

Abstract
Aminoacylase was identified in cell extracts of the hyperthermophilic archaeon Pyrococcus furiosus by its ability to hydrolyze N-acetyl-l-methionine and was purified by multistep chromatography. The enzyme is a homotetramer (42.06 kDa per subunit) and, as purified, contains 1.0 ± 0.48 g-atoms of zinc per subunit. Treatment of the purified enzyme with EDTA resulted in complete loss of activity. This was restored to 86% of the original value (200 U/mg) by treatment with ZnCl2 (and to 74% by the addition of CoCl2). After reconstitution with ZnCl2, the enzyme contained 2.85 ± 0.48 g-atoms of zinc per subunit. Aminoacylase showed broad substrate specificity and hydrolyzed nonpolar N-acylated l amino acids (Met, Ala, Val, and Leu), as well as N-formyl-l-methionine. The high Km values for these compounds indicate that the enzyme plays a role in the metabolism of protein growth substrates rather than in the degradation of cellular proteins. Maximal aminoacylase activity with N-acetyl-l-methionine as the substrate occurred at pH 6.5 and a temperature of 100°C. The N-terminal amino acid sequence of the purified aminoacylase was used to identify, in the P. furiosus genome database, a gene that encodes 383 amino acids. The gene was cloned and expressed in Escherichia coli by using two approaches. One involved the T7 lac promoter system, in which the recombinant protein was expressed as inclusion bodies. The second approach used the Trx fusion system, and this produced soluble but inactive recombinant protein. Renaturation and reconstitution experiments with Zn2+ ions failed to produce catalytically active protein. A survey of databases showed that, in general, organisms that contain a homolog of the P. furiosus aminoacylase (≥50% sequence identity) utilize peptide growth substrates, whereas those that do not contain the enzyme are not known to be proteolytic, suggesting a role for the enzyme in primary catabolism.
Aminoacylases (N-acyl amino acid amidohydrolases; EC 3.5.1.14) catalyze the hydrolysis of N-acyl amino acids to yield the corresponding organic acid and amino acid according to the following equation (2):
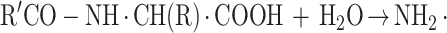 |
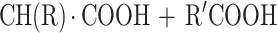 |
They have been purified from various bacteria, including species of Bacillus and Pseudomonas, as well as from plants and animals (34, 37, 38, 41, 44, 52, 53), although the role of the enzyme is typically ill defined at best. In mammals, aminoacylases are thought to function in the detoxification of xenobiotic-derived amino acid derivatives and they are of great interest to the pharmaceutical industry (2, 13, 50). Indeed, there is evidence that these enzymes play a role in the development of lung cancer (17), and aminoacylase activity has been used as a monitor of hepatic dysfunction (35). Moreover, based on the economic value of the products and their intrinsic industrial importance, aminoacylases are among the top 10 enzymes used in biotechnology (47). Due to their chiral specificity, chemically synthesized mixtures of d and l forms of N-acyl amino acids can be optically resolved with l or d aminoacylases to yield the l or d amino acids. Hence, they are used industrially in the synthesis of certain amino acids, such as phenylalanine and alanine, as well as in the production of acylated amino acids (15, 44, 47).
The aminoacylases that have been characterized so far all consist of a single subunit of approximately 45,000 Da. Most are dimeric enzymes, although homotetrameric forms have been reported (14, 21, 40, 61). Some have a broad substrate specificity, whereas others are highly specific and hydrolyze, for example, only the acetylated form of aspartate, proline, or one of the aromatic amino acids (2, 44). One factor common to all of the enzymes is a requirement of a divalent cation for maximal activity. This is typically zinc, but cobalt, manganese, and nickel are also usually effective. The exact role of the metal is not clear, although in some of these enzymes, it is thought to have a structural role, as well as a catalytic one (59). Unfortunately, biochemical analyses of this group of enzymes have been limited mainly to kinetic studies (11, 55, 59, 60) and detailed structural information, such as that obtained by crystallography, is not yet available (46, 54). Because of their biotechnological potential, there have been many attempts to stabilize various types of aminoacylases, either by immobilization or by obtaining the enzyme from thermophilic sources (5, 6, 7). The most thermostable reported so far is the enzyme from Bacillus stearothermophilus. This has an optimal temperature for catalysis near 70°C, but the time required for a 50% loss of activity is less than 1 min at 80°C (56).
As yet, an enzyme that can hydrolyze N-acylated amino acids has not been characterized from either an archaeon or a hyperthermophilic microorganism. Herein, we report on the purification, biochemical properties, and sequence of such an enzyme from the hyperthermophilic archaeon Pyrococcus furiosus. This organism grows optimally at 100°C, utilizing proteins and peptides as substrates, and it produces organic acids, CO2, and H2. Several enzymes involved in the catabolism of amino acids have been purified from P. furiosus (1, 22), including aminotransferases, glutamate dehydrogenase (3), 2-keto acid oxidoreductases (25), and acetyl coenzyme A synthetases (36). We have now found that cell extracts contain significant aminoacylase activity using N-acetyl-l-methionine as the substrate. From the related organism P. horikoshii, an acyl amino acid-releasing enzyme (AARE) was recently characterized (26). This enzyme catalyzes the hydrolysis of N-acyl peptides to release N-acylated amino acids and does not hydrolyze N-acylated amino acids like the aminoacylase of P. furiosus. The physiological role of these two enzymes in heterotrophic hyperthermophiles is also discussed herein.
MATERIALS AND METHODS
Growth of microorganisms.
P. furiosus (DSM 3638) was grown at 95°C in a 600-liter fermentor with maltose as the carbon source as previously described (9). Escherichia coli strains were grown in Luria-Bertani (LB) medium or M9 glucose medium supplemented with 0.5% Casamino Acids. Ampicillin (100 μg/ml) and chloramphenicol (35 μg/ml) were added as needed for plasmid maintenance.
Enzyme assay.
Aminoacylase activity was measured by determining the production of l-methionine from N-acetyl-l-methionine using the colorimetric ninhydrin method described by Rosen (43). The assay mixture (500 μl) containing the enzyme sample in 50 mM MOPS (morpholine propanesulfonic acid) buffer (pH 6.5) and 30 mM N-acetyl-l-methionine (Sigma Chemical Co., St. Louis, Mo.) was incubated at 100°C for 5 min, and 500 μl of trichloroacetic acid (15%, wt/vol) was added to stop the reaction. Precipitated protein was removed by centrifugation, and an aliquot (500 μl) of the supernatant solution was mixed with 250 μl of ninhydrin reagent (3% [wt/vol] ninhydrin in 2-methoxyethanol) and 250 μl of acetate-cyanide buffer (0.2 mM NaCN in 250 mM acetic acid) and incubated at 100°C for 15 min. The mixture was cooled to ambient temperature by the addition of 1.5 ml of isopropanol (50%, vol/vol), and the A570 was measured. The amount of methionine produced was determined from a standard curve. One unit of aminoacylase activity is defined as the amount of enzyme that liberates 1 μmol of l-methionine per min under these assay conditions.
Purification of P. furiosus aminoacylase.
