

Abstract
Purpose:
The purpose of this study is to evaluate ponatinib for advanced gastrointestinal stromal tumors (GIST).
Patients and Methods:
This single-arm phase II trial enrolled patients with metastatic and/or unresectable GIST with failure of prior tyrosine kinase inhibitor (TKI) treatment into two cohorts based on presence or absence of KIT exon 11 (ex11) primary mutations. Patients initially received ponatinib 45 mg once daily. Following a temporary clinical hold in October 2013, dose reductions were implemented to reduce risk of arterial occlusive events (AOE). Primary endpoint was 16-week clinical benefit rate (CBR) in KIT ex11–positive cohort. KIT mutations in circulating tumor DNA (ctDNA) were assessed.
Results:
Forty-five patients enrolled (30 KIT ex11–positive and 15 KIT ex11–negative); median follow-up was 14.7 and 13.6 months, respectively, as of August 1, 2016. Sixteen-week CBR was 36% (KIT ex11–positive; primary endpoint) and 20% (KIT ex11–negative). ctDNA analyses (n = 37) demonstrated strong concordance of primary KIT mutations between plasma and tumor. At least two secondary mutations were detected in 35% of patients overall and 54% of KIT ex11–positive patients. Changes from baseline in mutated ctDNA levels were consistent with clinical activity. Ponatinib was ineffective in patients with KIT exon 9 primary mutations. Resistance was associated with emergence of V654A. AOEs and venous thromboembolic events occurred in three and two patients, respectively. Six patients died; two deaths (pneumonia and pulmonary embolism) were considered possibly ponatinib-related.
Conclusions:
Ponatinib demonstrated activity in advanced GIST, particularly in KIT ex11–positive disease. ctDNA analysis confirmed heterogeneous resistance mutations in TKI-pretreated advanced GIST. Safety was consistent with previous studies.
Translational Relevance.
Ponatinib demonstrated antitumor activity in patients with advanced gastrointestinal stromal tumors (GIST), particularly in those with KIT exon 11 mutations. Analysis of circulating tumor DNA (ctDNA) confirmed heterogeneous resistance mutations in patients with advanced GIST previously treated with tyrosine kinase inhibitors (TKI). To the best of our knowledge, this is the first report of a clinical trial in advanced GIST in which patient enrollment was prospectively guided by mutational status. Our results demonstrate that trials evaluating TKI efficacy based on primary GIST mutations are feasible and informative. There was concordance between primary mutations detected in plasma ctDNA using beads, emulsions, amplification, and magnetics (BEAMing) and in tumor biopsies. The majority of patients had primary and/or secondary KIT mutations detected in plasma. The incomplete spectrum of KIT mutations detectable by BEAMing may be overcome by use of next-generation sequencing. Future GIST trials should continue to evaluate ctDNA using sequencing-based approaches.
Introduction
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal malignancy of the gastrointestinal tract, with an estimated annual incidence of 5,000 to 6,000 new cases in the United States (1, 2). Oncogenic, gain of function mutations in KIT and PDGFRA are found in approximately 85% and 5% of GIST, respectively (3, 4). Although the currently approved tyrosine kinase inhibitors (TKI) for KIT-mutant GIST (first-line: imatinib; second-line: sunitinib; third-line: regorafenib, fourth-line: ripretinib; refs. 5–7) are typically initially effective in the treatment of unresectable disease, the emergence of secondary mutations and subsequent disease progression is a major challenge in the treatment of GIST (8–10). Patients with GIST refractory to approved TKIs typically experience rapid disease progression; median progression-free survival (PFS) and overall survival (OS) generally decrease with each additional line of treatment (11–15).
Ponatinib, a potent BCR::ABL1 inhibitor, is a third-generation TKI designed to potently inhibit BCR::ABL1 with or without any single resistance mutation, including T315I. In the United States, ponatinib is approved for the treatment of adults with chronic phase (CP) chronic myeloid leukemia (CML) with resistance or intolerance to at least two prior kinase inhibitors; accelerated phase (AP) or blast phase (BP) CML or Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL) for whom no other kinase inhibitor is indicated; and for T315I-positive CML or Ph+ ALL (16). Ponatinib has potent preclinical activity against mutated KIT and platelet-derived growth factor-α (PDGFRA), including clinically relevant mutations found in GIST that confer resistance to available TKIs (17, 18). Detailed in vitro analysis has shown that ponatinib has a broad, but imperfect, inhibitory profile against various KIT mutations (19). Ponatinib demonstrated strong potency against KIT with primary mutations in exon 11 (ex11) and most secondary mutations, including mutations in the activation loop (exon 17/18) and at the T670 gatekeeper residue (exon 14). However, ponatinib had reduced activity against KIT with an exon 13 (V654A) secondary mutation or with an exon 9 (ex9) primary activating mutation (19).
The strong preclinical activity of ponatinib against KIT and PDGFRA supported its clinical investigation in patients with GIST. The objective of the current phase II study (NCT01874665) was to evaluate the efficacy and safety of ponatinib in patients with advanced GIST after failure of prior TKI therapy; biomarker assessments were also performed. Given the known preclinical profile of ponatinib, patients were prospectively enrolled into two cohorts based on the presence or absence of primary KIT ex11 mutations; ctDNA analysis was performed to explore the relationship between tumor mutation status and ponatinib activity in this heavily pretreated population.
