

Abstract
Pulmonary hypertension (PH) complicates the treatment of interstitial lung disease (ILD) patients resulting in poor functional status and worse outcomes. Early recognition of PH in ILD is important for initiating therapy and considering lung transplantation. However, no standard exists regarding which patients to screen for PH‐ILD or the optimal method to do so. The aim of this study was to create a risk assessment tool that could reliably predict PH in ILD patients. We developed a PH‐ILD Detection tool that incorporated history, exam, 6‐min walk distance, diffusion capacity for carbon monoxide, chest imaging, and cardiac biomarkers to create an eight‐component score. This tool was analyzed retrospectively in 154 ILD patients where each patient was given a score ranging from 0 to 12. The sensitivity (SN) and specificity (SP) of the PH‐ILD Detection tool and an area‐under‐the‐curve (AUC) were calculated. In this cohort, 74 patients (48.1%) had PH‐ILD. A score of ≥6 on the PH‐ILD Detection tool was associated with a diagnosis of PH‐ILD (SN: 86.5%; SP: 86.3%; area‐under‐the‐curve: 0.920, p < 0.001). The PH‐ILD Detection tool provides high SN and SP for detecting PH in ILD patients. With confirmation in larger cohorts, this tool could improve the diagnosis of PH in ILD and may suggest further testing with right heart catheterization and earlier intervention with inhaled treprostinil and/or lung transplant evaluation.
Keywords: idiopathic pulmonary fibrosis, interstitial lung disease, prostacyclin, pulmonary hypertension, treprostinil
Abbreviations
- 6MWD
6‐min walk distance
- 6MWT
6‐min walk test
- AUC
area‐under‐the‐curve
- BNP
B‐type natriuretic peptide
- CTD
connective tissue disease
- DLCO
diffusion capacity for carbon monoxide
- ICD‐10
International Classification of Diseases 10th Revision
- ILD
interstitial lung disease
- IPF
idiopathic pulmonary fibrosis
- JVP
jugular venous pressure
- mPAP
mean pulmonary artery pressure
- NT‐proBNP
N‐terminal proBNP
- P2
pulmonic component of the second heart sound
- PA
pulmonary artery
- PAH
pulmonary arterial hypertension
- PA:A
pulmonary artery to aorta ratio
- PFT
pulmonary function test
- PH
pulmonary hypertension
- PVD
pulmonary vascular disease
- PVR
pulmonary vascular resistance
- RHC
right heart catheterization
- ROC
receiver operating characteristics
- SAPH
sarcoidosis‐associated pulmonary hypertension
- SN
sensitivity
- SP
specificity
- WHO
World Health Organization
BACKGROUND
Pulmonary hypertension (PH) is a frequent complication in patients with interstitial lung disease (ILD) and is associated with poor functional status, need for supplemental oxygen, and worse outcomes. 1 , 2 , 3 , 4 , 5 Over 40 studies have reported concomitant PH in idiopathic pulmonary fibrosis (IPF) patients; prevalence ranges from 3% to 86%. 6 , 7 This variability in prevalence depends on when PH was investigated during the ILD course, as well as the method of diagnosis. Mean pulmonary artery pressure (mPAP) is elevated in 8%–15% of patients when IPF is initially diagnosed; the prevalence increases with the progression of lung disease, complicating up to 30%–50% of advanced cases and more than 60% of end‐stage disease. 8 , 9 , 10 , 11 PH can also develop in other ILDs, besides IPF, and a severe phenotype of PH‐ILD including depressed cardiac index has been described by large European studies. 12 , 13 The development of PH in ILD patients is associated with decreased survival and increased morbidity with Song et al. 14 reporting 1‐year mortality of 61.2% for patients with IPF and PH detected on echocardiogram compared to 19.9% in those without PH. Similarly, Nadrous et al. 15 reported a median survival of 0.7 years for patients with IPF and PA systolic pressure >50 mmHg on echocardiogram.
Thus, early recognition of PH in ILD is important in planning diagnostic tests, initiating therapy, and considering referral for lung transplantation. For example, in regard to therapy, results from the INCREASE trial demonstrated clinical and functional improvement with inhaled treprostinil in PH‐ILD patients, thus increasing the importance of making the diagnosis of PH. 16 However, no standard currently exists regarding which patients to screen for PH‐ILD nor the optimal method to do so. 17 Furthermore, the diagnosis of PH in the context of ILD is often difficult because of the overlap in symptoms and diagnostic testing. 18 To obviate these issues, there have been multiple attempts to incorporate various noninvasive parameters into a clinical prediction tool, but none has been widely adopted. 19 , 20 , 21 Currently, the most common recommendation is an echocardiogram annually or sooner if there is a significant change in symptoms. 17
In an attempt to address these problems, our study determined whether a PH‐ILD Detection tool incorporating eight common variables could provide useful information for screening for PH in ILD patients. To this end, we developed a simple, noninvasive tool based on patient history and symptoms, physical exam, 6‐min walk test (6MWT), pulmonary function tests (PFTs), chest imaging, and cardiac biomarkers, all of which are routinely monitored in ILD patients.
METHODS
We performed a retrospective analysis of 162 ILD patients who underwent evaluation between October 2016 and February 2022 at Hartford Hospital (Hartford, CT). Patients were identified by the International Classification of Diseases 10th Revision (ICD‐10). Of these 162 patients, 8 patients receiving background PH therapy were excluded. Consequently, we examined a total of 154 ILD patients.
