

Abstract
Circulating proteins associated with transforming growth factor-β (TGF-β) signaling are implicated in the development of diabetic kidney disease (DKD). No study has comprehensively examined which of these proteins are involved in the pathogenesis of DKD and its progression to end-stage kidney disease (ESKD) in humans. Using the SOMAscan proteomic platform, we measured concentrations of 25 TGF-β-signaling family proteins, in 4 different cohorts comprised of 754 Caucasian and Pima Indian subjects with type 1 and type 2 diabetes. Of these 25 circulating proteins, we identified neuroblastoma suppressor of tumorigenicity 1 (NBL1, aliases DAN & DAND1), a small, secreted protein known to inhibit members of the bone morphogenic protein family, to be most strongly and independently associated with progression to ESKD during 10-year follow-up in all cohorts. The extent of damage to podocytes and other glomerular structures measured morphometrically in 105 research kidney biopsies correlated strongly with circulating NBL1 concentrations. Also, in vitro exposure to NBL1 induced apoptosis in podocytes. In conclusion, concentration of NBL1 in circulation may identify subjects with diabetes at increased risk of progression to ESKD and may serve as a target for pharmacological intervention of the TGF-β signaling pathway to slow the development and progression of DKD.
One Sentence Summary:
Proteomic profiling of circulating TGF-β signaling proteins in subjects with diabetes identified a robust association of NBL1 with 10-year risk of ESKD
Introduction
The transforming growth factor- β (TGF-β) superfamily of signaling proteins plays an important role in the development and progression of diabetic kidney diseases (DKD) (1–10). This superfamily consists of proteins active in intra- and extracellular compartments. In the latter, three sub-families of proteins play a role: TGF-β subfamily, bone morphogenic proteins (BMP), and activins (11). Interaction among these proteins regulates intracellular TGF-β signaling and impacts many processes including tissue differentiation during development and tissue repair in various diseases including DKD (1, 2). TGF-β superfamily proteins have been investigated in the context of DKD but mainly in in vitro and animal studies and without consideration for their extracellular vs. intracellular interactions (12–21). We hypothesize that circulating proteins involved in the extracellular regulation of TGF- β signaling are associated with DKD progression. Of 33 TGF- β signaling proteins recently reviewed (11), 25 were measured on the first version of the SOMAscan proteomics platform. Supplementary Table S1 lists the proteins and related publications (12–57). Supplementary Fig. S1 shows their possible interactions.
The present study was conducted in patients participating in two follow-up studies: the Joslin Kidney Study (JKS) and the Pima Indian Kidney Study. Subjects in these studies had baseline concentration of circulating proteins (1,129 proteins in 1st stage and 564 in 2nd stage) measured by the SOMAscan proteomics platform and were followed to ascertain progression to end-stage kidney disease (ESKD) during 10-year follow-up (58–60). The SOMAscan measurements were used to examine 25 pre-specified circulating proteins modulating TGF-β signaling as predictors for progression to ESKD. This targeted approach was used in our recent studies (61–63). In this study we identified increased concentration of neuroblastoma suppressor of tumorigenicity 1 (NBL1, aliases DAN & DAND1), a presumed inhibitor of BMP proteins, as a strong and independent predictor of progression to ESKD in all study cohorts (64–70). Furthermore, to explore possible mechanisms through which circulating NBL1 may damage kidney we used single nucleus sequencing (snRNA-seq) to examine NBL1 expression in kidney cells, immunostaining to examine NBL1 expression in kidney and 10 other tissues and assessed the in vitro effects of NBL1 on apoptosis of kidney cells.
Results
Study cohorts
Four independent cohorts of subjects with type 1 diabetes (T1D) and type 2 diabetes (T2D) at various DKD stages were followed to ascertain progression to ESKD. Clinical characteristics of the study cohorts are shown in Table 1. By design, the cohorts differed at baseline by race, diabetes type, age, sex, diabetes duration, glycemic control, albuminuria, and kidney function. Two cohorts recruited from the Joslin Kidney Study with late DKD at baseline, defined by albuminuria and impaired kidney function, were designated as the T1D Discovery and the T2D Replication cohorts. The late DKD T1D cohort was selected as the Discovery cohort since it had the greatest statistical power to detect circulating proteins associated with risk of ESKD due to the large number of cases. Positive findings from this cohort were investigated in the T2D Replication cohort and in two Validation cohorts with early DKD. The first Validation cohort included subjects with T1D recruited from the Joslin Kidney Study with albuminuria and normal kidney function. The second Validation cohort included subjects with T2D recruited from the Pima Indian Study with either normoalbuminuria or albuminuria but normal kidney function. In the Joslin cohorts, 92% of subjects were of European ancestry. All subjects in the Pima cohort were American Indians. A subgroup of 105 Pima Indians had research kidney biopsies performed in conjunction with the baseline examination.
Table. 1 |.
Clinical characteristics of subjects included in the study cohorts.
| Late DKD cohorts | Early DKD cohorts | ||||
|---|---|---|---|---|---|
| Characteristics | Joslin | Joslin | Joslin | Pima | Pima |
| T1D Discovery cohort | T2D Replication cohort | T1D Validation cohort 1 | T2D Validation cohort 2 | T2D cohort for auxiliary study* | |
| N | 219 | 144 | 238 | 153 | 105 |
| Male/Female (n) | 106/113 | 94/50 | 131/107 | 43/110 | 29/76 |
| Age (years) | 45 ± 10 | 60 ± 6 | 39 ± 9 | 46 ± 9.8 | 45 ± 10 |
| Diabetes duration (years) | 30 ± 9 | 16 ± 9 | 26 ± 9 | 16 ± 6 | 15 ± 6 |
| HbA1c (DCCT, %) | 8.8 ± 1.7 | 7.6 ± 1.6 | 9.0 ± 1.7 | 9.3 ± 2.3 | 9.2 ± 2.3 |
| SBP (mmHg) | 135 ± 20 | 140 ± 20 | 131 ± 18 | 124 ± 14 | 122 ± 13 |
| DBP (mmHg) | 77 ± 11 | 75 ± 11 | 78 ± 11 | 77 ± 9 | 77 ± 9 |
| ACR (mg/g) ¶ | 757 (214, 1792) | 255 (56, 1100) | 591 (250, 1195) | 55 (13, 357) | 40 (12, 124) |
| eGFR (ml/min/1.73m2)┼ | 43 ± 11 | 49 ± 11 | 98 ± 21 | 150 ± 47 | 151 ± 47 |
| During Follow-up | |||||
| GFR slope (ml/min/y) | −3.8 (−7.5, −1.9) | −3.2 (−6.4, −0.9) | −3.1 (−7.7, −1.3) | −4.0 (−8.9, −1.3) | −4.4 (−10.3, −1.6) |
| New cases of ESKD within 10 years (n (%)) | 108 (49%) | 35 (24%) | 50 (21%) | 34 (22%) | 15 (14%) |
Abbreviations: DKD, Diabetic kidney disease; Late DKD, individuals with impaired kidney function; Early DKD, individuals with normal kidney function; T1D, Type 1 Diabetes; T2D, Type 2 diabetes; DM, diabetes mellitus; HbA1c, hemoglobin A1c; SBP, Systolic blood pressure; DBP, Diastolic blood pressure; ACR, urine albumin to creatinine ratio; eGFR, estimated glomerular filtration; ESKD, End Stage Kidney Disease, i.e. dialysis or kidney transplant.
Subgroup of T2D Pima validation cohort for kidney structural study.
In Pima cohort, GFR (ml/min) was measured directly using urinary clearance of iothalamate.
Data are expressed as mean ± standard deviation or ¶median (25%, 75%).
Of the 754 subjects included in the study cohorts, 227 progressed to ESKD during the first 10 years of follow-up. In combined cohorts, follow-up was 10 (median) and 7–12 (25%, 75%) years in subjects who did not develop ESKD, and 4 (median) and 3–7 (25%, 75%) years in subjects who progressed to ESKD (Supplementary Fig. S2).
Circulating TGF-β signaling proteins and risk of ESKD
Baseline concentrations of the 25 circulating TGF-β signaling proteins were evaluated in the Discovery cohort for association with risk of ESKD. Seven of these proteins belonged to the TGF-β sub-family proteins, 13 to BMP sub-family proteins, and 5 to activin sub-family proteins. The names of these proteins and magnitude (Hazard Ratio [HR]) of their effects on the risk of ESKD in univariate Cox regression analysis are shown in Fig. 1. Six proteins were associated with 10-year risk of ESKD statistically significant after Bonferroni adjustment. These proteins included TGF-β receptor III (TGF-βRIII), Anti-Muellerian hormone type-2 receptor (AMHR2), repulsive guidance molecule BMP co-receptor A (RGMA), repulsive guidance molecule BMP co-receptor B (RGMB), NBL1, and Follistatin like 3 (FSTL3). Of the 6 proteins associated with risk of ESKD in the Discovery cohort, AMHR2 was not measured in the 2nd stage due to technical reasons and RGMA was not confirmed in the other cohorts (Supplementary Table S2).
Fig. 1 |. Association of ESKD risk in Discovery cohort with baseline concentration of 25 circulating candidate proteins. Results of univariate Cox regression analysis.
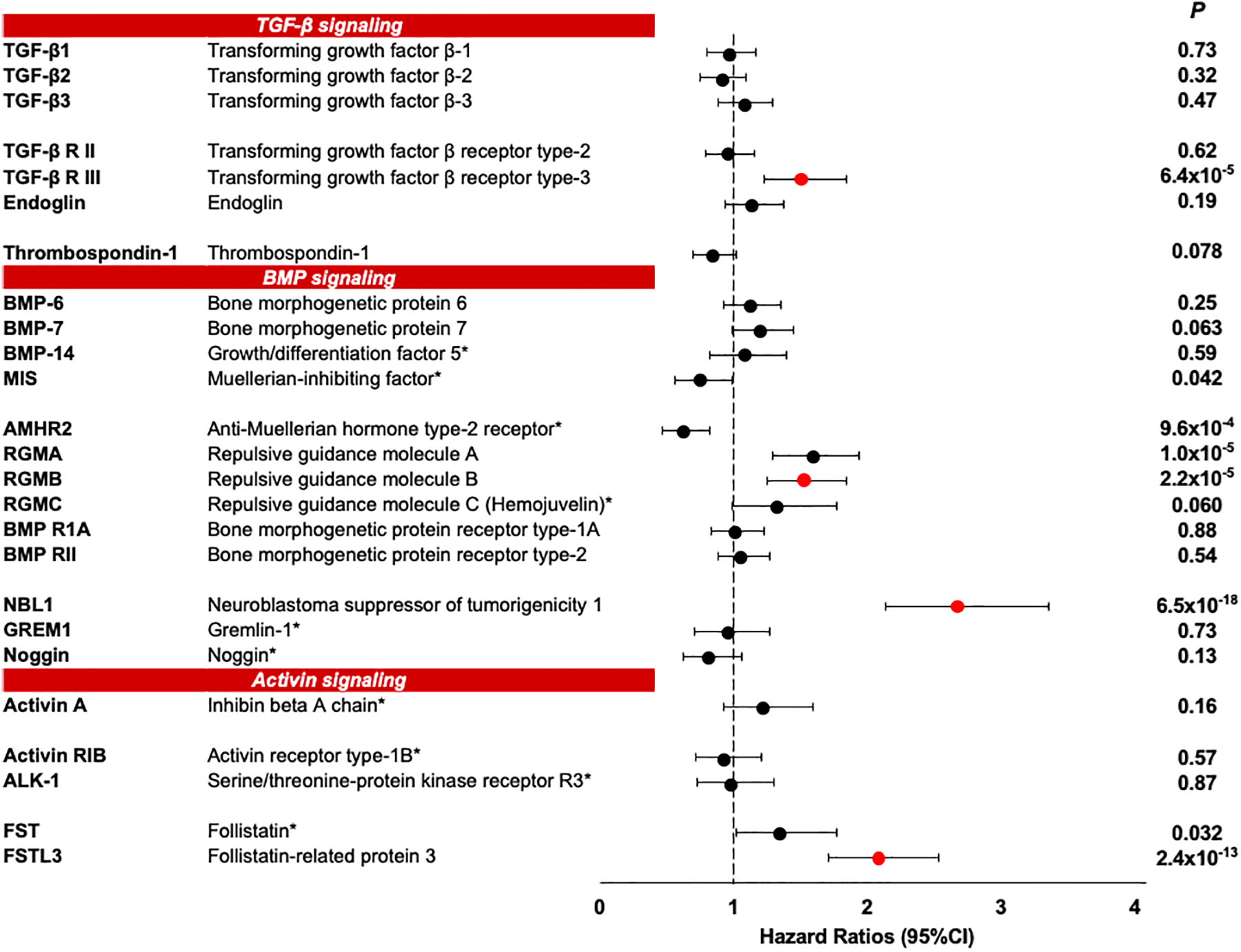
Effect size (Hazard Ratio and 95% CI) in Discovery cohort is presented per standard deviation change in protein concentration. Proteins are grouped to TGF-β signaling, BMP signaling, and Activin signaling. Bonferroni correction for n=25 independent tests (the number of examined proteins by the study design) yielded a threshold of P < 2.0×10−3. Bonferroni correction for n=1,129 independent tests (the number of proteins on SOMAscan) yielded a threshold of P < 4.0×10−5. Red dots show proteins that were replicated and validated in other study cohorts. See Supplementary Table S2
* Proteins which were measured only in 113 out of 219 subjects in the Discovery cohort in the 1st stage of SOMAscan screening (see methods).
Anti-Muellerian hormone type-2 receptor (AMHR2) was significant (Bonferroni corrected P=0.024) in the 1st stage, but was not measured in the 2nd stage due to technical reasons.