Aminoacylase was purified from P. furiosus under anaerobic conditions at 23°C. Frozen cells (200 g, wet weight) were thawed in 600 ml of 50 mM Tris-HCl buffer (pH 8.0) containing DNase I (10 μg/ml) and 2 mM sodium dithionite (DT) and were lysed by incubation at 37°C for 2 h. A cell extract was obtained by ultracentrifugation at 18,000 × g for 2 h. The supernatant (600 ml) was loaded onto a column (10 by 14 cm) of Q-Sepharose Fast Flow (Pharmacia, Piscataway, N.J.) equilibrated with 50 mM Tris (pH 8.0) containing 2 mM DT (Tris-DT buffer). The column was eluted at a flow rate of 12 ml/min with a 2.5-liter linear gradient of 0 to 1.0 M NaCl in the same Tris-DT buffer. Aminoacylase activity was eluted in fractions as 0.25 to 0.35 M NaCl. The active fractions were combined (300 ml), and solid sodium sulfate was added to a final concentration of 0.5 M. This solution was applied to a column (3.5 by 10 cm) of phenyl Sepharose (Pharmacia) equilibrated with Tris-DT buffer containing 0.5 M sodium sulfate. The column was eluted with a gradient (1 liter) of 0.5 to 0 M sodium sulfate in the Tris-DT buffer at a flow rate of 7 ml/min. Aminoacylase eluted as 0.30 to 0.40 M sodium sulfate. The aminoacylase-containing fractions (200 ml) were applied to a column (3.5 by 10 cm) of Q-Sepharose High Performance (Pharmacia) equilibrated with 25 mM bis-Tris buffer (pH 6.5). The column was eluted with a gradient (1 liter) of 0 to 0.5 M NaCl in the same Tris-DT buffer at a flow rate of 8 ml/min. Aminoacylase activity was eluted as 0.23 to 0.28 M NaCl. The active fractions (186 ml) were applied to a column (1 by 10 cm) of hydroxyapatite (Pharmacia) equilibrated with 25 mM bis-Tris buffer (pH 7.0). The column was eluted at a flow rate of 5 ml/min with a 100-ml linear gradient of 0 to 0.5 M potassium phosphate buffer. The active fractions (35 ml) from the hydroxyapatite were applied to a column of HiTrap-Q (1.6 by 2.5 cm; Pharmacia) equilibrated with 25 mM bis-Tris (pH 6.5), and the enzyme was eluted with a gradient (75 ml) of 0 to 0.5 M NaCl in the same buffer at a flow rate of 2 ml/min. Fractions containing aminoacylase activity (17 ml) eluted as 0.14 to 0.20 M NaCl was applied and were stored frozen as pellets in liquid nitrogen until required.
Characterization of recombinant aminoacylase.
The recombinant form of P. furiosus aminoacylase was obtained by PCR amplification of the gene encoding the enzyme and its subsequent cloning into the T7 polymerase-driven expression vector pET-21b (Novagen, Milwaukee, Wis.). For amplification, the forward primer (GGATCCTTCGGAGGACCAAATGTTCAACCCCCTTGAGGAGG; Stratagene, La Jolla, Calif.) contained an engineered BamHI site and spanned positions −19 to +22 on the coding strand, while the reverse primer (ATGCGGCCGCAATCCCAACTGTATAACCCATTACGAATATGA; Stratagene) had an engineered NotI site and corresponded to the sequence ranging from +1406 to +1438 on the noncoding strand. PCR amplification was performed with native P. furiosus DNA polymerase and a Robocycler 40 (Stratagene) programmed for 1 cycle of denaturation at 95°C for 5 min; 2 cycles of denaturation at 95°C for 1 min, annealing at 50°C for 2 min, and extension at 72°C for 5 min; 39 cycles of denaturation at 95°C for 1 min, annealing at 61°C for 1.5 min, and extension at 72°C for 2 min; and 1 cycle of extension at 72°C for 7 min. The resultant 1.4-kb gene encoding aminoacylase was subcloned into the blunt-end SrfI site of plasmid pCR-Script (Stratagene) to yield plasmid pAA. The insert DNA was then sequenced to ensure that no mutations were present in the gene. The gene was then excised from plasmid pAA by restriction digestion with the enzymes BamHI and NotI (Stratagene) and cloned into the BamHI and NotI sites in expression plasmid pET-21b, resulting in plasmid pET-AA2.
The gene encoding aminoacylase was initially expressed by using E. coli BL21(λDE3)/pET-AA2 cultures (1 liter in 2.8- liter Fernbach flasks) grown in LB medium with and without ZnCl2 supplementation (100 μM) that were also coexpressing the rare Arg, Ile, and Leu tRNAs from plasmid pRIL (Stratagene). The cultures were incubated at 37°C with shaking (250 rpm) until an optical density of 0.6 to 0.8 was reached. Production of recombinant aminoacylase was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM. The cultures were then maintained at 37°C for 3 h. This expression resulted in the production of insoluble, inactive protein. The expression using plasmid pET-AA2 was repeated with different media (glucose-M9 medium supplemented with Casamino Acids at 0.5%), with and without the addition of ZnCl2, NiCl2, or FeSO4 (each at a final concentration of 100 μM). Expression of the gene was also attempted by induction over a range of temperatures, including 4°C for 16 h, 15°C for 12 h, 25°C for 8 h, and 30°C for 6 h. None of these modifications yielded soluble, active protein.
In an attempt to generate active, soluble protein, the insoluble recombinant aminoacylase was dissolved in 8.0 M urea (in 50 mM Tris-HCl, pH 8.0) and renaturation buffer (50 mM Tris-HCl, pH 8.0; 1 mM ZnCl2) was then slowly added with stirring. A second method involved dissolving the insoluble aminoacylase in 6.0 M guanidinium-HCl (in 50 mM MOPS, pH 7.0, containing 20 mM dithiothreitol [DTT]). In this case, the solution was incubated with stirring for 2 h at room temperature and then spun at 5,000 × g for 10 min. The supernatant was diluted 1:100 with chilled renaturation solution (50 mM Tris-HCl, pH 8.0; 0.5 M l-arginine; 10 mM DTT; 100 μM ZnCl2). The renaturation solution was stirred gently overnight at 4°C. The renaturation solution (15 ml) was then dialyzed for 12 h against 4 liters of 50 mM MOPS buffer, pH 7.0, at 4°C. In a third approach, insoluble aminoacylase (0.33 mg/ml, 86 ml) was initially suspended in 50 mM Tris-HCl, pH 8.0, containing 6 M guanidinium-HCl, 10 mM DTT, and 20 mM EDTA. This solution was dialyzed overnight against 500 ml of acidic-denaturation solution (2 M guanidinium-HCl, 10 mM DTT, 10 mM EDTA, 5% acetic acid, pH 2.7). The denatured protein solution was then separated into two samples and dialyzed overnight at 4°C against 500 ml of renaturing solution I (50 mM Tris-HCl, pH 8.0; 100 μM ZnCl2) or solution II (50 mM Tris-HCl, pH 8.0; 100 μM CoCl2). The partially renatured protein solutions were then dialyzed overnight at 4°C against 1 liter of renaturation solution I to II, respectively.
The gene encoding aminoacylase was also cloned into thioredoxin fusion expression plasmid pET-32a (Novagen). The forward primer for PCR (CCATGGTCAACCCCCTTGAGGAGGCCATGA) contained an engineered NcoI site and spanned positions +1 to +28 on the coding strand, and the reverse primer (AAGAGGATCCACTGGCTAACCTCTAAAGTT) had an engineered BamHI site and corresponded to positions +1134 to +1163 on the noncoding strand. The conditions of amplification were as described above, except that an annealing temperature of 48°C was employed. A resulting 1.2-kb gene was cloned into plasmid pCR-Script as described above to give plasmid pTrx-aa and was sequenced to ensure that there were no PCR-induced errors. The gene was removed from plasmid pTrx-aa by digestion with the restriction enzymes NcoI and BamHI and cloned into the corresponding sites in the thioredoxin fusion expression plasmid pET-32a, yielding plasmid pET-trx/aa. Recombinant aminoacylase produced from this system carries both a His tag and an S-protein tag and is fused to thioredoxin. By using enterokinase, the thioredoxin and affinity tags can be cleaved from the aminoacylase protein, leaving no extra amino acids. For expression of the thioredoxin-aminoacylase fusion protein, two 1-liter cultures of BL21(λDE3)/pET-trx/aa were grown in LB medium at 37°C with shaking (250 rpm). Protein expression was induced by the addition of IPTG (0.4 mM) once the optical density of the cultures had reached 0.6 to 0.8. The cultures were then incubated at 28°C for 2 h before harvesting of the cells. Soluble thioredoxin-aminoacylase fusion protein was detected both by visualization of a Coomassie-stained sodium dodecyl sulfate (SDS)–12.5% polyacrylamide gel electrophoresis (PAGE) gel and by Western antibody detection in accordance with the manufacturer's (Novagen) instructions.
