

Abstract
Over the past fifteen years, we have unveiled a new mechanism by which cells achieve greater efficiency in de novo purine biosynthesis. This mechanism relies on the compartmentalization of de novo purine biosynthetic enzymes into a dynamic complex called the purinosome. In this review, we highlight our current understanding of the purinosome with emphasis on its biophysical properties and function and on the cellular mechanisms that regulate its assembly. We propose a model for functional purinosomes in which they consist of at least ten enzymes that localize near mitochondria and carry out de novo purine biosynthesis by metabolic channeling. We conclude by discussing challenges and opportunities associated with studying the purinosome and analogous metabolons.
Keywords: metabolism, de novo purine biosynthesis, channeling, protein complex, metabolon, purinosome
1. INTRODUCTION
Purines are generated either through the recycling of bases via the salvage pathway or through the de novo purine biosynthesis (DNPB) pathway (1–4). Purine salvage is predominantly used under normal physiological growth conditions; however, when purine demand exceeds the capacity of the salvage pathway, DNPB is activated (5–8). Activation of the DNPB pathway can occur in response to signaling through oncogenes (e.g., MYC, AKT) and tumor suppressors in cancer cells and as a result of deficiencies in activity among salvage enzymes (9–11). In addition, pathway utilization differs between the various phases of the cell cycle; a fivefold increase in the rate of purine production through the DNPB pathway was noted between mid-G1-phase and early S-phase in HCT116 human colon carcinoma cells (12).
The DNPB is an energy-intensive pathway that consists of a series of ten metabolic transformations that convert phosphoribosyl pyrophosphate (PRPP), generated from the pentose phosphate pathway, into inosine 5′-monophosphate (IMP). In humans, these reactions are catalyzed by six enzymes and require various amino acid substrates (Gln, Gly, Asp), ATP, carbon dioxide, and the N10-formyltetrahydrofolate (10-formyl-THF) cofactor (Figure 1). The first enzyme in the pathway, PRPP amidotransferase (PPAT) converts PRPP into 5-phosphoribosylamine (5-PRA). Since PRPP is utilized by both the salvage and the DNPB enzymes for the formation of purines, as well as pyrimidines, the action of PPAT commits PRPP to the DNPB pathway (13, 14). The second pathway enzyme, phosphoribosylglycinamide formyltransferase (GART), is trifunctional and catalyzes nonsequential steps in the pathway. The second and third transformations incorporate a glycyl and a formyl group onto 5-PRA to generate formylglycinamide ribonucleotide (FGAR). Then, FGAR is handed off to phosphoribosyl formylglycinamide synthase (PFAS or FGAMS) to produce the substrate, formylglycinamidine ribonucleotide (FGAM), for the last transformation catalyzed by GART. The challenges posed by the thermodynamic instability of the pathway intermediate 5-PRA and the need for the enzymes GART and PFAS to coordinate the efficient hand off of the intermediates FGAR and FGAM between the two enzymes to generate 5-aminoimidazole ribonucleotide (AIR) led us to consider the advantages of multi-enzyme complexation to coordinate the activities of different DNPB enzymes in human cells. It was not until intracellular fluorescence microscopy could be applied to study the localization of DNPB enzymes within the cell that a level of coordination between the pathway enzymes was discovered. Using transient expression of pathway enzymes tagged with a fluorescent protein reporter, we showed by confocal microscopy that each of the five pathway enzymes coclustered with PFAS in purine-depleted HeLa cells, a growth condition shown to activate the DNPB pathway (15). We termed these discrete punctate structures of enzymes purinosomes. The process of purinosome formation is reversible. Upon purine supplementation, purinosomes are quickly destabilized, as indicated by a loss of discrete PFAS foci, and they reemerge upon subsequent purine depletion (15). Since their discovery, purinosomes have been detected in a wide variety of cancer cell lines such as breast cancer (15–17), ovarian cancer (15), and liver cancer cells (18, 19). In addition, PFAS-containing bodies have been observed in neurons (20–22) and primary dermal fibroblasts from patients diagnosed with Lesch-Nyhan syndrome (10, 23). Subsequent investigations revealed that the six enzymes of the classical DNPB pathway not only colocalize within a cluster but also channel pathway intermediates without their loss from the cluster, fitting the definition of a metabolon (33, 42).
Figure 1.
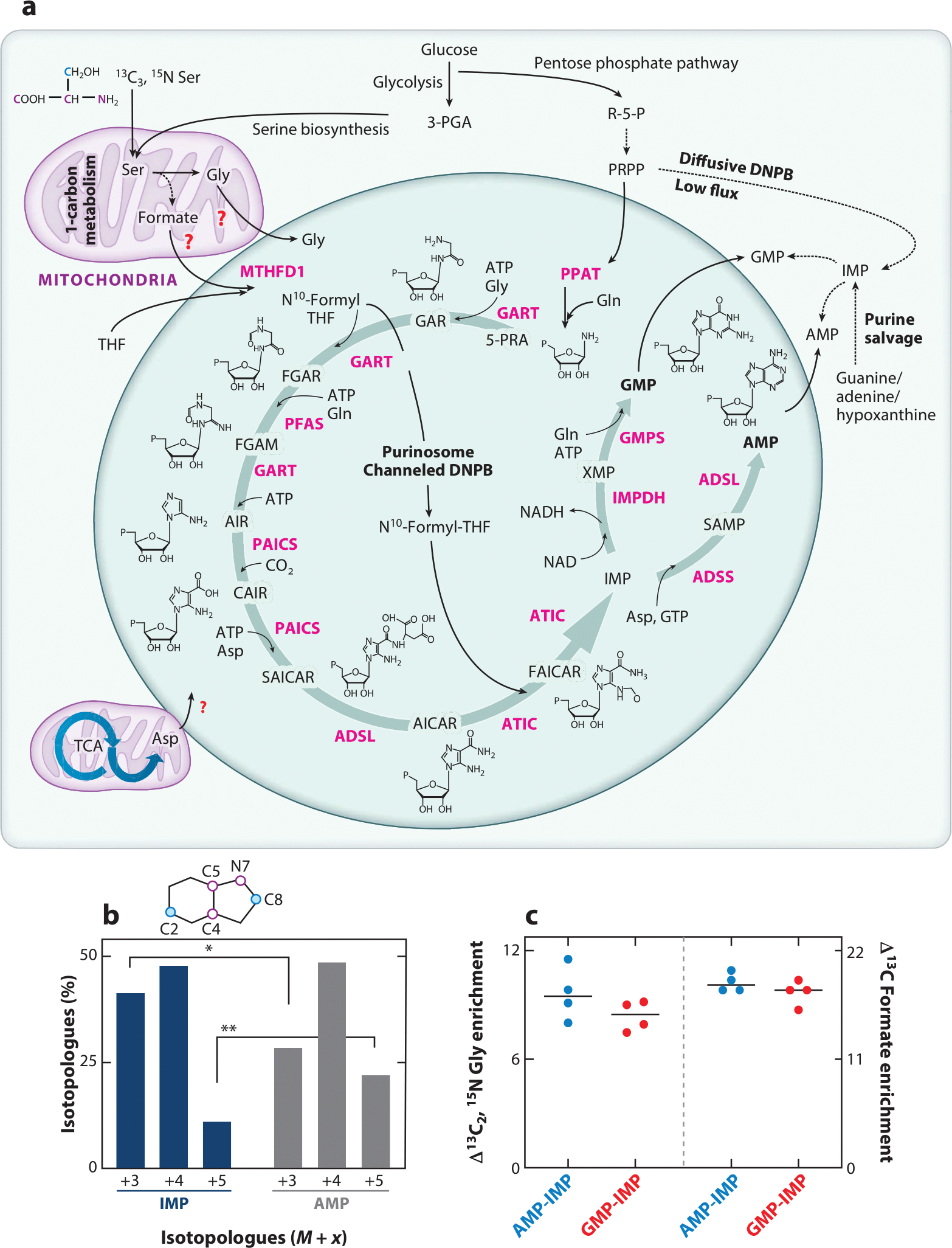