The 4 proteins confirmed in all cohorts had strong associations with risk of ESKD in models adjusted for clinical covariates (Fig. 2a). Since these proteins were highly inter-correlated (Supplementary Fig. S3), we examined which protein had a dominant and independent effect on the 10-year risk of ESKD using multivariable Cox model that included the 4 proteins and all clinical covariates. As shown in Fig. 2b, NBL1 was the only protein that had a strong and independent association with risk of ESKD in both late and early DKD. Fig. 2c showed cumulative risk of ESKD according to duration of follow-up and quartiles of baseline concentration of NBL1. The cumulative risk of ESKD at 10 years in the late DKD cohorts was 85% in subjects in the highest NBL1 quartile and 12% in subjects in the lowest quartile. Subjects in the intermediate NBL1 quartiles had cumulative risks in-between the two extremes. A similar pattern of risk was found in the early DKD cohorts; risk of ESKD was 51% in the subjects with highest quartile versus 6.7% in the lowest quartile of NBL1.
Fig. 2 |. Association of baseline circulating NBL1 with risk of ESKD in the study cohorts during 10-year follow-up.
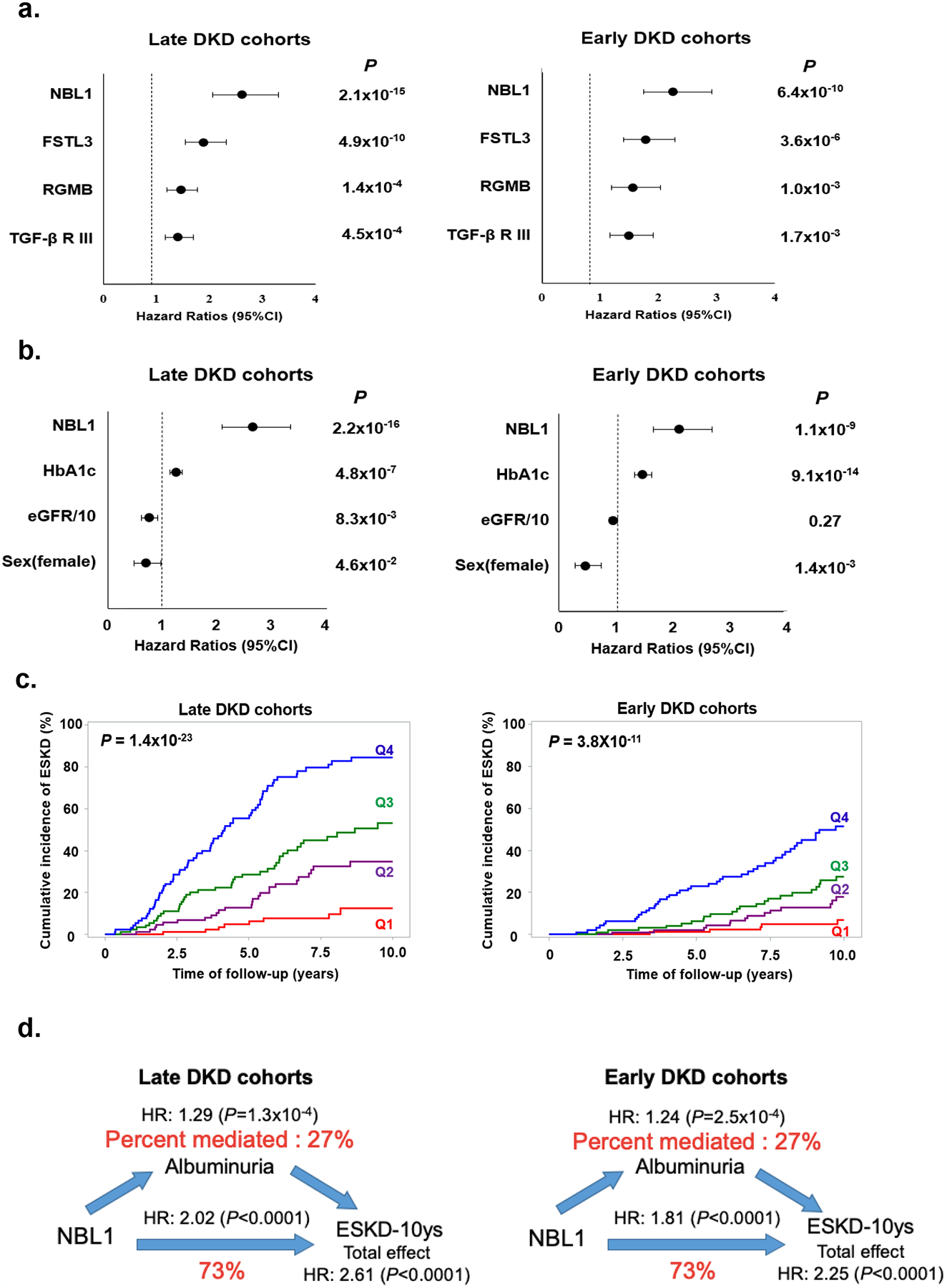
a. Risk of ESKD according to baseline circulating concentration of each of 4 confirmed proteins in late and early DKD cohorts. Results of Cox regression analysis are shown. Effect measures were expressed as HR and 95% CI per standard deviation (SD) increase in protein concentration. Effect for each protein was adjusted for sex, duration of diabetes, HbA1c, systolic blood pressure, diastolic blood pressure, and baseline eGFR with stratification of type of diabetes.
b. Effect of baseline circulating concentration of NBL1 on risk of ESKD after adjustment for other candidate proteins and baseline clinical characteristics. Results of Cox regression analysis with backward elimination of covariates are shown. Factors considered were NBL1, FSLT3, RGMB, TGF-βRIII, sex, duration of diabetes, HbA1c, systolic blood pressure, diastolic blood pressure, and baseline eGFR. Results are shown for late and early DKD cohorts. Effect measures were expressed as HR and 95% CI per SD increase in NBL1 concentration, 1% increase in HbA1c, 10 ml change in baseline eGFR, and 1 for women and 0 for men. P<0.05 was used to retain variables.
c. Cumulative incidence of ESKD according to quartiles of baseline circulating concentration of NBL1 in late and early DKD cohorts. Q1, First quartile; Q2, Second quartile; Q3, Third quartile; Q4, Fourth quartile.
d. Mediation analysis of association between baseline circulating concentrations of NBL1 and risk of ESKD during 10-year follow-up according to baseline albuminuria (ACR) in late and early DKD cohorts. NBL1 concentration was considered as exposure, risk of ESKD was outcome and ACR was mediator. In the analyses we used Cox regression model for 10-year risk of ESKD adjusted for sex, duration of diabetes, HbA1c, systolic blood pressure, diastolic blood pressure, and baseline eGFR with stratification of type of diabetes. Effect measures were expressed as the HR per SD increase in NBL1 concentration. The effect of a NBL1 on ESKD (total effect) is split into a natural indirect effect (through ACR) and natural direct effect, which is independent from the ACR.
Circulating NBL1 and risk of ESKD according to albuminuria
To determine whether the effect of circulating NBL1 on the risk of ESKD varied according to albuminuria, we performed a mediation analysis. As shown in Fig. 2d, 73% of the effect of NBL1 on risk of ESKD was independent of albuminuria, with a significant albuminuria-independent HR of 2.02 (P <0.0001) for 10-year risk of ESKD per 1- standard deviation (SD) increase of NBL1 in late and HR of 1.81 (P <0.0001) in early DKD cohorts.
Circulating NBL1 and kidney structural lesions in early DKD
We next evaluated the correlation between circulating NBL1 and the severity of the lesions in research kidney biopsies performed in 105 of the 153 subjects in the T2D Pima Indian cohort. The biopsies were performed within a median of 1.3 years of the examination in which the serum was obtained for SOMAscan measurments. Circulating NBL1 concentrations correlated negatively with podocyte number, fractional volume of podocyte cells per glomerulus, percent glomerular capillary fenestrated endothelium, glomerular filtration surface density, and positively with mesangial fractional volume, glomerular basement membrane (GBM) width, and cortical interstitial fraction volume (Supplementary Fig. S4).
For comparison, correlations were evaluated between kidney structural lesions and circulating concentrations of several other proteins (Fig. 3). The correlations were absent for podocytes damage and weaker for other structural lesions for tumor necrosis factor receptor 1 (TNFR1) than for NBL1 and none of the lesions were correlated with bone morphogenetic protein 7 (BMP-7) or TGF-β1 (71–74).
Fig. 3 |. Association between NBL1 and kidney structural lesions.
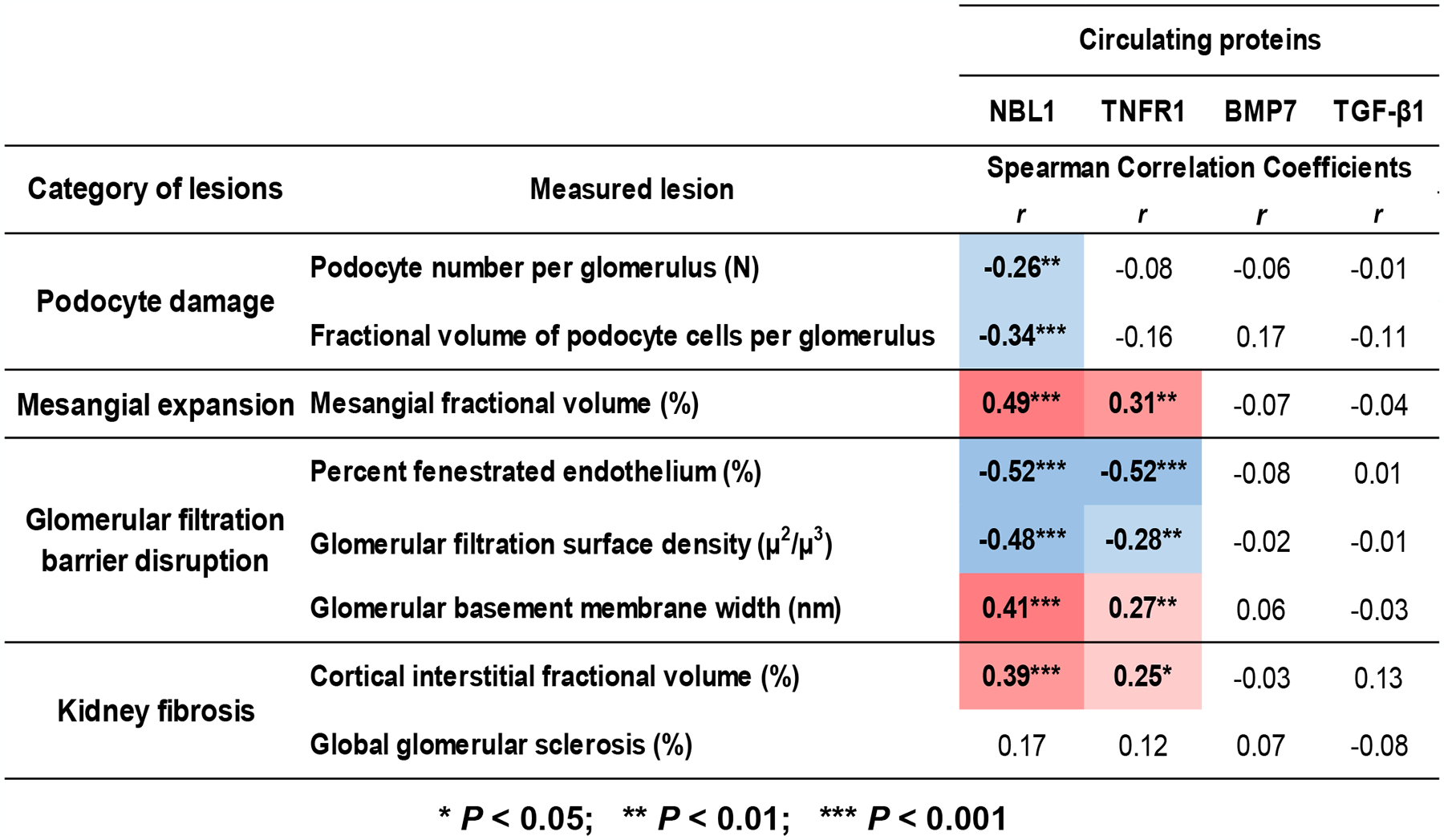
Association between baseline serum concentrations of NBL1 and structural lesions observed in research kidney biopsies obtained from 105 subjects in the T2D Pima Indian Validation cohort. Biopsies were obtained on average 1 year after baseline examination. For comparison, associations with three additional circulating proteins are shown: TNFR1- strong predictor of ESKD (71, 72), BMP7 and TGF-β1 both proteins not associated with risk of ESKD. Correlation was analyzed using Spearman rank correlation. See methods in Appendix.
NBL1 expression in single nucleus RNA sequencing of kidney cells
To further study the relationship between NBL1 and kidney structural lesions in DKD, we reanalyzed our previously-published kidney cortex snRNA-seq libraries obtained from three healthy adults and three subjects with DKD (75). The DKD subjects had estimated glomerular filtration rate (eGFR) ranging from 56 to 85 mL/min/1.73m2 and two of three had proteinuria and moderate interstitial fibrosis and glomerulosclerosis on biopsy. Kidney cell types showed minimal NBL1 expression (Fig. 4). In contrast, snRNA-seq detected a larger proportion of kidney cell types expressing TNFRSF1A (TNFR1) (Fig. 4).
Fig. 4 |. Single Nucleus RNA Sequencing of Kidney Cortex.