To purify recombinant aminoacylase, 7 g (wet weight) of cell paste was resuspended in 20 ml of buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.1% Triton X-100) and cells were broken by two passages of the suspension through a French pressure cell at 20,000 lb/in2. The suspension was spun at 7,000 × g for 1.5 h, and the supernatant was applied to a 6-ml S-protein affinity agarose column (Novagen). The column was washed with 30 ml of buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.1% Triton X-100; 1 mM DTT). Bound fusion protein was eluted from the column with 12 ml of elution buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.1% Triton X-100; 1 mM DTT; 3 M MgCl2). The eluted protein was concentrated to 1.3 ml using Centricon-30 concentrators (Amicon Inc., Beverly, Mass.). The concentrated protein solution was dialyzed against 2 liters of enterokinase buffer (20 mM Tris-HCl, pH 8.0; 50 mM NaCl; 2 mM CaCl2; 1 mM DTT). To remove the fusion part of the protein, 20 U of enterokinase (Stratagene) was added and the reaction mixture was incubated at room temperature for 15 h. The enterokinase and cleaved S-protein tag were sequentially removed from the recombinant aminoacylase protein by passage of the protein sample through enterokinase capture STI-agarose (Stratagene) and S-protein affinity resin. The protein sample was applied to a 0.75-ml STI-agarose column equilibrated with enterokinase-binding buffer (50 mM Tris-HCl, pH 8.0; 200 mM NaCl). Recombinant aminoacylase was eluted with 4 ml of buffer and applied to the 6-ml S-protein affinity agarose column, from which it was eluted with 12 ml of wash buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.1% Triton X-100; 1 mM DTT). Recombinant aminoacylase was concentrated to 0.9 ml (0.88 mg/ml) and stored as pellets in liquid nitrogen.
Other methods.
Molecular weights were estimated by gel filtration with a column (1 by 27 cm) of Superdex 200 (Pharmacia LKB) with amylase (Mr, 200,000), alcohol dehydrogenase (Mr, 150,000), bovine serum albumin (Mr, 66,000), and carbonic anhydrase (Mr, 29,000) as standard proteins. SDS-PAGE was performed with 12% polyacrylamide by the method of Laemmli (33). Molecular weights were estimated by using a 10-kDa (10 to 120 kDa) standard molecular size protein ladder derived from T4 gene 32 (GIBCO BRL) also containing myosin (200 kDa). Protein concentrations were determined by the Bradford method (8), with bovine serum albumin as the standard. To determine metal content, exogenous metal ions were removed from the aminoacylase by gel filtration using a G-25 column equilibrated with 50 mM Tris-HCl, pH 8.0, containing 0.5 M NaCl. A complete metal analysis (31 elements) was obtained by plasma emission spectroscopy with a Jarrel Ash Plasma Comp 750 instrument at the Chemical Analysis Laboratory of the University of Georgia. Amino-terminal sequences were determined by using an Applied Biosystems model 477 sequencer in the Molecular Genetics Instrumentation Facility of the University of Georgia. Samples were electroblotted onto polyvinylidene difluoride protein-sequencing membranes (Stratagene) from SDS-PAGE gels by using a Bio-Rad electroblotting system. Electroblotting was carried out with 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) buffer, pH 11.0, containing methanol (10% [vol/vol]) for 1 h at 50 V. DNA sequences were analyzed by using the Genetics Computer Group (University of Wisconsin, Madison) and Mac Vector (International Biotechnologies, Inc., New Haven, Conn.) computer software programs.
RESULTS
Purification of P. furiosus aminoacylase.
Extracts of P. furiosus cells grown with maltose as the primary carbon source contained a significant amount of aminoacylase activity (approximately 0.34 U/mg at 100°C) using N-acetyl-l-methionine as the substrate. The enzyme appeared not to be regulated, as the specific activities of extracts of cells grown with yeast extract (5.0 g/liter) and maltose (1.0 g/liter) or with yeast extract (5.0 g/liter), tryptone (5.0 g/liter), and maltose (1.0 g/liter) as the primary carbon sources, were similar. All cells used for purification were obtained from cell cultures grown on a 500-liter scale. Since maltose-grown cells are routinely used in this laboratory to purify various oxygen-sensitive, oxidoreductase-type enzymes from P. furiosus, such cells were also used for aminoacylase purification. In addition, the procedure was carried out under anaerobic conditions, not because the aminoacylase was sensitive to oxygen but to allow the purification of both aminoacylase and enzymes that are oxygen sensitive from the same batch of P. furiosus cells.
Aminoacylase activity was not detected in the culture supernatant during the log phase of cell growth or in the membrane fraction of the cell extract. The activity was found only in the soluble fraction, indicating that the enzyme is a cytoplasmic protein. The results of a typical purification are summarized in Table 1. The enzyme was purified 580-fold with a yield of 7% and a specific activity of approximately 200 U/mg. When the aminoacylase was treated with SDS sample buffer at 100°C for 10 min prior to electrophoresis, it migrated as a single major band corresponding to a molecular mass of approximately 45 kDa (Fig. 1). The aminoacylase was eluted from a gel filtration column, corresponding to a molecular mass of 190 ± 10 kDa. This result, taken together with the electrophoretic data, suggests that the enzyme is a homotetramer.
TABLE 1.
Purification of aminoacylase from P. furiosus
| Step | Activitya (U) | Protein (mg) | Sp act (U/mg) | Purification (fold) | Recovery (%) |
|---|---|---|---|---|---|
| Cell extract | 8,800 | 26,000 | 0.3 | 1 | 100 |
| QFF Sepharose | 5,800 | 1,600 | 3.7 | 11 | 65 |
| Phenyl Sepharose | 4,100 | 160 | 25.4 | 75 | 46 |
| QHP Sepharose | 1,050 | 27 | 39.0 | 115 | 12 |
| Hydroxyapatite | 700 | 7 | 100 | 290 | 8 |
| HiTrap Q | 660 | 3 | 200 | 580 | 7 |
Activity was measured at 100°C by using N-acetyl-l-methionine (30 mM) as the substrate.
FIG. 1.

SDS–12% PAGE of the aminoacylase purified from P. furiosus. Lanes: 1, standard molecular size markers; 2, native aminoacylase (4 μg).
The N-terminal sequence of the purified aminoacylase is MFNPLEEAMKIKDEI-. This sequence was used to search the genomic sequence database of P. furiosus (http://comb5-156.umbi.umd.edu/). A gene was located whose translated N-terminal region matched exactly the sequence obtained from the purified enzyme. It consists of 1,149 bp and encodes a protein of 383 residues with a calculated molecular mass of 42.06 kDa (Fig. 2). The latter value is in good agreement with that (∼45 kDa) obtained by SDS-PAGE analysis. The enzyme appears to show nonideal behavior when subjected to gel filtration, however, since the molecular mass estimated by that method (190 kDa) is higher than that expected (168 kDa) for a homotetrameric protein. A mass of 42.06 kDa for the aminoacylase subunit was used in all calculations.