Patients and Methods
Study design
In this phase II, single-arm, open-label, multicenter study, patients were enrolled in one of two cohorts, the KIT ex11–positive (KIT ex11–pos) or the KIT ex11–negative (KIT ex11-neg) as reported by investigators based on historical tumor analysis. Tumor KIT/PDGFRA mutations were recorded for all patients enrolled in the study. If the patient did not have a genetic analysis of a tumor sample performed at a Clinical Laboratory Improvement Amendments–certified laboratory, mutational analysis of KIT/PDGFRA genes in at least 1 archival tumor specimen was performed. Patients who did not have an archival specimen were required to undergo a tumor biopsy for genetic analysis. Blood samples were obtained at baseline and at times of tumor assessment for detection of mutations in KIT and PDGFRA in circulating DNA.
All patients started on 45 mg/day ponatinib; treatment continued until the occurrence of disease progression or unacceptable toxicity. Between October 8, 2013, and May 21, 2014, a temporary clinical hold was imposed by the FDA on all ponatinib trials in order to assess the risk of vascular occlusive events and to provide dose reduction instructions for management of these events. A total of 45 patients were enrolled prior to the hold; of those, 13 patients were censored. After the October 2013 temporary clinical hold, eligibility was amended to include only those patients who had received all three TKIs approved for GIST at that time (imatinib, sunitinib, and regorafenib); enrolled patients who had not received at least 3 prior therapies were discontinued if they had not achieved at least stable disease (SD) lasting ≥16 weeks during ponatinib treatment. Accordingly, patients with SD or better at the end of Cycle 6 (1 cycle = 4 weeks) had ponatinib reduced to 30 mg/day if the dose was not already reduced for other reasons; doses could be escalated to 45 mg/day upon disease progression. Reduction of ponatinib dose to as low as 15 mg/day was permitted to manage drug-related adverse events (AE) or drug–drug interactions, and the dose could be re-escalated up to 45 mg/day once events resolved. Ponatinib was supplied as oral tablets in either the 45-mg or 15-mg strengths.
This study was conducted in accordance with the ethical standards consistent with the Declaration of Helsinki and with Good Clinical Practice guidelines and all applicable regulatory requirements. The protocol and the informed consent received institutional review board/ethics committee approval. All patients signed the informed consent.
Patients
Patients ≥18 years of age with histologically confirmed metastatic and/or unresectable GIST who experienced progression on prior TKIs were eligible. At study initiation, patients with failure of at least one prior TKI treatment were eligible. After the October 2013 temporary clinical hold, eligibility was amended to include only those patients who had received all three approved TKIs for GIST at that time (imatinib, sunitinib, and regorafenib); enrolled patients who had not received at least three prior therapies were discontinued if they had not achieved at least SD lasting ≥16 weeks during ponatinib treatment. Eligible patients had Eastern Cooperative Oncology Group performance status ≤2, measurable disease per modified RECIST version 1.1 (12), and adequate hepatic, renal, and pancreatic function. Patients with clinically significant, uncontrolled, or active cardiovascular disease, uncontrolled hypertension, uncontrolled hypertriglyceridemia, a bleeding disorder, ongoing or active infection, or those taking medications with risk of serious cardiac ventricular arrhythmias (e.g., torsades de pointes) were excluded, as were patients with history of chronic pancreatitis or acute pancreatitis within 1 year prior to study commencement. Women who were pregnant or lactating were not eligible. The use of approved TKIs or investigational agents within 2 weeks or six half-lives of the agent prior to start of treatment (whichever was longer) was prohibited.
Efficacy outcomes
The primary outcome measure was investigator-assessed clinical benefit rate (CBR) at 16 weeks, defined as the composite of complete response (CR), partial response (PR), and SD according to modified RECIST version 1.1 (12), in the KIT ex11–pos cohort. Secondary outcome measures included investigator-assessed CBR at 16 weeks in the KIT ex11–neg cohort and in the total patient population and objective response rate (ORR), defined as the composite of CR and PR according to modified RECIST version 1.1 (12), PFS, and OS in both cohorts and in the total patient population. Similar endpoints were used in phase II studies of regorafenib and sunitinib in patients with GIST (20, 21).
Tumor response was assessed by CT or magnetic resonance imaging every two cycles for the first six cycles and every three cycles thereafter. PFS was defined as the time from start of ponatinib administration to objective disease progression or death due to any cause. OS data were collected every 6 months following discontinuation for up to 24 months after enrollment of the last patient.
Correlative assessments
The association between KIT mutations detected using ctDNA and antitumor activity of ponatinib was assessed as an exploratory outcome. Blood was drawn for detection of KIT mutations in ctDNA (liquid biopsy) at baseline and at the time of tumor assessment. Samples were analyzed for KIT mutations using beads, emulsions, amplification, and magnetics (BEAMing) digital polymerase chain reaction technology (Sysmex Inostics, Inc.; refs. 22, 23), which allowed detection of seven primary (exons 9, 11, 13) and 20 secondary (exons 13/14, 17/18) mutations (ref. 24; Supplementary Table S1).