Enzymatic composition and function of the purinosome. (a) Channeled DNPB is carried out by the purinosome metabolon, which is composed of at least ten enzymes (pink): PPAT, GART, PFAS, PAICS, ADSL, ATIC, IMPDH, ADSS, GMPS, and MTHFD1. Structures of DNPB intermediates are shown; “P” denotes a phosphate group. Glycolysis, the serine biosynthesis pathway, one-carbon metabolism, the pentose phosphate pathway, and the TCA cycle generate the building block substrates utilized in DNPB. Involvement of mitochondrial Gly, formate, and Asp transporters for the direct uptake of these substrates for DNPB by purinosomes has been proposed though not confirmed yet (red question marks). The stable isotope–labeled positions of Ser are shown for the backbone atoms (pink) and side chain carbon (blue); the positions of these labeled atoms upon incorporation into the purine ring are shown as pink and blue circles in the purine ring diagram (shown in panel b). Alternatively, low-flux DNPB is carried out by the diffusive pool of DNPB enzymes outside purinosomes and by purine salvage enzymes that produce IMP, GMP, and AMP. (b) Upon 13C3, 15N Ser–mediated labeling of de novo synthesized purines, a different isotopologue distribution was discovered in the newly synthesized IMP (blue bars) and AMP (dark gray bars), revealing the two parallel purine-generating pathways (33). The purine ring positions replaced with isotope-labeled atoms are shown as pink and blue circles in the purine ring schematic. The tightly channeled, high-flux DNPB pathway that primarily generates AMP and GMP is carried out by mitochondria-associated purinosomes. Alternatively, the diffusive, low-flux DNPB primarily generates the free IMP pool. (c) The end nucleotides AMP and GMP showed higher incorporation of the mitochondrially derived substrates Gly and formate (33). AMP and GMP showed ~10% higher 13C2, 15N Gly enrichment and ~20% higher 13C formate enrichment compared with diffusively synthesized IMP. The plot shows the difference in isotope incorporation between AMP and IMP (blue circles) and GMP and IMP (red circles). Abbreviations: 3-PGA, 3-phosphoglyceric acid; 5-PRA, 5- phosphoribosylamine; ADSL, adenylosuccinate lyase; ADSS, adenylosuccinate synthetase; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; AIR, 5-aminoimidazole ribonucleotide; ATIC, 5-aminoimidazole-4-carboxamide nucleotide formyltransferase/IMP cyclohydroxylase; CAIR, carboxyaminoimidazole ribonucleotide; DNPB, de novo purine biosynthesis; FAICAR, 5-formamidoimidazole-4-carboxamide ribonucleotide; FGAM, formylglycinamidine; FGAR, formylglycinamide ribonucleotide; GAR, glycinamide ribonucleotide; GART, phosphoribosylglycinamide formyltransferase; GMPS, GMP synthetase; IMP, inosine monophosphate; IMPDH, IMP dehydrogenase; MTHFD1, cytosolic methylenetetrahydrofolate dehydrogenase; NAD, nicotinamide adenine dinucleotide oxidized; NADH, nicotinamide adenine dinucleotide reduced; PAICS, phosphoribosyl aminoimidazole succinocarboxamide synthase; PFAS, phosphoribosyl formylglycinimidine transferase; PPAT, PRPP amidotransferase; PRPP, phosphoribosyl pyrophosphate; R-5-P, ribose-5-phosphate; SAICAR, phosphoribosyl aminoimidazole succinocarboxamide; SAMP, succinyladenosine monophosphate; TCA, tricarboxylic acid; THF, tetrahydrofolate; XMP, xanthosine monophosphate.
2. BIOPHYSICAL PROPERTIES OF THE PURINOSOME
Heavy reliance on using fluorescence microscopy in transient protein overexpression models to detect purinosome formation has drawn criticism over the years (24, 25). The main critique is centered on our interpretation of what the observed PFAS assemblies represent. Arguments include the fact that experimental conditions used to promote purinosome formation, such as nutrient depletion and transient protein overexpression, might also result in increased cell stress and favor protein aggregation (25, 26). These conflicting reports have led us to carefully characterize the biophysical properties of the purinosome to help differentiate it from protein aggregates and other stress-induced bodies.
In models expressing PFAS–EGFP as a marker for the purinosome, we observed 50–300 submicron particles per purine-depleted HeLa cell (Figure 2a) (27, 28). The average cluster size and number of purinosomes did not agree with those associated with other cytosolic bodies such as processing bodies (P-bodies), stress granules, and aggresomes (27). These PFAS-containing assemblies were spherical, as determined by lattice light-sheet fluorescence microscopy (Figure 2b), and their roundness was perturbed upon increasing cell stress and inducing protein aggregation by heat shock protein 90 (HSP90) inhibition (29). Further, the lack of colocalization of purinosomes with G3BP1, an intracellular marker for stress granules, and poly(A)+ mRNA confirms that these bodies are not traditional stress granules (17, 18, 29). We have also ruled out the possibility that the purinosome is similar to an early-stage protein aggregate based on the lack of colocalization with the aggresome markers GFP250 and GFP170* (18). In addition, we have shown that these bodies are not due to repressed protein translation, a consequence of extended nutrient depletion (29). These features suggest that the purinosome is likely a unique subcellular organization distinct from what typically defines commonly observed P-bodies and stress granules.
Figure 2.
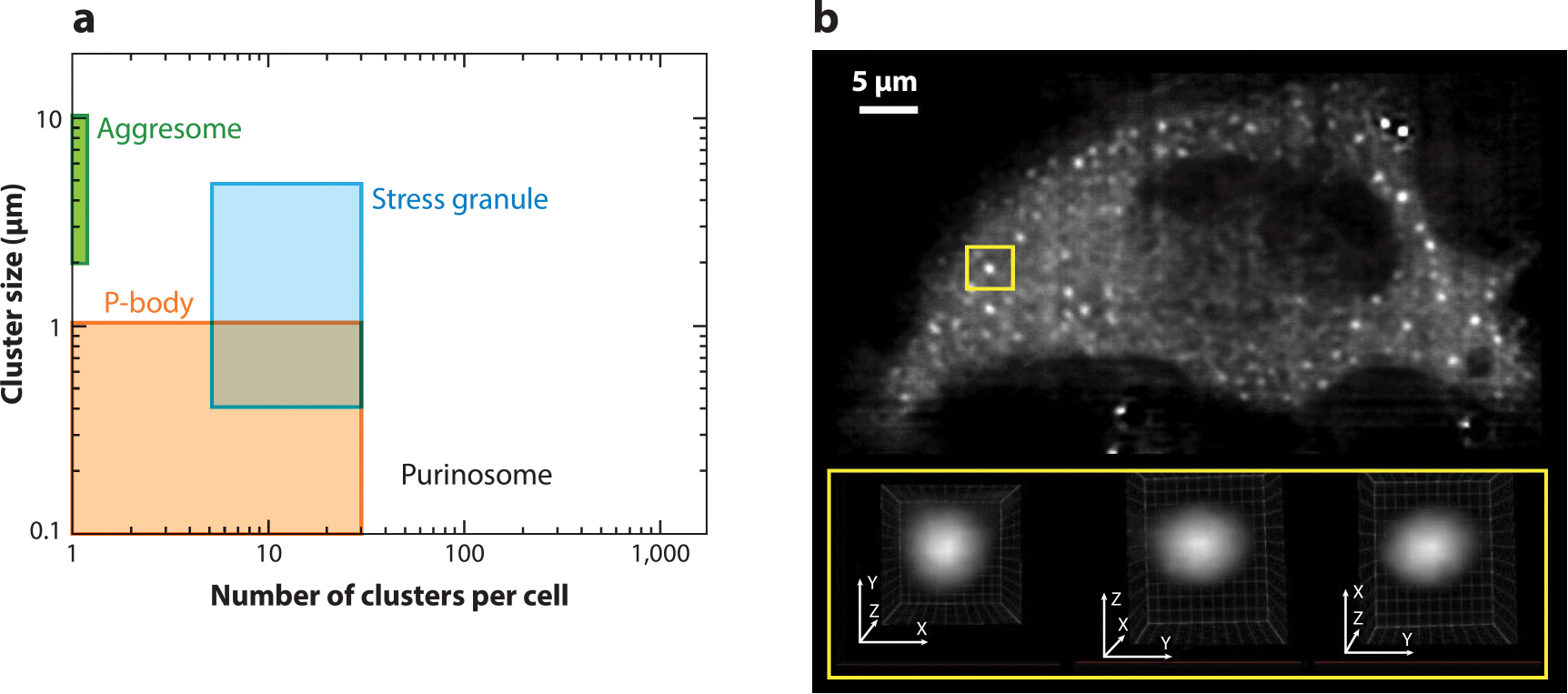
Biophysical properties of purinosomes. (a) The size and number of PFAS–EGFP clusters in purine-depleted HeLa cells compared to other reported biomolecular condensates. (b) A representative lattice light-sheet fluorescence microscopy image of a purinosome-positive HeLa cell transfected with PFAS–mCherry. Analysis of these clusters showed that they are spherical, as demonstrated by the region of interest (yellow boxes). Panel b modified from the original image published in Pedley et al. (29) with permission from E.L. Kennedy and M. Kyoung. Abbreviations: EGFP, enhanced green fluorescent protein; P-body, processing body; PFAS, phosphoribosyl formylglycinamide synthase.