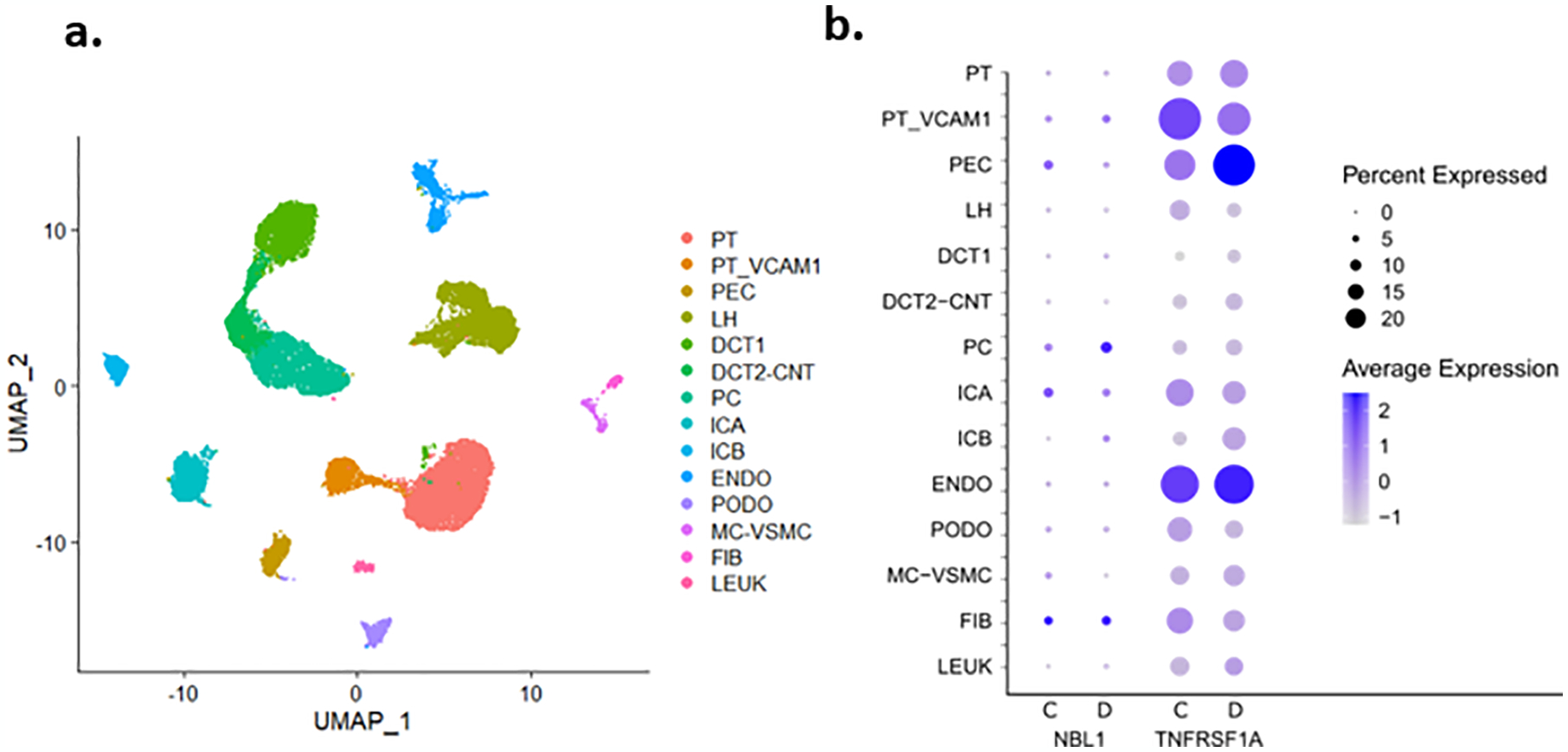
Healthy control (n=3) and patients with early diabetic kidney disease (n=3) underwent nuclear dissociation and single nucleus RNA sequencing (snRNA-seq) with 10X Genomics 5’ chemistry. Libraries were processed with cellranger and Seurat. (a) UMAP of all kidney cell types identified in the aggregated dataset. PT_VCAM1=proximal tubule cells that express VCAM1, PT=proximal tubule, PEC=parietal epithelial cells, TAL=thick ascending limb, DCT1=early distal convoluted tubule, DCT2-CNT=late distal convoluted tubule and connecting tubule, PC=principal cells, ICA=type A intercalated cells, ICB=type B intercalated cells, PODO=podocytes, ENDO=endothelial cells, MC-VSMC mesangial and vascular smooth muscle cells, FIB=fibroblasts, LEUK=leukocytes (b) NBL1 is not significantly expressed in kidney cell types in either control (C) or diabetes (D) specimens. TNFRSF1A (TNFR1) is significantly expressed in multiple kidney cell types. Scale represents normalized log-fold-change and was prepared with the Seurat DotPlot function. See Supplementary Table S3 and methods in Appendix.
To validate these findings, we reanalyzed a previously published bulk RNA-seq dataset obtained from patients with DKD (76). This dataset consists of 37 kidney tissue samples from subjects with biopsy proven early DKD (N=6), advanced DKD (N=22), and healthy controls (N=9). Differential expression analysis of the bulk RNA-seq dataset showed that subjects with advanced DKD had increased expression of NBL1 (log fold change=1.16, P adj. =3.4 × 10−10) compared to healthy controls. In contrast, subjects with early DKD had no increase in NBL1 expression.
Bulk RNA-seq datasets are an aggregate measurement of the transcriptional changes in multiple cell types. Deconvolution of this bulk RNA-seq dataset showed a significant increase in the number of leukocytes in kidney specimens obtained from subjects with advanced but not from early DKD or healthy controls (77). Thus, infiltration of leukocytes during progression of DKD may account for the increased expression of NBL1 seen in the bulk RNA sequencing of kidney specimens obtained from subjects with advanced DKD. Overall, the snRNA-seq and bulk RNA-seq data did not indicate that expression of NBL1 in kidney preceded the development of DKD or that it is the source of elevated concentrations of NBL1 in circulation.
Immunostaining for NBL1 in kidney biopsy specimens
Immunohistochemical and immunofluorescent analyses examined the distribution of NBL1 in healthy and DKD kidneys. DKD histological sections included glomeruli with mesangial thickening, glomerulosclerosis, interstitial fibrosis/inflammation and tubular atrophy.
In healthy kidney biopsies only minimal NBL1 positivity was found in cytoplasmic and nuclear compartments of tubular epithelial and interstitial cells (Fig. 5A i) and moderate positivity in nuclei of podocytes (Fig. 5A iii). In DKD kidney biopsies proximal tubule epithelial cells were stained at high intensity (Fig. 5A ii).
Fig. 5. Localization of NBL1 in kidney from healthy and DKD subjects.
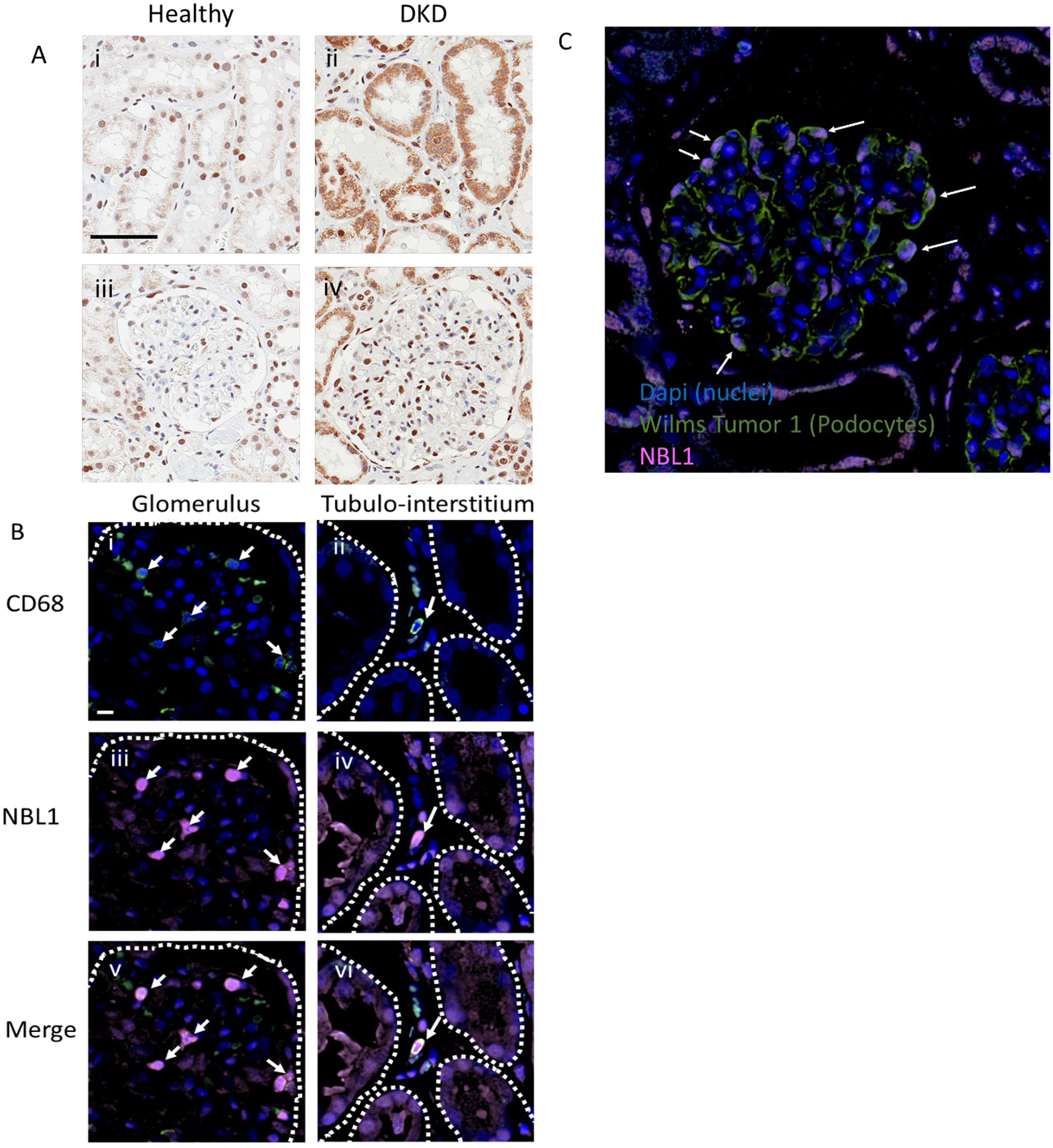
Panel A. Representative 40x IHC images show proximal tubule (i, ii) and glomerular (iii, iv) localization of NBL1 (brown stain) in healthy (i, iii), and DKD kidney (ii, iv). The magnification Bar in panel i represents 60μm. Both podocyte and proximal tubule epithelial cells exhibit strong NBL1 staining in diabetic kidney disease tissue tested from 32 patients.
Panel B. Representative 60x images show co-localization of NBL1 (iii and iv, pink) with CD68 positive macrophages (i and ii, green) in a glomerulus (left panels) and interstitium (right panels) of kidney tissue from a patient with diabetic kidney disease. The NBL1 positive macrophages are marked by white arrows. NBL1 staining is also present in tubular epithelial cells (iv and vi). Cell nuclei are stained with Dapi (blue). Glomerular Bowman’s capsule (i, iii, and v) and tubules (ii, iv, and vi) are outlined with dashed white line. The magnification bar in panel B represents 20μm.
Panel C. Representative 40x image showing glomerular colocalization of NBL1 (pink) and Wilms Tumor-1 (WT-1, green, podocyte cell specific marker). Characteristic WT-1 positive podocytes in a diabetic kidney are labeled with arrows indicating colocalization of NBL1 signal in podocytes.
To study the spatial localization in multiple cell types, additional immunofluorescence studies were employed with cell-specific markers of macrophages and podocytes. NBL1 colocalized with CD68, a macrophage cell marker, in glomerular (Fig. 5B I, iii, and v) and tubulo-interstitial compartments (Fig. 5B ii, iv, and vi) of DKD kidney. In addition, NBL1 staining was observed in podocytes (Fig. 5C).
Immunostaining for NBL1 in other organs/tissues
A comprehensive study of immunostaining of NBL1 in healthy human tissues (NBL1 Atlas) was performed (Supplementary Fig. S5). NBL1 expression was strong in cells of small and large intestine, as shown in Panel #1, with NBL1 being detectable in both cytoplasm and nuclei. Similar pattern was seen for prostate and testis. In contrast, only nuclear staining was evident in ovary (Panel #2), while it was weaker in the nuclei of skeletal/smooth and heart muscles (Panel #3). NBL1 staining was absent in mesenchymal tissues (Panel #4). On flow cytometry analysis, white blood cells revealed a high percentage of NBL1 positive cells, with T cells and monocytes the predominant subpopulations with the highest NBL1 expression (Panel #5, Supplementary Fig. S5b).
Effect of NBL1 on apoptosis of kidney cells in in vitro study.
We examined the effect of circulating NBL1 on apoptosis of kidney cells in in vitro experiments. Human podocytes, mesangial cells, tubular cells and umbilical vein endothelial cells were cultured for 48 hours with and without NBL1 at increasing concentrations (Fig. 6 a–e). The culture media did not contain any of the circulating proteins that may modulate/regulate TGF-β signaling. The addition of NBL1 to culture media increased apoptosis/death of human podocytes, and to a lesser degree, mesangial and tubular cells, while cell death was undetectable in NBL1-cultured umbilical vein endothelial cells even at higher concentrations (Fig. 6 a–e and f1–2). A transcriptome analysis performed in NBL1-treated human podocytes confirmed the upregulation of several pro-apoptotic-related genes, including BCL2A1, CASP2, TNFRSF10A and FADD (Fig. 6g).
Fig. 6 |. Effect of NBL1 exposure on apoptosis in kidney cells.