Development of the PH‐ILD Detection tool
We developed a PH‐ILD Detection tool that incorporated patient history and symptoms, physical exam, 6MWT results, PFTs, chest imaging, and cardiac biomarkers to create an eight‐component score. Physical exam findings included increased jugular venous pressure (JVP), pedal edema, ascites, accentuated pulmonic component of the second heart sound (P2), and/or parasternal heave. Six‐min walk distance (6MWD) in meters (m) and the need for supplemental oxygen were also included in the PH‐ILD Detection tool. For 6MWD, we used the cut‐point of 350 m to delineate decreased exercise capacity. For PFTs, we used a diffusion capacity for carbon monoxide (DLCO, % predicted) cut‐point of 40%. We also incorporated a history of syncope, concomitant diagnosis of connective tissue disease (CTD) or sarcoidosis, elevated B‐type natriuretic peptide (BNP) or N‐terminal proBNP (NT‐proBNP) levels, and abnormal findings on computed tomography scan of the chest (CT chest) such as pulmonary artery (PA) to aorta ratio (PA:A) >0.9 or PA enlargement >30 mm.
Based on multivariate analysis, the candidate variables that were significantly associated with the PH‐ILD subgroup were a physical exam, 6MWD < 350 m, and DLCO < 40%. Based on the regression coefficients of the covariates in the multivariate model, we assigned a weighted score of 2 points to each of these covariates as well as to oxygen supplementation because of its high combined sensitivity (SN) and specificity (SP). We assigned 1 point to the other four covariates: syncope, CTD or sarcoidosis, elevated cardiac biomarkers, and PA enlargement on CT chest. The individual points were totaled to obtain a composite score ranging from 0 to 12 (Table 1).
Table 1.
Metrics of the detection tool for screening for PH in ILD patients
| Clinical finding | Score |
|---|---|
| 6MWD < 350 m | 2 |
| Physical exam for PHa | 2 |
| DLCO < 40% | 2 |
| Supplemental oxygen | 2 |
| Elevated BNP or NT‐ProBNPb | 1 |
| Syncope or presyncope | 1 |
| PA enlargement on CT chestc | 1 |
| CTD or sarcoidosis | 1 |
Abbreviations: 6MWD, 6‐min walk distance; BNP, B‐type natriuretic peptide; CTD, connective tissue disease; DLCO, diffusion capacity for carbon monoxide; ILD, interstitial lung disease; NT‐ProBN, N‐terminal proBNP; PA, pulmonary artery; PH, pulmonary hypertension.
Increased JVP, peripheral edema, ascites, accentuated P2, TR murmur, parasternal heave.
BNP > 50 pg/ml, NT‐ProBNP > 300 pg/ml.
Ratio of pulmonary artery (PA) to aorta (A) > 0.9, enlargement of main PA > 30 mm.
The PH‐ILD Detection tool was designed for use in patients with an established diagnosis of ILD. Depending on the total score for a given patient, patients were then stratified into low, intermediate, or high‐risk with the assumption that low‐risk patients were unlikely to have PH‐ILD and intermediate and high‐risk patients had a greater probability of PH‐ILD (Table 2). Continued risk assessment at follow‐up visits would be recommended for low‐risk patients and further screening with an echocardiogram for intermediate‐risk patients. For high‐risk patients, an echocardiogram and immediate referral to a PH specialist for further evaluation and management, including right heart catheterization (RHC), would be recommended.
Table 2.
Low‐, intermediate‐, and high‐risk category scores and clinical recommendations
| Score | Risk category | Recommendations |
|---|---|---|
| ≤3 | Low | Reassess during follow‐up visit |
| 4–5 | Intermediate | Echocardiogram and short‐term reassessment |
| ≥6 | High | Echocardiogram and immediate referral to PH center for RHC |
Abbreviations: PH, pulmonary hypertension; RHC, right heart catheterization.
Statistics
After evaluating continuous data for normality of distribution, descriptive statistics comprised means and standard deviations. Categorical data were presented as frequencies, using percentages. Inferential statistics comprised a Student's t‐test, for comparisons of continuous variables between ILD and ILD‐PH groups, and a χ 2 test, for comparisons of categorical variables. Variables that showed statistically significant differences upon univariate testing were included in a forward, conditional logistic regression model. A receiver operating characteristics (ROC) curve was generated, and an area‐under‐the‐curve (AUC) was calculated from the values of SN and SP. All analyses were conducted with SPSS v. 26 (IBM) using an a priori α level of 0.05.
RESULTS
Patients
Patient demographics and other clinical data, dichotomized by the presence or absence of PH, are shown in Table 3. The average duration between the evaluation of all components of the PH‐ILD Detection tool and RHC was 6 months.
Table 3.
