, for unchallenged (mock) controls. Flies were always infected at 3:00 – 4:00 pm to ensure diurnal reproducibility, and subsequently incubated at 25 °C and 65% humidity on a 12 hr light/dark cycle for required infection times (16–20h for P.e.) while feeding.
Drosophila immunostaining, Nile Red staining, and microscopy
For fat body/adipose immunostaining, carcass was dissected (with all eggs and intact intestines removed) in 1X PBS and fixed with 4% paraformaldehyde for 20 min at room temperature, washed 3 times with 1X PBS containing 0.2% Triton X-100 (1X PBST) and then blocked in blocking buffer (5% BSA in 1X PBST) for 1h. Primary antibodies; anti-ATP5A (ab14748, 1:500, Abcam) was applied overnight at 4 °C. Samples were then incubated with Alexa Flour-conjugated secondary antibodies (Jackson Immunoresearch, 1:500), and fresh Nile Red solution (1 μL 0.004% Nile Red Solution in 500 μL 1X PBS) with Hoechst (DAPI; 1:500), overnight at 4 °C, followed by rinsing with 1X PBS. Confocal images were collected using a Nikon Eclipse Ti confocal system (utilizing a single focal plane) and processed using the Nikon software and Adobe Photoshop. ATP5A Staining intensity was quantified using ImageJ. The image scale was set according the image size (Analyze > set scale). The ATP5A signal was marked using freehand selection tools. The area of marked signal was measured using the measure tool (Analyze > measure). At least 3–5 individual fat body whole images (using a global scale) were measured and averaged.
Bacterial load (CFUs) in fly midguts
Mock-treated or P.e. infected flies were sterilized with 70% ethanol and washed with sterile 1X PBS along with dissecting forceps and dissecting dish. Midguts from flies were then dissected individually in 1X PBS. Each gut was homogenized using a sterile pestle in 1.5 mL Eppendorf tubes containing 200 μL 1X PBS. Homogenates were diluted 1:1000 with 1X PBS. 50 μL of the diluted homogenates were plated on LB plates and incubated at 29 °C overnight. The number of bacterial colonies (colony forming units; CFUs) in each plate were counted (only counting separated, well defined single colonies). At least 10 individual guts were measured for each treatment. Colonies derived from midgut dissections were routinely genotyped (using P.e. specific primers) for accuracy and reproducibility of experiments. All plates from mock-treated controls were negative for bacterial colonies. CFU/fly for every plate was calculated using the following formulas: CFU/ml = ((total number of colonies on a plate) * (dilution factor (1000)) / (plated volume: 0.05ml). CFU/fly = ((CFU/ml) * total volume Eppendorf tube: 0.2ml)) / (number of flies per condition: 1 gut).
Survival analysis