FIG. 2.

The 1,178-bp gene encoding the P. furiosus aminoacylase and the deduced amino acid sequence (383 amino acids). A putative TATA box is indicated in bold print, and the ribosomal binding site is underlined.
Characterization of recombinant P. furiosus aminoacylase.
The production of aminoacylase in recombinant E. coli cells was assessed by the appearance of a protein band corresponding to its subunit size (approximately 42 kDa) after SDS-PAGE analysis of cell extracts (data not shown). However, detectable levels of active aminoacylase were not produced after induction of the recombinant gene by IPTG. Analysis of the gene and translated amino acid sequence revealed that the gene contains 44 codons (17% of the total) that are rarely used in E. coli (a combination of the Arg codons, AGA and AGG; one Leu codon; CUA; and one Ile codon, AUA). This dramatically affected the expression of the gene, since when the same plasmid was expressed in conjunction with the genes encoding the relevant tRNAs (present on plasmid pRIL), the amount of recombinant aminoacylase produced was easily visible after SDS-PAGE analysis. However, aminoacylase activity (at 100°C) could not be detected in the cell extract, and the recombinant protein appeared to be insoluble, as it was removed by centrifugation. Attempts to produce a soluble recombinant protein by varying the induction temperature (4 to 37°C) or growth medium composition (LB medium or Casamino Acid-supplemented M9-glucose medium with or without ZnCl2, NiCl2, or FeSO4) proved unsuccessful, as no soluble recombinant aminoacylase was detectable by SDS-PAGE analysis of the cytoplasmic fraction. Furthermore, the aminoacylase present in the insoluble fraction could not be solubilized into a form with detectable activity by using a variety of denaturation-renaturation strategies. Similarly, while a soluble form of recombinant aminoacylase was produced as part of an enterokinase-cleavable thioredoxin fusion protein, no activity could be detected either before or after cleavage of the thioredoxin fusion.
Physical properties of P. furiosus aminoacylase.
Of the 31 metals analyzed, the aminoacylase purified from P. furiosus cells contained only zinc and magnesium in significant amounts (>0.1 g-atoms/subunit). The values were 1.0 ± 0.48 g-atoms of Zn2+/subunit and 0.11 ± 0.03 g-atoms of Mg2+/subunit, respectively. The purified enzyme has a specific activity of 200 U/mg. When the enzyme (0.21 mg/ml in 50 mM bis-Tris buffer, pH 6.5) was treated with EDTA (20 mM) for 1 h at 23°C and then dialyzed against the same buffer (lacking EDTA), no activity could be detected by using the routine assay at 100°C. As shown in Fig. 3, activity could be restored by the addition of millimolar concentrations of Zn2+ ions and, to a lesser extent, Co2+ ions. Other divalent (Mg2+, Ni2+, Fe2+, Mn2+, or Cu2+) or monovalent (Na+ or K+) cations were ineffective. Maximal recoveries of activity for the metal-reconstituted enzyme were obtained by using 3.5 mM ZnCl2 (86% of original activity recovered) and 4.6 mM CoCl2 (74%), although both cations caused some inhibition when added at concentrations greater than their optimal concentrations (Fig. 3). When the zinc-reconstituted enzyme (using 3.5 mM ZnCl2) was passed through a Superdex 200 column equilibrated with 50 mM bis-Tris buffer, pH 6.5, or dialyzed against 50 mM bis-Tris buffer (1,000 volumes), pH 6.5, the enzyme preparations contained 3.0 ± 0.21 and 2.7 ± 0.48 g-atoms of Zn2+/subunit, respectively. From Fig. 3, the apparent association constants (concentrations giving half-maximal activities) for Zn2+ and Co2+ were 1.7 and 1.1 mM, respectively.
FIG. 3.

The effects of Co2+ and Zn2+ ions on the activity of the P. furiosus aminoacylase after EDTA treatment. The enzyme (29 μg in 50 mM bis-Tris, pH 6.5), was incubated at 80°C for 30 min with various concentrations of either ZnCl2 (solid symbols) or CoCl2 (square symbols) and then assayed under standard conditions (in the absence of added metal ions). The assay mixture contained aminoacylase (2.9 μg), N-acetyl-l-methionine (30 mM), and 50 mM bis-Tris buffer, pH 6.5. The 100% activity level corresponds to 200 U/mg.
The aminoacylase as purified from P. furiosus was not very thermostable. When a sample (0.35 mg/ml in 50 mM bis-Tris buffer containing 0.5 M NaCl, pH 6.5) was incubated at 100°C, the time required for 50% loss of activity was 25 min. Under the same conditions, but in the presence of 100 μM ZnCl2 (the concentration in the assay mixture giving the highest activity of the pure enzyme; Fig. 4), there was no detectable loss of activity after a 7 h of incubation at 100°C.
FIG. 4.

The effects of metal ions on the catalytic activity of the P. furiosus aminoacylase under standard assay conditions. The assay mixtures contained aminoacylase (0.78 μg), N-acetyl-l-methionine (30 mM), and various concentrations of either ZnCl2 (solid circles), CoCl2 (open squares), MnCl2 (open circles), or NiCl2 (solid squares). A level of 200 U/mg corresponds to 100% specific (Sp.) activity.
Catalytic properties of P. furiosus aminoacylase.
As shown in Fig. 5, the enzyme showed a sharp pH optimum at 6.5 (at 100°C) and a temperature optimum of at least 100°C (at pH 6.5). There was no detectable activity below 40°C (at pH 6.5). From the temperature-dependent data, the calculated activation energy for aminoacylase is 12.1 kcal/mol. The standard assay mixture for aminoacylase did not contain any metal ions, but activity could be increased by addition of Zn2+, Co2+, Mg2+, or Ni2+ ions, although other divalent (Ca2+, Fe2+, or Cu2+) and monovalent (Na+ or K+) cations had no effect (Fig. 4). For Co2+, Mg2+, or Ni2+ ions, concentrations above 250 μM were required, but for ZnCl2, concentrations lower than 200 μM resulted in increased activity (Fig. 4).
FIG. 5.

Effects of temperature and pH on the activities of P. furiosus aminoacylase. The assay mixture contained aminoacylase (0.64 mg/ml) and N-acetyl-l-methionine (30 mM) in 50 mM bis-Tris, pH 6.5. For the effects of pH, the following buffers (each at 50 mM) were used at the indicated pHs: MES (morpholineethanesulfonic acid), pHs 5.5 and 6.0; bis-Tris, pH 6.5; MOPS, pH 7.0; EPPS (N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid), pH 8.4; CHES [2-(N-cyclohexylamino)ethanesulfonic acid], pH 8.6; CAPS, pHs 10, 10.5, and 11.0. For effects of temperature, the buffer used was 50 mM bis-Tris, pH 6.5. A 100% activity level corresponds to 200 U/mg.
Aminoacylase was identified in cell extracts of P. furiosus by detecting its ability to hydrolyze N-acetyl-l-methionine, and this substrate was used in all routine assays. The activity of the enzyme with other N-acetylated amino acids, N-chloroacetylated amino acids, and N-formylated amino acids is summarized in Table 2. No activity was detected with the following compounds: N-acetyl-dl-phenylglycine, N-acetylglycine, N-acetyl-d-methionine, N-acetyl-l-phenylalanine, N-acetyl-l-proline, N-acetyl-l-tryptophan, N-acetyl-l-tyrosine, N-formyl-dl-tryptophan, N-formyl-methionine-phenylalanine, N-acetylmethionine-alanine and N-acetylmethionine-leucine-phenylalanine. Notably, N-acetyl-d-methionine was not hydrolyzed, showing the stereospecificity of the aminoacylase, while the potential physiological substrate N-formyl-l-methionine was hydrolyzed, although N-formyl-Met-Phe was not. Table 3 shows the results from kinetic analyses with three substrates. All exhibited normal Michaelis-Menten-type kinetics over the range of 0.5 to 10 mM substrate concentrations, and the kinetic constants were calculated from linear double-reciprocal plots. The apparent Km value for N-formyl-l-methionine was very high (13 mM) and about twice that of N-acetylmethionine. The best (nonphysiological) substrate was N-chloroacetyl-l-valine, where the kcat/Km value was almost threefold that measured with N-acetyl-l-methionine.