Baseline characteristics
| Characteristic | All | ILD | PH‐ILD | p Value |
|---|---|---|---|---|
| Sample size—number (%) | 154 | 80 (51.9) | 74 (48.1) | — |
| Female sex—number (%) | 78 (50.6) | 43 (53.8) | 35 (47.3) | 0.424 a |
| Age, years (mean ± SD) | 70.0 ± 12.1 | 67.0 ± 11.5 | 73.2 ± 12.0 | 0.001 b |
| Race—number (%) | ||||
| White | 85 (55.2) | 32 (40.0) | 53 (71.6) | 0.001 b |
| Black/African American | 21 (13.6) | 13 (16.3) | 8 (10.8) | |
| Asian | 2 (1.3) | 2 (2.5) | 0 (0.0) | |
| Other (undefined) | 46 (29.9) | 33 (41.3) | 13 (17.6) | |
| Hispanic/Latinx—number (%) | 49 (31.8) | 36 (45.0) | 13 (17.6) | <0.001 b |
| Cause of lung disease—number (%) | ||||
| Idiopathic pulmonary fibrosis | 63 (40.9) | 31 (38.8) | 32 (43.2) | 0.014 b |
| Nonspecific interstitial pneumonia | 46 (29.9) | 32 (40.0) | 14 (18.9) | |
| Combined pulmonary fibrosis and emphysema | 22 (14.3) | 6 (7.5) | 16 (21.2) | |
| Post‐Coronavirus‐2019 lung disease | 8 (5.2) | 5 (6.3) | 3 (4.1) | |
| Respiratory bronchiolitis with ILD | 4 (2.6) | 3 (3.8) | 1 (1.4) | |
| Cryptogenic organizing pneumonia | 4 (2.6) | 3 (3.8) | 1 (1.4) | |
| Drug‐related lung disease | 3 (1.9) | 0 | 3 (4.1) | |
| Sarcoidosis‐related lung disease | 2 (1.3) | 0 | 2 (2.7) | |
| Occupational lung disease | 1 (1.0) | 0 | 1 (1.4) | |
| Pulmonary Langerhans cell histiocytosis | 1 (1.0) | 0 | 1 (1.4) | |
| Antifibrotic therapy—number (%) | ||||
| No therapy | 136 (88.3) | 75 (93.8) | 61 (82.4) | 0.029 b |
| On therapy | 18 (11.7) | 5 (6.3) | 13 (17.6) | |
| Left heart dysfunction—number (%) | 22 (14.2) | 10 (12.5) | 12 (16.2) | |
Note: Values in bold are statistically significant at p < 0.05.
χ 2.
Student's t‐test.
Patients underwent RHC to determine the presence or absence of PH. PH was diagnosed by RHC with an mPAP ≥ 20 mmHg, pulmonary capillary wedge pressure (PCWP) ≤15 mmHg, and pulmonary vascular resistance (PVR) >3 Wood units. The diagnosis of ILD was confirmed by the identification of diffuse parenchymal lung disease on the CT chest.
In the cohort, PH was diagnosed in 74 patients (48.1%) with a mean age of 73.2 years; 47.3% (n = 35) were female. The most common cause of ILD was IPF in both the PH‐ILD (n = 32; 43.2%) and non‐PH ILD (n = 31; 38.8%) cohorts. There was a significantly larger portion of patients with nonspecific interstitial pneumonia in the non‐PH ILD group (n = 32; 40.0%) compared to the PH‐ILD group (n = 14; 18.9%) and conversely, a higher prevalence of combined pulmonary fibrosis and emphysema in the PH‐ILD group (n = 16; 21.2%) versus the non‐PH ILD cohort (n = 6; 7.5%).
Results of individual components to predict PH‐ILD
Four of the eight components had a combined SN and SP greater than 1.4; however, the two highest components, 6MWD and physical exam, had a much higher SP than SN (Table 4). The component with the highest SN was supplemental oxygen, although that did not contribute significantly to the multivariate model in comparison to 6MWD, physical exam, and DLCO.
Table 4.
Sensitivity and specificity for each scoring tool component
| Component | SN (%) | SP (%) | SN + SP |
|---|---|---|---|
| 6MWD < 350 m | 78.4 | 90.0 | 1.684 |
| Physical exam for PH | 52.7 | 95.0 | 1.477 |
| DLCO < 40% | 71.6 | 71.3 | 1.429 |
| Supplemental oxygen | 85.1 | 55.0 | 1.401 |
| NT‐ProBNP >300 pg/ml | 75.7 | 61.3 | 1.369 |
| Syncope or presyncope | 29.7 | 86.3 | 1.160 |
| PA enlargement on CT chest | 36.5 | 78.8 | 1.152 |
| CTD or sarcoidosis | 35.1 | 60.0 | 0.951 |
Abbreviations: 6MWD, 6‐min walk distance; CTD, connective tissue disease; DLCO, diffusion capacity for carbon monoxide; NT‐ProBN, N‐terminal proBNP; PA, pulmonary artery; SN, sensitivity; SP, specificity.
Accuracy and validation of the PH‐ILD Detection tool
We created a cumulative graph and plotted the risk scores using the ROC curve. A score of ≥6 was associated with the greatest likelihood that a patient would have a diagnosis of PH‐ILD (Figure 1). The SN was 86.5% and SP was 86.3%, resulting in an AUC of 0.920 (95% confidence interval [CI]: 0.878–0.962, p < 0.001). Using the detection tool to identify cases of PH‐ILD, the resulting false positives were 11 (7.1%) and false negatives were 10 (6.5%).
Figure 1.
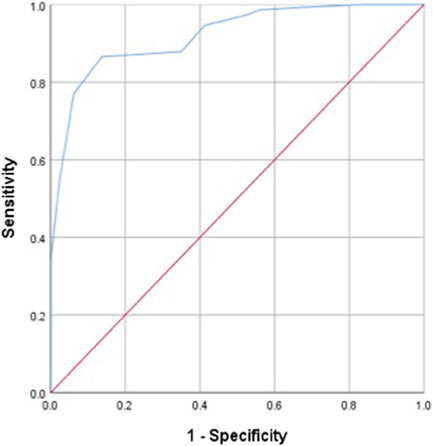
ROC curve for scoring tool to identify PH‐ILD. ILD, interstitial lung disease; PH, pulmonary hypertension; ROC, receiver operating characteristics.
Prognostic implications of the PH‐ILD Detection tool
A score of ≥8 on the PH‐ILD Detection tool resulted in an AUC of 0.680 (95% CI: 0.581–0.778, p < 0.001) for mortality with SN of 53.3% and SP of 82.6% (Figure 2), suggesting a score on the risk assessment tool below this cut‐off was a strong identifier of patients who survived.
Figure 2.