Flies were infected with P.e. at OD600=30–40 as described above and kept at 25 °C for 16 hrs. After overnight feeding (flies were always orally infected at 3:00 – 4:00 pm), bacterial infected flies were transferred from bacterial infection vials to vials containing standard lab food. Flies were transferred every day to a fresh vial for the first two days, and every two days after, and dead flies were counted (and removed) when changing vials.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are representative of two or more independent experiments with n=3 or greater unless specifically noted in the figure legends. For all quantifications, n represents the number of biological replicates, either number of wells containing cells, number of mice, or number of flies. Error bars represent SEM. For in vitro assays, statistical significance was determined using either a two-tailed Student’s unpaired T test, one-way ANOVA with Sidak’s post hoc test, or two-way ANOVA with Tukey’s post hoc test. For in vivo mouse infections, significance was determined using a Mann-Whitney U test based on the assumption that samples (mice) followed a non-normal distribution. The specific statistical test used to determine significance for each experiment is listed at the end of the figure legends. All calculations of significance were determined using GraphPad Prism 7 Software expressed as P values The threshold for significance was determined by a p value of < 0.05, and annotated as * = p<0.05, ** = p<0.01, *** = p<0.001. For in vivo infection mouse experiments, we estimated that detecting a significant effect requires two samples to differ in CFUs by 0.7ê10. Using a standard deviation of 0.3ê10 for each population, we calculated that a minimum size of 4 age- and sex-matched mice per group per time point is necessary to detect a statistically significant difference by a t-test with alpha (2-sided) set at 0.05 and a power of 80%. Therefore, we used a minimum of 4 mice per genotype per time point to assess infection-related readouts.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-GSDMD-PE | Abcam | ab246713 |
| anti-mouse CD16/CD32 Fc shield | Tonbo | 2.4G2, 70-0161 |
| anti-CD3 | BioLegend | APC-fire |
| anti-CD11b | Tonbo | APC Cy7 |
| anti-CD19 | BioLegend | BV605 |
| anti-Ly6C | BioLegend | BV650 |
| anti-MHCII | BioLegend | BV711 |
| anti-Ly6G | Tonbo | PE |
| anti-F4/80 | Tonbo | PerCP-C5.5 |
| anti-TNF-α | BioLegend | PEC7 |
| anti-CD16/CD32 monoclonal | eBiosciences | 56-0161-82 |
| anti-CD11b | BD Biosciences | BV421 |
| anti-CD11c | BioLegend | BV605 |
| anti-CD45 | BioLegend | BV785 |
| anti-CD170 | eBiosciences | eFluor-488 |
| anti-MHCII | BD Biosciences | PE |
| anti-Ly6G | eBiosciences | PerCP-Cy5.5 |
| anti-Ly6C | eBiosciences | APC |
| anti-CD206 | eBiosciences | APCeFLuor-700 |
| anti-B220 | eBiosciences | APCeFluor-780 |
| anti-phospho-MLKL (Ser358) (D6H3V) | Cell Signaling Technology | #91689 |
| anti-GSDMD [EPR20859] | Abcam | ab219800 |
| anti-Tom20 | Millipore Sigma | MABT166 |
| anti-Beta Actin | Abcam | ab6276 |
| anti-DRP1 | Cell Signaling | ab184247 |