TABLE 2.
Substrate specificity of P. furiosus aminoacylase
| Substratea | % Relative activityb |
|---|---|
| N-Acetyl-l-methionine | 100 |
| N-Chloroacetyl-l-valine | 325 |
| N-Chloroacetyl-l-leucine | 287 |
| N-Chloroacetylglycine | 233 |
| N-Formyl-l-methionine | 155 |
| N-Acetyl-l-valine | 77 |
| N-Chloroacetyl-l-tyrosine | 75 |
| N-Acetyl-dl-allylglycine | 47 |
| N-Acetyl-l-alanine | 36 |
| N-Acetyl-l-asparagine | 33 |
| N-Acetyl-l-cysteine | 15 |
| N-Acetyl-dl-serine | 15 |
| N-Acetyl-l-glutamic acid | 13 |
| N-Chloroacetyl-l-phenylalanine | 13 |
| N-Acetyl-l-leucine | 9 |
| N-Acetyl-dl-norleucine | 6 |
| N-Chloroacetyl-l-tryptophan | 5 |
| N-Formyl-l-leucine | 4 |
All substrates were used at a final concentration of 30 mM.
The rate of hydrolysis is expressed as a percentage of the activity compared to that obtained by using N-acetyl-l-methionine as the substrate at 100°C, where 100% activity corresponds to 200 U/mg.
TABLE 3.
Kinetic parameters for substrates of P. furiosus aminoacylase
| Substratea | Km (mM) | Vmax (μmol/ min/mg) | kcatb (s−1) | kcat/km (mM−1s−1) |
|---|---|---|---|---|
| N-Acetyl-l-methionine | 6.6 | 500 | 2,720 | 412 |
| N-Formyl-l-methionine | 13.0 | 99 | 538 | 41 |
| N-Chloroacetyl-l-valine | 2.5 | 500 | 2,720 | 1,090 |
All assays were carried out at 100°C in 50 mM Bis-Tris HCl, pH 6.5.
Based on a minimum molecular mass of 42.06 kDa.
DISCUSSION
Although the gene encoding P. furiosus aminoacylase was successfully expressed in E. coli, surprisingly, the recombinant form was not catalytically active, and the enzyme would not have been characterized by a cloning-expression approach. The production of what appears to be incorrectly folded, recombinant apoprotein may be due to the inability of E. coli to insert the appropriate metal ion, as the enzyme, when purified from P. furiosus, contains 1 g-atom of Zn/subunit. In this regard, the P. furiosus enzyme is similar to other members of the aminoacylase family, many of which have also been shown to contain Zn. For example, the Bacillus stearothermophilus (56), B. thermoglucosidius (14), pig (23), and human (17) enzymes contain one zinc ion per catalytic subunit, while the enzymes from Alcaligenes denitrificans DA181 (61) and Aspergillus oryzae (21) contain two and three zinc ions per subunit, respectively. Like the P. furiosus enzyme, some of these aminoacylases require the addition of a divalent cation (typically Zn, although Co, Mn, and Mg are also effective in some cases) for maximal catalytic activity in vitro (14, 21, 61), but this is not true for all of them (23, 56). However, in all cases, incubation in the presence of EDTA results in complete loss of activity, and this can be restored by the addition of Zn2+ ions. The precise nature of the metal sites in these enzymes is not clear. Both the reconstituted P. furiosus enzyme and the aminoacylase from A. oryzae (21) contain three Zn2+ ions per subunit, suggesting that this group of enzymes represents a novel class of zinc-containing protein that is distinct from members of the metallohydrolase family that contain binuclear metal sites (58).
All of the aminoacylases that have been characterized have subunits of comparable size (37 to 50 kDa), but the majority are dimers (21, 40, 52, 56, 61) and only one (14) is a homotetramer, like the P. furiosus enzyme. The crystal structure of an aminoacylase has not yet been reported (46), although complete amino acid sequences are available for the B. stearothermophilus (44), pig (37), and human (38) enzymes and sequences of putative aminoacylases can be identified in the genome sequences of P. horikoshii (29), P. abyssi (http://www.genoscope.cns.fr/Pab), B. subtilis (31), Deinococcus radiodurans (57), Lactococcus lactis (18), Synechocystis sp. (2, 8), and Streptomyces coelicolor (42). The P. furiosus enzyme has 34, 15, 15, 80, 81, 26, 38, 32, 41, and 14% identity, respectively, with these enzymes but shows no sequence similarity with that of the AARE found in P. horikoshii (26). It does show high similarity, however, to the sequences of Sulfolobus solfataricus carboxypeptidase (16), Thermotoga maritima hippurate hydrolase (39), Campylobacter jejuni hippuricase (24), Arabidopsis thaliana (ILR1) indole-3-acetic acid amino acid hydrolase (4), and Arabidopsis thaliana JR3 protein (identities of 38, 28, 35, 38, and 40%, respectively). The sequences of several of these enzymes are aligned with that of the P. furiosus enzyme in Fig. 6. Most of these enzymes contain a conserved region near the N terminus (MHACGHDXHTAMLLG-, residues 136 to 153 in the P. furiosus sequence) that has putative zinc-binding residues (one Cys, one Asp, and three His residues). There are also other potential zinc-binding residues (51) (His-51, Glu-60, Glu-69, Glu-177, Glu-178, His-205, and His-239 in the P. furiosus enzyme) that are conserved in all of these enzymes (Fig. 6). It therefore appears that these enzymes are part of a metallohydrolase family (58) that might contain three Zn atoms per mole, although the true nature of the metal sites will likely be apparent only from crystallographic analyses.
FIG. 6.

Alignment of the amino acid sequence of the P. furiosus aminoacylase with those of the aminoacylases from various sources and with the carboxypeptidases (CP) from Sulfolobus solfataricus and P. horikoshii and the hippurate hydrolase (HH) from Thermotoga maritima. The sources of the data are as follows: P. furiosus aminoacylase, this work; P. horikoshii aminoacylase, accession no. BAA29813; P. horikoshii carboxypeptidase, accession no. P54955; P. abysii aminoacylase, accession no. CAB50230; T. maritima hippurate hydrolase, accession no. AAD36583; S. solfataricus carboxypeptidase, accession no. CAA88397; B. stearothermophilus aminoacylase, accession no. P37112; D. radiodurans aminoacylase, accession no. AAF11266; Streptomyces coelicolor aminoacylase, accession no. T35974; A. thaliana ILR1, accession no. P54968; Syncechocystis sp. accession no., BAA18770; pig aminoacylase, accession no. JN0584. Identical residues are outlined, while similar residues are designated by gray shading.