Safety outcomes
Safety was assessed by routine physical examination, laboratory evaluations, electrocardiograms, and echocardiograms as defined prospectively in the protocol. The laboratory evaluations included serum analyses performed at baseline and prespecified time points for the following labs: serum chemistry, fasting serum lipid panel, hemoglobin A1c (HbA1c), C-reactive protein, cardiac troponin (cTn), and N-terminal pro-brain natriuretic peptide (NT-proBNP). AEs were assessed throughout the study and categorized by the U.S. National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.
Statistical analysis
For CBR and ORR, 95% exact confidence intervals (CI) were calculated. For PFS and OS, medians were estimated using the Kaplan–Meier method. Descriptive statistics were used to summarize exposure to ponatinib over time, AE incidence rates, and the incidence of occurrence of overall toxicity, categorized by toxicity grades.
Determination of sample size
The planned enrollment for the study was 45 patients (30 in the KIT ex11–pos cohort and 15 in the KIT ex11–neg cohort). For the primary analysis population (KIT ex11–pos cohort), an exact binomial test determined that a one-stage design with at least 30 patients with the ex11 mutation could distinguish a favorable true CBR of 30% from a null rate of 10% with more than 80% power and a one-sided type 1 error rate of 0.025. Ponatinib would be considered promising in this population if at least seven of 30 patients experienced clinical benefit.
Data availability
The data sets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results reported in this article, will be made available within three months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
Results
Demographics and baseline characteristics
A total of 45 patients were enrolled and initiated ponatinib treatment at three centers in the United States between June 5, 2013, and October 1, 2014. The demographics and baseline characteristics of patients (30 with GIST harboring KIT ex11 mutations and 15 with GIST without KIT ex11 mutations) are summarized in Table 1. Of the 15 patients with KIT ex11–neg tumors, seven cases harbored KIT ex9 mutations, one harbored a primary KIT exon 13 mutation, one harbored a primary KIT exon 17 mutation, and six were wild type at the KIT and PDGFRA loci. Of the six patients with wild-type KIT tumors, two patients had tumors that were succinate dehydrogenase complex iron sulfur subunit B (SDHB)–intact (wild-type) and one was SDHB-negative by IHC; no tissue was available for testing in tumors from three patients. Overall, patients were heavily pretreated (with 82% having undergone treatment with at least three prior anticancer regimens) and typically had multiple lesions (with 73% having three or more lesions).
Table 1.
Demographics and baseline characteristics.
| Characteristics | KIT exon 11–positive (n = 30) | KIT exon 11–negative (n = 15) | Total (N = 45) |
|---|---|---|---|
| Median age (range), years | 62 (40–81) | 56 (24–77) | 59 (24–81) |
| Sex, male, n (%) | 19 (63) | 7 (47) | 26 (58) |
| Race,a white, n (%) | 29 (97) | 15 (100) | 44 (98) |
| Median time since diagnosis (range), years | 6 (1–30) | 7 (1–25) | 6 (1–30) |
| ECOG performance status, n (%) | |||
| 0 | 17 (57) | 6 (40) | 23 (51) |
| 1 | 12 (40) | 9 (60) | 21 (47) |
| 2 | 1 (3) | 0 | 1 (2) |
| Primary tumor site, n (%) | |||
| Stomach | 10 (33) | 5 (33) | 15 (33) |
| Small intestine | 7 (23) | 7 (47) | 14 (31) |
| Esophagus | 2 (7) | 0 | 2 (4) |
| Other/unknown | 11 (37) | 3 (20) | 14 (31) |
| Number of target lesions, n (%) | |||
| 1 | 0 | 0 | 0 |
| 2 | 7 (23) | 5 (33) | 12 (27) |
| 3 | 12 (40) | 3 (20) | 15 (33) |
| 4 | 8 (27) | 5 (33) | 13 (29) |
| 5 | 3 (10) | 2 (13) | 5 (11) |
| Number of prior anticancer regimens, n (%) | |||
| ≤2 | 6 (20) | 2 (13) | 8 (18) |
| 3–5 | 16 (53) | 10 (67) | 26 (58) |
| ≥6 | 8 (27) | 3 (20) | 11 (24) |
| Median (range) | 4 (1–10) | 5 (2–7) | 4 (1–10) |
| Number of prior approved TKIs, n (%) | |||
| 1 | 2 (7) | 1 (7) | 3 (7) |
| 2 | 10 (33) | 6 (40) | 16 (36) |
| 3 | 18 (60) | 8 (53) | 26 (58) |
| Among patients with KIT ex11–negative tumors (n = 15) | |||
| KIT exon 9 mutations | NA | 7 (47) | 7 (16) |
| KIT exon 13 mutations | NA | 1 (7) | 3 (7) |
| KIT exon 17 mutations | NA | 2 (13) | 5 (11) |
| Wild-type KIT and PDGFRA | NA | 6 (40) | 6 (13) |
| Among patients with wild-type KIT tumors (n = 6) | |||
| SDHB-intact | NA | 2 (13) | 2 (4) |
| SDHB-negative | NA | 1 (7) | 1 (2) |
| Unknown (tissue not available for testing) | NA | 3 (20) | 3 (7) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; SDHB, succinate dehydrogenase complex iron sulfur subunit B.
aRace was unknown in one patient in cohort A.