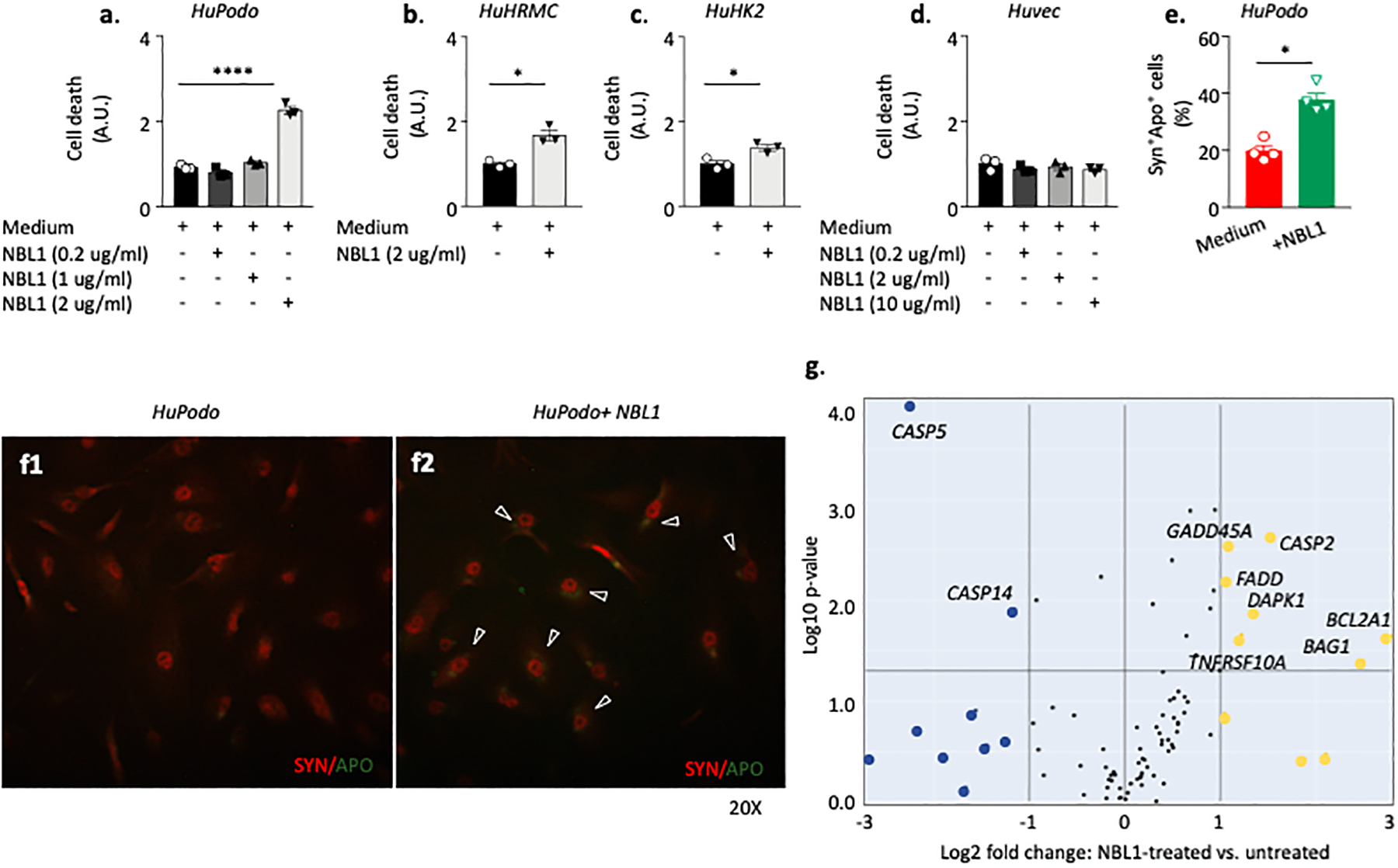
(a, b, c, d). Bar graph representing cell death analysis in human podocytes (HuPodo) cultured in a dose-dependent manner with recombinant NBL1 (a), in human tubular (HuK2) and mesangial (HuMRC) cell lines (b, c) and in human umbilical vein endothelial cells (Huvec,(d) cultured with NBL1 2 ug/ml. (e). Bar graph showing the cell count of Apoptag+Synaptopodyn+ HuPodo detected in the presence/absence of NBL1 at the confocal analysis and presented as percentage of double positive cells (n=4). (f1-f2). Representative images of confocal analysis conducted on human podocytes cultured with NBL1 2 ug/ml or left untreated and stained with Apoptag and Synaptopodin. Merge pictures are presented. Magnification 20X. (g). Transcriptome analysis of apoptotic-related genes of human podocytes cultured with NBL1 2 ug/ml or left untreated. Three independent experiments were run in duplicates (a, b, c, d, e). Data are presented as mean ±SEM. *P <0.05 and ****P <0.0001 by two-sided t-test, One-way ANOVA followed adjusted for multiple comparisons. A.U., arbitrary unit; Syn, synaptopodin; APO, apoptag; HuPodo, human podocytes; HuK2, human tubular cells; HuMRC, human mesangial cells; Huvec, human umbilical vein endothelial cells; SEM, standard error of the mean. See methods in Appendix.
Circulating NBL1 and clinical characteristics
To examine whether increased concentration of NBL1 in circulation was a consequence of impaired kidney function, we measured NBL1 urinary excretion and compared it with plasma concentrations in two nested case control studies. Clinical characteristics of participants in both studies are shown in Supplementary Table S3. Baseline plasma and urine specimens were measured using the SOMAscan platform. Fold-changes for NBL1 in plasma and urinary concentrations in subjects who progressed to ESKD over those who did not progress are shown in Fig. 7a. Subjects at risk of ESKD had higher plasma and urinary concentrations of NBL1 at baseline examination than those who did not progress. Similar findings were observed in the T1D Discovery and T2D Replication cohorts and the increased fold changes were highly statistically significant. This pattern indicates systemic overproduction as a cause of increased plasma and urine NBL1, rather than retention due to impaired kidney function. A similar pattern was seen for TNFR1.
Fig. 7 |. Association of circulating NBL1 with clinical characteristics.
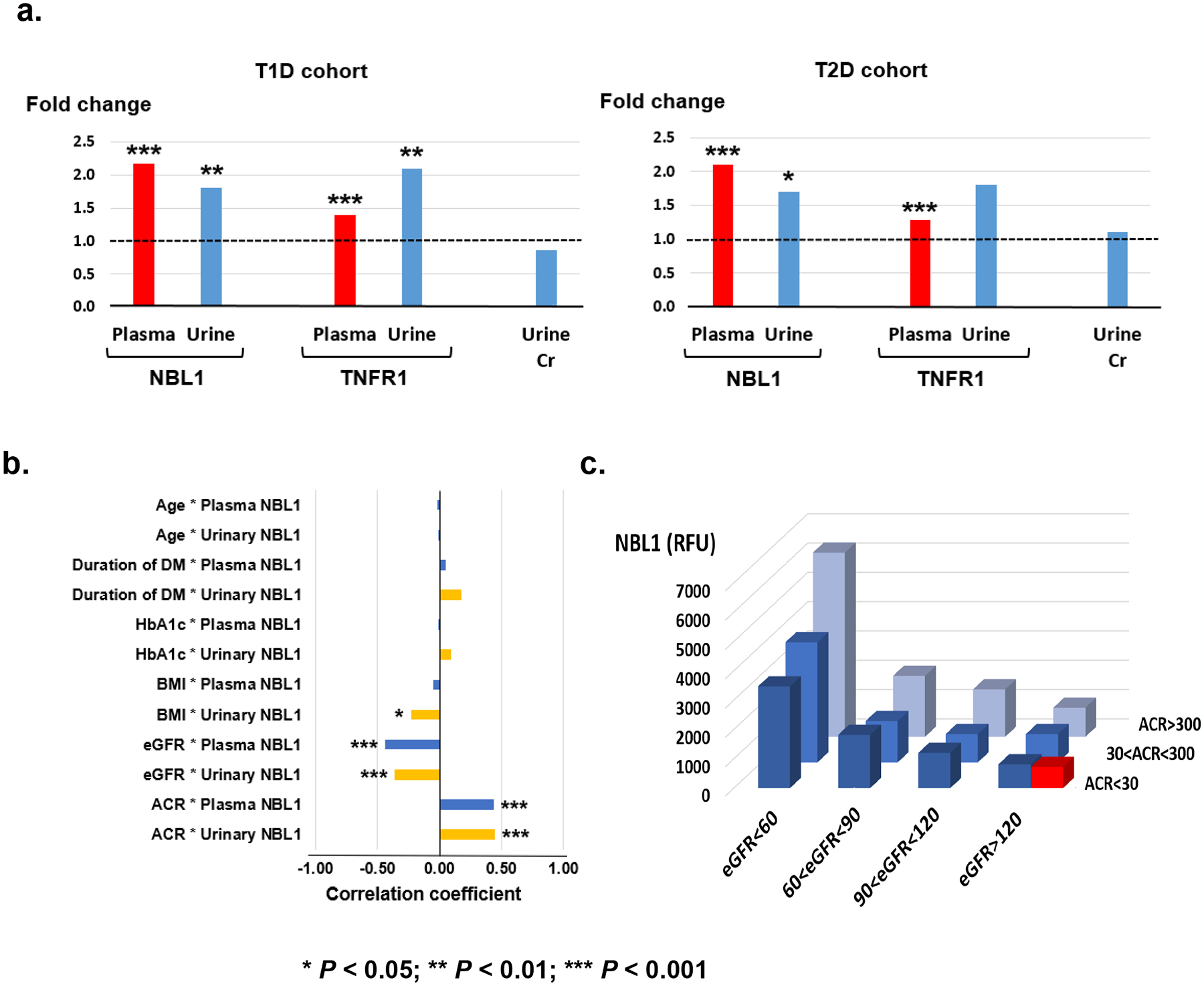
a. Fold changes in plasma concentrations of NBL1 and TNFR1 in cases over controls in T1D discovery (n=219) and T2D replication (n=144) cohorts. Fold changes in urine concentrations of NBL1 and TNFR1 normalized by urinary creatinine in cases over controls in nested case-control study of T1D (n=60) and T2D (n=52) subjects selected from the Joslin late DKD cohorts.
b. Spearman rank correlation coefficient between baseline plasma NBL1 concentrations and clinical characteristics in Joslin T1D with early DKD (n=238), and urinary NBL1 concentrations and clinical characteristics in T1D (n=60) and T2D (n=52) subjects selected from the Joslin late DKD cohorts.
c. Mean circulating NBL1 concentrations in relative fluorescence units (RFU) according to categories of baseline eGFR (ml/min/1.73m2) and ACR (mg/g) in Joslin T1D combined cohorts (n=457) and healthy controls (n=79). Red box shows mean RFU in healthy controls (see also Supplementary Fig. 7). There was no significant difference between concentrations of NBL1 in subjects with diabetes (ACR <30 mg/g and eGFR>120 ml/min/1.73m2) and in healthy controls (813 ± 393 vs 723 ± 150, P > 0.05).
To search for possible determinants of systemic overproduction of circulating NBL1, we examined correlations between plasma concentration of NBL1 and clinical characteristics of subjects in the T1D Validation cohort with early DKD (Fig. 7b). No correlation was found with age, diabetes duration, body mass index (BMI) and HbA1c. However, there were moderate correlations with eGFR and albumin-to-creatinine ratio (ACR). A similar pattern was found between urinary NBL1 and clinical characteristics in subjects in the T1D and T2D with late DKD cohorts (Fig. 7b). The highest concentration of circulating NBL1 was observed in subjects with highest concentration of albuminuria (ACR >300 mg/g) and greatest impairment of kidney function (eGFR <60 ml/min/1.73m2) and the lowest concentration in subjects with normoalbuminuria (ACR <30 mg/g) and normal kidney function (eGFR >120 ml/min/1.73 m2) (Fig. 7c). The concentration of NBL1 in subjects with normoalbuminuria (ACR <30 mg/g) and eGFR >60 ml/min/1.73 m2 were almost twice as high as in non-diabetic subjects (1208 ± 452 relative fluorescent units [RFU] vs 723 ± 150, P=1.1×10−4). Supplementary Fig. S6 shows similar concentrations of circulating NBL1 in men and women in all cohorts, although there were systematic differences among study cohorts.
Circulating NBL1 as biomarker to identify subjects at risk of ESKD
To examine the performance of circulating NBL1 as a predictor of ESKD, four multivariable Cox regression models were developed using combined data from the 4 cohorts (n=754, Table 2). The 1st model included only clinical variables selected by Cox regression analysis with backward elimination. Of the baseline variables: sex, diabetes duration, HbA1c, systolic blood pressure (SBP), diastolic blood pressure (DBP), eGFR, and ACR, only three contributed significantly to 10-year ESKD risk: eGFR, ACR and HbA1c. The 2nd model added TNFR1, the 3rd model added NBL1, and the 4th model added both variables. In each of these additional models, the effects of all variables were highly statistically significant except for baseline eGFR. In the 4th model, HRs for both TNFR1 (HR 1.76; 95% CI 1.46, 2.13) and NBL1 (HR 1.58; 95% CI 1.30, 1.92) were highly statistically significant, suggesting that the effect of NBL1 is independent from clinical covariates and circulating concentrations of TNFR1.
Table. 2 |.
