

Abstract
Knee osteoarthritis (KOA) is one of the most common degenerative diseases, and its core feature is the degeneration and damage of articular cartilage. The cartilage degeneration of KOA is due to the destruction of dynamic balance caused by the activation of chondrocytes by various factors, with oxidative stress playing an important role in the pathogenesis of KOA. The overproduction of reactive oxygen species (ROS) is a result of oxidative stress, which is caused by a redox process that goes awry in the inherent antioxidant defence system of the human body. Superoxide dismutase (SOD) inside and outside chondrocytes plays a key role in regulating ROS in cartilage. Additionally, synovitis is a key factor in the development of KOA. In an inflammatory environment, hypoxia in synovial cells leads to mitochondrial damage, which leads to an increase in ROS levels, which further aggravates synovitis. In addition, oxidative stress significantly accelerates the telomere shortening and ageing of chondrocytes, while ageing promotes the development of KOA, damages the regulation of redox of mitochondria in cartilage, and stimulates ROS production to further aggravate KOA. At present, there are many drugs to regulate the level of ROS, but these drugs still need to be developed and verified in animal models of KOA. We discuss mainly how oxidative stress plays a part in the development of KOA. Although the current research has achieved some results, more research is needed.
Keywords: knee osteoarthritis, oxidative stress, reactive oxygen species, cartilage, synovitis
Introduction
Osteoarthritis is the most common joint degenerative disease among adults in the world, affecting approximately 78 million people worldwide by 2040 (Hootman et al., 2016; Sharma, 2021). Due to the increase in stress in the weight-bearing part of the joint, long-term strain will lead to cartilage exfoliation, hyperosteogeny, synovial hyperplasia, degeneration, etc. This series of changes is called joint degenerative disease (Abbasi, 2017). Most of a person’s weight is distributed across their knees, making the knee a very important joint, so knee osteoarthritis (KOA) is also one of the most common degenerative diseases (Michael et al., 2010). At present, there is no effective treatment for KOA, but various methods are used to delay the progression of KOA. When KOA develops to the end stage, total knee arthroplasty is generally used to improve the living conditions of patients (Quinn et al., 2018). However, total knee arthroplasty is expensive, so with the development of the ageing world, the prevention and treatment of KOA have increased the burden on society and patients.
Redox biological reactions have the two characteristics of promoting physiological signal responses as well as promoting pathophysiological cues (Espinosa-Diez et al., 2015; Zhang et al., 2022). Oxidative stress is a state of imbalance that causes more reactive oxygen species (ROS) to be produced or reduces the body’s natural antioxidant defences (Kimball et al., 2021). ROS are a class of substances containing oxygen free radicals, which have unpaired electrons that make them unstable and highly reactive; hydrogen peroxide (H2O2), hydroxyl (OH−) radicals, superoxide (O2 −) anions, and nitric oxide (NO) are all examples of reactive oxygen species (Trachootham et al., 2008). Mechanical and chemical stress can lead to an increase in the production of oxygen free radicals, resulting in oxidative damage to tissue (Cannizzo et al., 2011). The excessive production of ROS leads to damage to macromolecules such as protein, fat and DNA (Dröge, 2002; Cutler, 2005; Espinosa-Diez et al., 2015). When free radical production exceeds cellular scavenging, lipid peroxidation, for instance, can be brought on by an overabundance of hydroxyl radicals and peroxynitrite, which destroys cell membranes and lipoproteins (Pizzino et al., 2017).
Articular cartilage deterioration and destruction is the hallmark of KOA, which affects all tissues of the knee joint (Loeser et al., 2012). Cartilage degeneration in KOA is caused by the destruction of the dynamic balance of chondrocytes caused by activation based on other different aspects, in which the matrix degrades enzyme production and exceeds the ability of chondrocytes to secrete matrix components (Bolduc et al., 2019). Elevated levels of ROS and oxidative stress in chondrocytes play a role in the development of KOA (Blanco et al., 2011). This article reviews which pathological reactions are mainly involved in the pathogenesis of KOA by oxidative stress.
Pathogenesis of knee osteoarthritis
Among the various structures that make up the knee joint, damage to hyaline articular cartilage is the main cause of osteoarthritis. One of the most obvious risk factors for KOA is ageing, in which ageing of cartilage and chondrocytes plays an important role in the pathogenesis and development of KOA (Rahmati et al., 2017). At first, the surface of the cartilage becomes worn, and with the continuation of the pathological process, deep cracks related to the shedding of cartilage fragments gradually forms, causing delamination and exposure of the calcified cartilage and bone below (Burr and Schaffler, 1997; Burr, 2004). In addition to cartilage lesions, KOA is accompanied by changes in subchondral trabecular structure and bone mass, osteophytes, bone marrow lesions, and the development of cysts (Sandell, 2012) (Figure 1). In addition, the decrease in bone tissue hardness may lead to cartilage deformation and cartilage pathology, which are associated with osteoarthritis (Stewart and Kawcak, 2018).
FIGURE 1.
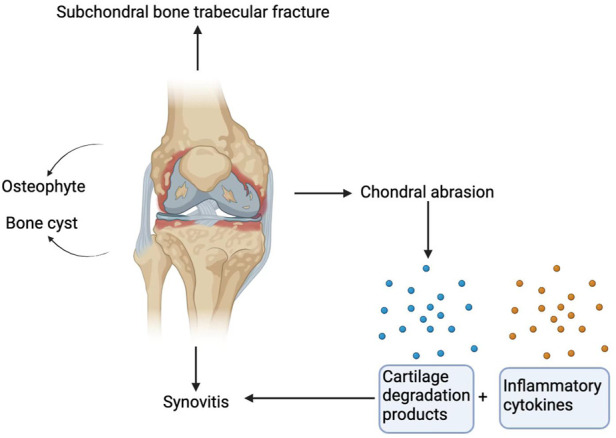
Pathological manifestations of osteoarthritis of the knee joint. Damage to hyaline articular cartilage is the main cause of osteoarthritis. In addition to cartilage lesions, KOA is accompanied by subchondral trabecular fracture, and bone cysts and osteophytes are also characteristics of KOA. Cartilage degradation products are produced after cartilage injury. These cartilage degradation products and other inflammatory factors act on the synovium to release preinflammatory products to induce synovitis.
Additionally, synovitis is crucially involved in the development of KOA. Synovitis is characterized by synovial hyperplasia and diffuse infiltration of T and B lymphocytes (Scanzello and Goldring, 2012). Magnetic resonance imaging and ultrasound imaging studies have confirmed that synovitis is positively correlated with the risk of osteoarthritis progression (Baker et al., 2010). Additionally, cartilage damage and malfunctioning chondrocytes contribute significantly to the onset of synovitis. Chondrocytes release matrix metalloproteinases to degrade the cartilage matrix and release cartilage degradation products, which, together with other proinflammatory cell derivatives, act on preinflammatory products of synovium. These preinflammatory products are fed back to chondrocytes to further affect the regulation of their function (Glyn-Jones et al., 2015) (Figure 1).