Patient disposition
At study termination (data cutoff: August 1, 2016), median follow-up was 14.7 months (range, 0.4–34.3) in the KIT ex11–pos cohort and 13.6 months (0.6–32.0) in the KIT ex11–neg cohort. Two patients (4.4%) continued ponatinib treatment after study termination at the discretion of their physician. Primary reasons for discontinuation included progressive disease (50% KIT ex11–pos cohort; 67% KIT ex11–neg cohort) and AEs (20% KIT ex11–pos; 13% KIT ex11–neg cohort; Supplementary Table S2).
Efficacy
Tumor response data are summarized in Table 2, which excludes two patients who were discontinued from the study per FDA request as they had not already responded to ponatinib, nor had they been treated with all other TKIs approved for GIST. Clinical benefit at 16 weeks was reported in 10 of 28 patients (36%) in the KIT ex11–pos cohort (primary outcome, Fig. 1) and 3 of 15 patients (20%) in the KIT ex11–neg cohort, with a CBR of 30% (13/43) in the total patient population.
Table 2.
Investigator-reported tumor response.
| KIT exon 11–positive | KIT exon 11–negative | Total | |
|---|---|---|---|
| Endpoint | n = 28a | n = 15 | N = 43a |
| Response at 16 weeks, n (%) | |||
| CBR at 16 weeks, n (%) | 10 (36) | 3 (20) | 13 (30) |
| (95% CI) | (19–56) | (4–48) | (17–46) |
| CR | 0 | 0 | 0 |
| PR | 1 (4) | 0 | 1 (2) |
| SD | 9 (32) | 3 (20) | 12 (28) |
| PD | 9 (32) | 7 (47) | 16 (37) |
| Not evaluableb | 9 (32) | 5 (33) | 14 (33) |
| Best response, n (%) | |||
| ORR (CR + PR), n (%) | 2 (7) | 0 | 2 (5) |
| (95% CI) | (1–24) | (0–22) | (1–16) |
| CR | 0 | 0 | 0 |
| PR | 2 (7) | 0 | 2 (5) |
| SD | 14 (50) | 7 (47) | 21 (49) |
| PD | 5 (18) | 5 (33) | 10 (23) |
| Not evaluableb | 7 (25) | 3 (20) | 10 (23) |
Abbreviation: PD, progressive disease.
aExcludes two patients who were discontinued from study per FDA request.
bNot evaluable group are patients whose tumor was not evaluable but who remained on study.
Figure 1.

Clinical response by duration of treatment. *Patient was discontinued per FDA request.
In patients with KIT ex11–pos tumors treated with two and three prior TKIs, CBR at 16 weeks was 44% and 33%, respectively. Clinical benefit was not experienced by patients with KIT ex9–positive tumors. PR as best response was reported in two patients, both with KIT ex11–pos tumors, resulting in an ORR of 7% in the KIT ex11–pos cohort, and 0% in the KIT ex11–neg cohort. Best response data for individual patients are summarized in Fig. 2. In the patient with SDHB-negative GIST as assessed by IHC, the best response was SD; total time on treatment at study termination was 29.4 months; the patient continued ponatinib 45 mg/day after study termination.
Figure 2.

Tumor response in evaluable patients (n = 36), mutation status, and prior therapies. Dotted line at −30% indicates threshold for PR. Two patients with 0% change from baseline and one patient with 0.5% change from baseline do not have visible bars. *Patients with KIT exon 11–negative tumors. NL, new lesion; WT, wild-type.
Kaplan–Meier plots of PFS and OS are shown in Supplementary Fig. S1. Median PFS was 4.0 months (95% CI, 2–9) in the KIT ex11–pos cohort and 2.0 months (2–18) in the KIT ex11–neg cohort; median OS was 14.7 months (95% CI, 8–30) and 14.3 months (3–not reached), respectively.
Association of KIT mutations detected in ctDNA with antitumor activity of ponatinib
At the time of ctDNA analyses (March 2, 2015), a total of 105 plasma samples from 37 patients were analyzed (Supplementary Table S3); 28 patients had baseline and postbaseline samples. In 100% (17/17) of cases in which the primary KIT mutation detected in the tumor was included in the BEAMing panel, the same mutation was also detected in the plasma (Supplementary Fig. S2). In the 12 cases in which the primary KIT mutation detected in the tumor was not included in the BEAMing panel, no primary mutations were detected in the plasma.
Analysis of ctDNA revealed a high degree of tumor heterogeneity, with at least two (and up to eight) unique secondary KIT mutations detected in 35% (13/37) of all patients and 54% (13/24) of patients in the KIT ex11–pos cohort (Fig. 3). In order to understand the heterogeneity and dynamics of resistance mutations post-ponatinib treatment, mutation dynamics in individual patients and the dynamics of the most common secondary mutations [V654A (exon 13), N822K, Y823D, D820A/G (all in exon 17/18)] were assessed (Supplementary Figs. S3 and S4). Although V654A was only detectable at baseline in one patient, it frequently emerged during treatment with ponatinib, becoming detectable in eight additional patients (range, 60–480 days of ponatinib treatment). Sustained or transient decreases in ctDNA levels were seen in the majority of patients with N822K, Y823D, or D820A/G.
Figure 3.