Predictive performance of the Cox regression models evaluating 10-year ESKD risk in combined 4 cohorts (n=754).
| Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variable | HR (95%CI) | P | HR (95%CI) | P | HR (95%CI) | P | HR (95%CI) | P | HR (95%CI) | P |
| eGFR/10 | 0.85 (0.79, 0.92) | 3.8× 10−5 | 0.95 (0.89 1.02) | 0.15. | 0.96 (0.89, 1.03) | 0.27. | 1.00 (0.93, 1.07) | 0.94. | 1.01 (0.95, 1.09) | 0.69 |
| log2ACR | 1.38 (1.29, 1.47) | 3.2× 10−22 | 1.30 (1.22, 1.39) | 4.2× 10−16 | 1.26 (1.18, 1.35) | 3.1× 10−12 | 1.26 (1.18, 1.34) | 8.2× 10−12 | 1.25 (1.17, 1.34) | 6.2× 10−11 |
| HbA1c | 1.23 (1.15, 1.33) | 1.4× 10−8 | 1.27 (1.18, 1.36) | 2.0× 10−10 | 1.27 (1.18, 1.36) | 1.0× 10−10 | 1.28 (1.19, 1.37) | 2.4× 10−11 | 1.27 (1.18, 1.37) | 8.4× 10−11 |
| TNFR1 | 2.15 (1.83, 2.52) | 9.7× 10−21 | 1.76 (1.46, 2.13) | 3.9× 10−9 | 1.58 (1.29, 1.93) | 6.6× 10−6 | ||||
| NBL1 | 2.09 (1.76, 2.48) | 2.6× 10−17 | 1.58 (1.30, 1.92) | 4.4× 10−6 | 1.38 (1.07, 1.78) | 1.3× 10−2 | ||||
| TNFRSF27 | 1.25 (1.06, 1.48) | 9.9× 10−3 | ||||||||
| CCL15 | 1.17 (0.99, 1.39) | 6.4× 10−2 | ||||||||
| IL17F | 1.15 (0.96, 1.39) | 0.13 | ||||||||
| TNFSF15 | 0.97 (0.80, 1.18) | 0.77 | ||||||||
| C-statistics | 0.797 ± 0.012 | 0.825 ± 0.011 | 0.824 ± 0.011 | 0.836 ± 0.011 | 0.840 ± 0.013 | |||||
| P (vs model 1) # | - | 1.5×10−4 | 6.4×10−4 | 1.4×10−5 | ||||||
| P (vs model 2) # | - | - | 0.87 | 1.5×10−2 | ||||||
| P (vs model 4) # | 0.12 | |||||||||
| NRI (vs model 1) ## | - | 0.183 (0.124, 0.242) *** | 0.177 (0.121, 0.234) *** | 0.230 (0.168, 0.292) *** | ||||||
| NRI (vs model 2) ## | - | - | - | 0.076 (0.032, 0.120) *** | ||||||
| NRI (vs model 4) ## | 0.0016 (−0.052, 0.055) | |||||||||
| -2 log likelihood (2LL) | 2567 | 2483 | 2495 | 2462 | 2447 | |||||
| Akaike Information Criterion (AIC) | 2579 | 2497 | 2509 | 2478 | 2471 | |||||
Cox regression models:
Model 1: HbA1c + ACR + baseline eGFR + cohort indicator
Model 2: HbA1c + ACR + baseline eGFR + TNFR1 + cohort indicator
Model 3: HbA1c + ACR + baseline eGFR + NBL1 + cohort indicator
Model 4: HbA1c + ACR + baseline eGFR + TNFR1 + NBL1 + cohort indicator
Model 5: HbA1c + ACR + baseline eGFR + TNFR1 + NBL1 + 4 other KRIS proteins + cohort indicator
Uno’s concordance statistics with two-sided P value.
Risk categories for ESKD during 10 years are defined by < 5.0%, 5.0–9.9%, 10.0–19.9%, and 20% and more.
P for NRI <0.001.
HR, Hazard ratios; CI, Confidence intervals; NRI, Net reclassification improvement; AIC, Akaike information criterion.
Prognostic performance of these models is summarized in Table 2. The C-statistic for the clinical model was 0.797; it increased to 0.825 after adding TNFR1 and to 0.836 after adding NBL1. Models with TNFR1 and NBL1 were characterized by improvements in the Uno’s concordance statistic (model 3 vs 1: P=6.4×10−4; model 4 vs model 2: P=1.5×10−2). Adding NBL1 to model 2 resulted in significant improvement of the net reclassification index (NRI: 0.076; P<0.001) (Table 2). The Akaike Information Criterion (AIC) values also decreased in the more comprehensive models. All metrics demonstrated the added value of circulating NBL1 in predicting 10-year risk of ESKD.
We previously identified 17 circulating KRIS (kidney risk inflammatory signature) proteins significantly associated with progression to ESKD in univariate analyses (61). Since the proteins were highly inter-correlated, only 5 of them had an independent effect on progression to ESKD in multivariable analysis with TNFR1 having a dominant effect and the others (TNFRSF27, CCL15, IL17F, and TNFSF15) contributing much less. When these 5 KRIS proteins were added to model #4, NBL1, TNFR1, and TNFRSF27 remained statistically significant with CCL15 and IL17F having only marginal significance (see model #5). The predictive power of this model increased slightly relative to model #4 but there was no statistically significant improvement of C-statistic or NRI.
Discussion
Of the 25 circulating proteins that modulate TGF-β signaling in the extracellular space, measured on the SOMAscan proteomic platform, circulating NBL1 (aliases DAN & DAND1) was robustly associated with high risk of progression to ESKD during 10-year follow-up. This finding was validated in multiple cohorts, i.e., in subjects with T1D and T2D, and in Caucasian as well as in Pima Indians. The mechanisms accounting for the observed association are unknown. However, circulating NBL1 may play an important etiological role in progressive kidney functional decline leading to ESKD in diabetes and may be a therapeutic target for reno-protective drugs. The concentration of circulating NBL1 can also be used as a prognostic marker to identify subjects at increased risk of progression.
NBL1 is a 165 amino acid secretory protein originally identified as a tumor suppressor in neuroblastoma cell lines (64, 65). Subsequently, it was demonstrated to have BMP inhibitory activity, especially for BMP-2 and BMP-7 (66–70). Since then, NBL1 has been investigated in numerous biological and in vitro assays as a BMP antagonist (78–80). NBL1 is one of 7 members of the differential screening-selected gene in neuroblastoma (DAN) family comprising a diverse group of BMP inhibitors (Supplementary Fig. S7). Two of them, USAG-1 and Gremlin 1, contribute to kidney fibrosis in cellular and animal studies (18, 19, 48, 49, 81, 82). However, we found no association between circulating Gremlin 1 concentration and progression to ESKD; USAG-1 was not measured on the SOMAscan platform.
Elevated concentrations of NBL1 in circulation and in urine were associated with risk of progression to ESKD. This suggests that overproduction, rather than impaired kidney excretion of NBL1, was responsible for the increased concentration of NBL1 in circulation and increased risk of ESKD. Organs/tissues responsible for such NBL1 overproduction are presently unknown. Our negative findings of snRNA sequencing of kidney cells for NBL1 from healthy subjects and subjects with advanced DKD suggest that the kidney is an unlikely source of circulating NBL1. This inference is supported by the absence or minimal NBL1 immunostaining of kidney biopsies from healthy subjects. The significant NBL1 staining of tubules in kidney biopsies from subjects with DKD could be secondary to the development of DKD. We found strong NBL1 immunostaining in the intestine, which releases numerous factors into circulation, and could be a major source of NBL1 in circulation. NBL1 was also highly expressed in immune cells, such as monocytes, CD4 and CD8 T cells, suggesting another plausible non-kidney source of circulating NBL1.
Subjects with diabetes had higher concentrations of circulating NBL1 than healthy individuals. Through correlation analysis, we excluded the effect of HbA1c, age and BMI on concentration of NBL1. Statistically significant correlations existed, however, between baseline concentration of NBL1 and baseline measurements of ACR, and eGFR. The results of mediation analyses and multivariable Cox regression, however, showed that association between baseline circulating concentration of NBL1 and risk of ESKD was largely independent from the baseline ACR and eGFR.
Three possible mechanisms through which elevated concentrations of circulating NBL1 may impact progression to ESKD are outlined in Fig. 8. In the 1st mechanism, NBL1 quenches Bone Morphogenic Proteins (BMPs) and prevents BMPs interaction with BMP-receptors (BMP-Rs) thereby favoring pro-fibrotic signaling pathways (66, 73). In the 2nd mechanism, NBL1 binds the BMPs/BMP-Rs complex, thereby blocking its intracellular signaling and unlocking the TGF-β-mediated profibrotic action (74). The 3rd mechanism postulates that circulating NBL1 damages podocytes through a yet unspecified process that is independent from the 1st and 2nd mechanism. This hypothesis is supported by our results. The extent of early structural kidney lesions in the Pima Indians was strongly correlated with concentration of circulating NBL1. Interestingly, the loss of podocyte number and reduction of fractional volume of podocyte cells per glomerulus were associated with elevated concentrations of circulating NBL1 but not with circulating TNFR1, another protein strongly associated with progression to ESKD. The results of in vitro experiments are also consistent with these findings as elevated concentrations of NBL1 in media had a toxic effect on podocytes leading to increased apoptosis. We did not observe an apoptotic effect of NBL1 on HUVECs. Since the media did not contain any BMPs, the action of NBL1 on the podocytes could not be mediated through inhibition of BMPs and activation of TGF-β/Smad signaling.
Fig. 8 |. Postulated mechanisms through which elevated concentrations of circulating NBL1 impact progression to ESKD.
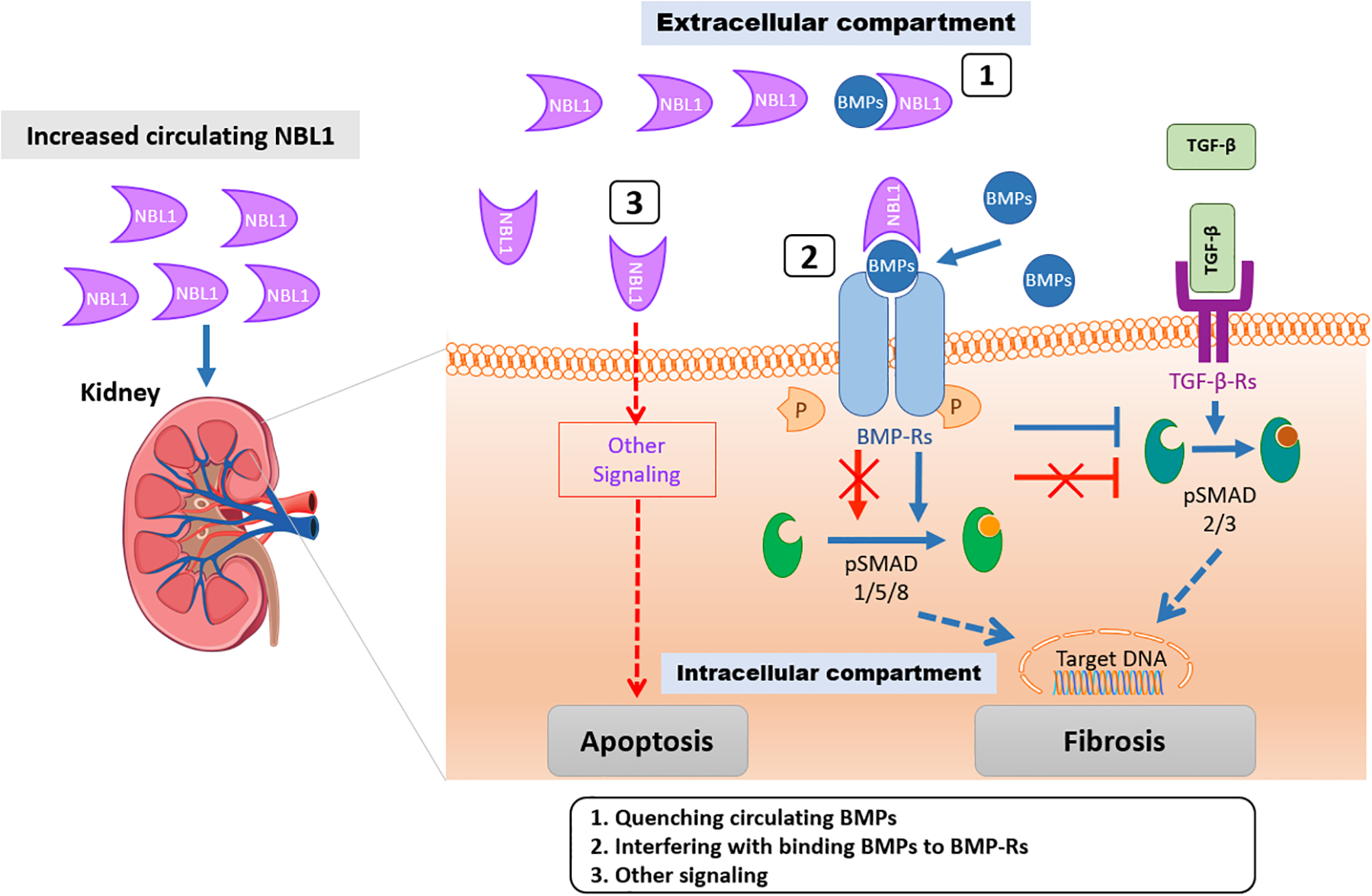
Excessive circulating NBL1 is associated with progression to ESKD. We propose three mechanisms whereby NBL1 exerts its negative effects on kidney cells. In mechanism 1, NBL1 quenches Bone Morphogenic Proteins (BMPs) and prevents BMPs interaction with BMP-receptors (BMP-Rs) thereby favoring pro-fibrotic signaling pathways (66, 73). In mechanism 2, NBL1 binds the BMPs/BMP-Rs complex, thereby blocking its intracellular signaling and unlock the TGF-β-mediated profibrotic action (74). In mechanism 3, a NBL1-mediated BMPs-independent apoptosis of podocytes cells is postulated.
Effect of normal concentration of NBL1 in circulation is indicated as blue arrows. Negative effect of elevated concentration of NBL1 in circulation is indicated as red arrows.