Overview of oxidative stress
O2 − is the most abundant oxygen free radical under physiological conditions, and mitochondria are thought to be the primary source. O2 − is a potent reactive oxygen species that has a strong effect on the redox state of cells. Not only is O2 − crucial for a normal immune response, but its direct oxidation of proteins also has far-reaching effects on signal transduction, gene expression, and the cell cycle (Turrens, 2003). To convert O2 − to H2O2, superoxide dismutase (SOD) is needed. O2 −, produced by the mitochondrial electron transport chain or by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOx) activity. By converting O2 − to OH- in the presence of Fenton reactive metals, SOD can also generate a more reactive and destructive ROS, hydroxyl radical (OH−). (Dickinson and Chang, 2011). NO is produced by three different NO synthases (NOSs): inducible NOS (iNOS), endothelial NOS (eNOS) and neuronal NOS (nNOS). NO derivatives can cause macromolecular cell damage and nitrosation stress; for example, O2 − combines with NO to produce ONOO-, which leads to the synthesis of nitrotyrosine, another posttranslational alteration signalling oxidative stress and injury (Oh et al., 2017). One of the illnesses of ageing is osteoarthritis, and nitrotyrosine is found in joints where there is wear and tear (Carlo and Loeser, 2003). Superoxide (O2 −) and hydrogen peroxide (H2O2) are the most typical ROS types in chondrocytes with ageing and osteoarthritis. Peroxynitrite (ONOO-), an NO product, is also present in cartilage and helps control how chondrocytes work (Bolduc et al., 2019). As ROS production rises, the defence system of the cell against oxidative stress is triggered, leading to efficient clearance of ROS molecules. Multiple enzymes, such as mitochondrial catalase (MCAT), peroxidases (Prxs), glutathione peroxidase (GPx), SOD and nonenzymes, such as glutathione and ascorbic acid (vitamin C) (Pisoschi and Pop, 2015), work together to form the antioxidant defence mechanism of the cell. Glutathione is a small molecular mercaptan that plays a key role in oxidative metabolism (Figure 2). Sufficient levels of glutathione must be maintained to exert protective and biosynthetic functions (Wang et al., 2021). Glutathione is essential for maintaining proper cellular redox potential and protecting against oxidative damage. It is important to note that glutathione can be either reduced (GSH) or oxidized (GSSG). Maintaining redox homeostasis and delivering antioxidant stress protection are dependent on the total glutathione concentration and the GSH/GSSG ratio in cells (Diaz-Vivancos et al., 2015). Prxs receive new oxygen at the mercaptan active site to protect proteins from oxidation by hydrogen peroxide (Rhee et al., 2012). By oxidizing GSH, GPx protects membrane lipids from H2O2-induced oxidation (Lubos et al., 2011).
FIGURE 2.

Generation and regulation of ROS. Superoxide (O2 −) is produced by incomplete reduction of molecular oxygen in the mitochondrial electron transport chain (ETC) or through NADPH oxidase (NOX) activity. In general, SOD disproportionates O2 − to form H2O2, and peroxidase (Prxs) further reduces H2O2 to water, catalase (CAT) or glutathione peroxidase (GPx). SOD can also be converted into OH−, and OH− is a more reactive and destructive ROS. NO is produced by three different NO synthases (NOSs). O2 − reacts with NO to produce ONOO−, which leads to the formation of nitrotyrosine. Reduced glutathione (GSH) and ascorbic acid (vitamin C) can reduce ROS levels.
The role of oxidative stress in articular cartilage degeneration
Biochemical analysis of degenerative cartilage from patients with OA showed that there was a pathological relationship between the downregulation of SOD2 and cartilage degeneration in the progression of OA, suggesting that the redox balance centred on SOD2 in mitochondria plays a central role in the pathogenesis of KOA (Ruiz-Romero et al., 2009; Scott et al., 2010). We all know that increased weight loading is one of the main risk factors for KOA. Koike et al. have shown that mechanical load in vivo promotes the production of O2 − in mitochondria of chondrocytes, and the expression of SOD1 and SOD2 in mitochondria decreases, while mitochondrial dysfunction induced by superoxide in mitochondria will further lead to cartilage degeneration (Koike et al., 2015). In addition, the expression of all three SODs was shown to be high in human cartilage but dramatically reduced in advanced OA cartilage (34), which further aggravated the oxidative stress response in chondrocytes and promoted the degeneration of chondrocytes (Scott et al., 2010). The decrease in SOD2 in OA chondrocytes is related to the increase in promoter methylation (Scott et al., 2010). Extracellular SOD has also been shown to decrease in human OA cartilage, suggesting that extracellular SOD also plays a key role in regulating ROS in cartilage (Regan et al., 2005). (Figure 3) Some studies have shown that Nox is involved in macrophage phagocytosis and neutrophil bactericidal activity, so Nox has a firmly established importance in immune function (Dang et al., 2002; Brandes et al., 2014). In addition, O2 − and H2O2 produced by Noxs in many cell types are essential for normal signal transduction of growth factors and cytokines (Holmström and Finkel, 2014). For example, ROS produced by Nox2 and Nox4 is involved in chondrocyte differentiation (Kim K. S. et al., 2010). But other researchers have found Nox4 is considered to be the main active subtype of chondrocytes in OA cartilage, in which the activation of Nox4 by proinflammatory cytokines increases the production of O2 −and H2O2 (Morel et al., 2015). The formation of ROS caused by the activation of Nox4 aggravates the decomposition of cartilage (Rousset et al., 2015).
FIGURE 3.

Oxidative stress is involved in the injury and senescence of chondrocytes. Weight loading promotes the production of O2 − in mitochondria and reduces the expression of SOD, while mitochondrial dysfunction induced by ROS in mitochondria will further lead to cartilage degeneration. The production of NO by activating iNOS is initiated by signals from proinflammatory cytokines, including IL-1β, IL-17, tumour necrosis factor-α (TNF-α) and interferon-γ (INF-γ). O2 − reacts with NO to produce ONOO−. Peroxynitrite can induce mitochondrial dysfunction, resulting in chondrocyte damage. Reduced glutathione (GSH) can reduce the level of ROS.
NO in cartilage and synovium is produced by iNOS, which mediates the expression of inflammatory factors, inhibits the synthesis of collagen and proteoglycan, and induces chondrocyte apoptosis and pain (Suantawee et al., 2015). The selective inhibition of iNOS reduces the tissue level of catabolic factors, so NO plays an inflammatory role in OA (Ahmad et al., 2020). The production of NO in OA cartilage is unusually high because OA chondrocytes produce high levels of NO. NO regulates ECM homeostasis and cytokine expression, leading to oxidative damage and chondrocyte apoptosis, thus promoting the pathogenesis of OA (Scher et al., 2007). In addition, excessive NO produced by iNOS leads to cartilage injury by enhancing matrix metalloproteinase (MMP) activity and down regulating proteoglycan and collagen biosynthesis (Lepetsos and Papavassiliou, 2016). In addition, by reacting with oxidants such as superoxide anions, NO promotes cell injury and makes chondrocytes vulnerable to apoptosis induced by cytokines (Amin et al., 2000).