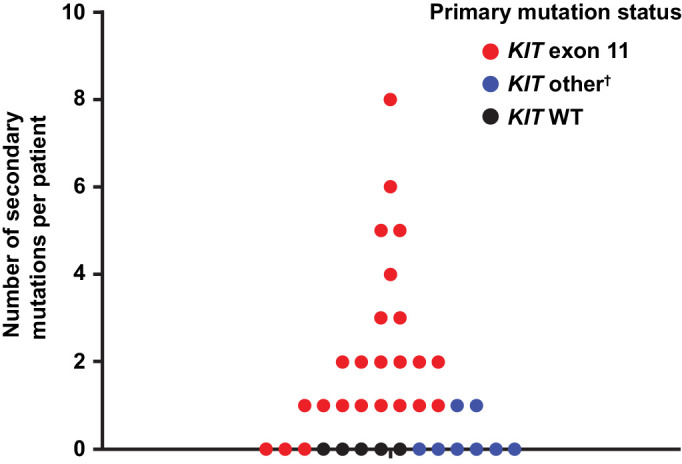
Number of secondary mutations detected per patient in ctDNA.* n = 37 patients. *Number of unique secondary mutations per patient observed across all samples analyzed (pre– and post–treatment initiation). Each dot represents a unique patient. †Includes exon 9 (n = 6), exon 13 (n = 1), and exon 17 (n = 1). WT, wild-type.
A summary of CBR and mutational status at baseline is included in Supplementary Table S4. The CBR was 43% (95% CI, 18%–71%) and median PFS was 6.54 months (range, 0.03 months–10.58 months) in patients with KIT ex11–pos tumors with secondary exon 17/18 mutations detected at baseline (n = 24). Following ponatinib treatment, a correlation between the change in tumor burden and change in plasma mutation load was observed (Spearman correlation = 0.6298; P = 0.0039; n = 19; Fig. 4). Changes in target lesion size were generally associated with changes in ctDNA levels. Decreases in target lesion size and ctDNA levels were observed in patients with primary ex11, but not exon 9 mutations. Exon 9 primary and V654A (exon 13) secondary mutation ctDNA levels generally increased during ponatinib treatment. ctDNA levels of the most common exon 17/18 mutations decreased in a majority of patients during ponatinib treatment.
Figure 4.
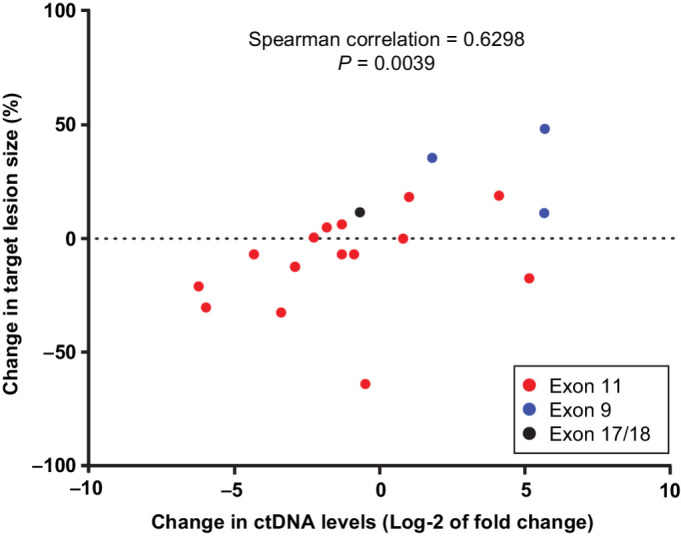
Association between changes in target lesion size and KIT-mutated ctDNA levels (n = 19). Target lesion size (sum of all target lesions) and levels of primary mutation (in ctDNA) were compared at baseline and first post-baseline visits.
Post hoc analyses
Correlation of CT scan and liver biopsy in a single patient
In one patient who had been treated with two prior TKIs, tumor regression was observed after two cycles of ponatinib on a CT scan (Supplementary Fig. S5A and S5B). Pretreatment liver biopsy showed presence of viable GIST cells with abundant mitotic figures in 100% of this region (Supplementary Fig. S5C and S5E). In contrast, liver biopsy on day 19 of cycle 1 of ponatinib treatment was markedly hypocellular, with a myxoid background and rare viable GIST cells and an absence of mitotic figures (Supplementary Fig. S5D and S5F).
Safety
Exposure
Time on treatment is shown in Supplementary Fig. S6. The median time on treatment for all patients was 3.0 months (range, 0.1–32.0) with a median time of 3.8 months (0.1–29.9) in the KIT ex11–pos cohort and 2.0 months (0.2–32.0) in the KIT ex11–neg cohort. Median dose intensity values were 41 mg/day (range, 23–45) in all patients, 42 mg/day (29–45) in the KIT ex11–pos cohort, and 40 mg/day (23–45) the KIT ex11–neg cohort. Dose reductions (any) and dose interruptions (≥3 days) were reported in 12 patients (40%) and 15 patients (50%), respectively, in the KIT ex11–pos cohort and in eight patients (53%) and seven patients (47%), respectively, in the KIT ex11–neg cohort.
AEs
Treatment-emergent AEs reported in ≥20% of patients and serious treatment-emergent AEs reported in ≥2% of patients are included in Table 3. Three patients had arterial occlusive events [AOE; myocardial ischemia (n = 1), cerebrovascular accident (n = 1), and peripheral artery stenosis (n = 1)]. Two patients had venous thromboembolic events [pulmonary embolism (n = 2); in one of these patients, deep vein thrombosis was also reported].