Recently, purinosomes were shown not to be lipid encapsulated (16), and fluorescently tagged DNPB enzymes within the purinosome were shown to rapidly exchange between the assembly and bulk solvent (15, 30). These characteristics, in combination with their spherical nature, gave us an indication that purinosomes might exhibit liquid-like properties. Unlike many condensates, purinosomes have not been reconstituted outside of the cell. Therefore, we have further substantiated the claim that the purinosome exhibits properties of a liquid droplet by looking at their spatio-temporal intracellular dynamics. Purinosomes, as denoted by PFAS clusters, are not static assemblies (23, 29), and multiple small clusters show a tendency to coalesce when they are in close proximity (29). This process results in the formation of larger spherical clusters inside the cell. Combined, these studies support an argument that the purinosome arises from a liquid–liquid phase separation.
3. ENZYMATIC COMPOSITION AND SUBCELLULAR LOCALIZATION OF PURINOSOMES
While early research indicated that the DNPB enzymes transiently associate to form purinosomes, detection of such a complex on the endogenous level and direct evidence for its function has been achieved only recently (15, 17, 19, 31–33). Discrepancies in the abundance, size, and composition of DNPB enzyme clusters detected by different techniques stem from both the natural heterogeneity and experimental artifacts arising from DNPB enzyme aggregation and self-assembly upon transient overexpression (Figure 3). We also questioned whether partial assembly of pathway complexes would preexist under conditions of sufficient as well as restricted purine supply and explored various techniques to obtain decisive evidence.
Figure 3.
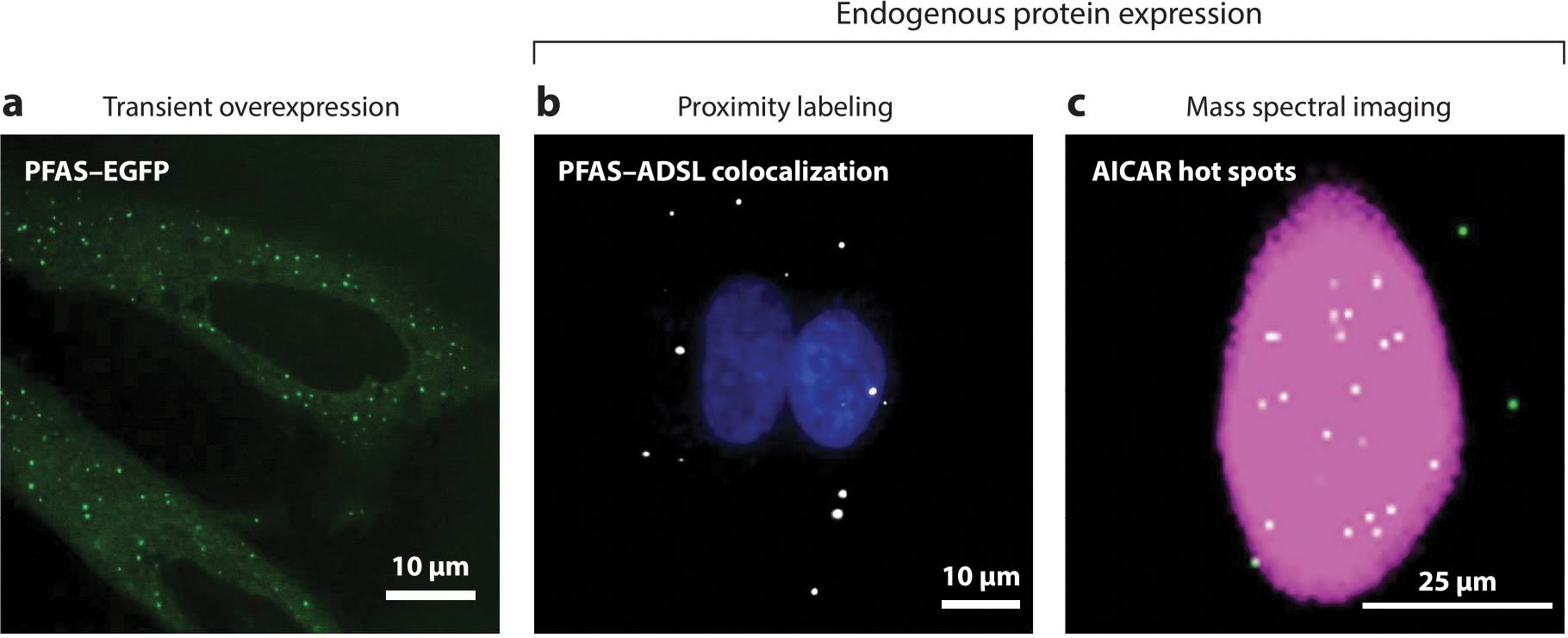
Imaging methods for purinosome detection. Purinosomes can be visualized by three different imaging methods. (a) Initial studies used the transient overexpression of DNPB enzymes tagged with a fluorescent protein such as PFAS–EGFP in purine-depleted HeLa cells to characterize these enzyme assemblies (green punctate staining) (64). The misidentification of purinosomes due to the possibility of DNPB enzyme self-assembly and aggregation resulted in the development of new methods for detecting the complex using endogenous protein expression. These methods include proximity labeling assays (PLAs) and mass spectral imaging. (b) PLAs detect purinosomes through the colocalization of two DNPB enzymes, such as PFAS and ADSL (white) in purine-depleted HeLa cells (17). Nuclei are shown in blue. (c) A representative GCIB-SIMS image of a HeLa cell showing the total ion current (pink) and the pixels with high AICAR abundance (white) (33). These AICAR hot spots also show high levels of ATP, arising as a result of purinosome activity. Abbreviations: AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; ADSL, adenylosuccinate lyase; DNPB, de novo purine biosynthesis; EGFP, enhanced green fluorescent protein; GCIB-SIMS, gas cluster ion beam–time of flight–secondary ion mass spectrometry; PFAS, phosphoribosyl formylglycinamide synthase; PLA, proximity labeling assay.
Colocalization of various pairs of DNPB enzymes was first discovered by transfecting HeLa cells with pairs of chimeric DNPB enzymes tagged with fluorescent proteins under both purine-rich and purine-depleted conditions (15, 34). While dysfunction of the salvage pathway promotes this colocalization, knocking out any one of the DNPB enzymes, transfection with disease-related mutants of adenylosuccinate lyase (ADSL) and 5-aminoimidazole-4-carboxamide nucleotide formyltransferase/IMP cyclohydroxylase (ATIC), and small molecule–mediated disruption of native dimeric ATIC (the last enzyme in the DNPB pathway) led to purinosome disassembly (17, 19, 31, 32). In situ proximity of the DNPB enzymes was probed under purine-rich and -depleted conditions using a bimolecular fluorescence complementation assay with a split-SYFP2, an optimized monomeric form of enhanced yellow fluorescent protein (EYFP) (35). In this system, different pairs of DNPB enzymes are genetically fused with different nonfluorescent parts of SYFP2, and reconstitution of the mature fluorescent proteins reports on the interaction of the tagged enzymes. These interactions were supported by coimmunoprecipitation, which detected interactions between different DNPB enzymes and PFAS, and have been verified by experiments in which DNPB enzyme expression was kept at or near endogenous levels (35). In addition, an affinity-tagged version of phosphoribosyl aminoimidazole succinocarboxamide synthase (PAICS) was stably integrated into a PAICS-knockout HeLa cell line. PAICS is an octameric enzyme that has been proposed to act as a protein interaction hub in the purinosome assembly (21). Using buffer conditions that stabilize the assembly, several DNPB enzymes were isolated with PAICS (35). The detected associations included not only the enzymes that catalyze the reaction steps directly upstream (GART) and downstream (ADSL) of PAICS but also ones farther apart in the reaction cascade [PFAS, ATIC, and methylenetetrahydrofolate dehydrogenase (MTHFD1)]. These associations have been complemented and confirmed by cofractionation, immunofluorescence, and proximity labeling assays (PLAs) with the endogenous enzymes (17, 31, 32, 36) (Tables 1 and 2).