We now turn to the possible physiological role of P. furiosus aminoacylase. Unfortunately, as yet, none of the aminoacylases that have been characterized have well-defined functions. The P. furiosus enzyme hydrolyzes N-formylmethionine, as well as a broad range of N-acetylated amino acids and is absolutely specific for the naturally occurring l isomers. The relatively high Km values for these substrates, however, suggest that such amino acid derivatives must be present at significant intracellular concentrations in vivo if they are the physiological substrates. Such compounds could be generated either by cellular protein degradation or by the metabolism of protein growth substrates. The latter is a possible source, as P. furiosus grows well with peptides as the carbon source (20). Similarly, other organisms from which aminoacylases have been purified, such as B. stearothermophilus (44), A. denitrificans (61), and Pseudomonas maltophilia (52), also are capable of growing on peptides. In fact, of the completed microbial genome sequences available, homologs of the P. furiosus enzyme (showing ≥50% sequence similarity) are present in 21 organisms (including archaea and bacteria), and 19 of them are capable of peptidolytic growth. They include P. horikoshii, P. abysii, B. stearothermophilus, B. subtilis, Clostridium acetobutylicum, Pseudomonas aeruginosa, Bordetella pertussis, D. radiodurans, Staphylococcus aureus, Campylobacter jejuni, and Treponema denticola. The other two organisms are the photosynthetic bacteria Synechocystis sp. and Chlorobium tepidum, which are not known to use peptides. Homologs of aminoacylase are not found in hyperthermophilic archaea that are not capable of peptidolytic growth, including Archaeoglobus fulgidus (32), Methanobacterium thermoautotrophicum (45), and Methanococcus jannaschii (10) nor in the hyperthermophilic bacterium Aquifex aeolicus (19). On the other hand, there is no obvious aminoacylase homolog in the hyperthermophilic archaeon Aeropyrum pernix (30), an organism that does grow on peptides.
The microbial genomic data, therefore, generally support the notion that aminoacylases represented by the P. furiosus enzyme are involved in metabolizing protein growth substrates rather than the cell's own proteins. Figure 7 shows a possible pathway for the initial steps of protein degradation in P. furiosus. It is proposed that the substrates for the aminoacylase are generated by the AARE. AARE generates N-acyl amino acids from short N-acyl peptides (four or fewer residues), but it does not hydrolyze N-acetyl- or N-formylmethionine (26). The AARE of P. horikoshii has been characterized (26), and a homolog is present in P. furiosus (http://comb5-156.umbi.umd.edu/). P. furiosus also contains methionine aminopeptidase (48), but this enzyme is not known to use N-acetylated proteins or peptides. The small N-acyl peptides used by AARE are presumably produced from larger N-acylated proteins by an acyl aminopeptidase. The nature of this enzyme is not clear, since P. furiosus contains an enzyme of this type, but it was reported to be extracellular (49). It is also not known whether N-acylated proteins themselves serve as growth substrates for P. furiosus or if proteins and/or peptides are acylated intracellularly, perhaps as a signal for subsequent digestion (Fig. 7). N-Formylmethionine also serves as a substrate for P. furiosus aminoacylase (Table 3), although it remains to be seen if N-formylated rather than N-acetylated derivatives play a role in protein degradation. Growth studies with such substrates are currently under way.
FIG. 7.

Proposed pathway of catabolism of N-acetylated proteins in P. furiosus. See the text for details. CoA, coenzyme A.
While the present paper was under review, an article was published describing the cloning and expression of a gene encoding a bifunctional carboxypeptidase-aminoacylae from P. horikoshii, a close relative of P. furiosus (27). As was mentioned above, this organism contains a gene that encodes a protein (a putative amidohydrolase; 29) with extremely high similarity (80% identity) to P. furiosus aminoacylase (Fig. 6), but this gene has not been expressed. The carboxypeptidase that was characterized from P. horikoshii (27) has a much lower sequence identity (57%) to the P. furiosus enzyme, although, like the carboxypeptidase from S. solfataricus (16) noted above, it is clearly a member of the aminoacylase family of enzymes (Fig. 6). From its substrate specificity (27), the P. horikoshii enzyme appears to have dipeptidase rather than carboxypeptidase activity, and it also hydrolyzes acetylated aromatic amino acids, unlike the P. furiosus enzyme described herein. The genome of P. furiosus does not contain any other gene analogous to that encoding the bifunctional carboxypeptidase of P. horikoshii. P. furiosus does contain a conventional carboxypeptidase (12), as does P. horikoshii (29), but these enzymes show no significant sequence similarity to the sequences of the aminoacylase family (Fig. 6). Clearly, much remains to be understood about the diversity and physiological roles of this group of enzymes.
ACKNOWLEDGMENT
This work was supported by a grant from the National Science Foundation (BES-0004257).
REFERENCES
- 1.Adams M W W, Kletzin A. Oxidoreductase-type enzymes and redox proteins involved in the fermentative metabolisms of hyperthermophilic archaea. Advs Protein Chem. 1996;48:101–180. doi: 10.1016/s0065-3233(08)60362-9. [DOI] [PubMed] [Google Scholar]
- 2.Anders M W, Dekant W. Aminoacylases. Adv Pharmacol. 1994;27:431–448. doi: 10.1016/s1054-3589(08)61042-x. [DOI] [PubMed] [Google Scholar]
- 3.Andreotti G, Cubellis M V, Nitti G, Sannia G, Mai X, Adams M W W, Marino G. An extremely thermostable aromatic aminotransferase from the hyperthermophilic archaeon Pyrococcus furiosus. Biochim Biophys Acta. 1995;1247:90–96. doi: 10.1016/0167-4838(94)00211-x. [DOI] [PubMed] [Google Scholar]
- 4.Bartel B, Fink G R. ILR1, an amidohydrolase that releases active indole-3-acetic acid from conjugates. Science. 1995;268:1745–1748. doi: 10.1126/science.7792599. [DOI] [PubMed] [Google Scholar]
- 5.Bodalo A, Bastida J, Gomez J L, Gomez E, Alcaraz I, Asanza M L. Stabilization of L-aminoacylase-producing Pseudomonassp. BA2 immobilized in calcium alginate gel. Enzyme Microb Technol. 1997;21:64–69. [Google Scholar]
- 6.Bodalo A, Rodriguez J, Gomez J L, Gomez C, Teruel M L, Alcaraz Rojo R R. Preliminary studies on the growth of Pseudomonassp. BA2 and the production of L-aminoacylase. Biotechnol Lett. 1996;17:859–862. [Google Scholar]
- 7.Boross L, Kosary J, Stefanovits-Banyai E, Sisak C, Szajani B. Studies on kinetic parameters and stability of aminoacylase in non-conventional media. J Biotechnol. 1998;66:69–73. doi: 10.1016/s0168-1656(98)00158-8. [DOI] [PubMed] [Google Scholar]
- 8.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 9.Bryant F O, Adams M W W. Characterization of hydrogenase from the hyperthermophilic archaebacterium, Pyrococcus furiosus. J Biol Chem. 1989;264:5070–5079. [PubMed] [Google Scholar]