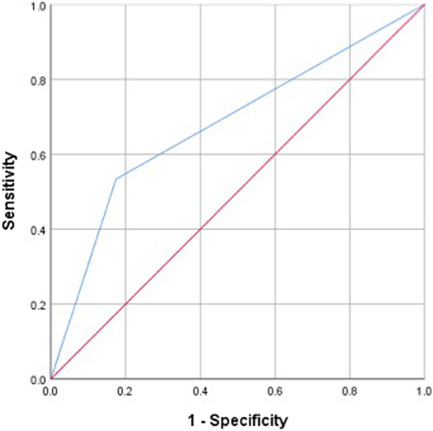
ROC curve for scoring tool to identify mortality. ROC, receiver operating characteristics.
Summary of PH‐ILD Detection tool metrics
A score of ≥6 using the metrics in the PH‐ILD Detection tool was associated with a high likelihood of PH and a score of ≥8 was associated with poorer survival.
DISCUSSION
The presence of PH exerts a significant negative impact on survival in ILD patients. 6 , 22 , 23 , 24 A study from the Giessen registry demonstrated that patients with World Health Organization (WHO) Group 3 PH had a worse prognosis than patients with WHO Group 1 pulmonary arterial hypertension (PAH). 25 Early detection of PH‐ILD is important because even mild elevations in mPAP can be a significant predictor of mortality in patients with ILD. 26 , 27
PH commonly intercedes at various stages of ILD so a regular, noninvasive, and affordable screening tool is imperative for the ongoing management and decision‐making in these patients. Although RHC is the gold standard to diagnose PH, it is not a convenient screening tool because it is invasive, time‐consuming, costly, and the risk of moderate sedation is higher in those with baseline hypoxemia. 19 There have been several unsuccessful attempts to develop a reliable screening tool for PH‐ILD and most have used components of PFTs and/or oxygen saturation; as such, recent authors have concluded that noninvasive testing alone is not sufficiently accurate to screen these patients. 20 , 28 , 29 , 30
Justification for eight components of PH‐ILD Detection tool
In our study, eight variables routinely monitored in ILD patients were incorporated into the PH‐ILD Detection tool in an attempt to create a useful and reliable method for screening for PH in this population. Each individual component has previously been utilized in the evaluation and prognostication of PH patients.
6MWD < 350 m
The development of concomitant PH poses an additional burden on exercise capacity in ILD patients. 31 In ILD or PH patients individually, the 6MWT has proven to be highly reproducible and easy to perform while providing information on functional capacity and need for supplemental oxygen with exertion. 32 Multiple studies have demonstrated that patients with PH‐ILD have a lower 6MWD than ILD patients without PH. 10 , 11 , 13 , 19 , 33 Not only does a reduced 6MWD suggest the diagnosis of concomitant PH in ILD patients, but it is also associated with decreased survival. 2 However, there is conflicting data demonstrating that shorter distances on a 6MWT were actually not predictive of PH in ILD. 20
Physical exam findings suggestive of PH
Physical exam findings, including increased JVP, pedal edema, ascites, P2, and/or parasternal heave are suggestive of right heart dysfunction associated with PH. 34 In fact, ILD patients are often evaluated for concomitant PH only after such signs are detected; at that point, pulmonary vascular disease (PVD) may have significantly progressed. 35 , 36 However, there is no consensus whether physical exam findings alone have sufficient SN to predict PH‐ILD accurately. 34
Percent predicted DLCO < 40%
Decreased DLCO reflects fibrosis of the alveoli and perfusion inhomogeneity in ILD and PH patients, respectively. 37 , 38 Studies have proposed that PH should be suspected when DLCO is disproportionately low compared with functional and radiological impairment in ILD patients. 3 Several reports have suggested that DLCO ranging from 30% to 45% can predict PH. 10 , 18 , 39 , 40 Additionally, worsening DLCO in the setting of preserved lung volumes could suggest the possibility of underlying PVD. 17 Therefore, forced vital capacity to DLCO ratio has been incorporated into a multi‐component scoring tool to improve the accuracy of PH prediction. 19 , 28 , 29 , 30 , 41 The fact that DLCO not only serves as a diagnostic clue in ILD patients who develop concomitant PH, but also as a prognosticator denotes the potential importance of this metric in this subset of patients. 2 , 28 , 29 , 39 , 42
Supplemental oxygen
The need for supplemental oxygen has been evaluated as a predictor of PH in ILD patients. 10 In ILD and sarcoidosis, recent studies suggest that the need for oxygen supplementation is an independent predictor of PH. 10 , 11 , 40 , 43 Hypoxic pulmonary vasoconstriction, an innate physiologic response to alveolar hypoxia resulting in constriction of intrapulmonary arteries and diversion of blood to better oxygenated lung areas, explains why supplemental oxygen would be predictive of underlying PH in chronically hypoxemic patients. 10 , 44
Elevated cardiac biomarkers
Although cardiac biomarkers, such as BNP and NT‐proBNP, are controversial as independent predictors of PH in ILD patients, the bulk of available data suggests that they are potentially very useful. 31 For example, Sonti et al. 19 found a statistically significant difference in BNP levels between PH‐ILD patients (738 pg/ml) and non‐PH ILD patients (263 pg/ml). Other studies have not only shown the potential diagnostic utility of these biomarkers, but also a prognostic value as well. To wit, BNP values have correlated with transplant‐free survival in ILD patients and cardiac biomarker levels have been predictive of prognosis in this subset of patients as well. 31 , 36 , 45 , 46 , 47 And, a cross‐sectional study demonstrated that BNP levels were able to detect PH in IPF patients and not only showed a correlation of BNP with invasive hemodynamics, but also a significant association with WHO functional class and 6MWD. 31
Yet, there are several factors that make the measurements unreliable on their own including left heart dysfunction, renal failure, and obesity. 36 , 48 Therefore, other authors have suggested that the predictive value of cardiac biomarkers in newly diagnosed ILD is overall low. 48
Syncope or presyncope
Syncope in PH patients suggests depressed right heart function. 49 The REVEAL registry further highlighted the incidence and clinical implications of syncope in the PH population. 50 Although the effect of syncope in PH‐ILD has not been extensively evaluated, there is hemodynamic evidence to suggest that syncope is a poor prognostic indicator in this entity as well. 13 , 14