Loss of podocytes is a mechanism of disease initiation and progression in many kidney diseases (83). In diabetes, podocytes are injured early in DKD (84–86), and that podocyte injury may precede the changes seen in the GBM and mesangial area by several years (87). Importantly, animal models show that enhancing podocyte injury early in the disease promotes the development of mesangial expansion, interstitial fibrosis, and increased GBM thickness (88). All these lesions were strongly correlated with circulating concentrations of NBL1. Taken together, our findings suggest that circulating NBL1 causes early kidney structural lesions through apoptosis of kidney cells, especially podocytes, which in turn may contribute to progressive kidney function decline and progression to ESKD.
Our findings regarding circulating NBL1 suggests that this protein may be an accessible target for pharmaceutical modification of TGF-β signaling to slow DKD progression to ESKD. Inhibition of NBL1 is predicted to free BMP proteins to enable their interaction with and inhibition of TGF-β signaling, therefore diminishing inflammation and fibrosis in kidney tissue and podocyte loss in DKD. A previous attempt to inhibit TGF-β1 directly with a therapeutic monoclonal antibody failed to slow eGFR loss, reduce albuminuria, or modulate DKD biomarkers of inflammation and disease, presumably because the antibody lacked access to TGF-β in kidney cells (28). NBL1 may be a more appropriate target in this pathway than TGF-β directly. NBL1 knockdown mice are viable and have normal phenotypes, suggesting that inhibition of NBL1 will not lead to significant off-target safety issues (68, 89).
Independent of its role in the etiology of DKD, the concentration of NBL1 in circulation can be used as a prognostic marker for identifying individuals at risk of progression to ESKD. In our study, the prognostic performance of NBL1 in circulation was identical to the prognostic performance of circulating TNFR1. Measuring both proteins and combining the results with clinical markers such as HbA1c, eGFR and ACR significantly increased the ability of a multi-marker prognostic model to identify individuals at risk of progression to ESKD. However, for such a multi-marker prognostic test to be useful in clinical practice, additional studies of large cohorts will be required to estimate the probabilities of ESKD during specific intervals of follow-up (i.e. 5, 10 or 15 years).
Strengths of our study include: 1) prospective long-term follow-up in multiple cohorts, different ethnicities, different types of diabetes and different stages of DKD, 2) use of gold standard definitions of DKD, i.e. structural lesions assessed by quantitative morphometry in research kidney biopsies and 10-year risk of ESKD, 3) unbiased and standardized measurements of key proteins on TGF-β related signaling, 4) robust results consistent in all cohorts. There are also some limitations. The study is observational, and the causal relationship of NBL1 with kidney disease progression will need to be established through animal studies and clinical trials. In addition, since the study was conducted only for DKD in Caucasians and Pima Indians, we cannot assess the generalizability of these findings to Black subjects with diabetes or to individuals with other kidney diseases.
Materials and Methods
Study Design
Subjects were selected from participants of the Joslin Kidney Study and the Pima Indian Kidney Study. Subjects were followed for 7–12 years (25% and 75% in those who did not develop ESKD) to examine changes in kidney function and onset of ESKD. To identify circulating TGF-β signaling proteins associated with the development of ESKD, we selected 4 different cohorts: a Discovery cohort (Joslin cohort with T1D CKD stage 3), a Replication cohort (Joslin cohort with T2D CKD stage 3), and two Validation cohorts; Validation cohort 1 (Joslin cohort with T1D CKD stage 1–2) and Validation cohort 2 (Pima cohort with T2D with CKD stage 1–2). The Joslin cohort with T2D CKD stage 3 was used to replicate the findings obtained in subjects with T1D CKD stage 3. The Joslin cohort with T1D CKD stage 1–2 and the Pima Indian cohort were used to further validate the findings in subjects with early stage DKD and in those of different ethnicities.
Joslin Kidney Study
The Joslin Diabetes Center Committee on Human Studies approved the JKS. The JKS is a longitudinal observational study that investigates the determinants and natural history of kidney function decline in T1D and T2D. Approximately 2,000 subjects with T1D and 1,500 subjects with T2D were recruited into the JKS from among 20,000 subjects attending the Joslin Clinic between 1991 and 2006 (61, 90). All the JKS participants were queried every two years against the United States Renal Data System (USRDS) and the US National Death Index (NDI) rosters to ascertain individuals who developed ESKD or died. The last inquiries were in 2017.
Discovery and Replication Cohorts with late DKD:
We selected the T1D Discovery (n=219), and the T2D Replication (n=144) cohorts from participants enrolled in the JKS between 1991 and 2006 with albuminuria and impaired kidney function (eGFR 20–60 ml/min/1.73 m2) at baseline. These subjects were monitored for 10 (median) years for changes in kidney function and ESKD onset (61, 90). The late DKD T1D cohort was selected as the Discovery cohort since it had the greatest statistical power to detect circulating proteins associated with risk of ESKD due to the large number of cases.
Validation Cohort with early DKD:
The T1D Validation cohort (n= 238) included JKS participants with albuminuria and eGFR 60 – 170 ml/min/1.73 m2ml/min (median eGFR 97 ml/min) at enrollment (between 1991 and 2006). They were also followed for 7–15 years to monitor kidney function changes and for ascertainment of ESKD onset (61, 90).
Healthy controls:
The Joslin Kidney Study also examined parents of subjects with T1D. The group of non-diabetic parents of T1D subjects was derived from our genetic study on determinants of DKD in T1D. Parents had baseline examinations performed according to the same protocols as participants of the Joslin Kidney Study. Biospecimens obtained at examinations were stored at −85°C. For this study, 79 white non-diabetic parents aged 50–69 years at baseline examination were selected to be used as non-diabetic controls. Plasma specimens obtained at baseline examination underwent SOMAscan analysis.
Pima Indian Kidney Study
Pima Indians from the Gila River Indian Community in Arizona have a high prevalence of T2D and a high incidence of ESKD due to diabetes (91). This population participated in a longitudinal study of diabetes and its complications for more than 40 years. Beginning in the late 1980s, informative subsets of subjects from this population were selected for more detailed longitudinal studies of DKD (92, 93). A subset of 153 subjects from this kidney study cohort with baseline examinations between 1994 and 2007 was selected for our previous study on the role of circulating inflammatory proteins in ESKD development (61). For the current T2D Validation cohort, we selected only individuals with a baseline GFR of 60 – 240 ml/min (median GFR 150 ml/min). ESKD onset was ascertained in all participants through December 31, 2017. For the present study, we right censored at the 10-year of follow-up. Within this timeframe, 34 subjects developed ESKD. The study was approved by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Disease.
Determination of kidney function
In the Joslin Kidney Study, GFR was estimated by the CKD Epidemiology Collaboration equation using serum creatinine concentrations and was expressed as mL/min/1.73m2 (94). In the Pima Indian Study, GFR was measured by the urinary clearance of iothalamate. For each participant, we used all available GFR determinations performed during follow-up to determine GFR slopes by general linear model procedure. ESKD was defined either by a match with USRDS roster or a listing of kidney failure among the causes of death on an NDI death certificate (95, 96).
SOMAscan platform to measure concentration of circulating proteins
Concentration of proteins in plasma and urine in the Joslin Cohorts and in serum in the Pima Indian cohort were measured using the SOMAscan proteomics platform (SomaLogic, Inc.; Boulder, CO, USA). (58, 59). We used it in our previous studies (61–63). The present study evaluated assay results for 25 proteins present on the SOMAscan array. The 25 circulating TGF-β proteins include families of TGF-β related signaling, BMP signaling, and Activin signaling according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and literature review (1–11, 97). Samples in this study were run in batches balanced by the prospective case status. Twelve anchored samples were also applied across the batches to allow for universal calibration based on intercept and beta estimates from the regression model drawn using PROC GLM in SAS software.
Two stage study design to measure circulating proteins by SOMAscan platform
Due to the high cost of SOMAscan assays, we performed measurements in two stages. In the 1st stage, all 1,129 plasma proteins available on the SOMAscan platform were measured in 113 (56 ESKD cases) subjects selected randomly from the 219 included in the T1D Discovery (late DKD) Joslin cohort. At this stage, we identified 232 (20%) proteins “potentially” associated with risk of ESKD using univariate logistic regression analysis and liberal statistical threshold (P< 0.01). In the 2nd stage, the 232 “potentially” associated proteins together with 332 (30%) proteins not associated with ESKD selected randomly from among all SOMAscan proteins, were measured in individuals from the second half of the discovery cohort as well as in the three other cohorts. In total, 564 proteins were measured.
Validation of circulating NBL1 concentration measured by SOMAscan proteomics platform
To validate the aptamer-based measurement of NBL1 obtained by SOMAscan proteomics platform, we measured circulating NBL1 concentration using an antibody based custom built Single-Plex Meso Scale Discovery (MSD) assay. The protocol for this assay is described in Appendix.
Measurement of kidney structural lesions in kidney biopsies
Kidney structural lesions were studied in research kidney biopsies obtained from individuals in the T2D Pima Indian cohort. Of the 153 individuals in this cohort, 105 had kidney biopsies performed within 1.26 years (range 0.04–4.49) of the baseline examinations. The morphometric techniques used in this study were described previously (93). See also Appendix.
Single nucleus RNA sequencing of kidney biopsy specimens for NBL1 expression and reanalysis of bulk RNA-seq from patients with DKD
In this study we reanalyzed data previously published (75). Detailed protocols of these analyses are described in Appendix.
Immunostaining for NBL1 in kidney biopsy specimens
Formalin fixed paraffin embedded human kidney tissue from healthy donors and subjects with T2D were analyzed using NBL1 immunostaining with Sigma HPA007394 antibody (St. Louis, MO). For colocalization studies, the sections were incubated with CD68 (ThermoFisher, Waltham, MA) and WT1 (MAB13453, Abnova, Taipei City, Taiwan) antibodies. Detailed protocols are described in Appendix.
Effect of NBL1 on apoptosis of kidney cells in in vitro study
Detailed protocols of in vitro studies are described in Appendix.
Statistical Analyses
Clinical characteristics and summary of outcomes were expressed as counts and percentages (proportions) for categorical variables, means (standard deviations) for normally distributed continuous variables, and as medians (25%, 75%) for variables with skewed distributions.
Effects of baseline proteins on the risk of ESKD were estimated using a Cox proportional hazards regression model and were expressed as HR and their CIs. We applied a Bonferroni correction for n=25 independent tests (the number of examined proteins by the study design), which yielded a threshold of P < 2.0×10−3. This threshold for significance was applied to the results obtained in the Discovery cohort. Nominal significance P < 0.05 was used to evaluate the findings obtained in the other study cohorts. Multivariable Cox regression analyses were performed to further examine the association of candidate proteins with the risk of ESKD. In these analyses, the effects of protein concentrations on risk of ESKD were expressed in terms of hazard ratios per one SD increase for easier interpretability and uniform scaling. The models were adjusted for sex, duration of diabetes, HbA1c, SBP, DBP, and baseline eGFR (measured GFR in the Pima Indians) and stratified by subject cohort. ACR was not considered as a covariate, since it was an outcome measure and a part of the disease presentation.
We selected the best predictors in Cox regression models, applying backward variable elimination at the 5% significance concentration in the combined analysis of the late and early DKD cohorts. Four candidate proteins, sex, duration of diabetes, HbA1c, SBP, DBP, and baseline eGFR were included in these models. We used the Kaplan-Meier method to estimate 10-year cumulative risk of ESKD and to plot survivor function estimates according to quartiles of baseline NBL1 concentrations.
To evaluate the effect of baseline concentration of circulating NBL1 on risk of progression to ESKD controlling for ACR as an intermediate outcome variable, we performed a mediation analysis. In our model of kidney disease progression, NBL1 was considered an upstream exposure whose effect was mediated by elevated ACR. In the analyses we used Cox regression model for 10-year risk of ESKD adjusted for sex, duration of diabetes, HbA1c, SBP, DBP, and baseline eGFR with stratification of type of diabetes. Effect measures were expressed as the HR per SD increase in NBL1 concentration. The effect of NBL1 on ESKD (total effect) is split into an indirect effect (through ACR) and direct effect, which is independent from the ACR. The SAS MEDIATE Macro was used for the analysis (98).
The correlation between candidate protein concentrations, and clinical factors, kidney structural lesions and transcription in kidney were evaluated by Spearman rank correlation coefficients.
In the in vitro study, continuous variables are presented as means with standard errors. We used independent sample t-tests to compare continuous variables. For multiple comparisons, one-way ANOVA followed by Sidak post hoc test between the group of interest and all other groups were used. Two-tailed P values of less than 0.05 were considered statistically significant.
Performance of prognostic models was compared with measures of calibration (AIC) and discrimination (NRI) in the data set comprising the four cohorts. Considerations were inclusive for significant clinical covariates (sex, HbA1c, GFR or eGFR and log2 transformed ACR), cohort indicator, 5 KRIS proteins and NBL1. Fit of the consecutive nested models was tested with likelihood ratio tests and by the AIC values (the lower the better). Uno’s C-statistic was calculated as proportions of pairs for subjects whose observed and predicted outcomes were concordant. In addition, the NRI methodology was applied for categorical risk estimates. Risk categories for ESKD during 10 years are defined by < 5.0%, 5.0–9.9%, 10.0–19.9%, and 20% and more (63, 99). Statistical analyses were performed with SAS version 9.4 (SAS Institute, Cary, NC), R statistical software versions 3.2.4 and 4.0.2.