| anti-pDRP1 Ser616 | Cell Signaling | #3455S |
| anti-pDRP1 Ser637 | Cell Signaling | #4867S |
| anti-TFAM | Millipore | ABE483 |
| anti-VDAC | Protein Tech | #820702 |
| anti-ATP5A | Abcam | ab14748 |
| anti-phospho-MLKL | Abcam | ab196436 |
| anti-MLKL | Sigma-Aldrich | MABC604 |
| anti-IFNb | Santa Cruz Biotechnology | sc-57201 |
| anti-RIP3 | Cell Signaling Technology | #95702 |
| anti-RIP1 | Cell Signaling Technology | #3493 |
| anti-IL-IB | Cell Signaling Technology | #12507 |
| Bacterial and virus strains | ||
| M. tuberculosis | Erdman | |
| M. marinum | BD Biosciences | |
| L. monocytogenes | Robert Watson | strain 10304 |
| S. enterica (ser. Typhimirium) | Denise Monack, Stanford | SL1344 |
| P. entomophila | Jason Karpac | |
| Chemicals, peptides, and recombinant proteins | ||
| BSA Fraction V | Thermo Fisher | BP1600-100 |
| MCSF conditioned media | Watson lab | |
| Dispase II | Sigma | D4693 |
| DNASE I | Worthington | LS002138 |
| Horse serum | Gibco | 16050-130 |
| HCS LipidTOX Green Neutral Lipid Stain | Thermo Fisher | H34475 |
| NucBlue | Thermo Fisher | R37605 |
| mitoSOX | Thermo Fisher | M36008 |
| cellROX green | Thermo Fisher | C10444 |
| TMRE | Invitrogen | 11560796 |
| JC-1; CB1C2 | Invitrogen | T3168 |
| Propidium Iodide | Invitrogen | P1304MP |
| Annexin-V AF647 | Biolegend | 640912 |
| Annexin-V FITC | Biolegend | 640906 |
| Annexin binding buffer | Biolegend | 422201 |
| etoposide | Fisher | E0675 |
| ABT737 | ChemCruz | Sc-207242 |
| Mdivi-1 | abcam | Ab144589 |
| Nigericin, free acid | Molecular Probes | N1495 |
| Poly(dA:dT) naked | InvivoGen | tlrl-patn-1 |
| CL097 | InvivoGen | tlrl-c97 |
| LPS | InvivoGen | tlrl-pb5lps |
| cGAMP | InvivoGen | tlrl-nacga23-02 |
| Pam3CSK4 | InvivoGen | tlrl-pms |
| ISD | Watson lab | |
| AC-YVAD-CMK | InvivoGen | 10014 |
| Dapansutrile | Cayman Chemicals | 24671 |
| GNE9605 | Cayman Chemicals | 23446 |
| Z-IETD-FMK | BD biosciences | 550380 |
| necrostatin-1 (CAS 4311-88-0) | Calbiochem | 480065 |
| GSK2982772 | Cayman Chemicals | 29230 |
| GSK872 | Cayman Chemicals | 23300 |
| 2’,3’-Dideoxycytidine (ddC) | abcam | Ab142240 |
| necrox-5 | Cayman chemicals | 17278 |
| menadione | Sigma | M2518 |
| disulfiram | Sigma | PHR1690 |
| necrosulfamide | EMD millipore | 480073 |
| Fugene SI | Fugene | SI-1000 |
| Lipofectamine 2000 | Thermo Fisher | 52887 |
| GeneJuice | Novagen | 70967 |
| Full length murine GSDMD | Pingwei Li | |
| 100-373aa murine CASP11 | Pingwei Li | |
| 100-373aa (C254A) murine CASP11 | Pingwei Li | |
| Critical commercial assays | ||
| Seahorse XF mito stress test kits | Agilent | 103015-100 |
| Direct-zol RNAeasy kit | Zymogen | R2052 |
| iScript Direct Synthesis kit | BioRad | 1708891 |
| Foxp3/Transcription Factor Staining Buffer Kit | Tonbo | TNB-0607-KIT |
| Mitochondria/Cytosol Fractionation Kit | Abcam | ab65320 |
| Bicinchoninic acid assay (BCA) | Thermo Fisher | #23225 |