- 10.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, Sutton G G, Blake J A, FitzGerald L M, Clayton R A, Gocayne J D, Kerlavage A R, Dougherty B A, Tomb J F, Adams M D, Reich C I, Overbeek R, Kirkness E F, Weinstock K G, Merrick J M, Glodek A, Scott J L, Geoghagen N S M, Venter J C. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 11.Chen R, Xu W, Zhou H M. The essential tryptophan residues of pig kidney amino acylase. Biochem Mol Biol Int. 1997;43:1277–1283. doi: 10.1080/15216549700205101. [DOI] [PubMed] [Google Scholar]
- 12.Cheng T C, Ramakrishnan V, Chan S I. Purification and characterization of a cobalt-activated carboxypeptidase from the hyperthermophilic archaeon Pyrococcus furiosus. Protein Sci. 1999;8:2474–2486. doi: 10.1110/ps.8.11.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cherry J L, Young H H, Disera L J, Ferguson F M, Kimball A W, Dunn D M, Gesteland R F, Weiss R B. Enzyme-linked fluorescent detection for automated multiplex DNA-sequencing. Genomics. 1994;20:68–74. doi: 10.1006/geno.1994.1128. [DOI] [PubMed] [Google Scholar]
- 14.Cho H Y, Tanizawa K, Tanaka H, Soda K. Thermostable aminoacylase from Bacillus thermoglucosidius: purification and characterization. Agric Biol Chem. 1987;51:2793–2800. [Google Scholar]
- 15.Cho H Y, Tanizawa K, Tanaka H, Soda K. Thermostable dipeptidase from Bacillus stearothermophilus: its purification, characterization, and comparison with aminoacylase. J Biochem. 1988;103:622–628. doi: 10.1093/oxfordjournals.jbchem.a122317. [DOI] [PubMed] [Google Scholar]
- 16.Colombo S, Toietta G, Zecca L, Vanoni M, Tortora P. Molecular cloning, nucleotide sequence, and expression of a carboxypeptidase-encoding gene from the archaebacterium Sulfolobus solfataricus. J Bacteriol. 1995;177:5561–5566. doi: 10.1128/jb.177.19.5561-5566.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cook R M, Burke B J, Buckhagen D L, Minna J D, Miller Y E. Human aminoacylase I: cloning, sequencing, and expression analysis of a chromosome 3p21 gene inactivated in small cell lung cancer. J Biol Chem. 1993;268:17010–17017. [PubMed] [Google Scholar]
- 18.Curley P, van Sinderen D. Identification and characterization of a gene encoding aminoacylase activity from Lactococcus lactisMG1363. FEMS Microbiol Lett. 2000;183:177–182. doi: 10.1111/j.1574-6968.2000.tb08954.x. [DOI] [PubMed] [Google Scholar]
- 19.Deckert G, Warren P V, Gaasterland T, Young W G, Lenox A L, Graham D E, Overbeek R, Snead M A, Keller M, Aujay M, Huber R, Feldman R A, Short J M, Olsen G J, Swanson R V. The complete genome sequence of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 20.Fiala G, Stetter K O. Pyrococcus furiosussp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100°C. Arch Microbiol. 1986;145:56–61. [Google Scholar]
- 21.Gentzen I, Loffler H G, Schneider F. Aminoacylase from Aspergillus oryzae. Comparison with the pig kidney enzyme. Z Naturforsch. 1980;35c:544–550. doi: 10.1515/znc-1980-7-804. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh M, Grunden A M, Dunn D M, Weiss R, Adams M W W. Characterization of native and recombinant forms of an unusual cobalt-dependent proline dipeptidase (prolidase) from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol. 1998;180:4781–4789. doi: 10.1128/jb.180.18.4781-4789.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giardina T, Biagini A, Dalle Ore F, Ferre E, Reynier M, Puigserver A. The hog intestinal mucosa acylase I: subcellular localization, isolation, kinetic studies and biological function. Biochimie. 1997;79:265–273. doi: 10.1016/s0300-9084(97)83514-6. [DOI] [PubMed] [Google Scholar]
- 24.Hani E K, Chan V L. Expression and characterization of Campylobacter jejuni benzoylglycine amidohydrolase (hippuricase) gene in Escherichia coli. J Bacteriol. 1995;177:2396–2402. doi: 10.1128/jb.177.9.2396-2402.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heider J, Mai X, Adams M W W. Characterization of 2-ketoisovalerate ferredoxin oxidoreductase, a new and reversible coenzyme A-dependent enzyme involved in peptide fermentation by hyperthermophilic archaea. J Bacteriol. 1996;178:780–787. doi: 10.1128/jb.178.3.780-787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishikawa K, Ishida H, Koyama Y, Kawarabayasi Y, Kawahara J, Matsui E, Matsui I. Acylamino-acid releasing enzyme from the thermophilic archaeon Pyrococcus horikoshii. J Biol Chem. 1998;273:17726–17731. doi: 10.1074/jbc.273.28.17726. [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa K, Ishida H, Matsui I, Kawarabayasi Y, Kikuchi H. Novel bifunctional hyperthermostable carboxypeptidase/aminoacylase from Pyrococcus horikoshiiOT3. Appl Environ Microbiol. 2001;67:673–679. doi: 10.1128/AEM.67.2.673-679.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, Kimura T, Hosouchi T, Matsuno A, Muraki A, Nakazaki N, Naruo K, Okumura S, Shimpo S, Takeuchi C, Wada T, Watanabe A, Yamada M, Yasuda M, Tabata S. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystissp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- 29.Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, Nagai Y, Sakai M, Ogura K, Otsuka R, Nakazawa H, Takamiya M, Ohfuku Y, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Yoshizawa T, Nakamura Y, Robb F, Horikoshi K, Masuchi Y, Shizuya H, Kikuchi H. Complete sequence and gene organization of the genome of a hyperthermophilic archaebacterium, Pyrococcus horikoshiiOT3. DNA Res. 1998;5:147–155. doi: 10.1093/dnares/5.2.147. [DOI] [PubMed] [Google Scholar]
- 30.Kawarabayasi Y, Hino Y, Horikawa H, Yamazaki J, Haikawa Y, Jin-no K, Takahashi M, Sekine M, Baba S, Ankai A, Kosugi H, Hosoyama A, Fukui S, Nagai Y, Nishijima K, Nakazawa H, Takamiya M, Masuda S, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Kikuchi H, et al. Complete genome sequence of an aerobic hyperthermophilic crenarchaeon, Aeropyrum pernixK1. DNA Res. 1999;6:145–152. doi: 10.1093/dnares/6.2.83. [DOI] [PubMed] [Google Scholar]
- 31.Kempf B, Bremer E. A novel amidohydrolase gene from Bacillus subtiliscloning: DNA-sequence analysis and map position of amhx. FEMS Microbiol Lett. 1996;141:129–137. doi: 10.1111/j.1574-6968.1996.tb08374.x. [DOI] [PubMed] [Google Scholar]
- 32.Klenk H P, Clayton R A, Tomb J F, White O, Nelson K E, Ketchum K A, Dodson R J, Gwinn M, Hickey E K, Peterson J D, Richardson D L, Kerlavage A R, Graham D E, Kyrpides N C, Fleischmann R D, Quackenbush J, Lee N H, Sutton G G, Gill S, Kirkness E F, Dougherty B A, McKenney K, Adams M D, Loftus B, Venter J C, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Linder H, Hopfner S, Tafler-Naumann M, Miko M, Konrad L, Rohm K H. The distribution of aminoacylase I among mammalian species and localization of the enzyme in porcine kidney. Biochimie. 2000;82:129–137. doi: 10.1016/s0300-9084(00)00191-7. [DOI] [PubMed] [Google Scholar]
- 35.Lorentz K, Flatter B. Clinical application of a new method for the determination of aminoacylase in human serum. Clin Chim Acta. 1975;63:271–274. doi: 10.1016/0009-8981(75)90047-9. [DOI] [PubMed] [Google Scholar]