PA enlargement on CT chest
PA enlargement on CT chest can be due to a variety of conditions including PH. A normal PA diameter has been designated as <27–29 mm. 51 Along with PA dilation, the PA:A ratio has been utilized to suggest PVD. 19 Several studies have evaluated the utility of PA diameter in ILD patients to predict PH; many have shown that a PA:A >0.9 or >1.1 was predictive of an mPAP > 20 mmHg as well as predictive of decreased survival. 30 , 52 Others have shown that a PA:A ratio >1.0 can predict PH in COPD and IPF patients. 20 , 30 , 52 , 53 King et al. incorporated this ratio into a multi‐score predictor tool to establish an association with increased PVR and mortality in patients with ILD. 54 As such, it has been suggested that PA:A combined with other metrics could improve the accuracy of PH prediction in ILD patients. 19 However, other authors remain skeptical about the overall utility of PA size in predicting PH‐ILD and in a cross‐sectional study of 65 IPF patients, there was no correlation between PH and increased PA diameter or PA:A ratio. 55 , 56
CTD or sarcoidosis
The existence of CTD, especially scleroderma, or sarcoidosis can often cloud the clinical picture in PH‐ILD. 3 , 47 Scleroderma is associated with both PAH and ILD individually. 57 Differentiating the contribution of PVD and parenchymal lung disease in scleroderma patients who have both PAH and ILD is challenging. 17 , 47 Nevertheless, the presence of PAH or ILD are two major causes of morbidity and mortality in patients with scleroderma. 58 Even in CTD patients with WHO Group 1 PAH, the presence of minor parenchymal lung disease affects survival. 59 However, it remains difficult to determine the contributory role of ILD in the development of PH in CTD patients since they are already at risk for WHO Group 1 PAH independent of parenchymal lung disease. 60
The prevalence of PH in sarcoidosis ranges from 6% to 74% with a 5‐year survival of 50%–60%. 5 , 43 , 61 The complex mechanisms of sarcoidosis‐associated PH (SAPH) include lung fibrosis and obliteration of pulmonary vessels, extrinsic compression of pulmonary vasculature by lymphadenopathy or fibrosing mediastinitis, pulmonary veno‐occlusive disease, granulomatous involvement of pulmonary vessels, left ventricular dysfunction, and portopulmonary hypertension. 61 However, despite these complexities, the need for supplemental oxygen was shown to be an independent predictor of PH in a multivariate analysis, suggesting that hypoxic pulmonary vasoconstriction is a major contributor to the pathophysiology. 47 , 62 This is further suggested by the fact that advanced stage disease has a higher prevalence of both hypoxemia and concomitant PH. 63
Clinical implications of PH‐ILD detection tool
Our study assessed the accuracy of a PH‐ILD Detection tool, incorporating eight independent factors to predict PH‐ILD. In this cohort, the PH‐ILD Detection tool stratified PH‐ILD patients by level of risk for PH; surprisingly, it also seemingly was able to predict prognosis in this subset of patients. From these data, we would suggest that for low‐risk individuals (i.e., lower score), reassessment at follow‐up visits is warranted unless another metric develops in the interim. Those patients in the intermediate‐risk category should be considered for further evaluation including screening echocardiogram and closer follow‐up. ILD patients classified as high‐risk warrant prompt evaluation for concomitant PH, including echocardiogram and referral to a PH specialist where PH can formally be evaluated and RHC performed.
Strengths and limitations of this study
The present study has several limitations: (1) this was a single‐center, retrospective study; (2) there were 12 patients (16.2%) in the PH‐ILD group with depressed ejection fraction and/or abnormal diastolic indices on echocardiogram; however, during RHC, PCWP was ≤15 mmHg in these patients and therefore, they were included in the study; (3) eight metrics were tested and incorporated into the PH‐ILD Detection tool but other potential risk factors, such as age, were not included; and (4) the PH‐ILD Detection tool was validated internally, but does require external validation with an independent cohort of patients.
The strengths of our study are that it is a relatively large cohort of patients with a diverse demographic background that was investigated over a several‐year period. Moreover, the effectiveness of this detection tool is quite plausible since many of the individual variables have been independently associated with the development of PH in ILD patients.
CONCLUSION
There is currently no available detection tool widely accepted to suggest PH‐ILD. Our study developed a noninvasive detection tool that not only incorporated several variables recently published in a consensus statement for PH screening in ILD but also had both a high SN and SP for detecting PH in ILD within our study cohort. 64
The development of the PH‐ILD Detection tool and its further refinement has important implications in the evaluation and treatment of ILD patients in clinical practice: concomitant PH may be diagnosed sooner and more accurately in these patients, allowing for earlier interventions such as initiation of inhaled therapies or referral for lung transplant evaluation.
AUTHOR CONTRIBUTIONS
Raj Parikh: Conceptualization, data curation, investigation, methodology, validation, writing–original draft, writing–review & editing, guarantor. Ippokratis Konstantinidis: Conceptualization, data curation. David M. O'Sullivan: Conceptualization, data curation, formal analysis, methodology, writing–original draft, writing–review & editing. Harrison W. Farber: Conceptualization, investigation, methodology, validation, writing–original draft; writing–review & editing.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
This study was approved by the Hartford HealthCare Institutional Review Board (HHC‐2022‐0014).