| Duo-set IL1B ELISA | R&D Systems | DY401-05 |
| DNeasy Blood & Tissue Kit | Qiagen | 69504 |
| Experimental models: Organisms/strains | ||
| B6.Cg-Tg(Lrrk2*G2019S)2Yue/J mice | Jackson Laboratories | stock # 012467 |
| B6.Cg-Lrrk2tm1.1Hlme/J | Jackson Laboratories | stock # 030961 |
| B6.Cg-Tg(Lrrk2)6Yue/J | M. Tansey University of Florida | |
| Drosophila w1118 | Bloomington Drosophila Stock Center | |
| Drosophila DaGal4 | Bloomington Drosophila Stock Center | |
| Drosophila UAS-hLRRK2-G2019S | C. Elliott and W. Smith | |
| Drosophila UAS-hLRRK2-G2019S-K1906M | C. Elliott and W. Smith | |
| Drosophila CGGal4 | C. Thummel | |
| Oligonucleotides | ||
| Mouse beta Actin RT-qPCR Forward:GGTGTGATGGTGGGAATGG Reverse:GCCCTCGTCACCCACATAGGA |
This paper | N/A |
| Mouse Ifnb1 RT-qPCR Forward:TCCGAGCAGAGATCTTCAGGAA Reverse:TGCAACCACCACTCATTCTGA |
This paper | N/A |
| Mouse Isg15 RT-qPCR Forward:GAGCTAGAGCCTGCAGCAAT Reverse:TTCTGGGCAATCTGCTTCTT |
This paper | N/A |
| Mouse Irf7 RT-qPCR Forward:CTTCAGCACTTTCTTCCGAGA Reverse:TGTAGTGTGGTGACCCTTGC |
This paper | N/A |
| Mouse Ifit1 RT-qPCR Forward:CGTAGCCTATCGCCAAGATTTA Reverse:AGCTTTAGGGCAAGGAGAAC |
This paper | N/A |
| Mouse TNF RT-qPCR Forward:ATGGCCTCCCTCTCATCAGT Reverse:GTTTGCTACGACGTGGGCTA |
This paper | N/A |
| Mouse Il1b RT-qPCR Forward:GGTGTGTGACGTTCCCATTA Reverse:ATTGAGGTGGAGAGCTTTCAG |
This paper | N/A |
| Mouse Il18 RT-qPCR Forward:GAAGGACACTTTCTTGCTTGC Reverse:GTGAGAGTGAACATTACAGATTTATCC |
This paper | N/A |
| Mouse Il1a RT-qPCR Forward:CTCTGAGAACCTCTGAAACGTC Reverse:GAAACTCAGCCGTCTCTTCTT |
This paper | N/A |
| Mouse Il6 RT-qPCR Forward:CCAGAGTCCTTCAGAGAGATACA Reverse:CCTTCTGTGACTCCAGCTTATC |
This paper | N/A |
| Mouse Aim2 RT-qPCR Forward:AGGCAGTGGGAACAAGACAG Reverse:GAAAACTTCCTGACGCCACC |
This paper | N/A |
| Mouse Nlrp3 RT-qPCR Forward:AAAATGCCTTGGGAGATCCA Reverse:AAGTAAGGCCAGAATTCACC |
This paper | N/A |
| Mouse Pycard RT-qPCR Forward:ACTGTGCTTAGAGACATGGGC Reverse:TGGTCCACAAAGTGTCCTGTT |
This paper | N/A |
| Mouse Gsdmd RT-qPCR Forward:CAAGGTTCTGGAAACCCCGTT Reverse:CCAAAACACTCCGGTTCTGGTTC |
This paper | N/A |
| Mouse Zbp1 RT-qPCR Forward:CCCAGCCTAGCCTTGATGAAA Reverse:TTTGGCTGTCGTCATTCCCA |
This paper | N/A |
| Mouse Nfe212 RT-qPCR Forward:TCCATTCCCGAATTACAGTGTC Reverse:TCCAGCGAGGAGATCGATGA |
This paper | N/A |
| Mouse Nq01 RT-qPCR Forward:CAACGGTCCTTTCCAGAATAAGAA Reverse:GAAGCCACAGAAACGCAGGA |
This paper | N/A |
| Mouse Hmox1 RT-qPCR Forward:GCCACCAAGGAGGTACACAT Reverse:AGGAAGCCATCACCAGCTTAAA |
This paper | N/A |
| Mouse Mlkl RT-qPCR Forward: TCTTTCTGGCAGAGAACGAATCT Reverse: TCTTACACCTTCTTGTCCGTGG |
This paper | N/A |
| Mouse Lrrk2 RT-qPCR Forward:GATCTCTGCACTCAGCTGTTTA Reverse: GCTTCTCACTGTCTTCCTCTTC |
This paper | N/A |
| Mouse Ifng RT-qPCR Forward:CTCTTCCTCATGGCTGTTTCT Reverse: CGCTTATGTTGCTGATGG |
This paper | N/A |
| Mouse Cxcl1 RT-qPCR Forward: CCGAAGTCATAGCCACACTCA Reverse: CTCCGTTACTTGGGGACACC |
This paper | N/A |