Table 1.
Interenzyme and enzyme–subcellular structure interactions involving DNPB enzymes in human cell lines detected by various techniques. Evidence for interactions detected in transiently overexpressed enzymes
| DNPB enzyme interactions probed | Technique | |||
|---|---|---|---|---|
| Fluorescence imaging of tagged enzymes | Coimmunoprecipitation | Luminescence-based mammalian interactome (LUMIER) assay | Bimolecular fluorescence complementation (BiFC) assay | |
| Enzyme participants | PPAT, GART, PFAS, PAICS, ADSL, ATIC, IMPDH, ADSS (15, 16, 34, 64) | PFAS, PAICS, ATIC (18) PFAS–PAICS, MTHFD1, GART, IMPDH2 (65) |
ND | GART, PFAS, PAICS, ADSL, ATIC (35) |
| Mitochondria and microtubule | Mitochondria–PFAS (17, 23, 28) Microtubule–PFAS (23,37) |
Mitochondria–PFAS (28) | ND | ND |
| Chaperone machinery | HSP90/HSP70/DNAJ-C7/DNAJ A1–PFAS/PPAT/PAICS (18) | HSP90/HSP70/DNAJ-C7–PFAS/PPAT (18, 56, 65) | HSP90–PPAT/PFAS (56) | ND |
Abbreviations: ADSL, adenylosuccinate lyase; ADSS, adenylosuccinate synthetase; ATIC, 5-aminoimidazole-4-carboxamide nucleotide formyltransferase/IMP cyclohydroxylase; DNPB, de novo purine biosynthesis; GART, phosphoribosylglycinamide formyltransferase; HSP, heat shock protein; IMPDH, inosine monophosphate dehydrogenase; MTHFD, methylenetetrahydrofolate dehydrogenase/cyclohydrolase (cytosolic isoform); ND, not determined; PAICS, phosphoribosyl aminoimidazole succinocarboxamide synthase; PFAS, phosphoribosyl formylglycinimidine transferase; PPAT, phosphoribosyl pyrophosphate amidotransferase.
Table 2.
Interenzyme and enzyme–subcellular structure interactions involving DNPB enzymes in human cell lines detected by various techniques. Evidence for interactions detected with endogenous expression of enzymes
| DNPB enzyme interactions probed | Technique | ||||
|---|---|---|---|---|---|
| Chromatographic cofractionation | Immunofluorescence imaging | Coimmunoprecipitation | Proximity ligation assay | In-cell isotope incorporation assay | |
| Enzyme participants | GART, PFAS, PAICS, ADSL, ATIC (36) | PPAT, GART, PFAS, PAICS, ADSL, ATIC (19, 20, 31, 32) | GART, PFAS, PAICS, ADSL, ATIC, MTHFD1 (35) | PFAS–ADSL (17) | PPAT, GART, PFAS, PAICS, ADSL, ATIC, MTHFD1, IMPDH, GMPS, ADSS (33) |
| Mitochondria and microtubule | ND | ND | ND | Tom20–PFAS (17) | Mitochondria purinosome (33) |
| Chaperone machinery | ND | ND | ND | HSP70–ADSL (17) | ND |
Abbreviations: ADSL, adenylosuccinate lyase; ADSS, adenylosuccinate synthetase; ATIC, 5-aminoimidazole-4-carboxamide nucleotide formyltransferase/IMP cyclohydroxylase; DNPB, de novo purine biosynthesis; GART, phosphoribosylglycinamide formyltransferase; GMPS, GMP synthetase; HSP, heat shock protein; IMPDH, inosine monophosphate dehydrogenase; MTHFD, methylenetetrahydrofolate dehydrogenase/cyclohydrolase (cytosolic isoform); ND, not determined; PAICS, phosphoribosyl aminoimidazole succinocarboxamide synthase; PFAS, phosphoribosyl formylglycinimidine transferase; PPAT, phosphoribosyl pyrophosphate amidotransferase; Tom20, translocase of outer membrane 20.
Another interesting aspect of the DNPB enzyme clusters is their intracellular localization. These assemblies colocalize with microtubules (23, 37) and mitochondria (23, 28). The importance of the microtubule network was substantiated by characterizing the intracellular movement of purinosomes. Purinosomes were observed to move directionally along microtubules toward mitochondria, suggesting that their trafficking is assisted by some helper or adapter protein via active transport (23). This observation was corroborated by experiments showing that nocodazole, an inhibitor of microtubule assembly, led to the disruption of DNPB enzyme clusters (23) and a reduction in purine monophosphate generation through the DNPB pathway (37). These discoveries support the hypothesis that purinosome function is maximized near mitochondria because of the pathway’s high reliance on mitochondria-derived substrates (Gly, formate, and Asp). Additionally, mitochondria rely heavily on the cytosolic pool of nucleotides to fulfill their nucleotide requirements, for both enzymatic functions as well as mitochondrial DNA replication. Thus, an interesting possibility is that localization of purinosomes near mitochondria might lead to an efficient uptake of purines by the mitochondria.
4. METABOLIC CHANNELING BY PURINOSOMES
Despite the mounting evidence supporting the interaction of DNPB enzymes in mammalian cells, many attempts to detect metabolite channeling in vitro by partial assembly of the complex were not fruitful. In one exemplary study, transformation of PRPP to 5-PRA by PPAT was studied (38). Under physiological conditions, 5-PRA exhibits a relatively short half-life (5 s at 37°C) in the absence of the subsequent pathway enzyme GART, thus disallowing accumulation of 5-PRA. This finding was recently confirmed in HeLa cells in which expression of GART, the 5-PRA processing enzyme, was knocked out, and 5-PRA accumulation was not observed (39). Although these studies provide a rationale for the sequestration of 5-PRA and its efficient conversion into the downstream stable metabolite glycinamide ribonucleotide (GAR) by GART, in vitro protein–protein interaction studies did not support this proposition (38). Also, GART catalyzes two additional discontinuous steps in the pathway, wherein FGAR, the product of GART, is utilized by PFAS and converted to FGAM, which in turn is acted upon by GART to form AIR. For effective catalysis of these nonsequential steps by GART, direct handover of the involved metabolites has been proposed for GART and PFAS (40). The last two reactions in the DNPB pathway are catalyzed by different active sites on ATIC, but no evidence for metabolic channeling has been found despite their proximity (41). While these data rule out the possibility of direct and proximity channeling in the DNPB pathway, a metabolon assembly supported by transient multivalent interactions involving all the participating enzymes could provide a general channeling solution for all pathway intermediates by cluster channeling (see the sidebar titled Metabolic Channeling) (42). Cluster channeling arises because of restricted diffusion and efficient processing of intermediates generated within the metabolon constituted by multiple copies of a pathway’s enzymes. This creates biochemical hot spots of high intermediate concentration within a metabolon and high localized pathway activity. The fragile and transient nature of purinosomes and the limitations posed by overexpression systems necessitated the functional characterization of purinosomes within cells at endogenous protein expression levels.
METABOLIC CHANNELING.