ACKNOWLEDGMENT
There is no funding for this study.
Parikh R, Konstantinidis I, O'Sullivan DM, Farber HW. Pulmonary hypertension in patients with interstitial lung disease: a tool for early detection. Pulm Circ. 2022;12:e12141. 10.1002/pul2.12141
REFERENCES
- 1. Nathan SD, Hassoun PM. Pulmonary hypertension due to lung disease and/or hypoxia. Clin Chest Med. 2013;34:695–705. [DOI] [PubMed] [Google Scholar]
- 2. Lettieri CJ, Nathan SD, Browning RF, Barnett SD, Ahmad S, Shorr AF. The distance‐saturation product predicts mortality in idiopathic pulmonary fibrosis. Respir Med. 2006;100:1734–41. [DOI] [PubMed] [Google Scholar]
- 3. Caminati A, Cassando R, Harari S. Pulmonary hypertension in chronic interstitial lung diseases. Eur Respir Rev. 2013;22(129):292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oliveira RK, Pereira CA, Ramos RP, Ferreira EV, Messina CM, Kuranishi LT, Gimenez A, Campos O, Silva CM, Ota‐Arakaki JS. A hemodynamic study of pulmonary hypertension in chronic hypersensitivity pneumonitis. Eur Respir J. 2014;44(2):415–24. [DOI] [PubMed] [Google Scholar]
- 5. Baughman RP, Shlobin OA, Wells AU, Alhamad EH, Culver DA, Barney J, Cordova FC, Carmona EM, Scholand MB, Wijsenbeek M, Ganesh S, Birring SS, Kouranos V, O'Hare L, Baran JM, Cal JG, Lower EE, Engel PJ, Nathan SD. Clinical features of sarcoidosis associated pulmonary hypertension: results of a multi‐national registry. Respir Med. 2018;139:72–8. [DOI] [PubMed] [Google Scholar]
- 6. Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J. 2015;46:1113–30. [DOI] [PubMed] [Google Scholar]
- 7. Andersen CU, Mellemkjær S, Nielsen‐Kudsk JE, Bendstrup E, Simonsen U, Hilberg O. Diagnostic and prognostic role of biomarkers for pulmonary hypertension in interstitial lung disease. Respir Med. 2012;106:1749–55. [DOI] [PubMed] [Google Scholar]
- 8. Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, Olsson KM, Peacock AJ, Pepke‐Zaba J, Provencher S, Weissmann N, Seeger W. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019;53(1):1801914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, Burton N, Leslie K. Serial development of pulmonary hypertension in patients with IPF. Respiration. 2008;76:288–94. [DOI] [PubMed] [Google Scholar]
- 10. Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129(3):746–52. [DOI] [PubMed] [Google Scholar]
- 11. Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30(4):715–21. [DOI] [PubMed] [Google Scholar]
- 12. Hoeper MM, Behr J, Held M, Grunig E, Vizza CD, Vonk‐Noordegraaf A, Lange TJ, Claussen M, Grohé C, Klose H, Olsson KM, Zelniker T, Neurohr C, Distler O, Wirtz H, Opitz C, Huscher D, Pittrow D, Gibbs JS. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS One. 2015;10(12):e0141911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chebib N, Mornex JF, Traclet J, Philit F, Khouatra C, Zeghmar S, Turquier S, Cottin V. Pulmonary hypertension in chronic lung diseases: comparison to other pulmonary hypertension groups. Pulm Circ. 2018;8(2):2045894018775056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song JW, Song JK, Kim DS. Echocardiography and brain natriuretic peptide as prognostic indicators in idiopathic pulmonary fibrosis. Respir Med. 2009;103(2):180–6. [DOI] [PubMed] [Google Scholar]
- 15. Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, Ryu JH. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005;128(4):2393–9. [DOI] [PubMed] [Google Scholar]
- 16. Waxman A, Restrepo‐Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K, Deng C, Peterson L, Bell H, Tapson V, Nathan SD. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325–34. [DOI] [PubMed] [Google Scholar]
- 17. King CS, Shlobin OA. The trouble with group 3 pulmonary hypertension in interstitial lung disease: dilemmas in diagnosis and the conundrum of treatment. Chest. 2020;158:1651–64. [DOI] [PubMed] [Google Scholar]
- 18. Nathan SD, Shlobin OA, Ahmad S, Urbanek S, Barnett SD. Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest. 2007;131(3):657–63. [DOI] [PubMed] [Google Scholar]
- 19. Sonti R, Gersten RA, Barnett S, Brown AW, Nathan SD. Multimodal noninvasive prediction of pulmonary hypertension in IPF. Clin Respir J. 2019;13(9):567–73. [DOI] [PubMed] [Google Scholar]
- 20. Furukawa T, Kondoh Y, Taniguchi H, Yagi M, Matsuda T, Kimura T, Kataoka K, Johkoh T, Ando M, Hashimoto N, Sakamoto K, Hasegawa Y. A scoring system to predict the elevation of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis. Eur Respir J. 2018;51(1):1701311. [DOI] [PubMed] [Google Scholar]
- 21. Nathan SD, Shlobin OA, Barnett SD, Saggar R, Belperio JA, Ross DJ, Ahmad S, Saggar R, Libre E, Lynch JP, Zisman DA. Right ventricular systolic pressure by echocardiography as a predictor of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2008;102(9):1305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K, Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Res Int Rev Thoracic Dis. 2013;85:456–63. [DOI] [PubMed] [Google Scholar]