Table 3.
Treatment-emergent AEs.
| Treatment-emergent AEs reported in ≥20% of patients (N = 45) | Any grade, n (%) | Grades 3–5, n (%) |
|---|---|---|
| Rash | 27 (60) | 2 (4) |
| Fatigue | 24 (53) | 3 (7) |
| Abdominal pain | 22 (49) | 4 (9) |
| Headache | 20 (44) | 0 |
| Constipation | 19 (42) | 0 |
| Myalgia | 18 (40) | 0 |
| Hypertension | 17 (38) | 4 (9) |
| Peripheral edema | 16 (36) | 1 (2) |
| Decreased appetite | 15 (33) | 1 (2) |
| Dry skin | 15 (33) | 1 (2) |
| Increased blood alkaline phosphatase | 13 (29) | 2 (4) |
| Cough | 12 (27) | 0 |
| Vomiting | 11 (24) | 3 (7) |
| Nausea | 11 (24) | 2 (4) |
| Diarrhea | 10 (22) | 2 (4) |
| Increased aspartate aminotransferase | 10 (22) | 1 (2) |
| Pyrexia | 10 (22) | 0 |
| Serious treatment-emergent AEs reported in ≥2 patients (N = 45) | n (%) | |
| Abdominal pain | 4 (9) | |
| Pneumonia | 3 (7) | |
| Small intestine obstruction | 3 (7) | |
| Fatigue | 2 (4) | |
| Nausea | 2 (4) | |
| Pulmonary embolism | 2 (4) | |
| Vomiting | 2 (4) | |
Deaths within 30 days of the last dose and treatment-related deaths at any time were reported in six patients: hepatic failure (n = 1), pneumonia (n = 1), pulmonary embolism (n = 1), respiratory failure (n = 1), neoplasm progression (n = 2); deaths due to pneumonia and pulmonary embolism were considered possibly related to ponatinib treatment by the investigator. Hepatic failure was considered not related to treatment by the investigator; this patient with hepatic failure received ponatinib at a dose of 45 mg.
Discussion
In this phase II study in patients with advanced GIST who progressed on prior TKIs, ponatinib showed evidence of clinical activity in patients with activating mutations in KIT ex11. Clinical benefit (43% CBR) was observed in patients with primary KIT ex11 mutations and secondary exon 17/18 mutations detected at baseline. Ponatinib had no clinical activity in patients with primary mutations in KIT ex9. Consistent with the observed clinical activity, ctDNA levels of the most common exon 17 and exon 18 mutations typically decreased during ponatinib treatment, whereas ctDNA levels of KIT ex9 primary and V654A (exon 13) secondary mutations generally increased during treatment, the latter of which may represent a key acquired resistance mechanism. Associations between KIT mutation status assessed using ctDNA and ponatinib antitumor activity correlated with data from preclinical models that demonstrated that ponatinib has potent activity against secondary mutations such as N822K, Y823D, and D820A/G but not against V654A (19). ctDNA analysis indicated the presence of at least two secondary mutations in 54% of patients with KIT ex11 mutations. The reduced activity of ponatinib in KIT ex11–negative patients could be attributed to the presence of other mutations such as KIT ex9. Previous preclinical studies demonstrated limited activity of ponatinib in ex9 mutant models (19). In addition, the lack of activity in patients with KIT ex9 mutations is not unique and was previously reported in the phase III trial of nilotinib versus imatinib, in which nilotinib demonstrated reduced efficacy compared with imatinib (25). These findings suggest that the presence of a KIT ex11 mutation may serve as a response biomarker for ponatinib. Furthermore, the presence of KIT ex9 mutation may serve as a marker of resistance to ponatinib. However, additional studies are needed because the observed resistance to ponatinib could be due to the presence of other mutations, such as KIT exon 17 or exon 13 mutations.
The safety profile of ponatinib in this study is consistent with that reported in leukemia studies (26, 27), with the exception that myelosuppression occurred less frequently in patients with GIST. Based on experience in phase I and phase II clinical studies, the starting dose of ponatinib used was 45 mg/day in order to maximize potential efficacy in this heavily pretreated patient population. To reduce the potential risk of AOEs due to long-term exposure, the ponatinib dose was reduced after six cycles of treatment to 30 mg/day in patients with SD or better, if it had not already been reduced for other reasons. In this heavily pretreated population, ponatinib showed modest response and clinical benefit rates with limited durability of response. Based on the dose-dependent toxicities observed in this study, lower dosing (30 mg) was used in the POETIG phase II trial in pretreated patients with GIST (NCT03171389; ref. 28).
To the best of our knowledge, this is the first report of a clinical trial in advanced GIST in which patient enrollment was prospectively guided by mutational status and ctDNA analysis was incorporated to better understand acquired resistance to ponatinib. Our results support the utility of this approach and demonstrate that clinical trials evaluating the efficacy of TKIs based on primary tumor mutation in GIST are feasible and informative. ctDNA analysis by BEAMing further defined the primary and secondary resistance landscape in this trial. There was concordance between primary mutations detected in ctDNA using BEAMing and in tumor biopsies. BEAMing detected primary and/or secondary KIT mutations in plasma from the majority of patients enrolled; the incomplete spectrum of KIT mutations detectable by BEAMing may be overcome by use of next-generation sequencing approaches (29–31). Future GIST trials should continue to evaluate ctDNA using sequencing-based approaches. The utility of ctDNA analysis in the management of patients with GIST was demonstrated in a study that included 243 patients with GIST. In comparison with analysis of tumor mutations, plasma ctDNA analysis had a positive predictive value of 100%. ctDNA analysis provides a rapid, noninvasive approach to characterize current mutations, thereby allowing tailored, personalized therapeutic strategies based on resistance mutations (32).