In the crowded and viscous cellular milieu, enzyme diffusion is hindered, thereby reducing the probability of a substrate–enzyme encounter. In the latter half of the twentieth century, various enzyme complexes involving two or more enzymes in a metabolic pathway were detected, and these complexes localized within or in proximity to intracellular membrane compartments, often stabilized by cytoskeletal elements. These enzyme complexes were proposed to ensure the sequestration of the pathway intermediates through metabolic channeling, and thus provide for their more effective processing. Different researchers termed these multienzyme complexes (59), quinary complexes (60), ambiquitous enzyme systems (61), or metabolons (62, 63). While these complexes vastly differed in their localization, composition, and size, the manifestations of metabolic channeling became a defining feature of such enzyme organizations (42). Metabolic channeling can be achieved through direct, proximity, or clustered mechanisms (42, 66). Direct channeling involves sequestration and transfer of a reaction intermediate between its site of production on one enzyme to another through molecular tunnels connecting the two active sites. In proximity channeling, the reaction intermediate(s) show restricted diffusion and are processed by physically proximal active sites. The intermediates are electrostatically bound on the enzyme surfaces, trapped in molecular cages, or tethered to mobile enzyme domains. In cluster channeling, a higher local concentration of reaction intermediate(s) is achieved within the cluster to allow for their processing by any of the multiple copies of each enzyme instead of dependence on the nearest one.
As a first step, we developed an in-cell isotope-incorporation assay to selectively detect and quantify the DNPB flux by mitochondria-associated purinosomes (33). Under limited supplementation with 13C3, 15N Ser in the growth media, incorporation of the labeled Gly and formate derived from the mitochondrial metabolism of Ser was traced (Figure 1a). A homogenous isotopic incorporation in all the DNPB intermediates and end products would be expected, assuming that the ten-step conversion of PRPP to IMP occurred in a diffusive manner (i.e., all steps are mutually independent events) with complete equilibration of all the intermediates with the bulk cytosolic pool and that the distribution of all the enzymes, substrates, and cofactors was homogenous (i.e., no purinosome formation or localization proximal to mitochondria). Contrary to this, the isotopologue distribution in the newly synthesized IMP was found to be significantly different than that in the end product nucleotides AMP and GMP (Figure 1b). Thus, the method allowed distinct labeling and quantification of DNPB flux from the mitochondria-associated purinosome versus the diffusive cytosolic flux. The newly synthesized end products, AMP and GMP, showed preferential incorporation of mitochondria-derived, isotopically labeled substrates, Gly and formate. AMP and GMP showed ~10% higher Gly enrichment and 15–20% higher formate enrichment compared to IMP (Figure 1c) (33). We infer that the purinosome-mediated synthesis of AMP and GMP is accomplished in a highly channeled manner, preventing bulk equilibration of the intermediates with the cytosol, thus increasing the pathway flux at least sevenfold compared to a diffusive synthesis in HeLa cells. Under purine depletion, the diffusive DNPB pathway has only a limited flux contribution that might arise from a separate pool of enzymes outside of the purinosomes, such as free enzymes or partial complexes lacking one or more enzymes that allow intermediate mixing with their bulk cytosolic pools. These data provide strong evidence for the presence of the purinosome at the endogenous level that channels all the pathway intermediates.
MTHFD1 recruitment to the purinosomes, as detected by protein–protein interaction studies, further supports purinosome-mediated channeled DNPB and preferential incorporation of mitochondria-derived formate by generating 10-formyl-THF at the site of its utilization. Note that the cytosolic NADH/NAD+ ratio has been shown to drive MTHFD1-mediated reduction of 10-formyl-THF into 5,10-methylene-THF, thus limiting the availability of 10-formyl-THF for DNPB (43, 44). Cancer cells show an overexpression of the mitochondrial one-carbon metabolism enzymes, making mitochondria the primary source of formate to propel nucleotide production under these conditions (45, 46). The localization of MTHFD1 within purinosomes presents an elegant way to avoid reduction of 10-formyl-THF to 5,10-methylene-THF and its consumption in pyrimidine biosynthesis. Our data suggest direct uptake of mitochondrially generated formate by the purinosome prior to the mixing of formate with the bulk cytosol. While formate transport by specific transporters across the mitochondrial membrane and their proximity to purinosomes is anticipated to aid purinosome function, mitochondrial formate transporters have not been discovered yet. Also, the proteins involved in making contact between the purinosome and mitochondria are unknown, although the mitochondrial transporters are speculative candidates.
One would anticipate a higher residence time of intermediates inside a metabolon due to their restricted diffusion. This implies localized accumulation of an intermediate when its consumption rate is lower than its production. To investigate this possibility, we grew purine-depleted HeLa cells under limiting Ser supplementation. This in turn limits formate production by mitochondria and insufficient availability of 10-formyl-THF to purinosomes. This imbalance in 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) production and utilization results in accumulation of AICAR by purinosomes (33). Under these conditions, a monolayer of cells was flash frozen for single-cell in situ submicron 3D molecular scanning by gas cluster ion beam–time of flight–secondary ion mass spectrometry (GCIB-SIMS) imaging with a 1-μm beam focal diameter. GCIB-SIMS allows the generation and detection of molecular ions at high resolution and low chemical damage, thus making it uniquely suitable for multiplexed molecular analysis of single cells. Purinosomes were identified as the cellular hot spots of purine biosynthesis, as indicated by distinct loci with high isotope-labeled AICAR abundance (Figure 3) (0.3–1×106 15N-AICAR molecules per purinosome) and elevated isotope incorporation in ATP (33). In these AICAR synthesis hot spots, the observed AICAR abundance was ~300–8,000-fold higher than that expected for a homogenous distribution of AICAR throughout the cellular volume. This finding supports a purinosome model of cluster channeling where the intermediate abundance appears to be significantly higher than the estimated enzyme abundance per purinosome (see the sidebar titled Metabolic Channeling).
From our collective studies, we present a revised definition of a purinosome. Purinosomes are functional enzyme clusters composed of at least ten enzymes: six DNPB enzymes (PPAT, GART, PFAS, PAICS, ADSL, and ATIC); three enzymes shared with the salvage pathway [adenylosuccinate synthetase (ADSS), IMP dehydrogenase (IMPDH), and GMP synthetase (GMPS)]; and MTHFD1 (Figure 1a). Using in-cell isotope incorporation assays and PLA, the methods designed to detect purinosomes at endogenous protein expression levels, purinosomes have been found in several cancer cell lines (17). The enzymes in purinosomes function synergistically to convert PRPP to the purine nucleotides AMP and GMP. Detection of active purinosomes at endogenous protein expression levels by GCIB-SIMS imaging and PLA provides an estimate of ~10–30 active purinosomes per cell (17, 33), a number much lower than that observed for cells with transient overexpression of the DNPB enzymes (greater than 100 DNPB enzyme clusters per cell) (17, 27) (Figure 3). Fluorescence imaging gives a size range of ~0.1–1 μm in diameter for the DNPB enzyme clusters formed after transient overexpression of the enzyme PFAS (17, 23, 27). However, at the endogenous level, the total abundance of the rate limiting enzyme, PPAT, seems to greatly restrict this range. A calculation based on the upper limit of the protein density inside cells shows that realistically, a functional purinosome with equimolar distribution of all ten enzymes may reach only 200–300 nm in diameter, and the number of total enzyme molecules may range from ~103 to 105 per purinosome (33, 42).
5. PURINOSOME REGULATION
Purinosome formation is not a consequence of enhanced gene or protein expression or substrate availability (17, 27). This begs the questions, what might trigger functional purinosome formation and how does the complex maintain its biophysical properties? Our understanding of what regulates purinosome assembly under conditions that promote pathway activation is still in its infancy. Insights gathered have been driven by the outcomes of loss-of-function kinome screening, small molecule agonist profiling, and proteomic analyses.
5.1. Signaling Pathways
The influence of cell signaling on purinosome formation was first probed through the development of a label-free dynamic mass redistribution (DMR) assay (47, 48). This assay uses a resonant waveguide biosensor to measure changes in the distribution of cellular biomass, as detected by refractive index, upon gene perturbation or treatment with a small molecule (49–51). Using small molecule activators, the platform was calibrated to detect changes in biomass distribution as a result of purinosome formation. Then, a short hairpin RNA loss-of-function kinome screen was performed using the DMR assay to identify those kinases that might affect purinosome formation (28). Based on the kinases identified, the mitogen-activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR) signaling pathways appear to promote the formation of purinosomes.