- 23. Margaritopoulos GA, Antoniou KM, Wells AU. Comorbidities in interstitial lung diseases. Eur Respir Rev Offic J Eur Res Soc. 2017;26:160027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pitsiou G, Papakosta D, Bouros D. Pulmonary hypertension in idiopathic pulmonary fibrosis: a review. Res Int Rev Thoracic Dis. 2011;82:294–304. [DOI] [PubMed] [Google Scholar]
- 25. Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, Franco OH, Hofman A, Schermuly RT, Weissmann N, Grimminger F, Seeger W, Ghofrani HA. The Giessen pulmonary hypertension registry: survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017;36(9):957–67. [DOI] [PubMed] [Google Scholar]
- 26. Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K, Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85:456–63. [DOI] [PubMed] [Google Scholar]
- 27. Teramachi R, Taniguchi H, Kondoh Y, Ando M, Kimura T, Kataoka K, Suzuki A, Furukawa T, Sakamoto K, Hasegawa Y. Progression of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis with mild to moderate restriction. Respirology. 2017;22:986–90. [DOI] [PubMed] [Google Scholar]
- 28. Zisman DA, Karlamangla AS, Kawut SM, Shlobin OA, Saggar R, Ross DJ, Schwarz MI, Belperio JA, Ardehali A, Lynch JP, Nathan SD. Validation of a method to screen for pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2008;133:640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zisman DA, Ross DJ, Belperio JA, Saggar R, Lynch JP, Ardehali A, Karlamangla AS. Prediction of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2007;101:2153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alkukhun L, Wang XF, Ahmed MK, Baumgartner M, Budev MM, Dweik RA, Tonelli AR. Non‐invasive screening for pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2016;117:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leuchte HH, Neurohr C, Baumgartner R, Holzapfel M, Giehrl W, Vogeser M, Behr J. Brain natriuretic peptide and exercise capacity in lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2004;170:360–5. [DOI] [PubMed] [Google Scholar]
- 32. Eaton T, Young P, Milne D, Wells AU. Six‐minute walk, maximal exercise tests: reproducibility in fibrotic interstitial pneumonia. Am J Respir Crit Care Med. 2005;171:1150–7. [DOI] [PubMed] [Google Scholar]
- 33. Sobiecka M, Lewandowska K, Kober J, Franczuk M, Skoczylas A, Tomkowski W, Kuś J, Szturmowicz M. Can a new scoring system improve prediction of pulmonary hypertension in newly recognised interstitial lung diseases? Lung. 2020;198(3):547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braganza M, Shaw J, Solverson K, Vis D, Janovcik J, Varughese RA, Thakrar MV, Hirani N, Helmersen D, Weatherald J. A prospective evaluation of the diagnostic accuracy of the physical examination for pulmonary hypertension. Chest. 2019;155(5):982–90. [DOI] [PubMed] [Google Scholar]
- 35. Handa T, Nagai S, Miki S, Ueda S, Yukawa N, Fushimi Y, Ito Y, Ohta K, Mimori T, Mishima M, Izumi T. Incidence of pulmonary hypertension and its clinical relevance in patients with interstitial pneumonias: comparison between idiopathic and collagen vascular disease associated interstitial pneumonias. Intern Med. 2007;46:831–7. [DOI] [PubMed] [Google Scholar]
- 36. Leuchte HH, Baumgartner RA, Nounou ME, Vogeser M, Neurohr C, Trautnitz M, Behr J. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med. 2006;173:744–50. [DOI] [PubMed] [Google Scholar]
- 37. van der Lee I, Zanen P, Grutters JC, Snijder RJ, van den Bosch J. Diffusing capacity for nitric oxide and carbon monoxide in patients with diffuse parenchymal lung disease and pulmonary arterial hypertension. Chest. 2006;129:378–83. [DOI] [PubMed] [Google Scholar]
- 38. Collard HR, King TE Jr., Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168:538–42. [DOI] [PubMed] [Google Scholar]
- 39. Trip P, Nossent EJ, de Man FS, van den Berk IA, Boonstra A, Groepenhoff H, Leter EM, Westerhof N, Grünberg K, Bogaard HJ, Vonk‐Noordegraaf A. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. Eur Respir J. 2013;42:1575–85. [DOI] [PubMed] [Google Scholar]
- 40. Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007;132:998–1006. [DOI] [PubMed] [Google Scholar]
- 41. Coghlan JG, Denton CP, Grünig E, Bonderman D, Distler O, Khanna D, Müller‐Ladner U, Pope JE, Vonk MC, Doelberg M, Chadha‐Boreham H, Heinzl H, Rosenberg DM, McLaughlin VV, Seibold JR, DETECT Study Group. Evidence‐based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014;73(7):1340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raghu G, Nathan SD, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Wells AU, Shao L, Zhou H, Henig N, Szwarcberg J, Gillies H, Montgomery AB, O'Riordan TG. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild‐to‐moderate restriction. Eur Respir J. 2015;46:1370–7. [DOI] [PubMed] [Google Scholar]
- 43. Shorr AF, Helman DL, Davies DB, Nathan SD. Pulmonary hypertension in advanced sarcoidosis: epidemiology and clinical characteristics. Eur Respir J. 2005;25:783–8. [DOI] [PubMed] [Google Scholar]