Supplementary Material
Table S1 | Proteins evaluated in the current study and relevant previous publications
Table S2 | Results of Cox regression analysis for time to ESKD for 25 circulating TGF-β signaling proteins that were evaluated in the current study
Table S3 | Characteristics of subjects with T1D and T2D selected from Joslin cohorts into the nested case-control studies to assess risk of ESKD according to baseline concentrations of urinary proteins measured by SOMAscan proteomics platform
Table S4 | QC metrics for the analysis of the snRNA-seq data
Fig. S1 | Circulating proteins involved in TGF-β signaling examined in the study
Fig. S2 | Distribution of follow-up duration in the study cohorts
Fig. S3 | Correlation matrix for 4 candidate proteins
Fig. S4 | Association between circulating NBL1 and kidney structural lesions
Fig. S5 | NBL1 Atlas - Expression of NBL1 protein in human tissues
Fig. S6 | Distribution of circulating NBL1 in healthy control and in the study cohorts according to sex
Fig. S7 | A phylogenetic representation of the DAN family proteins
Fig. S8 | Correlation of circulating NBL1 concentrations measured by Meso Scale Discovery assay and SOMAscan
Acknowledgments
Funding:
We acknowledge grants support from: the National Institutes of Health (NIH) (DK041526, DK110350 and DK126799) to A.S.K, the Novo Nordisk Foundation grant NNF14OC0013659 (PROTON) to A.S.K. The Uehara Memorial Foundation (Postdoctoral Fellowship), and the Japan Society for the Promotion of Science (Overseas Research Fellowship) to H.K. The Mary K. Iacocca Fellowship, the Sunstar Foundation, Japan (Hiroo Kaneda Scholarship) and the Foundation for Growth Science from Japan to E.S.This research was also supported by the American Diabetes Association (Clinical Science Award 1-08-CR-42) to R.G.N., by the Intramural Research Program of the NIH NIDDK to R.G.N., H.C.L., P.J.S., and by NIH DERC grant (P30 DK036836) to Joslin Diabetes Center, by the Fondazione Invernizzi to P.F., and F.D., the Italian Ministry of Health (RF-2016-02362512) to P.F., and the EFSD/JDRF/Lilly Programme 2019 to F.D.
Data and materials availability:
All data associated with this study are present in the paper or in the supplementary materials. Individual proteomics data may become available for collaborative research from Drs. A.S. Krolewski and R. Nelson.
References and Notes
- 1.Meng XM, Nikolic-Paterson DJ, Lan HY, TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol 12, 325–338 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Santibanez JF, Quintanilla M, Bernabeu C, TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin. Sci (Lond) 121, 233–251 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol 21, 212–222 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, Ten Dijke P, TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci 18, 2157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farris AB, Colvin RB, Renal interstitial fibrosis: mechanisms and evaluation. Curr. Opin. Nephrol. Hypertens 21, 289–300 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Böttinger EP, Bitzer M, TGF-beta signaling in renal disease. J. Am. Soc. Nephrol 13, 2600–2610 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Muñoz-Félix JM, González-Núñez M, Martínez-Salgado C, López-Novoa JM, TGF-beta/BMP proteins as therapeutic targets in renal fibrosis. Where have we arrived after 25 years of trials and tribulations? Pharmacol. Ther 156, 44–58 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Grgurevic L, Macek B, Healy DR, Brault AL, Erjavec I, Cipcic A, Grgurevic I, Rogic D, Galesic K, Brkljacic J, Stern-Padovan R, Paralkar VM, Vukicevic S, Circulating bone morphogenetic protein 1–3 isoform increases renal fibrosis. J. Am. Soc. Nephrol 22, 681–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta N, Krepinsky JC, The emerging role of activins in renal disease. Curr. Opin. Nephrol. Hypertens 29, 136–144 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, Bryer JS, Xu XX, Song WC, Palmer M, Hill J, Guarnieri P, Hawkins J, Boustany-Kari CM, Pullen SS, Brown CD, Susztak K, Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med 24, 1721–1731 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinck AP, Mueller TD, Springer TA, Structural Biology and Evolution of the TGF-beta Family. Cold Spring Harb. Perspect. Biol 8, a022103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K, Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. U S A 97, 8015–8020 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill C, Flyvbjerg A, Rasch R, Bak M, Logan A, Transforming growth factor-beta2 antibody attenuates fibrosis in the experimental diabetic rat kidney. J. Endocrinol 170, 647–651 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Daniel C, Schaub K, Amann K, Lawler J, Hugo C. Thrombospondin-1 is an endogenous activator of TGF-beta in experimental diabetic nephropathy in vivo. Diabetes 56, 2982–2989 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Lu A, Miao M, Schoeb TR, Agarwal A, Murphy-Ullrich JE. Blockade of TSP1-dependent TGF-β activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am. J. Pathol 178, 2573–2586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Chen Q, Simon TC, Strebeck F, Chaudhary L, Morrissey J, Liapis H, Klahr S, Hruska KA, Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int 63, 2037–2049 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Wang S, de Caestecker M, Kopp J, Mitu G, Lapage J, Hirschberg R, Renal bone morphogenetic protein-7 protects against diabetic nephropathy. J. Am. Soc. Nephrol 17, 2504–2512 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Roxburgh SA, Kattla JJ, Curran SP, O’Meara YM, Pollock CA, Goldschmeding R, Godson C, Martin F, Brazil DP, Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes 58, 1641–1650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchant V, Droguett A, Valderrama G, Burgos ME, Carpio D, Kerr B, Ruiz-Ortega M, Egido J, Mezzano S, Tubular overexpression of Gremlin in transgenic mice aggravates renal damage in diabetic nephropathy. Am. J. Physiol. Renal Physiol 309, F559–F568 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Lora Gil C, Henley N, Leblond FA, Akla N, Laurin LP, Royal V, Gerarduzzi C, Pichette V, Larrivée B, Alk1 haploinsufficiency causes glomerular dysfunction and microalbuminuria in diabetic mice. Sci. Rep 10, 13136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D, Gava AL, Van Krieken R, Mehta N, Li R, Gao B, Desjardins EM, Steinberg GR, Hawke T, Krepinsky JC, The caveolin-1 regulated protein follistatin protects against diabetic kidney disease. Kidney Int 96, 1134–1149 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature 346, 371–374 (1990). [DOI] [PubMed] [Google Scholar]
- 23.Isaka Y, Fujiwara Y, Ueda N, Kaneda Y, Kamada T, Imai E. Glomerulosclerosis induced by in vivo transfection of transforming growth factor-beta or platelet-derived growth factor gene into the rat kidney. J. Clin. Invest 92, 2597–2601 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Böttinger EP, Klotman PE, Thorgeirsson SS. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab. Invest 74, 991–1003 (1996). [PubMed] [Google Scholar]
- 25.Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Böttinger EP. Apoptosis in podocytes induced by TGF-beta and Smad7. J. Clin. Invest 108, 807–816 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suthanthiran M, Gerber LM, Schwartz JE, Sharma VK, Medeiros M, Marion R, Pickering TG, August P. Circulating transforming growth factor-beta1 levels and the risk for kidney disease in African Americans. Kidney Int 76, 72–80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong MG, Perkovic V, Woodward M, Chalmers J, Li Q, Hillis GS, Yaghobian Azari D, Jun M, Poulter N, Hamet P, Williams B, Neal B, Mancia G, Cooper M, Pollock CA. Circulating bone morphogenetic protein-7 and transforming growth factor-β1 are better predictors of renal end points in patients with type 2 diabetes mellitus. Kidney Int 83, 278–284 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Voelker J, Berg PH, Sheetz M, Duffin K, Shen T, Moser B, Greene T, Blumenthal SS, Rychlik I, Yagil Y, Zaoui P, Lewis JB, Anti-TGF-B1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol 28, 953–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ledbetter S, Kurtzberg L, Doyle S, Pratt BM. Renal fibrosis in mice treated with human recombinant transforming growth factor-beta2. Kidney Int 58, 2367–2376 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Petersen M, Thorikay M, Deckers M, van Dinther M, Grygielko ET, Gellibert F, de Gouville AC, Huet S, ten Dijke P, Laping NJ. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int 73, 705–715 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Gewin L, Bulus N, Mernaugh G, Moeckel G, Harris RC, Moses HL, Pozzi A, Zent R. TGF-beta receptor deletion in the renal collecting system exacerbates fibrosis. J. Am. Soc. Nephrol 21, 1334–1343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung S, Overstreet JM, Li Y, Wang Y, Niu A, Wang S, Fan X, Sasaki K, Jin GN, Khodo SN, Gewin L, Zhang MZ, Harris RC. TGF-β promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 3, e123563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy-Chaudhury P, Simpson JG, Power DA. Endoglin, a transforming growth factor-beta-binding protein, is upregulated in chronic progressive renal disease. Exp. Nephrol 5, 55–60 (1997). [PubMed] [Google Scholar]
- 34.Rodríguez-Peña A, Prieto M, Duwel A, Rivas JV, Eleno N, Pérez-Barriocanal F, Arévalo M, Smith JD, Vary CP, Bernabeu C, López-Novoa JM. Up-regulation of endoglin, a TGF-beta-binding protein, in rats with experimental renal fibrosis induced by renal mass reduction. Nephrol. Dial. Transplant 16, 34–39 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Rodríguez-Peña A, Eleno N, Düwell A, Arévalo M, Pérez-Barriocanal F, Flores O, Docherty N, Bernabeu C, Letarte M, López-Novoa JM. Endoglin upregulation during experimental renal interstitial fibrosis in mice. Hypertension 40, 713–720 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Docherty NG, López-Novoa JM, Arevalo M, Düwel A, Rodriguez-Peña A, Pérez-Barriocanal F, Bernabeu C, Eleno N. Endoglin regulates renal ischaemia-reperfusion injury. Nephrol. Dial. Transplant 21, 2106–2119 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Thakar CV, Zahedi K, Revelo MP, Wang Z, Burnham CE, Barone S, Bevans S, Lentsch AB, Rabb H, Soleimani M. Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J. Clin. Invest 115, 3451–3459 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohenstein B, Daniel C, Hausknecht B, Boehmer K, Riess R, Amann KU, Hugo CP. Correlation of enhanced thrombospondin-1 expression, TGF-beta signalling and proteinuria in human type-2 diabetic nephropathy. Nephrol. Dial. Transplant 23, 3880–3887 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui W, Maimaitiyiming H, Qi X, Norman H, Wang S. Thrombospondin 1 mediates renal dysfunction in a mouse model of high-fat diet-induced obesity. Am. J. Physiol. Renal Physiol 305, F871–880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dendooven A, van Oostrom O, van der Giezen DM, Leeuwis JW, Snijckers C, Joles JA, Robertson EJ, Verhaar MC, Nguyen TQ, Goldschmeding R. Loss of endogenous bone morphogenetic protein-6 aggravates renal fibrosis. Am. J. Pathol 178, 1069–1079 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falke LL, Kinashi H, Dendooven A, Broekhuizen R, Stoop R, Joles JA, Nguyen TQ, Goldschmeding R. Age-dependent shifts in renal response to injury relate to altered BMP6/CTGF expression and signaling. Am. J. Physiol. Renal Physiol 311, F926–F934 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J. Am. Soc. Nephrol 13, S14–21 (2002). [PubMed] [Google Scholar]
- 43.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9, 964–968 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Liu W, Li X, Zhao Y, Meng XM, Wan C, Yang B, Lan HY, Lin HY, Xia Y. Dragon (repulsive guidance molecule RGMb) inhibits E-cadherin expression and induces apoptosis in renal tubular epithelial cells. J. Biol. Chem 288, 31528–31539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu W, Chen B, Wang Y, Meng C, Huang H, Huang XR, Qin J, Mulay SR, Anders HJ, Qiu A, Yang B, Freeman GJ, Lu HJ, Lin HY, Zheng ZH, Lan HY, Huang Y, Xia Y. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc. Natl. Acad. Sci. U S A 115, E1475–E1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang JJ, Chi NH, Huang TM, Connolly R, Chen LW, Chueh SJ, Kan WC, Lai CC, Wu VC, Fang JT, Chu TS, Wu KD. Urinary biomarkers predict advanced acute kidney injury after cardiovascular surgery. Crit. Care 22, 108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kajimoto H, Kai H, Aoki H, Uchiwa H, Aoki Y, Yasuoka S, Anegawa T, Mishina Y, Suzuki A, Fukumoto Y, Imaizumi T. BMP type I receptor inhibition attenuates endothelial dysfunction in mice with chronic kidney disease. Kidney Int 87, 128–136 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Dolan V, Murphy M, Sadlier D, Lappin D, Doran P, Godson C, Martin F, O’Meara Y, Schmid H, Henger A, Kretzler M, Droguett A, Mezzano S, Brady HR, Expression of gremlin, a bone morphogenetic protein antagonist, in human diabetic nephropathy. Am. J. Kidney Dis 45, 1034–1039 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Church RH, Ali I, Tate M, Lavin D, Krishnakumar A, Kok HM, Hombrebueno JR, Dunne PD, Bingham V, Goldschmeding R, Martin F, Brazil DP, Gremlin1 plays a key role in kidney development and renal fibrosis. Am. J. Physiol. Renal Physiol 312, F1141–F1157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamashita S, Maeshima A, Kojima I, Nojima Y. Activin A is a potent activator of renal interstitial fibroblasts. J. Am. Soc. Nephrol 15, 91–101 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Gaedeke J, Boehler T, Budde K, Neumayer HH, Peters H. Glomerular activin A overexpression is linked to fibrosis in anti-Thy1 glomerulonephritis. Nephrol. Dial. Transplant 20, 319–328 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Ren XJ, Guan GJ, Liu G, Zhang T, Liu GH. Effect of activin A on tubulointerstitial fibrosis in diabetic nephropathy. Nephrology (Carlton) 14, 311–320 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Bian X, Griffin TP, Zhu X, Islam MN, Conley SM, Eirin A, Tang H, O’Shea PM, Palmer AK, McCoy RG, Herrmann SM, Mehta RA, Woollard JR, Rule AD, Kirkland JL, Tchkonia T, Textor SC, Griffin MD, Lerman LO, Hickson LJ. Senescence marker activin A is increased in human diabetic kidney disease: association with kidney function and potential implications for therapy. BMJ Open Diabetes Res. Care 7, e000720 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muñoz-Félix JM, López-Novoa JM, Martínez-Salgado C. Heterozygous disruption of activin receptor-like kinase 1 is associated with increased renal fibrosis in a mouse model of obstructive nephropathy. Kidney Int 85, 319–332 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Mehta N, Gava AL, Zhang D, Gao B, Krepinsky JC. Follistatin Protects Against Glomerular Mesangial Cell Apoptosis and Oxidative Stress to Ameliorate Chronic Kidney Disease. Antioxid. Redox Signal 31, 551–571. [DOI] [PubMed] [Google Scholar]