One study found that NO alone did not result in chondrocyte death but that NO triggered apoptosis when it reacted with O2- to produce chondrocytes (Blanco et al., 1995); therefore, it seems that NO can increase chondrocyte death through apoptosis (Del Carlo and Loeser, 2002). The flexibility of cartilage is due in large part to the presence of proteoglycans in the cartilage matrix. NO may disrupt cartilage homeostasis by inhibiting proteoglycan production (Clancy et al., 1997). The production of NO by activating iNOS is initiated by signals from proinflammatory cytokines, including interleukin (IL)-1β, IL-17, tumour necrosis factor-α and interferon-γ (Nathan and Xie, 1994). Martel-Pelletier et al. found that the expression of iNOS and the increased level of the downstream product NO in chondrocytes work together to maintain the role of inflammatory cytokines, which will further cause chondrocyte damage (Martel-Pelletier et al., 1999). O2 − reacts with NO to produce ONOO−. Peroxynitrite can induce mitochondrial dysfunction through a calcium-dependent process, which leads to chondrocyte apoptosis mediated by calpain (Whiteman et al., 2004).
Zhu et al. discovered that the redox balance and glutathione content were drastically altered depending on the presence and pattern of load-rest cycles. For example, loading without rest for 48 h under physiological cycle conditions caused significant net oxidation of glutathione in cartilage, which decreased the protection of glutathione against further oxidative stress (Zhu et al., 2020). One study found that Prx3 (mitochondrial Prx) is highly oxidized in the cartilage of elderly patients with osteoarthritis, indicating that oxidative stress is intensified in degenerative cartilage. Overproduction of ROS can be caused by mitochondrial malfunction or unchecked SOD2 expression, which in turn can irreparably damage chondrocytes and trigger cell death via apoptosis or necrosis (Lepetsos and Papavassiliou, 2016).
The role of oxidative stress in synovitis
The aetiology of KOA is heavily influenced by synovitis. Inflamed synovium produces prostaglandins, leukotrienes, ROS, cytokines, chemokines, and adipokines, all of which contribute to cartilage breakdown and further exacerbate inflammation (Scanzello and Goldring, 2012). Increased synovitis and angiogenesis are related to oxidative stress induced by hypoxia (Biniecka et al., 2010). To reduce oxidative stress and hypoxia-induced mitochondrial mutation in inflammatory arthritis, tumour necrosis factor (TNF) blocking treatment is effective (Biniecka et al., 2011b). Low synovial oxygen supply efficiency in an inflammatory environment is caused by a combination of factors, including an imbalanced network of synovial microvessels and the increased energy demand of activated infiltrating immune cells and resident inflammatory cells. This combination of factors, in turn, causes a hypoxic microenvironment and mitochondrial dysfunction (McGarry et al., 2018), promoting inflammation and oxidative damage by increasing the production of ROS (McGarry et al., 2018). During the process of oxidizing nutrients to generate adenosine triphosphate (ATP), mitochondria produce ROS (Kuksal et al., 2017). Changes in mitochondrial DNA (MtDNA) in somatic cells are caused by elevated oxidative stress because the mitochondrial genome is very susceptible to mutation (Biniecka et al., 2011a; Harty et al., 2012). The proinflammatory mitochondrial phenotype is related to mtDNA mutation and decreased oxygen partial pressure in the synovium (Du et al., 2020), suggesting that hypoxia and oxidative stress may play a significant role in causing joint inflammation. The inflammatory response of fibroblast-like synoviocytes in rheumatoid arthritis have been shown to be able to be reduced by reducing oxidative stress in peripheral blood mononuclear cells (Lee et al., 2021). Phagocytes produce large amounts of reactive oxygen species during respiratory outbursts, and T cells usually exist near phagocytes. Activated phagocytes produce H 2O 2 through NOX-2. H2O2 can oxidize mercaptan on the surface of T cells and enter into the interior of T cells. H2O2 in T cells can oxidize glutathione (GSH) and interfere with DNA synthesis (Belikov et al., 2015). Compared with traditional T cells, T reg cells have lower levels of intracellular ROS and can be protected from H 2O 2 induced death (Mougiakakos et al., 2009). In addition, T cell homeostasis requires the balance of redox reaction. Changing the level of ROS or antioxidants to disrupt this balance will lead to T cell hyperresponsiveness or hyporeactivity, which may lead to the development of various pathology (Gelderman et al., 2007). For example, increasing the level of mercaptan in T cells can lead to T cell-mediated arthritis in mice after collagen immunization (Gelderman et al., 2007).
Abnormal ROS signalling in OA synovial fibroblasts induced by cytokines, thrombin or stress is related to the increased activity of nuclear factor erythroid 2 p45-related Factor 2 (NFE2L2, also known as NRF2) (Bernard et al., 2017). Moreover, Balogh et al. found that oxidative stress in the synovium can promote glycolysis, which may help to accelerate the mechanism of inflammation (Balogh et al., 2018). In addition, Yao et al. found that magnesium ions (Mg2+) promote the synthesis of cartilage matrix mediated by hypoxia inducible factor-1α (HIF-1α) (Yao et al., 2019). However, oxidative stress can inhibit the expression of HIF-1α and magnify inflammation, which may impair the therapeutic effect of Mg2+ in OA (Mobasheri et al., 2017). The intraarticular combination of Mg2+ and vitamin C can reduce oxidative stress and synovitis in OA (Yao et al., 2021). Synovitis worsens, matrix components are destroyed, and apoptosis occurs as a result of oxidative stress-induced mitochondrial and nuclear DNA damage, lipid peroxidation, changes in cellular signal transduction and transcription, and epigenetic transcription factors (Marchev et al., 2017). Andrographis paniculata inhibits lipid peroxidation and nitrate levels to prevent neutrophils from gathering and passing through the cell membrane and reduces the levels of chemokines and inflammatory factors (Luo et al., 2020) .
The role of oxidative stress in aging of Knee osteoarthritis
Even while being older does not guarantee you will develop osteoarthritis, the changes that come with getting older lay the groundwork for OA to develop in the first place, such as cell senescence and telomere wear, which are thought to represent key mechanisms by which ageing leads to the development of age-related diseases (López-Otín et al., 2013). Mitochondrial failure is an age-related phenomenon that increases intracellular ROS and causes oxidative stress (Venkataraman et al., 2013). Because mitochondria control the ratios of NADH/NAD+, NADPH/NADP+, and GSH/GSSG, alterations in redox homeostasis have been proposed as a potential cause of ageing (Leeuwenburgh et al., 2011).