The MAPK signaling pathway is a cascade of intracellular protein kinases that amplifies the signal initiated from outside the cell to stimulate a variety of cellular processes such as proliferation, survival, and apoptosis (52). Within this signaling pathway, the DMR assay identified epidermal growth factor receptor (EGFR) and dual specificity mitogen-activated protein kinase 2 (MEK2) as possible regulators of purinosome formation (28). EGFR is a receptor tyrosine kinase that initiates the signaling cascade through the binding of its activating ligand, epidermal growth factor (EGF). In a recent study, treatment of serum-starved HeLa cells with EGF resulted in at least a twofold increase in the production of purine monophosphates through the DNPB pathway (53). Likewise, the enhanced activity of the DNPB pathway can also be a result of oncogenic activation of the small GTPase, Ras (53). These mechanisms have been linked to the activation of the protein kinase extracellular signal-regulated kinase 2 (ERK2) by MEK2. It is plausible that MEK2-mediated activation of ERK2 might result in increased purinosome formation.
Among other kinases identified in the DMR assay screen, mTOR was shown to stabilize purinosome colocalization with mitochondria (28). Inhibition of mTOR with rapamycin in purine-depleted HeLa cells decreased the number and localization of purinosomes with mitochondria. Further, rapamycin treatment also decreased the production of purines, further supporting the notion that the biological advantage prescribed to the purinosome is maximized near mitochondria. Additionally, mTOR mediates induction of ATF-4, a transcription factor that regulates expression of MTHFD2, a gene that encodes a mitochondrial enzyme important in the generation of one-carbon units and DNPB flux (54). These one-carbon units are exported from mitochondria and used to synthesize the 10-formyl-THF cofactor necessary for GART and ATIC transformylase activities in the purinosome. However, the time scale by which this transcriptional response is observed appears to be longer than the rapamycin-mediated destabilization of purinosome–mitochondria colocalization (28, 54). These observations raise the possibility that mTOR machinery may control purinosome assembly more directly rather than via modulation of expression of a mitochondrial one-carbon metabolism enzyme.
The DMR assay was also used to screen G protein–coupled receptor (GPCR) agonists to understand the connection between GPCR signaling and purinosome formation (47). Results from a screen of 113 GPCR agonists revealed that α2A adrenergic receptor ligands, such as epinephrine, perturb the biomass redistribution in HeLa cells that mimics purinosome formation. Further characterization of GPCR signaling pathways using the DMR platform and corroborated by fluorescence imaging of purinosomes suggested that purinosome formation can be induced through the activation of the Gαi signaling pathway.
5.2. Posttranslational Modifications
Given that purinosome formation appears to be under the regulation of various signaling pathways, it is no surprise that these enzymes are subject to being posttranslationally modified. A survey of the posttranslational modifications (PTMs) present on each of the DNPB enzymes was conducted in 293T cells under growth conditions that either promote or disfavor purinosome formation (55). Here, seven different PTMs (lysine acetylation; mono- and di-methylation; arginine mono-methylation; ubiquitination; Ser/Thr and Tyr phosphorylation) were detected on 135 residues among the six pathway enzymes.
One of the most notable observations was the degree to which these enzymes were Ser/Thr phosphorylated (55). Many of these phosphorylation events were observed only in one growth condition, a likely consequence of nutrient-dependent cellular signaling. For example, PFAS was shown to be highly phosphorylated under purine-depleted growth conditions, whereas ATIC phosphorylation was largely observed in cells that disfavor purinosome formation. One of the PFAS phosphorylation sites, Thr619, was recently shown to be a consequence of the EGF-stimulated or oncogenic Ras-dependent MAPK signaling cascade (53). This phosphorylation event is catalyzed by ERK2 and enhances A549 non–small cell lung cancer cell proliferation and tumor growth. Expression of a T619A PFAS mutant, devoid of endogenous PFAS, resulted in a decrease in DNPB, a 30–40% decrease in colony formation, and a substantial reduction in tumor growth when compared to those expressing wild-type PFAS. Interestingly, it was noted that a T619D PFAS mutation, observed in renal clear cell carcinoma and stomach adenocarcinoma, increased purine production through the DNPB pathway and cell proliferation when cultured in low-serum growth conditions.
5.3. Molecular Chaperones
A proteome-wide study of those proteins that copurify with the purinosome was performed in an effort to identify ancillary proteins that might regulate complexation (18). Using PFAS as a bait, we discovered an association with cochaperone proteins including Bcl-2–associated anthogene domain proteins, J-domain proteins (DNAJ/Hsp40) and the heat shock organizing protein (HOP/Stip1). These findings suggested that molecular chaperones might play a role in purinosome formation. Biochemical validation of chaperone interactions with enzymes in the DNPB pathway revealed that HSP90 and HSP70 interact with PPAT and PFAS (56). These associations were visualized by fluorescence microscopy, which showed that HSP70, HSP90, and their associated cochaperones colocalized with purinosomes (18).
HSP90 inhibition with STA9090 decreased its interaction with the proposed clients while maintaining their interactions with HSP70 (56). STA9090 treatment induced proteasome-mediated degradation of PPAT and PFAS while not perturbing the expression levels of the other pathway enzymes. Inhibition of HSP90 activity also resulted in PFAS readily forming intracellular granules with a morphology that differs from that of purinosomes, suggesting that these assemblies might have transformed into more aggregate-like bodies (29). Notably, unlike a functional purinosome, these PFAS granules formed by HSP90 inhibition did not colocalize with ADSL.
The roles that HSP90 and HSP70 play in the regulation of the purinosome are still largely unknown. Under hypoxic growth conditions, HSP70 isoform 2 (HSP70–2) is overexpressed and has been shown to colocalize with ADSL, a non-HSP90 client, by PLA (17). However, the positive influence that HSP70–2 has on purinosome formation under conditions that upregulate HIF-1α expression did not mirror its effect on purinosome formation under purine-depleted growth conditions. Overexpression of HSP70–2 did not induce purinosome formation, suggesting that HSP70–2 is likely to assist in the overall stability of the assembly. Recently, a mechanism was proposed in which the HSP90 chaperone machinery preserves active, multi-enzyme metabolic complexes in nonmembrane compartments (57). These intracellular assemblies leverage HSP90 as a nucleation site for metabolic enzyme condensation. Using an HSP90 inhibitor, PU-H71, as bait, associations between HSP90 and all pathway enzymes (except ATIC) were observed. In addition, HSP90 interactions with IMPDH and GMPS, enzymes that transform IMP into GMP, were also observed. These results help substantiate a model in which HSP90 regulates the biophysical and functional properties of the assembly by simply altering the conformation of PPAT and PFAS inside the purinosome.
6. CONCLUSION
Recent work has demonstrated the presence of an active DNPB metabolon, the purinosome, in human cells. We have endeavored to present a contemporary description of the purinosome, its assembly and disassembly, function, cellular loci, and regulation. The protocols and techniques that have been used to explore this unique metabolon are highlighted, especially those that led to our finding that the purinosome channels all the pathway intermediates. We hope that our summary provides the impetus for the identification of other similarly organized metabolic pathways. If metabolon formation is a common feature of many pathways, the next question concerns how different metabolons are organized relative to one another. The role of metabolons in the spatio-temporal regulation of metabolic pathways by membraneless compartmentalization is key to gaining a holistic view of cellular biochemistry.
SUMMARY POINTS.
The purinosome is an intracellular compartmentalization of metabolic enzymes that efficiently converts PRPP to AMP and GMP in a channeled manner. These metabolic enzymes extend beyond the six enzymes that comprise the DNPB pathway and include three additional enzymes that convert IMP to purine monophosphates and the 10-formyl-THF–cofactor generating enzyme, MTHFD1.
Purinosomes function by a clustered, channeled mechanism, which leads to at least a sevenfold increase in metabolite flux in purine-depleted HeLa cells. Localized metabolic hot spots that harbor increased pathway activity can be detected by mass spectral imaging.
Purinosomes are localized at the microtubule–mitochondria interface. Disruption of microtubule assembly destabilized purinosomes and decreased purine production through the DNPB pathway. This localization is hypothesized to facilitate the acquisition of substrates generated by mitochondrial processes.
Complexation alone does not guarantee that the complex is functional. Under hypoxic conditions, purinosome formation was induced. However, insufficient quantities of available substrates, through mitochondrial processes, did not allow for the assembly to exhibit enhanced purine production through the DNPB pathway.
The transient overexpression of enzymes within the DNPB pathway can lead to the formation of other intracellular assemblies that are distinct from the purinosomes observed on the endogenous level. Caution must be used to properly characterize purinosomes and differentiate them from those complexes generated as a result of self-assembly or aggregation.