- 56.Maeshima A, Mishima K, Yamashita S, Nakasatomi M, Miya M, Sakurai N, Sakairi T, Ikeuchi H, Hiromura K, Hasegawa Y, Kojima I, Nojima Y. Follistatin, an activin antagonist, ameliorates renal interstitial fibrosis in a rat model of unilateral ureteral obstruction. Biomed. Res. Int 2014, 376191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kralisch S, Hoffmann A, Klöting N, Bachmann A, Kratzsch J, Stolzenburg JU, Dietel A, Beige J, Anders M, Bast I, Blüher M, Zhang MZ, Harris RC, Stumvoll M, Fasshauer M, Ebert T. FSTL3 is increased in renal dysfunction. Nephrol. Dial. Transplant 32, 1637–1644(2017). [DOI] [PubMed] [Google Scholar]
- 58.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic N, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D, Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 5, e15004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tuerk C, Gold L, Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 (1990). [DOI] [PubMed] [Google Scholar]
- 60.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, Oliver-Williams C, Kamat MA, Prins BP, Wilcox SK, Zimmerman ES, Chi A, Bansal N, Spain SL, Wood AM, Morrell NW, Bradley JR, Janjic N, Roberts DJ, Ouwehand WH, Todd JA, Soranzo N, Suhre K, Paul DS, Fox CS, Plenge RM, Danesh J, Runz H, Butterworth AS, Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niewczas MA, Pavkov ME, Skupien J, Smiles A, Md Dom ZI, Wilson JM, Park J, Nair V, Schlafly A, Saulnier PJ, Satake E, Simeone CA, Shah H, Qiu C, Looker HC, Fiorina P, Ware CF, Sun JK, Doria A, Kretzler M, Susztak K, Duffin KL, Nelson RG, Krolewski AS, A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med 25, 805–813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Md Dom ZI, Satake E, Skupien J, Krolewski B, O’Neil K, Willency JA, Dillon ST, Wilson JM, Kobayashi H, Ihara K, Libermann TA, Pragnell M, Duffin KL, Krolewski AS, Circulating proteins protect against renal decline and progression to end-stage renal disease in patients with diabetes. Sci. Transl. Med 13, eabd2699 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Satake E, Saulnier PJ, Kobayashi H, Gupta MK, Looker HC, Wilson JM, Md Dom ZI, Ihara K, O’Neil K, Krolewski B, Pipino C, Pavkov ME, Nair V, Bitzer M, Niewczas MA, Kretzler M, Mauer M, Doria A, Najafian B, Kulkarni RN, Duffin KL, Pezzolesi MG, Kahn CR, Nelson RG, Krolewski AS, Comprehensive Search for Novel Circulating miRNAs and Axon Guidance Pathway Proteins Associated with Risk of ESKD in Diabetes. J. Am. Soc. Nephrol 32, 2331–2351 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ozaki T, Sakiyama S, Tumor-suppressive activity of N03 gene product in v-src-transformed rat 3Y1 fibroblasts. Cancer Res 54, 646–648 (1994). [PubMed] [Google Scholar]
- 65.Ozaki T, Enomoto H, Nakamura Y, Kondo K, Seki N, Ohira M, Nomura N, Ohki M, Nakagawara A, Sakiyama S, The genomic analysis of human DAN gene. DNA. Cell Biol 16, 1031–1039 (1997). [DOI] [PubMed] [Google Scholar]
- 66.Nolan K, Kattamuri C, Luedeke DM, Angerman EB, Rankin SA, Stevens ML, Zorn AM, Thompson TB, Structure of neuroblastoma suppressor of tumorigenicity 1 (NBL1): insights for the functional variability across bone morphogenetic protein (BMP) antagonists. J. Biol. Chem 290, 4759–4771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kattamuri C, Luedeke DM, Nolan K, Rankin SA, Greis KD, Zorn AM, Thompson ZB, Members of the DAN family are BMP antagonists that form highly stable noncovalent dimers. J. Mol. Biol 424, 313–327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dionne MS, Skarnes WC, Harland RM, Mutation and analysis of dan, the founding member of the DAN family of transforming growth factor beta antagonists. Mol. Cell Biol 21, 636–643 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pearce JJ, Penny G, Rossant J, A mouse Cerberus/Dan- related gene family. Dev. Biol 209, 98–110 (1999). [DOI] [PubMed] [Google Scholar]
- 70.Hung WT, Wu FJ, Wang CJ, Luo CW, Dan (NBL1) specifically antagonizes BMP2 and BMP4 and modulates the actions of GDF9, BMP2, and BMP4 in the rat ovary. Biol. Reprod 86, 158 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Gohda T, Niewczas MA, Ficociello LH, Walker WH, Skupien J, Rosetti F, Cullere X, Johnson AC, Crabtree G, Smiles AM, Mayadas TN, Warram JH, Krolewski AS, Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J. Am. Soc. Nephrol 23, 516–524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niewczas MA, Gohda T, Skupien J, Smiles AM, Walker WH, Rosetti F, Cullere X, Eckfeldt JH, Doria A, Mayadas TN, Warram JH, Krolewski AS, Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol 23, 507–515 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nolan K, Thompson TB, The DAN family: modulators of TGF-β signaling and beyond. Protein Sci 23, 999–1012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nolan K, Kattamuri C, Rankin SA, Read RJ, Zorn AM, Thompson TB, Structure of Gremlin-2 in Complex with GDF5 Gives Insight into DAN-Family-Mediated BMP Antagonism. Cell Rep 16, 2077–2086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, Welling PA, Waikar SS, Humphreys BD, The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc. Natl. Acad. Sci. U S A 116, 19619–19625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fan Y, Yi Z, D’Agati VD, Sun Z, Zhong F, Zhang W, Wen J, Zhou T, Li Z, He L, Zhang Q, Lee K, He JC, Wang N, Comparison of Kidney Transcriptomic Profile of Early and Advanced Diabetic Nephropathy Reveals Potential New Mechanisms for Disease Progression. Diabetes 68, 2301–2314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muto Y, Wilson PC, Ledru N, Wu H, Dimke H, Waikar SS, Humphreys BD, Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat. Commun 12, 2190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eimon PM, Harland RM, Xenopus Dan, a member of the Dan gene family of BMP antagonists, is expressed in derivatives of the cranial and trunk neural crest. Mech. Dev 107, 187–189 (2001). [DOI] [PubMed] [Google Scholar]
- 79.Ohtori S, Yamamoto T, Ino H, Hanaoka E, Shinbo J, Ozaki T, Takada N, Nakamura Y, Chiba T, Nakagawara A, Sakiyama S, Sakashita Y, Takahashi K, Tanaka K, Yamagata M, Yamazaki M, Shimizu S, Moriya H, Differential screening-selected gene aberrative in neuroblastoma protein modulates inflammatory pain in the spinal dorsal horn. Neuroscience 110, 579–586 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Kim AS, Pleasure SJ, Expression of the BMP antagonist Dan during murine forebrain development. Brain Res. Dev. Brain Res 145, 159–162 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Yanagita M, Okuda T, Endo S, Tanaka M, Takahashi K, Sugiyama F, Kunita S, Takahashi S, Fukatsu A, Yanagisawa M, Kita T, Sakurai T, Uterine sensitization-associated gene-1 (USAG-1), a novel BMP antagonist expressed in the kidney, accelerates tubular injury. J. Clin. Invest 116, 70–79 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tanaka M, Asada M, Higashi AY, Nakamura J, Oguchi A, Tomita M, Yamada S, Asada N, Takase M, Okuda T, Kawachi H, Economides AN, Robertson E, Takahashi S, Sakurai T, Goldschmeding R, Muso E, Fukatsu A, Kita T, Yanagita M, Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J. Clin. Invest 120, 768–777 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kriz W, Lemley KV, The role of the podocyte in glomerulosclerosis. Curr. Opin. Nephrol. Hypertens 8, 489–497 (1999). [DOI] [PubMed] [Google Scholar]
- 84.Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW, Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Invest 99, 342–348 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meyer TW, Bennett PH, Nelson RG, Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 42, 1341–1344 (1999). [DOI] [PubMed] [Google Scholar]
- 86.Lemley KV, Abdullah I, Myers BD, Meyer TW, Blouch K, Smith WE, Bennett PH, Nelson RG, Evolution of incipient nephropathy in type 2 diabetes mellitus. Kidney Int 58, 1228–1237 (2000). [DOI] [PubMed] [Google Scholar]
- 87.Jefferson JA, Alpers CE, Shankland SJ, Podocyte biology for the bedside. Am. J. Kidney Dis 58, 835–845 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang L, Tang Y, Eisner W, Sparks MA, Buckley AF, Spurney RF, Augmenting podocyte injury promotes advanced diabetic kidney disease in Akita mice. Biochem. Biophys. Res. Commun 444, 622–627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ohtori S, Isogai E, Hasue F, Ozaki T, Nakamura Y, Nakagawara A, Koseki H, Yuasa S, Hanaoka E, Shinbo J, Yamamoto T, Chiba H, Yamazaki M, Moriya H, Sakiyama S, Reduced inflammatory pain in mice deficient in the differential screening-selected gene aberrative in neuroblastoma. Mol. Cell Neurosci 25, 504–514 (2004). [DOI] [PubMed] [Google Scholar]
- 90.Krolewski AS, Niewczas MA, Skupien J, Gohda T, Smiles A, Eckfeldt JH, Doria A, Warram JH, Early progressive renal decline precedes the onset of microalbuminuria and its progression to macroalbuminuria. Diabetes Care 37, 226–234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nelson RG, Newman JM, Knowler WC, Sievers ML, Kunzelman CL, Pettitt DJ, Moffett CD, Teutsch SM, Bennett PH, Incidence of end-stage renal disease in type 2 (non-insulin-dependent) diabetes mellitus in Pima Indians. Diabetologia 31, 730–736 (1988). [DOI] [PubMed] [Google Scholar]
- 92.Nelson RG, Bennett PH, Beck GJ, Tan M, Knowler WC, Mitch WE, Hirschman GH, Myers BD, Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N. Engl. J. Med 335, 1636–1642 (1996). [DOI] [PubMed] [Google Scholar]
- 93.Weil EJ, Fufaa G, Jones LI, Lovato T, Lemley KV, Hanson RL, Knowler WC, Bennett PH, Yee B, Myers BD, Nelson RG, Effect of losartan on prevention and progression of early diabetic nephropathy in American Indians with type 2 diabetes. Diabetes 62, 3224–3231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration), A new equation to estimate glomerular filtration rate. Ann. Intern. Med 150, 604–612 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Centers for Disease Control and Prevention, National Center for Health Statistics: Data Access—National Death Index Available at: http://www.cdc.gov/nchs/ndi.htm. Queried September 1, 2013.
- 96.U.S. Renal Data System 2013 Annual Data Report.: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD: (2013). [Google Scholar]
- 97.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M, KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hertzmark E, Pazaris M, Spiegelman D, The SAS MEDIATE macro 2018.
- 99.Cook NR, Quantifying the added value of new biomarkers: how and how not. Diagn. Progn. Res 2, 14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ganz P, Heidecker B, Hveem K, Jonasson C, Kato S, Segal MR, Sterling DG, Williams SA, Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA 315, 2532–2541 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, Benson MD, O’Sullivan JF, Keshishian H, Farrell LA, Fifer MA, Vasan RS, Sabatine MS, Larson MG, Carr SA, Wang TJ, Gerszten RE, Aptamer-based proteomic profiling reveals novel candidate biomarkers and pathways in cardiovascular disease. Circulation 134, 270–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Williams SA, Murthy AC, DeLisle RK, Hyde C, Malarstig A, Ostroff R, Weiss SJ, Segal MR, Ganz P, Improving assessment of drug safety through proteomics: early detection and mechanistic characterization of the unforeseen harmful effects of torcetrapib. Circulation 137, 999–1010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weil EJ, Lemley KV, Mason CC, Yee B, Jones LI, Blouch K, Lovato T, Richardson M, Myers BD, Nelson RG, Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int 82, 1010–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P, A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol 13, 630–638 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Proteins evaluated in the current study and relevant previous publications
Table S2 | Results of Cox regression analysis for time to ESKD for 25 circulating TGF-β signaling proteins that were evaluated in the current study
Table S3 | Characteristics of subjects with T1D and T2D selected from Joslin cohorts into the nested case-control studies to assess risk of ESKD according to baseline concentrations of urinary proteins measured by SOMAscan proteomics platform
Table S4 | QC metrics for the analysis of the snRNA-seq data
Fig. S1 | Circulating proteins involved in TGF-β signaling examined in the study
Fig. S2 | Distribution of follow-up duration in the study cohorts
Fig. S3 | Correlation matrix for 4 candidate proteins
Fig. S4 | Association between circulating NBL1 and kidney structural lesions
Fig. S5 | NBL1 Atlas - Expression of NBL1 protein in human tissues
Fig. S6 | Distribution of circulating NBL1 in healthy control and in the study cohorts according to sex
Fig. S7 | A phylogenetic representation of the DAN family proteins
Fig. S8 | Correlation of circulating NBL1 concentrations measured by Meso Scale Discovery assay and SOMAscan
Data Availability Statement
All data associated with this study are present in the paper or in the supplementary materials. Individual proteomics data may become available for collaborative research from Drs. A.S. Krolewski and R. Nelson.