The gradual breakdown of extracellular matrix (ECM) is a hallmark of KOA (Loeser, 2017), which is caused by an imbalance of catabolic and anabolic signals in cartilage. Oxidative stress significantly accelerates the telomere shortening and ageing of chondrocytes (Brandl et al., 2011). In addition, in OA chondrocytes, the decrease in the activity of respiratory chain complexes I, II and III may affect several pathways related to cartilage degradation, including oxidative stress, biosynthesis of chondrocytes, increased inflammation and matrix catabolism induced by cytokines, calcification of cartilage matrix and increased apoptosis of cartilage cells (Blanco et al., 2011). Mutations in mtDNA or the direct impact of proinflammatory mediators such cytokines, prostaglandins, ROS, and nitric oxide may contribute to mitochondrial dysfunction in chondrocytes in KOA (Kim J. et al., 2010) (Figure 4). In addition, proteomic analysis showed that mitochondrial SOD2 in human chondrocytes decreases with age (Ruiz-Romero et al., 2006). Some studies have shown that ageing can lead to an imbalance in the anabolism and catabolism of chondrocytes and promote the senescence and apoptosis of chondrocytes (Loeser et al., 2002; Shane Anderson and Loeser, 2010). Both human and primate ageing cartilage and OA cartilage showed increased generation of hydrogen peroxide and active nitrogen (including NO) (Loeser et al., 2002). Human chondrocyte explants grown in the presence of hydrogen peroxide exhibited senescent features, including telomere shortening, decreased replication ability and decreased glycosaminoglycan production. Fu et al. found that Sirtuin 3 (SIRT3) protein is lost with ageing, which can damage the SOD2 activity of cartilage. Due to the decrease in SIRT3 expression and the impairment of SOD2-specific activity, ageing promotes the development of KOA, impairs the regulation of redox of mitochondria in cartilage and stimulates ROS production (Passos et al., 2010; Fu et al., 2016). Stress-induced chondrocyte senescence and OA may be the result of an increase in ROS generation in cartilage, which may be stimulated by damaging mechanical load (Yamazaki et al., 2003). Collins et al. found that MCAT inhibited the catabolism of chondrocytes induced by toluene diketone and inhibited the progression of age-related osteoarthritis in mouse models (Collins et al., 2016), pointing to the oxidative stress that comes with ageing being able to interfere with the regular physiological signal transduction of the body, which in turn can cause osteoarthritis (Figure 5).
FIGURE 4.
Oxidative stress is involved in the pathogenesis of synovitis. Hypoxia leads to mitochondrial dysfunction, which leads to an increase in the level of ROS and further promotes inflammation. Cytokines, thrombin or stress can cause an increase in nuclear factor erythroid 2 p45-related Factor 2 (NRF2), which leads to an increase in ROS. The combination of Mg2+ and vitamin C can reduce oxidative stress and synovitis in OA. At the cellular level, oxidative stress can cause DNA damage, lipid peroxidation and epigenetic changes in gene expression, leading to synovitis deterioration.
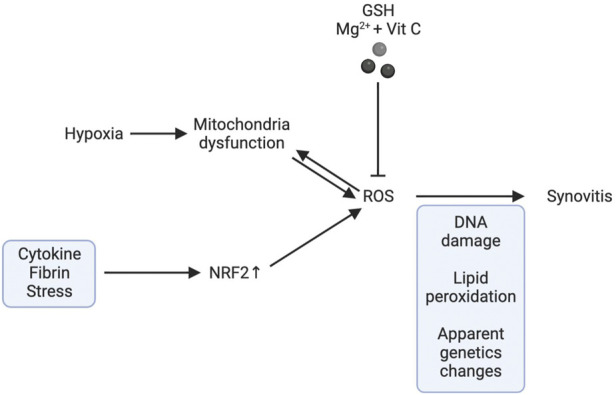
FIGURE 5.
Chondrocyte senescence and oxidative stress. Hydrogen peroxide can cause the ageing of chondrocytes, such as telomere shortening and mitochondrial dysfunction. Sirtuin 3 (SIRT3) protein is lost with ageing, which damages the SOD2 activity of cartilage, affects the regulation of mitochondrial redox in cartilage and stimulates ROS production. In addition, harmful mechanical loads can also stimulate an increase in ROS. Oxidative stress inactivates c-Jun N-terminal kinases (JNKs) and leads to cartilage degeneration. Reduced glutathione (GSH) can combat oxidative stress caused by ageing.

Members of the mitogen-activated protein kinase (MAPK) pathway have been shown in some research to play a role in regulating cell survival and oxidative stress tolerance. For example, oxidative stress inactivates c-Jun N-terminal kinases (JNKs) in human chondrocytes cultured in vitro (Nelson et al., 2018). In mice, the loss of JNK1 and JNK2 led to more severe age-related OA and ageing of cartilage and synovium (Loeser et al., 2020), an indication that JNK is a detrimental regulator of joint degeneration. Glutathione, as one of the reductants, is also a regulator of joint senescence in ageing-induced KOA. For example, Carlo et al. found that the ratio of GSH/GSSG in chondrocytes in elderly patients (age ≥50) is lower than the ratio of GSH/GSSG in chondrocytes in young patients (age 18–49), suggesting that oxidative stress increases with age, which increases the risk of oxidant-mediated cell death in chondrocytes through the imbalance of the glutathione antioxidant system (Carlo and Loeser, 2003). Therefore, damage to the glutathione system may lead to proinflammatory-induced oxidative stress in chondrocytes, especially in the process of senescence (McCutchen et al., 2017; Issa et al., 2018). The increase of ROS in chondrocytes caused by aging may be the cause of oxidative damage of genomic and mitochondrial DNA (McCulloch et al., 2017). Mitochondrial DNA damage in turn leads to the senescence of osteoarthritis chondrocytes. The stagnation of chondrocyte proliferation in this case is due to the accumulation of DNA damage after exposure to stress inducers (Minguzzi et al., 2018). Therefore, chronic oxidative stress and mitochondrial dysfunction may be the main causes of chronic degenerative diseases. In addition, researchers have found that the aging of cartilage is not caused by telomere wear and aging of mature chondrocytes, but is attributed to a group of progenitor cells (Fellows et al., 2017). Combining these results, we can see that there is a correlation between oxidative stress and cartilage ageing, which may promote the pathogenesis of KOA.
How to solve the oxidative stress in Knee osteoarthritis
Several different antioxidant treatment methods are being investigated, some of which are currently in clinical trials. These measures include removing O2 − before reacting with NO to form ONOO-, removing H2O2 before forming OH-, using precursors to increase GSH and increasing the synthesis of antioxidant enzymes.
SOD is essential for preventing oxidative stress since it is the sole enzyme that can remove O2 − from mammalian cells. Since it was discovered in 1969, SOD has generated interest due to its potential as a treatment. Numerous SOD simulations have been created since then. Metalloporphyrins, Mn cyclic polyamines, nitrogen oxides, and other compounds are included in these simulations. Previous research has listed a summary of their chemical characteristics (Batinić-Haberle et al., 2010; Bonetta, 2018). The most comprehensive SOD simulator studied is probably manganese porphyrin. At present, researchers have synthesized various manganese porphyrin compounds and evaluated their O2 −disproportionation activity (Batinic-Haberle et al., 2015). For example, the protective and therapeutic effects of MnTE-2-pYp 5 + and MnTDE-2-ImP5+ have been confirmed in animal models. (Mackensen et al., 2001; Gauter-Fleckenstein et al., 2008; Rabbani et al., 2009; Ganesh et al., 2016). The ability of GC4419 to remove superoxide anions selectively without interacting with other oxidants makes it a further intriguing SOD mimic (Aston et al., 2001), and GC4419 shows therapeutic effects in a mouse model of arthritis (Salvemini et al., 2001). In addition, a variety of GPX simulations have been developed, among which ebselenoline is the best known. Ebselenoline has been shown to reduce oxidative damage in inflammation-related carcinogenesis (Nakamura et al., 2002). However, there are no related experiments to prove that ebselenoline can effectively improve oxidative stress in KOA. One of the most researched medicinal antioxidants is N-acetylcysteine (NAC). There is some indication that NAC supplementation is particularly important in mediating the antioxidant effect of GSH (Rushworth and Megson, 2014). NAC has been used in the treatment of many diseases, including airway cystic fibrosis (Conrad et al., 2015) and nephropathy (Xu et al., 2016). However, exogenous GSH degrades quickly in plasma, and GSH cannot be delivered efficiently to most cells (Wendel and Cikryt, 1980). Therefore, the ester derivative of GSH is a more successful complementary strategy. Many studies have confirmed that GSH esters can efficiently increase GSH in cells and/or tissues in cells and animal models (Chen et al., 2000; Anderson et al., 2004).