| Mouse Cxcl5 RT-qPCR Forward: TGCCCTACGGTGGAAGTCAT Reverse: AGCTTTCTTTTTGTCACTGCCC |
This paper | N/A |
| Fly attA RT-qPCR Forward:TCGTTTGGATCTGACCAAGGGCAT Reverse: TTCCGCTGGAACTCGAAACCATTG |
This paper | N/A |
| Fly Drs RT-qPCR Forward:AAGTACTTGTTCGCCCTCTTCGCT Reverse:TCCTTCGCACCAGCACTTCAGACT |
This paper | NA |
| Nuclear Tert qPCR Foward:CTAGCTCATGTGTCAAGACCCTCTT Reverse:GCCAGCACGTTTCTCTCGTT |
This paper | N/A |
| Mitochondrial mt16s qPCR Forward:CACTGCCTGCCCAGTGA Reverse:ATACCGCGGCCGTTAAA |
This paper | N/A |
| Mitochondrial mtDloop qPCR Forward: CCCTTCTTTATTTGGTCT Reverse:TGGTTTCACGGAGGATGG |
This paper | N/A |
| Mitochondrial mtND4 Forward: AACGGATCCACAGCCGTA Reverse: AGTCCTCGGGCCATGATT |
This paper | N/A |
| Control Silencer Select Negative control siRNA | Thermo Fisher | #4390843 |
| Zbp1 siRNA | Thermo Fisher | #s233871 |
| MLKL siRNA | Thermo Fisher | #s92952 |
| GSDMD siRNA | Thermo Fisher | #s87492 |
| Ticam1 (Trif) siRNA | Thermo Fisher | #s98708 |
| CpG 2395 T*C*G*T*C*G*T*T*T*T*C*G*G*C*C*C*G* |
IDT | N/A |
| Software and algorithms | ||
| Image J | NIH | |
| FlowJo v10 | BD biosciences | |
| Nikon software | Nikon | |
| Prism v7 | Graph Pad | |
| Gen 3.5 | BioTek | |
| Adobe Photoshop CC 2018 | Adobe | |
| Adobe Illustrator CC 2018 | Adobe | |
| WAVE Desktop | Agilent | |
HIGHLIGHTS.
Altered mitochondrial homeostasis reprograms cell death modalities
Mitochondrial ROS directs GSDMD to mitochondria following inflammasome activation
Mito-GSDMD shifts cell death from pyroptosis to necroptosis in Lrrk2G2019S macrophages
Lrrk2G2019S elicits hyperinflammation and susceptibility to infection in flies and mice
ACKNOWLEDGEMENTS
We would like to thank C. Elliott at the University of York (U.K.) for providing us with hLRRK2-expressing Drosophila and Dr. Raquel Sitcheran and Michael Kamradt for technical guidance on mitochondrial flow cytometry. Thanks to Dr. Gerry Shadel for his invaluable advice about mitochondrial ROS. We are grateful for the technical expertise of Malea Murphy at the Integrated Microscopy and Imaging Laboratory and Robbie Moore at the COM-CAF core facility. We are supremely appreciative of Drs. Malu Tansey and Rebecca Wallings at the University of Florida for sharing femurs from B6.Cg-Tg(Lrrk2)6Yue/J mice. This work was supported by funds from the Michael J. Fox Foundation for Parkinson’s Research Grant 12185 (to ROW), the National Institutes of Health, grant R01 AI155621 (to ROW and APW), NIH grant R01 AI145287 (to PL and ROW), and the Texas A&M Clinical Science and Translational Research (CSTR) Pilot Grant Program (to ROW). JK was supported by NIH R01 DK108930 and NIH R56 DK108930–06. Additional funding was provided by the Parkinson’s Foundation Postdoctoral Fellowship (to CGW), NIH training grant 5T32OD011083–10 and Texas A&M CVM Postdoctoral Trainee Research Training Grant (to KJV), and NIH training grant T32GM135748 (to EM and AKC).