Our understanding of the mechanisms that drive purinosome formation is still limited. Assembly and stability of the purinosome have been correlated with activation of mTOR and the Gαi GPCR signaling pathways. Other plausible cell signaling pathways have been implicated by identifying the extent to which PTMs are present under purine-depleted growth conditions.
HSP90 and HSP70 have been identified as regulators of the biophysical properties that define a purinosome. PPAT and PFAS have been shown to be clients of HSP90. Inhibition of HSP90 decreases its interaction with the client proteins, induces proteasome-mediated degradation of the soluble client enzymes, and induces the formation of aggregated clusters that have a morphology that differs from the purinosome.
Purinosomes have been detected in a wide range of cell lines, suggesting that this phenomenon is a general mechanism for efficient purine nucleotide production.
FUTURE ISSUES.
To alleviate misidentification of purinosomes upon transient overexpression of any DNPB enzyme, application of alternative methods (58) and development of new techniques to characterize and study the purinosome at the endogenous level will be beneficial. Experiments where the purinosome has been studied at or near endogenous levels have uncovered some key protein–protein interactions by coimmunoprecipitation and proximity ligation assays, a feat previously not attained in transient overexpressed models. In addition, we have detected metabolic hot spots by mass spectral imaging. It remains to be seen if purinosomes have similar composition and metabolic output in different cell types.
Only the in vitro kinetic parameters of the various enzymes have been determined; however, we do not fully understand the effect multienzyme complexation might have on their individual activities. Further, the abundance of each of the pathway enzyme might vary in accordance with its catalytic activity to maximize pathway flux within purinosomes.
What are the biophysical properties of the endogenous purinosome? What is the microorganization of the purinosome? Are the individual enzymes mixed within the confines of the purinosome or are there localized pools of each enzyme?
Since the purinosome has been defined as a metabolon, it is advantageous to restrict diffusion of pathway intermediates, such as phosphoribosyl aminoimidazole succinocarboxamide (SAICAR) 5-aminoimidazole-4-carboxamide ribonucleotide and AICAR, outside of the assembly. However, substrates such as Gly, Gln, and ATP need to be brought in. So how does sequestration of SAICAR and AICAR by purinosome formation alter their function as signaling molecules? Is there a diffusion barrier to selectively allow certain substrates in, while preventing movement of pathway intermediates out? Is the import of the necessary substrates from mitochondria to purinosomes mediated by mitochondrial transporters?
IMP is further converted into either AMP or GMP. Although the enzymes that do this conversion are localized within purinosomes, the kinetic parameters for the IMP converting enzymes favor GMP production. However, the channeled DNPB pathway, via the purinosome, favors the generation of adenine nucleotides over guanine nucleotides. What are the deciding factors that direct the portioning of IMP within the purinosome?
HSP90 plays numerous roles inside a cell to maintain proteostasis. PFAS and PPAT are folded by HSP90 to promote interactions with other pathway enzymes and assist in purinosome formation. However, we do not understand exactly what HSP90 is doing to sustain purinosome formation. Is HSP90 acting as a scaffold to maintain a proper balance of active enzymes? And what role do the enzyme self-assemblies (IMPDH and PFAS) play in regulating pathway flux?
Several PTMs have been identified either under experimental conditions that favor or disfavor purinosome formation. We still do not understand which modifications control enzymatic activity and which ones play a regulatory role in purinosome formation. Thus, another outstanding question is, What are the different signaling pathways involved in purinosome assembly/disassembly?
Regulation of the DNPB pathway and the role of purinosomes in humans would benefit from more detailed structural and kinetic studies on the individual human DNPB enzymes. In its first catalytic step, mammalian PAICS carries out the carboxylation of AIR using CO2 as an electrophile in the absence of any known cofactors, metal ion, or activation by ATP. Recently, based on X-ray crystal structure analysis, a CO2 binding site has been proposed in the AIR binding pocket, though the mechanism of CO2 activation is still unclear.
ACKNOWLEDGMENTS
The authors thank Erin L. Kennedy and Minjoung Kyoung at the University of Maryland, Baltimore County for use of the fluorescence image presented in Figure 2b. Financial support is provided by Huck Institutes of the Life Sciences at Penn State (to V.P.) and the National Institutes of Health (R01 GM024129 to S.J.B.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Nyhan WL. 2014. Nucleotide synthesis via salvage pathway. In eLS. San Francisco: John Wiley and Sons Ltd. [Google Scholar]
- 2.Yin J, Ren W, Huang X, Deng J, Li T, Yin Y. 2018. Potential mechanisms connecting purine metabolism and cancer therapy. Front. Immunol. 9:1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashihara H, Ludwig IA, Crozier A. 2020. Salvage pathways of purine nucleotide biosynthesis. In Plant Nucleotide Metabolism—Biosynthesis, Degradation, and Alkaloid Formation, ed. Ashihara H, Ludwig IA, Crozier A, pp. 55–69. Hoboken, NJ: John Wiley & Sons Ltd. [Google Scholar]
- 4.Pareek V, Pedley AM, Benkovic SJ. 2021. Human de novo purine biosynthesis. Crit. Rev. Biochem. Mol. Biol. 56:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henderson JF, Khoo KY. 1965. On the mechanism of feedback inhibition of purine biosynthesis de novo in Ehrlich ascites tumor cells in vitro. J. Biol. Chem. 240:3104–9 [PubMed] [Google Scholar]
- 6.Lane AN, Fan TW. 2015. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43:2466–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murray AW. 1971. The biological significance of purine salvage. Annu. Rev. Biochem. 40:811–26 [DOI] [PubMed] [Google Scholar]
- 8.Yamaoka T, Kondo M, Honda S, Iwahana H, Moritani M, et al. 1997. Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. J. Biol. Chem. 272:17719–25 [DOI] [PubMed] [Google Scholar]
- 9.Villa E, Ali ES, Sahu U, Ben-Sahra I. 2019. Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers 11:688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu R, Sutcliffe D, Zhao H, Huang X, Schretlen DJ, et al. 2015. Clinical severity in Lesch-Nyhan disease: the role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 114:55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zoref E, Sperling O. 1979. Increased de novo purine synthesis in cultured skin fibroblasts from heterozygotes for the Lesch-Nyhan Syndrome: a sensitive marker for carrier detection. Hum. Heredity 29:64–68 [DOI] [PubMed] [Google Scholar]
- 12.Fridman A, Saha A, Chan A, Casteel DE, Pilz RB, Boss GR. 2013. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem. J. 454:91–99 [DOI] [PubMed] [Google Scholar]
- 13.Zalkin H, Dixon JE. 1992. De novo purine nucleotide biosynthesis. Prog. Nucleic Acid Res. Mol. Biol. 42:259–87 [DOI] [PubMed] [Google Scholar]
- 14.Hove-Jensen B, Andersen KR, Kilstrup M, Martinussen J, Switzer RL, Willemoës M. 2017. Phosphoribosyl diphosphate (PRPP): biosynthesis, enzymology, utilization, and metabolic significance. Microbiol. Mol. Biol. Rev. 81:e00040–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.An S, Kumar R, Sheets ED, Benkovic SJ. 2008. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320:103–6 [DOI] [PubMed] [Google Scholar]
- 16.Schmitt DL, Sundaram A, Jeon M, Luu BT, An S. 2018. Spatial alterations of de novo purine biosynthetic enzymes by Akt-independent PDK1 signaling pathways. PLOS ONE 13:e0195989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doigneaux C, Pedley AM, Mistry IN, Papayova M, Benkovic SJ, Tavassoli A. 2020. Hypoxia drives the assembly of the multienzyme purinosome complex. J. Biol. Chem. 295:9551–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.French JB, Zhao H, An S, Niessen S, Deng Y, et al. 2013. Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. PNAS 110:2528–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baresova V, Skopova V, Sikora J, Patterson D, Sovova J, et al. 2012. Mutations of ATIC and ADSL affect purinosome assembly in cultured skin fibroblasts from patients with AICA-ribosiduria and ADSL deficiency. Hum. Mol. Genet. 21:1534–43 [DOI] [PubMed] [Google Scholar]