Antioxidant enzyme induction of polyphenols is mediated by the NRF2 signal (Forman et al., 2014). Therefore, as a result, NRF2 activator is viewed as a promising medication for boosting antioxidant defences and reducing pathology. Some of the antioxidant enzymes utilized in clinical studies for treating and preventing disease are induced by extracts from foods such as tea, cocoa, and various vegetables and fruits (Pandurangan et al., 2015; Li et al., 2016). For example, for a variety of diseases, including chronic obstructive pulmonary disease, osteoarthritis, joint stiffness and diabetic nephropathy (Yagishita et al., 2019). NOXs, as the source of O2 − and H2O2, play an important role in redox signal transduction; however, a problem arises when NOXs are activated to an unhealthy degree and cause harm to healthy tissue. Inhibiting NOX1, NOX2, and NOX4 has been shown to be beneficial in animal models (Teixeira et al., 2017). By contributing an electron to neutralize free radicals, vitamin C is another key antioxidant that plays a role in lowering oxidative stress (Frei et al., 1989). The antioxidant properties of Vitamin E have been demonstrated by numerous other investigations (Hill et al., 2003; Bruno et al., 2006), especially in cases of oxidative stress or other antioxidant deficiencies (Traber and Atkinson, 2007). In animal models, the use of iNOS inhibitors significantly reduced cartilage degeneration and osteophyte formation (Pelletier et al., 1998).
Yamada et al. found that S. tuberculata can reduce the damage related to oxidative stress in serum and reduce the oxidative stress injury and pain caused by knee osteoarthritis in rats (Yamada et al., 2020). In addition, they found that S. tuberculata reduced the damage caused by oxidative stress and cytokines in follow-up studies (Yamada et al., 2022). And the combination of S. tuberculata and photobiologic therapy reduced the levels of cytokines and nitrite/nitrate (Yamada et al., 2022). Pan et al. found that Receptor-interacting protein 2 (RIP2) can regulate cartilage degradation and oxidative stress in IL-1 β-treated chondrocytes by regulating TRAF3 expression and p38-MAPK pathway activation (Pan et al., 2021). TERT-butylhydroquinone can effectively prevent oxidative stress and inhibit apoptosis of rat chondrocytes by activating Nrf2 pathway (Yang et al., 2021). Li et al. found that montelukast can effectively reduce oxidative stress and apoptosis in chondrocytes and improve the viability of chondrocytes (Li et al., 2021). Transforming growth factor β 1 can protect chondrocytes from oxidative stress by regulating autophagy (Kurakazu et al., 2021). Karim et al. found that iron overload in chondrocytes can induce oxidative stress, cell cycle arrest and apoptosis (Karim et al., 2022). Pang et al. found that Bardoxolonemethyl can inhibit chondrocyte apoptosis and ECM degradation induced by oxidative stress in vitro, and reduce OA in vivo (Pang et al., 2021).
Combined with the above research results, we can use these substances regulating oxidative stress as potential therapeutic drugs for KOA and verify the ability of these substances to regulate ROS in the pathogenesis of KOA in animal experiments.
Conclusion
Many studies have found that oxidative stress not only promotes the ageing and injury of chondrocytes but also drives the development of synovitis in the pathogenesis of KOA. In addition, with increasing age, the ROS level of chondrocytes increases, which further promotes cartilage injury. An increasing number of studies have found new means to regulate the level of ROS, which provides a new strategy for the prevention and treatment of KOA. Of course, more experiments are needed to study the effect of oxidative stress on KOA.
Author contributions
LL: writing-original draft. LL, WH, and PL: conceptualization, project administration, and writing-review and editing. PL, JW, and MY: data curation and methodology. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82072432).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Abbasi J. (2017). Can exercise prevent knee osteoarthritis? Jama 318 (22), 2169–2171. 10.1001/jama.2017.16144 [DOI] [PubMed] [Google Scholar]
- Ahmad N., Ansari M. Y., Haqqi T. M. (2020). Role of iNOS in osteoarthritis: Pathological and therapeutic aspects. J. Cell. Physiol. 235 (10), 6366–6376. 10.1002/jcp.29607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin A. R., Dave M., Attur M., Abramson S. B. (2000). COX-2, NO, and cartilage damage and repair. Curr. Rheumatol. Rep. 2 (6), 447–453. 10.1007/s11926-000-0019-5 [DOI] [PubMed] [Google Scholar]
- Anderson M. F., Nilsson M., Eriksson P. S., Sims N. R. (2004). Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci. Lett. 354 (2), 163–165. 10.1016/j.neulet.2003.09.067 [DOI] [PubMed] [Google Scholar]
- Aston K., Rath N., Naik A., Slomczynska U., Schall O. F., Riley D. P. (2001). Computer-aided design (CAD) of Mn(II) complexes: Superoxide dismutase mimetics with catalytic activity exceeding the native enzyme. Inorg. Chem. 40 (8), 1779–1789. 10.1021/ic000958v [DOI] [PubMed] [Google Scholar]
- Baker K., Grainger A., Niu J., Clancy M., Guermazi A., Crema M., et al. (2010). Relation of synovitis to knee pain using contrast-enhanced MRIs. Ann. Rheum. Dis. 69 (10), 1779–1783. 10.1136/ard.2009.121426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh E., Veale D. J., McGarry T., Orr C., Szekanecz Z., Ng C. T., et al. (2018). Oxidative stress impairs energy metabolism in primary cells and synovial tissue of patients with rheumatoid arthritis. Arthritis Res. Ther. 20 (1), 95. 10.1186/s13075-018-1592-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batinić-Haberle I., Rebouças J. S., Spasojević I. (2010). Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal. 13 (6), 877–918. 10.1089/ars.2009.2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batinic-Haberle I., Tovmasyan A., Spasojevic I. (2015). An educational overview of the chemistry, biochemistry and therapeutic aspects of Mn porphyrins--From superoxide dismutation to H2O2-driven pathways. Redox Biol. 5, 43–65. 10.1016/j.redox.2015.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belikov A. V., Schraven B., Simeoni L. (2015). T cells and reactive oxygen species. J. Biomed. Sci. 22, 85. 10.1186/s12929-015-0194-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard K., Logsdon N. J., Miguel V., Benavides G. A., Zhang J., Carter A. B., et al. (2017). NADPH oxidase 4 (Nox4) suppresses mitochondrial biogenesis and bioenergetics in lung fibroblasts via a nuclear factor erythroid-derived 2-like 2 (Nrf2)-dependent pathway. J. Biol. Chem. 292 (7), 3029–3038. 10.1074/jbc.M116.752261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biniecka M., Fox E., Gao W., Ng C. T., Veale D. J., Fearon U., et al. (2011a). Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheum. 63 (8), 2172–2182. 10.1002/art.30395 [DOI] [PubMed] [Google Scholar]