The trial had several limitations. Although patient enrollment was guided by KIT ex11 mutation status, grouping all KIT ex11–negative patients in one cohort posed challenges in interpreting the lack of activity due to the heterogeneity of the clinical impact of various mutations on response to treatment. In addition, most patients included in the study were white (98%), which may impact the broad applicability of the findings. Further mutational analysis is needed to mechanistically define the limited activity of ponatinib in KIT ex11–negative patients and to determine whether this was solely due to the presence of KIT ex9 mutation or combinations of other relevant mutations. Another key limitation of the trial is the limited number of patients with KIT exon 13 and exon 17 mutations.
The emergence of two or more secondary mutations in a large proportion of patients suggests a high degree of underlying tumor heterogeneity and indicates that effective treatment approaches for GIST may require earlier use of pan-KIT inhibitors or combination therapies that can suppress development of resistance mutations. Although ponatinib will not be developed further in GIST due to the risk/benefit profile, future studies in GIST should focus on informative study designs, based on strong preclinical data with robust correlatives together with broad coverage of known resistance mutations.
Supplementary Material
Acknowledgments
This study is sponsored by Takeda Development Center Americas, Inc. M.C. Heinrich received partial salary support from a Merit Review grant from the U.S. Department of Veterans Affairs (1IOBX005358-01) and a grant from the NCI (R21CA263400).
We thank all of the patients and their families, and the investigators and staff at all clinical sites, for their participation in the study. The authors acknowledge Duprane Young, PhD, of Peloton Advantage, an OPEN Health company, and Meenakshi Subramanian of Evidence Scientific Solutions for medical writing support for the development of this manuscript under the direction of the authors, which was funded by Takeda Pharmaceuticals, USA, Inc., and complied with the Good Publication Practice 3 ethical guidelines (Battisti WP, et al. Ann Intern Med 2015;163:461–4).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
S. George reports grants from ARIAD during the conduct of the study; grants from Blueprint Medicines, Deciphera, Daiichi Sankyo, Pfizer, Bayer, Merck, Eisai, and Springworks, personal fees from Blueprint Medicines, Deciphera, Immunicum, and Kayothera, and other support from WCG outside the submitted work. M. von Mehren reports personal fees and other support from Deciphera, Novartis, and Blueprint, and personal fees from Exelixis during the conduct of the study; grants from Novartis outside the submitted work. J.A. Fletcher reports a patent for Imatinib licensed to Novartis. S. Zhang reports other support from ARIAD and EMD Serono outside the submitted work. J.R. Pritchard reports other support from ARIAD Pharmaceuticals during the conduct of the study; grants from Theseus Pharmaceuticals, personal fees from Theseus Pharmaceuticals, Moma Therapeutics, Third Rock Ventures, and Takeda Pharmaceuticals, and other support from Theseus Pharmaceuticals and Moma Therapeutics outside the submitted work. D. Kerstein reports other support from ARIAD Pharmaceuticals during the conduct of the study and other support from Theseus Pharmaceuticals outside the submitted work. V.M. Rivera reports other support from ARIAD Pharmaceuticals and other support from Theseus Pharmaceuticals outside the submitted work. F.G. Haluska reports other support from ARIAD Pharmaceuticals during the conduct of the study. M.C. Heinrich reports grants from ARIAD during the conduct of the study; personal fees and other support from MolecularMD, personal fees from Novartis, and grants and personal fees from Blueprint Medicines and Deciphera Pharmaceuticals outside the submitted work; in addition, M.C. Heinrich has a patent for Treatment of GIST issued, licensed, and with royalties paid from Novartis. No disclosures were reported by the other authors.
Authors' Contributions
S. George: Conceptualization, resources, writing–original draft, writing–review and editing. M. von Mehren: Conceptualization, resources, writing–original draft, writing–review and editing. J.A. Fletcher: Conceptualization, resources, methodology, writing–original draft, writing–review and editing. J. Sun: Conceptualization, resources, methodology, writing–original draft, writing–review and editing. S. Zhang: Conceptualization, resources, methodology, writing–original draft, writing–review and editing. J.R. Pritchard: Conceptualization, resources, methodology, writing–original draft, writing–review and editing. J.G. Hodgson: Conceptualization, resources, methodology, writing–original draft, writing–review and editing. D. Kerstein: Conceptualization, resources, methodology, writing–original draft, writing–review and editing. V.M. Rivera: Conceptualization, resources, writing–original draft, writing–review and editing. F.G. Haluska: Conceptualization, project administration, writing–review and editing. M.C. Heinrich: Conceptualization, resources, writing–original draft, writing–review and editing.