- 20.Mangold CA, Yao PJ, Du M, Freeman WM, Benkovic SJ, Szpara ML. 2018. Expression of the purine biosynthetic enzyme phosphoribosyl formylglycinamidine synthase in neurons. J. Neurochem. 144:723–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamada S, Sato A, Sakakibara SI. 2020. Nwd1 regulates neuronal differentiation and migration through purinosome formation in the developing cerebral cortex. iScience 23:101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williamson J, Petralia RS, Wang YX, Mattson MP, Yao PJ. 2017. Purine biosynthesis enzymes in hippocampal neurons. Neuromol. Med. 19:518–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan CY, Pedley AM, Kim D, Xia C, Zhuang X, Benkovic SJ. 2018. Microtubule-directed transport of purine metabolons drives their cytosolic transit to mitochondria. PNAS 115:13009–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao A, Tsechansky M, Ellington AD, Marcotte EM. 2014. Revisiting and revising the purinosome. Mol. Biosyst. 10:369–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao A, Tsechansky M, Swaminathan J, Cook L, Ellington AD, Marcotte EM. 2013. Transiently transfected purine biosynthetic enzymes form stress bodies. PLOS ONE 8:e56203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O’Connell JD, et al. 2009. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. PNAS 106:10147–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan CY, Zhao H, Pugh RJ, Pedley AM, French J, et al. 2015. Purinosome formation as a function of the cell cycle. PNAS 112:1368–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.French JB, Jones SA, Deng H, Pedley AM, Kim D, et al. 2016. Spatial colocalization and functional link of purinosomes with mitochondria. Science 351:733–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedley AM, Boylan JP, Chan CY, Kennedy EL, Kyoung M, Benkovic SJ. 2022. Purine biosynthetic enzymes assemble into liquid-like condensates dependent on the activity of chaperone protein HSP90. J. Biol. Chem. In press. 10.1016/j.jbc.2022.101845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kyoung M, Russell SJ, Kohnhorst CL, Esemoto NN, An S. 2015. Dynamic architecture of the purinosome involved in human de novo purine biosynthesis. Biochemistry 54:870–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baresova V, Krijt M, Skopova V, Souckova O, Kmoch S, Zikanova M. 2016. CRISPR-Cas9 induced mutations along de novo purine synthesis in HeLa cells result in accumulation of individual enzyme substrates and affect purinosome formation. Mol. Genet. Metab. 119:270–77 [DOI] [PubMed] [Google Scholar]
- 32.Baresova V, Skopova V, Souckova O, Krijt M, Kmoch S, Zikanova M. 2018. Study of purinosome assembly in cell-based model systems with de novo purine synthesis and salvage pathway deficiencies. PLOS ONE 13:e0201432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pareek V, Tian H, Winograd N, Benkovic SJ. 2020. Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368:283–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H, Chiaro CR, Zhang L, Smith PB, Chan CY, et al. 2015. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J. Biol. Chem. 290:6705–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He J, Zou L-N, Pareek V, Benkovic SJ. 2022. Multienzyme interactions of the de novo purine biosynthetic protein PAICS facilitate purinosome formation and metabolic channeling. J. Biol. Chem. In press. 10.1016/j.jbc.2022.101853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wan C, Borgeson B, Phanse S, Tu F, Drew K, et al. 2015. Panorama of ancient metazoan macromolecular complexes. Nature 525:339–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.An S, Deng Y, Tomsho JW, Kyoung M, Benkovic SJ. 2010. Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. PNAS 107:12872–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudolph J, Stubbe J. 1995. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry 34:2241–50 [DOI] [PubMed] [Google Scholar]
- 39.Mazzarino RC, Baresova V, Zikanova M, Duval N, Wilkinson TG 2nd, et al. 2021. Transcriptome and metabolome analysis of crGART, a novel cell model of de novo purine synthesis deficiency: alterations in CD36 expression and activity. PLOS ONE 16:e0247227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedley AM, Benkovic SJ. 2017. A new view into the regulation of purine metabolism: the purinosome. Trends Biochem. Sci. 42:141–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulock KG, Beardsley GP, Anderson KS. 2002. The kinetic mechanism of the human bifunctional enzyme ATIC (5-amino-4-imidazolecarboxamide ribonucleotide transformylase/inosine 5′-monophosphate cyclohydrolase). A surprising lack of substrate channeling. J. Biol. Chem. 277:22168–74 [DOI] [PubMed] [Google Scholar]
- 42.Pareek V, Sha Z, He J, Wingreen NS, Benkovic SJ. 2021. Metabolic channeling: predictions, deductions, and evidence. Mol. Cell 81:3775–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diehl FF, Lewis CA, Fiske BP, Vander Heiden MG. 2019. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 1:861–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tibbetts AS, Appling DR. 2010. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30:57–81 [DOI] [PubMed] [Google Scholar]
- 45.Ducker GS, Rabinowitz JD. 2017. One-carbon metabolism in health and disease. Cell Metab. 25:27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman AC, Maddocks ODK. 2017. One-carbon metabolism in cancer. Br. J. Cancer 116:1499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verrier F, An S, Ferrie AM, Sun H, Kyoung M, et al. 2011. GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nat. Chem. Biol. 7:909–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang Y, French J, Zhao H, Benkovic S. 2013. G-protein-coupled receptor regulation of de novo purine biosynthesis: a novel druggable mechanism. Biotechnol. Genet. Eng. Rev. 29:31–48 [DOI] [PubMed] [Google Scholar]
- 49.Fang Y 2014. Label-free drug discovery. Front. Pharmacol. 5:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang Y 2014. Label-free cell phenotypic drug discovery. Comb. Chem. High Throughput Screen. 17:566–78 [DOI] [PubMed] [Google Scholar]
- 51.Ferrie AM, Goral V, Wang C, Fang Y. 2015. Label-free functional selectivity assays. Methods Mol. Biol. 1272:227–46 [DOI] [PubMed] [Google Scholar]
- 52.Zhang W, Liu HT. 2002. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12:9–18 [DOI] [PubMed] [Google Scholar]
- 53.Ali ES, Sahu U, Villa E, O’Hara BP, Gao P, et al. 2020. ERK2 phosphorylates PFAS to mediate posttranslational control of de novo purine synthesis. Mol. Cell 78:1178–91.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. 2016. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351:728–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C, Knudsen GM, Pedley AM, He J, Johnson JL, et al. 2019. Mapping post-translational modifications of de novo purine biosynthetic enzymes: implications for pathway regulation. J. Proteome Res. 18:2078–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pedley AM, Karras GI, Zhang X, Lindquist S, Benkovic SJ. 2018. Role of HSP90 in the regulation of de novo purine biosynthesis. Biochemistry 57:3217–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calvo-Vidal MN, Zamponi N, Krumsiek J, Stockslager MA, Revuelta MV, et al. 2021. Oncogenic HSP90 facilitates metabolic alterations in aggressive B-cell lymphomas. Cancer Res. 81:5202–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bassard JE, Halkier BA. 2018. How to prove the existence of metabolons? Phytochem. Rev. 17:211–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reed LJ. 1974. Multienzyme complexes. Acc. Chem. Res. 7:40–46 [Google Scholar]
- 60.McConkey EH. 1982. Molecular evolution, intracellular organization, and the quinary structure of proteins. PNAS 79:3236–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson JE. 1978. Ambiquitous enzymes: variation in intracellular distribution as a regulatory mechanism. Trends Biochem. Sci. 3:124–25 [Google Scholar]
- 62.Srere PA. 1985. The metabolon. Trends Biochem. Sci. 10:109–10 [Google Scholar]
- 63.Srere PA. 1987. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 56:89–124 [DOI] [PubMed] [Google Scholar]
- 64.Pedley AM, Benkovic SJ. 2018. Detecting purinosome metabolon formation with fluorescence microscopy. Methods Mol. Biol. 1764:279–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu A, Disoma C, Zhou Y, Chen Z, Zhang L, et al. 2019. Protein interactome of the deamidase phosphoribosylformylglycinamidine synthetase (PFAS) by LC-MS/MS. Biochem. Biophys. Res. Commun. 513:746–52 [DOI] [PubMed] [Google Scholar]
- 66.Huang X, Holden HM, Raushel FM. 2001. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 70:149–80 [DOI] [PubMed] [Google Scholar]