- Biniecka M., Kennedy A., Fearon U., Ng C. T., Veale D. J., O'Sullivan J. N. (2010). Oxidative damage in synovial tissue is associated with in vivo hypoxic status in the arthritic joint. Ann. Rheum. Dis. 69 (6), 1172–1178. 10.1136/ard.2009.111211 [DOI] [PubMed] [Google Scholar]
- Biniecka M., Kennedy A., Ng C. T., Chang T. C., Balogh E., Fox E., et al. (2011b). Successful tumour necrosis factor (TNF) blocking therapy suppresses oxidative stress and hypoxia-induced mitochondrial mutagenesis in inflammatory arthritis. Arthritis Res. Ther. 13 (4), R121. 10.1186/ar3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco F. J., Ochs R. L., Schwarz H., Lotz M. (1995). Chondrocyte apoptosis induced by nitric oxide. Am. J. Pathol. 146 (1), 75–85. [PMC free article] [PubMed] [Google Scholar]
- Blanco F. J., Rego I., Ruiz-Romero C. (2011). The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 7 (3), 161–169. 10.1038/nrrheum.2010.213 [DOI] [PubMed] [Google Scholar]
- Bolduc J. A., Collins J. A., Loeser R. F. (2019). Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 132, 73–82. 10.1016/j.freeradbiomed.2018.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetta R. (2018). Potential therapeutic applications of MnSODs and SOD-mimetics. Chemistry 24 (20), 5032–5041. 10.1002/chem.201704561 [DOI] [PubMed] [Google Scholar]
- Brandes R. P., Weissmann N., Schröder K. (2014). Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 76, 208–226. 10.1016/j.freeradbiomed.2014.07.046 [DOI] [PubMed] [Google Scholar]
- Brandl A., Hartmann A., Bechmann V., Graf B., Nerlich M., Angele P. (2011). Oxidative stress induces senescence in chondrocytes. J. Orthop. Res. 29 (7), 1114–1120. 10.1002/jor.21348 [DOI] [PubMed] [Google Scholar]
- Bruno R. S., Leonard S. W., Atkinson J., Montine T. J., Ramakrishnan R., Bray T. M., et al. (2006). Faster plasma vitamin E disappearance in smokers is normalized by vitamin C supplementation. Free Radic. Biol. Med. 40 (4), 689–697. 10.1016/j.freeradbiomed.2005.10.051 [DOI] [PubMed] [Google Scholar]
- Burr D. B. (2004). Anatomy and physiology of the mineralized tissues: Role in the pathogenesis of osteoarthrosis. Osteoarthr. Cartil. 12, S20–S30. 10.1016/j.joca.2003.09.016 [DOI] [PubMed] [Google Scholar]
- Burr D. B., Schaffler M. B. (1997). The involvement of subchondral mineralized tissues in osteoarthrosis: Quantitative microscopic evidence. Microsc. Res. Tech. 37 (4), 343–357. 10.1002/(sici)1097-0029(19970515)37:4<343::aid-jemt9>3.0.co;2-l [DOI] [PubMed] [Google Scholar]
- Cannizzo E. S., Clement C. C., Sahu R., Follo C., Santambrogio L. (2011). Oxidative stress, inflamm-aging and immunosenescence. J. Proteomics 74 (11), 2313–2323. 10.1016/j.jprot.2011.06.005 [DOI] [PubMed] [Google Scholar]
- Carlo M. D., Jr., Loeser R. F. (2003). Increased oxidative stress with aging reduces chondrocyte survival: Correlation with intracellular glutathione levels. Arthritis Rheum. 48 (12), 3419–3430. 10.1002/art.11338 [DOI] [PubMed] [Google Scholar]
- Chen T. S., Richie J. P., Nagasawa H. T., Lang C. A. (2000). Glutathione monoethyl ester protects against glutathione deficiencies due to aging and acetaminophen in mice. Mech. Ageing Dev. 120 (1-3), 127–139. 10.1016/s0047-6374(00)00214-1 [DOI] [PubMed] [Google Scholar]
- Clancy R. M., Rediske J., Tang X., Nijher N., Frenkel S., Philips M., et al. (1997). Outside-in signaling in the chondrocyte. Nitric oxide disrupts fibronectin-induced assembly of a subplasmalemmal actin/rho A/focal adhesion kinase signaling complex. J. Clin. Invest. 100 (7), 1789–1796. 10.1172/jci119706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J. A., Wood S. T., Nelson K. J., Rowe M. A., Carlson C. S., Chubinskaya S., et al. (2016). Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signaling in aging chondrocytes. J. Biol. Chem. 291 (13), 6641–6654. 10.1074/jbc.M115.693523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C., Lymp J., Thompson V., Dunn C., Davies Z., Chatfield B., et al. (2015). Long-term treatment with oral N-acetylcysteine: Affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J. Cyst. Fibros. 14 (2), 219–227. 10.1016/j.jcf.2014.08.008 [DOI] [PubMed] [Google Scholar]
- Cutler R. G. (2005). Oxidative stress profiling: Part I. Its potential importance in the optimization of human health. Ann. N. Y. Acad. Sci. 1055, 93–135. 10.1196/annals.1323.027 [DOI] [PubMed] [Google Scholar]
- Dang P. M., Cross A. R., Quinn M. T., Babior B. M. (2002). Assembly of the neutrophil respiratory burst oxidase: A direct interaction between p67PHOX and cytochrome b558 II. Proc. Natl. Acad. Sci. U. S. A. 99 (7), 4262–4265. 10.1073/pnas.072345299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Carlo M., Jr., Loeser R. F. (2002). Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 46 (2), 394–403. 10.1002/art.10056 [DOI] [PubMed] [Google Scholar]
- Diaz-Vivancos P., de Simone A., Kiddle G., Foyer C. H. (2015). Glutathione--linking cell proliferation to oxidative stress. Free Radic. Biol. Med. 89, 1154–1164. 10.1016/j.freeradbiomed.2015.09.023 [DOI] [PubMed] [Google Scholar]
- Dickinson B. C., Chang C. J. (2011). Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 7 (8), 504–511. 10.1038/nchembio.607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82 (1), 47–95. 10.1152/physrev.00018.2001 [DOI] [PubMed] [Google Scholar]
- Du J., Yu S., Wang D., Chen S., Chen S., Zheng Y., et al. (2020). Germline and somatic mtDNA mutation spectrum of rheumatoid arthritis patients in the Taizhou area, China. Rheumatol. Oxf. 59 (10), 2982–2991. 10.1093/rheumatology/keaa063 [DOI] [PubMed] [Google Scholar]
- Espinosa-Diez C., Miguel V., Mennerich D., Kietzmann T., Sánchez-Pérez P., Cadenas S., et al. (2015). Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 6, 183–197. 10.1016/j.redox.2015.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellows C. R., Williams R., Davies I. R., Gohil K., Baird D. M., Fairclough J., et al. (2017). Characterisation of a divergent progenitor cell sub-populations in human osteoarthritic cartilage: The role of telomere erosion and replicative senescence. Sci. Rep. 7, 41421. 10.1038/srep41421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman H. J., Davies K. J., Ursini F. (2014). How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo . Free Radic. Biol. Med. 66, 24–35. 10.1016/j.freeradbiomed.2013.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei B., England L., Ames B. N. (1989). Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. U. S. A. 86 (16), 6377–6381. 10.1073/pnas.86.16.6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Kinter M., Hudson J., Humphries K. M., Lane R. S., White J. R., et al. (2016). Aging promotes Sirtuin 3-dependent cartilage superoxide dismutase 2 acetylation and osteoarthritis. Arthritis Rheumatol. 