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Footnotes
DECLARATIONS OF INTEREST
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agalliu I, San Luciano M, Mirelman A, Giladi N, Waro B, Aasly J, Inzelberg R, Hassin-Baer S, Friedman E, Ruiz-Martinez J, et al. (2015). Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease: a pooled analysis. JAMA Neurol 72, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P, Cermelli S, Li Z, Kassan A, Bosch M, Sigua R, Huang L, Ouellette AJ, Pol A, Welte MA, et al. (2012). A novel role for lipid droplets in the organismal antibacterial response. Elife 1, e00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand VS, and Braithwaite SP (2009). LRRK2 in Parkinson’s disease: biochemical functions. FEBS J 276, 6428–6435. [DOI] [PubMed] [Google Scholar]
- Athanasopoulos PS, Heumann R, and Kortholt A (2018). The role of (auto)-phosphorylation in the complex activation mechanism of LRRK2. Biol Chem 399, 643–647. [DOI] [PubMed] [Google Scholar]
- Bae JR, and Lee BD (2015). Function and dysfunction of leucine-rich repeat kinase 2 (LRRK2): Parkinson’s disease and beyond. BMB Rep 48, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Smith KA, and Lavoie MJ (2010). Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Sanchez-Alvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, Moreira L, Lopez JA, Campo R, Mari M, et al. (2020). Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science 370. [DOI] [PubMed] [Google Scholar]
- Chan FK, Luz NF, and Moriwaki K (2015). Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol 33, 79–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, et al. (2015). Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 17, 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR (2012). Cellular effects of LRRK2 mutations. Biochem Soc Trans 40, 1070–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cording AC, Shiaelis N, Petridi S, Middleton CA, Wilson LG, and Elliott CJH (2017). Targeted kinase inhibition relieves slowness and tremor in a Drosophila model of LRRK2 Parkinson’s disease. NPJ Parkinsons Dis 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkinderen P, and Neunlist M (2018). Crohn’s and Parkinson disease: is LRRK2 lurking around the corner? Nat Rev Gastroenterol Hepatol 15, 330–331. [DOI] [PubMed] [Google Scholar]
- Di Paolo NC, and Shayakhmetov DM (2016). Interleukin 1alpha and the inflammatory process. Nat Immunol 17, 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, Oberbeck-Muller D, Duque-Correa MA, Reece ST, Ruland J, et al. (2012). Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol 42, 374–384. [DOI] [PubMed] [Google Scholar]
- Eberhardt EL, Ludlam AV, Tan Z, and Cianfrocco MA (2020). Miro: A molecular switch at the center of mitochondrial regulation. Protein Sci 29, 1269–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava VM, Manry J, Cobat A, Orlova M, Van Thuc N, Ba NN, Thai VH, Abel L, Alcais A, Schurr E, et al. (2016). A Missense LRRK2 Variant Is a Risk Factor for Excessive Inflammatory Responses in Leprosy. PLoS Negl Trop Dis 10, e0004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H, Seeger JM, et al. (2019). Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575, 683–687. [DOI] [PubMed] [Google Scholar]
- Fulda S (2016). Regulation of necroptosis signaling and cell death by reactive oxygen species. Biol Chem 397, 657–660. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, et al. (2005). The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J Med Genet 42, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosal D, Ross OA, Wiley J, Irvine GB, Johnston JA, Toft M, Mata IF, Kachergus J, Hulihan M, Taylor JP, et al. (2005). Clinical traits of LRRK2-associated Parkinson’s disease in Ireland: a link between familial and idiopathic PD. Parkinsonism Relat Disord 11, 349–352. [DOI] [PubMed] [Google Scholar]
- Hartlova A, Herbst S, Peltier J, Rodgers A, Bilkei-Gorzo O, Fearns A, Dill BD, Lee H, Flynn R, Cowley SA, et al. (2018). LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst S, Campbell P, Harvey J, Bernard EM, Papayannopoulos V, Wood NW, Morris HR, and Gutierrez MG (2020). LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J, e104494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst S, and Gutierrez MG (2019). LRRK2 in Infection: Friend or Foe? ACS Infect Dis 5, 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DH, Je AR, Lee H, Son I, Kweon HS, Kim HG, and Seol W (2018). LRRK2 Kinase Activity Induces Mitochondrial Fission in Microglia via Drp1 and Modulates Neuroinflammation. Exp Neurobiol 27, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DH, Lee H, Son I, and Seol W (2019). G2019s LRRK2 promotes mitochondrial fission and increases TNFalpha-mediated neuroinflammation responses. Anim Cells Syst (Seoul) 23, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schule B, Krainc D, Palmer TD, and Wang X (2016). Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 19, 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YL, Zhang GH, Zhu Q, Wu X, and Wu LG (2021). Expression levels of caspase-3 and gasdermin E and their involvement in the occurrence and prognosis of lung cancer. Cancer Rep (Hoboken), e1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, Chuang LS, Carmi S, Villaverde N, Li X, et al. (2018). Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, Massimino L, Rubio A, Morabito G, Caporali L, et al. (2018). Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep 22, 2066–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, and O’Neill LA (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, et al. (2005). Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128, 2786–2796. [DOI] [PubMed] [Google Scholar]
- Lei Y, Guerra Martinez C, Torres-Odio S, Bell SL, Birdwell CE, Bryant JD, Tong CW, Watson RO, West LC, and West AP (2021). Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, and Kong AN (2009). Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog 48, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, and Yue Z (2007). Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J Neurochem 103, 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, and Shirihai OS (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Park H, Heisler J, Maculins T, Roose-Girma M, Xu M, McKenzie B, van Lookeren Campagne M, Newton K, and Murthy A (2019). Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tsai PI, Wu RM, and Chien CT (2010). LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J Neurosci 30, 13138–13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Li Y, Choi HMC, Sarkar C, Koh EY, Wu J, and Lipinski MM (2018). Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis 9, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, et al. (2008). A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A 105, 2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobbestael E, Baekelandt V, and Taymans JM (2012). Phosphorylation of LRRK2: from kinase to substrate. Biochem Soc Trans 40, 1102–1110. [DOI] [PubMed] [Google Scholar]
- Loor G, Kondapalli J, Schriewer JM, Chandel NS, Vanden Hoek TL, and Schumacker PT (2010). Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic Biol Med 49, 1925–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtmann MHR, Kostic M, Horne A, Gandhi S, Sekler I, and Abramov AY (2019). LRRK2 deficiency induced mitochondrial Ca(2+) efflux inhibition can be rescued by Na(+)/Ca(2+)/Li(+) exchanger upregulation. Cell Death Dis 10, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzon-Toro B, Rubio de la Torre E, Delgado A, Perez-Tur J, and Hilfiker S (2007). Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum Mol Genet 16, 2031–2039. [DOI] [PubMed] [Google Scholar]
- MacDonald JA, Bothun AM, Annis SN, Sheehan H, Ray S, Gao Y, Ivanov AR, Khrapko K, Tilly JL, and Woods DC (2019). A nanoscale, multi-parametric flow cytometry-based platform to study mitochondrial heterogeneity and mitochondrial DNA dynamics. Commun Biol 2, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, and Bryant CE (2013). Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol 191, 5239–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinek P, Jha AN, Shinde V, Sundaramoorthy A, Rajkumar R, Suryadevara NC, Neela SK, van Tong H, Balachander V, Valluri VL, et al. (2013). LRRK2 and RIPK2 variants in the NOD 2-mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PLoS One 8, e73103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas DPA, Cerqueira DM, Pereira MSF, Castanheira FVS, Fernandes TD, Manin GZ, Cunha LD, and Zamboni DS (2017). Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog 13, e1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, and Tschopp J (2004). RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol 5, 503–507. [DOI] [PubMed] [Google Scholar]
- Moehle MS, Daher JP, Hull TD, Boddu R, Abdelmotilib HA, Mobley J, Kannarkat GT, Tansey MG, and West AB (2015). The G2019S LRRK2 mutation increases myeloid cell chemotactic responses and enhances LRRK2 binding to actin-regulatory proteins. Hum Mol Genet 24, 4250–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, and Muller DJ (2018). Mechanism of membrane pore formation by human gasdermin-D. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, and Lim KL (2009). Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci 29, 11257–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onate M, Catenaccio A, Salvadores N, Saquel C, Martinez A, Moreno-Gonzalez I, Gamez N, Soto P, Soto C, Hetz C, et al. (2020). The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ 27, 1169–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Lien E, and Fitzgerald KA (2019). Gasdermins and their role in immunity and inflammation. J Exp Med 216, 2453–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, et al. (2006). LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 354, 424–425. [DOI] [PubMed] [Google Scholar]