- 44. Dunham‐Snary KJ, Wu D, Sykes EA, Thakrar A, Parlow L, Mewburn JD, Parlow JL, Archer SL. Hypoxic pulmonary vasoconstriction: from molecular mechanisms to medicine. Chest. 2017;151(1):181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yogeswaran A, Tello K, Faber M, Sommer N, Kuhnert S, Seeger W, Grimminger F, Ghofrani HA, Richter MJ, Gall H. Risk assessment in severe pulmonary hypertension due to interstitial lung disease. J Heart Lung Transplant. 2020;39(10):1118–25. [DOI] [PubMed] [Google Scholar]
- 46. Andersen C, Mellemkjær S, Hilberg O, Bendstrup E. NT‐proBNP <95 ng/l can exclude pulmonary hypertension on echocardiography at diagnostic workup in patients with interstitial lung disease. Eur Clin Respir J. 2016;3:32027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Behr J, Ryu JH. Pulmonary hypertension in interstitial lung disease. Eur Respir J. 2008;31(6):1357–67. [DOI] [PubMed] [Google Scholar]
- 48. Andersen KH, Iversen M, Kjaergaard J, Mortensen J, Nielsen‐Kudsk JE, Bendstrup E, Videbaek R, Carlsen J. Prevalence, predictors, and survival in pulmonary hypertension related to end‐stage chronic obstructive pulmonary disease. J Heart Lung Transplant. 2012;31:373–80. [DOI] [PubMed] [Google Scholar]
- 49. Mar PL, Nwazue V, Black BK, Biaggioni I, Diedrich A, Paranjape SY, Loyd JE, Hemnes AR, Robbins IM, Robertson D, Raj SR, Austin ED. Valsalva maneuver in pulmonary arterial hypertension: susceptibility to syncope and autonomic dysfunction. Chest. 2016;149:1252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Le R, Kane GC, Frantz RP, Farber HW, Turner M, Barst RJ. REVEAL: syncope in pulmonary arterial hypertension. Chest. 2011;30(4):S14–5. [Google Scholar]
- 51. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yagi M, Taniguchi H, Kondoh Y, Ando M, Kimura T, Kataoka K, Furukawa T, Suzuki A, Johkoh T, Hasegawa Y. CT‐determined pulmonary artery to aorta ratio as a predictor of elevated pulmonary artery pressure and survival in idiopathic pulmonary fibrosis. Respirology. 2017;22(7):1393–9. [DOI] [PubMed] [Google Scholar]
- 53. Wells JM, Washko GR, Han MK, Abbas N, Nath H, Mamary AJ, Regan E, Bailey WC, Martinez FJ, Westfall E, Beaty TH, Curran‐Everett D, Curtis JL, Hokanson JE, Lynch DA, Make BJ, Crapo JD, Silverman EK, Bowler RP, Dransfield MT, COPDGene Investigators, ECLIPSE Study Investigators. Pulmonary arterial enlargement and acute exacerbations of COPD. N Engl J Med. 2012;367:913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bax S, Jacob J, Ahmed R, Bredy C, Dimopoulos K, Kempny A, Kokosi M, Kier G, Renzoni E, Molyneaux PL, Chua F, Kouranos V, George P, McCabe C, Wilde M, Devaraj A, Wells A, Wort SJ, Price LC. Right ventricular to left ventricular ratio at CT pulmonary angiogram predicts mortality in interstitial lung disease. Chest. 2020;157(1):89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zisman DA, Karlamangla AS, Ross DJ, Keane MP, Belperio JA, Saggar R, Lynch JP, Ardehali A, Goldin J. High resolution CT findings do not predict the presence of pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2007;132:773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Devaraj A, Wells AU, Meister MG, Corte TJ, Hansell DM. The effect of diffuse pulmonary fibrosis on the reliability of CT signs of pulmonary hypertension. Radiology. 2008;249:1042–9. [DOI] [PubMed] [Google Scholar]
- 57. Bazan IS, Mensah KA, Rudkovskaia AA, Adonteng‐Boateng PK, Herzog EL, Buckley L, Fares WH. Pulmonary arterial hypertension in the setting of scleroderma is different than in the setting of lupus: a review. Respir Med. 2018;134:42–6. [DOI] [PubMed] [Google Scholar]
- 58. Morrisroe K, Stevens W, Sahhar J, Rabusa C, Nikpour M, Proudman S, Australian Scleroderma Interest Group . Epidemiology and disease characteristics of systemic sclerosis‐related pulmonary arterial hypertension: results from a real‐life screening programme. Arthritis Res Ther. 2017;19:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Poms AD, Turner M, Farber HW, Meltzer LA, McGoon MD. Comorbid conditions and outcomes in patients with pulmonary arterial hypertension: a REVEAL registry analysis. Chest. 2013;144:169–76. [DOI] [PubMed] [Google Scholar]
- 60. Chang B, Wigley FM, White B, Wise RA. Scleroderma patients with combined pulmonary hypertension and interstitial lung disease. J Rheumatol. 2003;30:2398–405. [PubMed] [Google Scholar]
- 61. Nunes H, Humbert M, Capron F, Brauner M, Sitbon O, Battesti JP, Simonneau G, Valeyre D. Pulmonary hypertension associated with sarcoidosis: mechanisms, haemodynamics and prognosis. Thorax. 2006;61:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest. 2003;124:922–8. [PubMed] [Google Scholar]
- 63. Baughman RP, Engel PJ, Taylor L, Lower EE. Survival in sarcoidosis‐associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest. 2010;138:1078–85. [DOI] [PubMed] [Google Scholar]
- 64. Rahaghi FF, Kolaitis NA, Adegunsoye A, de Andrade JA, Flaherty KR, Lancaster LH, Lee JS, Levine DJ, Preston IR, Safdar Z, Saggar R, Sahay S, Scholand MB, Shlobin OA, Zisman DA, Nathan SD. Screening strategies for pulmonary hypertension in patients with interstitial lung disease: a multidisciplinary Delphi study. Chest. 2022;162(1):145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]