68 (8), 1887–1898. 10.1002/art.39618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh D., Kumarathasan P., Thomson E. M., St-Germain C., Blais E., Crapo J., et al. (2016). Impact of superoxide dismutase mimetic AEOL 10150 on the endothelin system of fischer 344 rats. PLoS One 11 (3), e0151810. 10.1371/journal.pone.0151810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauter-Fleckenstein B., Fleckenstein K., Owzar K., Jiang C., Batinic-Haberle I., Vujaskovic Z. (2008). Comparison of two Mn porphyrin-based mimics of superoxide dismutase in pulmonary radioprotection. Free Radic. Biol. Med. 44 (6), 982–989. 10.1016/j.freeradbiomed.2007.10.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderman K. A., Hultqvist M., Pizzolla A., Zhao M., Nandakumar K. S., Mattsson R., et al. (2007). Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J. Clin. Invest. 117 (10), 3020–3028. 10.1172/jci31935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glyn-Jones S., Palmer A. J., Agricola R., Price A. J., Vincent T. L., Weinans H., et al. (2015). Osteoarthritis. Lancet 386 (9991), 376–387. 10.1016/s0140-6736(14)60802-3 [DOI] [PubMed] [Google Scholar]
- Harty L. C., Biniecka M., O'Sullivan J., Fox E., Mulhall K., Veale D. J., et al. (2012). Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann. Rheum. Dis. 71 (4), 582–588. 10.1136/annrheumdis-2011-200245 [DOI] [PubMed] [Google Scholar]
- Hill K. E., Montine T. J., Motley A. K., Li X., May J. M., Burk R. F. (2003). Combined deficiency of vitamins E and C causes paralysis and death in Guinea pigs. Am. J. Clin. Nutr. 77 (6), 1484–1488. 10.1093/ajcn/77.6.1484 [DOI] [PubMed] [Google Scholar]
- Holmström K. M., Finkel T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15 (6), 411–421. 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- Hootman J. M., Helmick C. G., Barbour K. E., Theis K. A., Boring M. A. (2016). Updated projected prevalence of self-reported doctor-diagnosed arthritis and arthritis-attributable Activity limitation among US adults, 2015-2040. Arthritis Rheumatol. 68 (7), 1582–1587. 10.1002/art.39692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa R., Boeving M., Kinter M., Griffin T. M. (2018). Effect of biomechanical stress on endogenous antioxidant networks in bovine articular cartilage. J. Orthop. Res. 36 (2), 760–769. 10.1002/jor.23728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim A., Bajbouj K., Shafarin J., Qaisar R., Hall A. C., Hamad M. (2022). Iron overload induces oxidative stress, cell cycle arrest and apoptosis in chondrocytes. Front. Cell Dev. Biol. 10, 821014. 10.3389/fcell.2022.821014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Xu M., Xo R., Mates A., Wilson G. L., Pearsall A. W. t., et al. (2010a). Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr. Cartil. 18 (3), 424–432. 10.1016/j.joca.2009.09.008 [DOI] [PubMed] [Google Scholar]
- Kim K. S., Choi H. W., Yoon H. E., Kim I. Y. (2010b). Reactive oxygen species generated by NADPH oxidase 2 and 4 are required for chondrogenic differentiation. J. Biol. Chem. 285 (51), 40294–40302. 10.1074/jbc.M110.126821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball J. S., Johnson J. P., Carlson D. A. (2021). Oxidative stress and osteoporosis. J. Bone Jt. Surg. Am. 103 (15), 1451–1461. 10.2106/jbjs.20.00989 [DOI] [PubMed] [Google Scholar]
- Koike M., Nojiri H., Ozawa Y., Watanabe K., Muramatsu Y., Kaneko H., et al. (2015). Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci. Rep. 5, 11722. 10.1038/srep11722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuksal N., Chalker J., Mailloux R. J. (2017). Progress in understanding the molecular oxygen paradox - function of mitochondrial reactive oxygen species in cell signaling. Biol. Chem. 398 (11), 1209–1227. 10.1515/hsz-2017-0160 [DOI] [PubMed] [Google Scholar]
- Kurakazu I., Akasaki Y., Tsushima H., Sueishi T., Toya M., Kuwahara M., et al. (2021). TGFβ1 signaling protects chondrocytes against oxidative stress via FOXO1-autophagy axis. Osteoarthr. Cartil. 29 (11), 1600–1613. 10.1016/j.joca.2021.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. R., Yoo S. J., Kim J., Park C. K., Kang S. W. (2021). Reduction of oxidative stress in peripheral blood mononuclear cells attenuates the inflammatory response of fibroblast-like synoviocytes in rheumatoid arthritis. Int. J. Mol. Sci. 22 (22), 12411. 10.3390/ijms222212411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuwenburgh C., Pamplona R., Sanz A. (2011). Mitochondria and aging. J. Aging Res. 2011, 782946. 10.4061/2011/782946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepetsos P., Papavassiliou A. G. (2016). ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta 1862 (4), 576–591. 10.1016/j.bbadis.2016.01.003 [DOI] [PubMed] [Google Scholar]
- Li J., Sapper T. N., Mah E., Rudraiah S., Schill K. E., Chitchumroonchokchai C., et al. (2016). Green tea extract provides extensive Nrf2-independent protection against lipid accumulation and NFκB pro- inflammatory responses during nonalcoholic steatohepatitis in mice fed a high-fat diet. Mol. Nutr. Food Res. 60 (4), 858–870. 10.1002/mnfr.201500814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Wang J., Ma Y. (2021). Montelukast attenuates interleukin IL-1β-induced oxidative stress and apoptosis in chondrocytes by inhibiting CYSLTR1 (Cysteinyl Leukotriene Receptor 1) and activating KLF2 (Kruppel like Factor 2). Bioengineered 12 (1), 8476–8484. 10.1080/21655979.2021.1984003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser R. F., Carlson C. S., Del Carlo M., Cole A. (2002). Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 46 (9), 2349–2357. 10.1002/art.10496 [DOI] [PubMed] [Google Scholar]
- Loeser R. F., Goldring S. R., Scanzello C. R., Goldring M. B. (2012). Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 64 (6), 1697–1707. 10.1002/art.34453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser R. F., Kelley K. L., Armstrong A., Collins J. A., Diekman B. O., Carlson C. S. (2020). Deletion of JNK enhances senescence in joint tissues and increases the severity of age-related osteoarthritis in mice. Arthritis Rheumatol. 72 (10), 1679–1688. 10.1002/art.41312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser R. F. (2017). The role of aging in the development of osteoarthritis. Trans. Am. Clin. Climatol. Assoc. 128, 44–54. [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G. (2013). The hallmarks of aging. Cell 153 (6), 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubos E., Loscalzo J., Handy D. E. (2011). Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 15 (7), 1957–1997. 10.1089/ars.2010.3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S., Li H., Liu J., Xie X., Wan Z., Wang Y., et al. (2020). Andrographolide ameliorates oxidative stress, inflammation and histological outcome in complete Freund's adjuvant-induced arthritis. Chem. Biol. Interact. 319, 108984. 10.1016/j.cbi.2020.108984 [DOI] [PubMed] [Google Scholar]