- Patrick KL, and Watson RO (2021). Mitochondria: Powering the Innate Immune Response to Mycobacterium tuberculosis Infection. Infect Immun 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Carrion M, Pischedda F, Biosa A, Russo I, Straniero L, Civiero L, Guida M, Gloeckner CJ, Ticozzi N, Tiloca C, et al. (2018). The LRRK2 Variant E193K Prevents Mitochondrial Fission Upon MPP+ Treatment by Altering LRRK2 Binding to DRP1. Front Mol Neurosci 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, and Henry T (2012). AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ 19, 1709–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout HJ, and Re DB (2017). LRRK2 and the “LRRKtosome” at the Crossroads of Programmed Cell Death: Clues from RIP Kinase Relatives. Adv Neurobiol 14, 193–208. [DOI] [PubMed] [Google Scholar]
- Roca FJ, and Ramakrishnan L (2013). TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca FJ, Whitworth LJ, Redmond S, Jones AA, and Ramakrishnan L (2019). TNF Induces Pathogenic Programmed Macrophage Necrosis in Tuberculosis through a Mitochondrial-Lysosomal-Endoplasmic Reticulum Circuit. Cell 178, 1344–1361 e1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, and Alnemri ES (2019). Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10, 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, Yamamoto M, and Takeda K (2012). Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol 24, 637–644. [DOI] [PubMed] [Google Scholar]
- Samir P, Malireddi RKS, and Kanneganti TD (2020). The PANoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front Cell Infect Microbiol 10, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Kurz S, Manske K, Janas M, Heikenwalder M, Misgeld T, Aichler M, Weissmann SF, Zischka H, Knolle P, et al. (2019). Single organelle analysis to characterize mitochondrial function and crosstalk during viral infection. Sci Rep 9, 8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shindo R, Kakehashi H, Okumura K, Kumagai Y, and Nakano H (2013). Critical contribution of oxidative stress to TNFalpha-induced necroptosis downstream of RIPK1 activation. Biochem Biophys Res Commun 436, 212–216. [DOI] [PubMed] [Google Scholar]
- Shutinoski B, Hakimi M, Harmsen IE, Lunn M, Rocha J, Lengacher N, Zhou YY, Khan J, Nguyen A, Hake-Volling Q, et al. (2019). Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci Transl Med 11. [DOI] [PubMed] [Google Scholar]
- Srinivasan L, Ahlbrand S, and Briken V (2014). Interaction of Mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb Perspect Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YC, and Qi X (2013). Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet 22, 4545–4561. [DOI] [PubMed] [Google Scholar]
- Tait SW, and Green DR (2013). Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Lemoff A, Wang G, Zarek C, Lowe A, Yan N, and Reese TA (2020). Reactive oxygen species oxidize STING and suppress interferon production. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, et al. (2019). Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun 10, 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Goncalves A, Vande Walle L, Demon D, Matusiak M, Van Hauwermeiren F, et al. (2017). Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep 21, 3427–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallings R, Manzoni C, and Bandopadhyay R (2015). Cellular processes associated with LRRK2 function and dysfunction. FEBS J 282, 2806–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, and Zhu X (2012). LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 21, 1931–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, et al. (2015). Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 17, 799–810. [DOI] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, and Cox JS (2015). The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 17, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, and Watson RO (2020). LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, Chen X, Liang Y, Wu J, Zhao S, et al. (2018). RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol 20, 186–197. [DOI] [PubMed] [Google Scholar]
- Yuan J, Amin P, and Ofengeim D (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci 20, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, Cui Y, Yan XX, Yang HT, Yang RD, et al. (2009). Genomewide association study of leprosy. N Engl J Med 361, 2609–2618. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, Hong Z, Wu X, Zhao Y, and Ren J (2020). mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis 11, 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, and Karpac J (2021). Glutamate metabolism directs energetic trade-offs to shape host-pathogen susceptibility in Drosophila. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.