- 36.Mai X, Adams M W W. Purification and characterization of two reversible and ADP-dependent acetyl coenzyme A synthetases from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol. 1996;178:5897–5903. doi: 10.1128/jb.178.20.5897-5903.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitta M, Ohnogi H, Yanamoto A, Kato I, Sakiyama F, Tsunasawa S. The primary structure of porcine aminoacylase I deduced from cDNA sequence. J Biochem. 1992;112:737–742. doi: 10.1093/oxfordjournals.jbchem.a123968. [DOI] [PubMed] [Google Scholar]
- 38.Mitta M, Kato I, Tsunasawa S. The nucleotide sequence of human aminoacylase I. Biochim Biophys Acta. 1993;1174:201–203. doi: 10.1016/0167-4781(93)90116-u. [DOI] [PubMed] [Google Scholar]
- 39.Nelson K E, Clayton R A, Gill S R, Gwinn M L, Dodson R J, Haft D H, Hickey E K, Peterson J D, Nelson W C, Ketchum K A, McDonald L, Utterback T R, Malek J A, Linher K D, Garrett M M, Stewart A M, Cotton M D, Pratt M S, Phillips C A, Richardson D, Heidelberg J, Sutton G G, Fleischmann R D, Eisen J A, Fraser C M, et al. Evidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 40.Palm G J, Klaus-Heinrich Rohm Aminoacylase I from porcine kidney: identification and characterization of two major protein domains. Protein Chem. 1995;14:233–240. doi: 10.1007/BF01886764. [DOI] [PubMed] [Google Scholar]
- 41.Pittelkow S, Linder H, Rohm K H. Human and porccine aminoacylase I overproduced in a baculovirus expression vector system: evidence for structural and functional identity with enzymes isolated from kidney. Protein Expr Purif. 1998;12:269–276. doi: 10.1006/prep.1997.0816. [DOI] [PubMed] [Google Scholar]
- 42.Redenbach M, Kieser H M, Denapaite D, Eichner A, Cullum J, Kinashi H, Hopwood D A. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolorA3(2) chromosome. Mol Microbiol. 1996;21:77–96. doi: 10.1046/j.1365-2958.1996.6191336.x. [DOI] [PubMed] [Google Scholar]
- 43.Rosen H. A modified ninhydrin colorimetric analysis for amino acids. Arch Biochem Biophys. 1957;67:10–15. doi: 10.1016/0003-9861(57)90241-2. [DOI] [PubMed] [Google Scholar]
- 44.Sakanyan V, Desmarez L, Legrain C, Charlier D, Mett I, Kochikyan A, Savchenko A, Boyen A, Falmagne P, Pierard A, Glandsdorff N. Gene cloning, sequence analysis, purification, and characterization of a thermostable aminoacylase from Bacillus stearothermophilus. Appl Environ Microbiol. 1993;59:3878–3888. doi: 10.1128/aem.59.11.3878-3888.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith D R, Doucette-Stamm L A, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, Harrison D, Hoang L, Keagle P, Gibson R, Jiwani N, Caruso A, Bush D, Reeve J N, et al. Complete genome sequence of Methanobacterium thermoautotrophicumΔH: functional analysis and comparative genomics. J Bacteriol. 1997;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Y, Yu J Y, Zhou Q, He R, Wang Z F, Zhou H M. Secondary structure of holo- and apo-aminoacylase from prediction, circular dichroism, and FT-Raman spectroscopy. J Biochem. 1995;18:706–709. doi: 10.1093/oxfordjournals.jbchem.a124969. [DOI] [PubMed] [Google Scholar]
- 47.Tewari Y B. Thermodynamics of industrially-important, enzyme-catalyzed reactions. Appl Biochem Biotechnol. 1990;23:187–203. doi: 10.1007/BF02942054. [DOI] [PubMed] [Google Scholar]
- 48.Tsunasawa S, Izu Y, Miyagi M, Kato I. Methionine aminopeptidase from the hyperthermophilic archaeon Pyrococcus furiosus: molecular cloning and overexpression in Escherichia coliof the gene, and characteristics of the enzyme. J Biochem. 1997;122:843–850. doi: 10.1093/oxfordjournals.jbchem.a021831. [DOI] [PubMed] [Google Scholar]
- 49.Tsunasawa S. Purification and application of a novel N-terminal deblocking aminopeptidase (DAP) from Pyrococcus furiosus. J Protein Chem. 1998;17:521–522. [PubMed] [Google Scholar]
- 50.Uttamsingh V, Keller D M, Anders M W. Acylase I-catalyzed deacetylation of N-acetyl-L-cysteine and S-alkyl-N-acetyl-L-cysteine. Chem Res Toxicol. 1998;11:800–809. doi: 10.1021/tx980018b. [DOI] [PubMed] [Google Scholar]
- 51.Vallee B L, Auld D S. Zinc metallochemistry in biochemistry. In: Jolies P, Jornvall H, editors. Interface between chemistry & biochemistry. Basel, Switzerland: Birkhauser Verlag; 1995. pp. 259–277. [DOI] [PubMed] [Google Scholar]
- 52.Wakayama M, Shiiba E, Sakai K, Moriguchi M. Purification and characterization of L-aminoacylase from Pseudomonas maltophilaB1. J Ferment Bioeng. 1998;85:278–282. [Google Scholar]
- 53.Wakayama M, Hayashi S, Yatsuda Y, Katsuno Y, Sakai K, Moriguchi M. Overproduction of D-aminoacylase from Alcaligenes xylosoxidans subsp. xylosoxidans A-6 in Escherichia coliand its purification. Protein Expr Purif. 1996;7:395–399. doi: 10.1006/prep.1996.0059. [DOI] [PubMed] [Google Scholar]
- 54.Wakayama M, Ashka T, Miyamoto Y, Yoshikawa T, Sonoda Y, Sakai K, Moriguchi M. Primary structure of N-acyl-D-glutamate amidohydrolase from Alcaligenes xylosoxidanssubsp. xylosoxidans A-6. J Biochem. 1995;118:204–209. doi: 10.1093/oxfordjournals.jbchem.a124879. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z X, Wang H R, Zhou H M. Kinetics of inactivation of aminoacylase by 2-chloromercuric-4-nitrophenol: a new type of complexing inhibitor. J Biochem. 1995;34:6863–6868. doi: 10.1021/bi00020a033. [DOI] [PubMed] [Google Scholar]
- 56.Weiss H M, Palm J G, Rohm K H. Thermostable aminoacylase from Bacillus stearothermophilus: significance of the metal center for catalysis and protein stability. Biol Chem. 1995;376:643–649. doi: 10.1515/bchm3.1995.376.11.643. [DOI] [PubMed] [Google Scholar]
- 57.White O, Eisen J A, Heidelberg J F, Hickey E K, Peterson J D, Dodson R J, Haft D H, Gwinn M L, Nelson W C, Richardson D L, Moffat K S, Qin H, Jiang L, Pamphile W, Crosby M, Shen M, Vamathevan J J, Lam P, McDonald L, Utterback T, Zalewski C, Makarova K S, Aravind L, Daly M J, Minton K W, Fleischmann R D, Ketchum K A, Nelson K E, Salzberg S, Smith H O, Venter J C, Fraser C M. Genome sequence of the radioresistant bacterium Deinococcus radioduransR1. Science. 1999;286:1571–1577. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilcox D E. Binuclear metallohydrolases. Chem Rev. 1996;96:2435–2458. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- 59.Wu H B, Tsou C L. A comparison of Zn(II) and Co(II) in the kinetics of inactivation of aminoacylase by 1,10-phenanthroline and reconstitution of the apoenzyme. Biochem J. 1993;296:435–441. doi: 10.1042/bj2960435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang X, Wang H R, Zhou H M. Kinetics of inhibition of aminoacylase activity by dithiothreitol or 2-mercaptoethanol. Int J Pept Protein Res. 1996;48:532–538. doi: 10.1111/j.1399-3011.1996.tb00872.x. [DOI] [PubMed] [Google Scholar]
- 61.Yang Y B, Hu H L, Chang M C, Li H, Tsai Y C. Purification and characterization of L-aminoacylase from Alcaligenes denitrificansDA181. Biosci Biotechnol Biochem. 1994;58:204–205. doi: 10.1271/bbb.58.204. [DOI] [PubMed] [Google Scholar]