References
- 1. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol 2002;33:459–65. [DOI] [PubMed] [Google Scholar]
- 2. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001;438:1–12. [DOI] [PubMed] [Google Scholar]
- 3. Wasag B, Debiec-Rychter M, Pauwels P, Stul M, Vranckx H, Oosterom AV, et al. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol 2004;17:889–94. [DOI] [PubMed] [Google Scholar]
- 4. Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–9. [DOI] [PubMed] [Google Scholar]
- 5. Qinlock [package insert]. Waltham, MA: Deciphera Pharmaceuticals, LLC; 2020. [Google Scholar]
- 6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Soft tissue sarcoma v1.2021. Pennsylvania: National Comprehensive Cancer Network; 2020. Available from: https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf.
- 7. Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom H, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2020;21:923–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corless CL. Gastrointestinal stromal tumors: What do we know now? Mod Pathol 2014;27Suppl 1:S1–16. [DOI] [PubMed] [Google Scholar]
- 9. Schaefer IM, Marino-Enriquez A, Fletcher JA. What is new in gastrointestinal stromal tumor? Adv Anat Pathol 2017;24:259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008;26:620–5. [DOI] [PubMed] [Google Scholar]
- 12. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Montemurro M, Gelderblom H, Bitz U, Schutte J, Blay JY, Joensuu H, et al. Sorafenib as third- or fourth-line treatment of advanced gastrointestinal stromal tumour and pretreatment including both imatinib and sunitinib, and nilotinib: A retrospective analysis. Eur J Cancer 2013;49:1027–31. [DOI] [PubMed] [Google Scholar]
- 14. Italiano A, Cioffi A, Coco P, Maki RG, Schoffski P, Rutkowski P, et al. Patterns of care, prognosis, and survival in patients with metastatic gastrointestinal stromal tumors (GIST) refractory to first-line imatinib and second-line sunitinib. Ann Surg Oncol 2012;19:1551–9. [DOI] [PubMed] [Google Scholar]
- 15. Rutkowski P, Bylina E, Klimczak A, Switaj T, Falkowski S, Kroc J, et al. The outcome and predictive factors of sunitinib therapy in advanced gastrointestinal stromal tumors (GIST) after imatinib failure - one institution study. BMC Cancer 2012;12:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iclusig [package insert]. Cambridge, MA: Takeda Pharmaceutical Company Limited; 2020. [Google Scholar]
- 17. Gozgit JM, Wong MJ, Wardwell S, Tyner JW, Loriaux MM, Mohemmad QK, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol Cancer Ther 2011;10:1028–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lierman E, Smits S, Cools J, Dewaele B, Debiec-Rychter M, Vandenberghe P. Ponatinib is active against imatinib-resistant mutants of FIP1L1-PDGFRA and KIT, and against FGFR1-derived fusion kinases. Leukemia 2012;26:1693–5. [DOI] [PubMed] [Google Scholar]
- 19. Garner AP, Gozgit JM, Anjum R, Vodala S, Schrock A, Zhou T, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res 2014;20:5745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wagner AJ, Severson PL, Shields AF, Patnaik A, Chugh R, Tinoco G, et al. Association of combination of conformation-specific KIT inhibitors with clinical benefit in patients with refractory gastrointestinal stromal tumors: A phase 1b/2a nonrandomized clinical trial. JAMA Oncol 2021;7:1343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ben-Ami E, Barysauskas CM, von Mehren M, Heinrich MC, Corless CL, Butrynski JE, et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol 2016;27:1794–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A 2003;100:8817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. KIT Gene. Catalogue of Somatic Mutations in Cancer (COSMIC), 2019. Available from: https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=KIT.
- 25. Blay JY, Shen L, Kang YK, Rutkowski P, Qin S, Nosov D, et al. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): A randomised phase 3 trial. Lancet Oncol 2015;16:550–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med 2012;367:2075–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med 2013;369:1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Falkenhorst J, Hamacher R, Reichardt P, Ivanyi P, Kasper B, Hohenberger P, et al. Lower-dosing ponatinib in pre-treated GIST: Results of the POETIG phase II trial [abstract 11536]. J Clin Oncol 2020;38:11536. [Google Scholar]
- 29. Li B, Bai J, Zhu L, Zhang S, Wang G, Dai P, et al. Application of next generation sequencing (NGS) in gastrointestinal stromal tumor (GIST) [abstract]. J Clin Oncol 2020;38:e13675. [Google Scholar]
- 30. Vanden Bempt I, Vander Borght S, Sciot R, Spans L, Claerhout S, Brems H, et al. Comprehensive targeted next-generation sequencing approach in the molecular diagnosis of gastrointestinal stromal tumor. Genes Chromosomes Cancer 2021;60:239–49. [DOI] [PubMed] [Google Scholar]
- 31. Zhou Y, Zhang X, Wu X, Zhou Y, Zhang B, Liu X, et al. A prospective multicenter phase II study on the efficacy and safety of dasatinib in the treatment of metastatic gastrointestinal stromal tumors failed by imatinib and sunitinib and analysis of NGS in peripheral blood. Cancer Med 2020;9:6225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arshad J, Roberts A, Ahmed J, Cotta J, Pico BA, Kwon D, et al. Utility of circulating tumor DNA in the management of patients with GI stromal tumor: Analysis of 243 patients. JCO Precis Oncol 2020:66–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results reported in this article, will be made available within three months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.