- Mackensen G. B., Patel M., Sheng H., Calvi C. L., Batinic-Haberle I., Day B. J., et al. (2001). Neuroprotection from delayed postischemic administration of a metalloporphyrin catalytic antioxidant. J. Neurosci. 21 (13), 4582–4592. 10.1523/jneurosci.21-13-04582.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchev A. S., Dimitrova P. A., Burns A. J., Kostov R. V., Dinkova-Kostova A. T., Georgiev M. I. (2017). Oxidative stress and chronic inflammation in osteoarthritis: Can NRF2 counteract these partners in crime? Ann. N. Y. Acad. Sci. 1401 (1), 114–135. 10.1111/nyas.13407 [DOI] [PubMed] [Google Scholar]
- Martel-Pelletier J., Mineau F., Jovanovic D., Di Battista J. A., Pelletier J. P. (1999). Mitogen-activated protein kinase and nuclear factor kappaB together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: Possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (MAPKAPK). Arthritis Rheum. 42 (11), 2399–2409. 10.1002/1529-0131(199911)42:11<2399::AID-ANR19>3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]
- McCulloch K., Litherland G. J., Rai T. S. (2017). Cellular senescence in osteoarthritis pathology. Aging Cell 16 (2), 210–218. 10.1111/acel.12562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutchen C. N., Zignego D. L., June R. K. (2017). Metabolic responses induced by compression of chondrocytes in variable-stiffness microenvironments. J. Biomech. 64, 49–58. 10.1016/j.jbiomech.2017.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry T., Biniecka M., Veale D. J., Fearon U. (2018). Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 125, 15–24. 10.1016/j.freeradbiomed.2018.03.042 [DOI] [PubMed] [Google Scholar]
- Michael J. W., Schlüter-Brust K. U., Eysel P. (2010). The epidemiology, etiology, diagnosis, and treatment of osteoarthritis of the knee. Dtsch. Arztebl. Int. 107 (9), 152–162. 10.3238/arztebl.2010.0152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguzzi M., Cetrullo S., D'Adamo S., Silvestri Y., Flamigni F., Borzì R. M. (2018). Emerging players at the intersection of chondrocyte loss of maturational arrest, oxidative stress, senescence and low-grade inflammation in osteoarthritis. Oxid. Med. Cell. Longev. 2018, 3075293. 10.1155/2018/3075293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobasheri A., Rayman M. P., Gualillo O., Sellam J., van der Kraan P., Fearon U. (2017). The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 13 (5), 302–311. 10.1038/nrrheum.2017.50 [DOI] [PubMed] [Google Scholar]
- Morel F., Rousset F., Vu Chuong Nguyen M., Trocme C., Grange L., Lardy B. (2015). NADPH oxidase Nox4, a putative therapeutic target in osteoarthritis. Bull. de l'Academie. Natl. de Med. 199 (4-5), 673–687. 10.1016/s0001-4079(19)30941-0 [DOI] [PubMed] [Google Scholar]
- Mougiakakos D., Johansson C. C., Kiessling R. (2009). Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 113 (15), 3542–3545. 10.1182/blood-2008-09-181040 [DOI] [PubMed] [Google Scholar]
- Nakamura Y., Feng Q., Kumagai T., Torikai K., Ohigashi H., Osawa T., et al. (2002). Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant. Implication for inflammation-associated carcinogenesis. J. Biol. Chem. 277 (4), 2687–2694. 10.1074/jbc.M109641200 [DOI] [PubMed] [Google Scholar]
- Nathan C., Xie Q. W. (1994). Nitric oxide synthases: Roles, tolls, and controls. Cell 78 (6), 915–918. 10.1016/0092-8674(94)90266-6 [DOI] [PubMed] [Google Scholar]
- Nelson K. J., Bolduc J. A., Wu H., Collins J. A., Burke E. A., Reisz J. A., et al. (2018). H(2)O(2) oxidation of cysteine residues in c-Jun N-terminal kinase 2 (JNK2) contributes to redox regulation in human articular chondrocytes. J. Biol. Chem. 293 (42), 16376–16389. 10.1074/jbc.RA118.004613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh C. K., Sultan A., Platzer J., Dolatabadi N., Soldner F., McClatchy D. B., et al. (2017). S-nitrosylation of PINK1 attenuates PINK1/parkin-dependent mitophagy in hiPSC-based Parkinson's disease models. Cell Rep. 21 (8), 2171–2182. 10.1016/j.celrep.2017.10.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D., Lyu Y., Zhang N., Wang X., Lei T., Liang Z. (2021). RIP2 knockdown inhibits cartilage degradation and oxidative stress in IL-1β-treated chondrocytes via regulating TRAF3 and inhibiting p38 MAPK pathway. Clin. Immunol. 232, 108868. 10.1016/j.clim.2021.108868 [DOI] [PubMed] [Google Scholar]
- Pandurangan A. K., Saadatdoust Z., Esa N. M., Hamzah H., Ismail A. (2015). Dietary cocoa protects against colitis-associated cancer by activating the Nrf2/Keap1 pathway. Biofactors 41 (1), 1–14. 10.1002/biof.1195 [DOI] [PubMed] [Google Scholar]
- Pang Z., Jiang Z., Zhu R., Song C., Tang H., Cao L., et al. (2021). Bardoxolone-methyl prevents oxidative stress-mediated apoptosis and extracellular matrix degradation in vitro and alleviates osteoarthritis in vivo . Drug Des. devel. Ther. 15, 3735–3747. 10.2147/dddt.s314767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos J. F., Nelson G., Wang C., Richter T., Simillion C., Proctor C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347. 10.1038/msb.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J. P., Jovanovic D., Fernandes J. C., Manning P., Connor J. R., Currie M. G., et al. (1998). Reduced progression of experimental osteoarthritis in vivo by selective inhibition of inducible nitric oxide synthase. Arthritis Rheum. 41 (7), 1275–1286. 10.1002/1529-0131(199807)41:7<1275::AID-ART19>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- Pisoschi A. M., Pop A. (2015). The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 97, 55–74. 10.1016/j.ejmech.2015.04.040 [DOI] [PubMed] [Google Scholar]
- Pizzino G., Irrera N., Cucinotta M., Pallio G., Mannino F., Arcoraci V., et al. (2017). Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 8416763. 10.1155/2017/8416763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn R. H., Murray J. N., Pezold R., Sevarino K. S. (2018). Surgical management of osteoarthritis of the knee. J. Am. Acad. Orthop. Surg. 26 (9), e191–e193. 10.5435/jaaos-d-17-00424 [DOI] [PubMed] [Google Scholar]
- Rabbani Z. N., Spasojevic I., Zhang X., Moeller B. J., Haberle S., Vasquez-Vivar J., et al. (2009). Antiangiogenic action of redox-modulating Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin, MnTE-2-PyP(5+), via suppression of oxidative stress in a mouse model of breast tumor. Free Radic. Biol. Med. 47 (7), 992–1004. 10.1016/j.freeradbiomed.2009.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmati M., Nalesso G., Mobasheri A., Mozafari M. (2017). Aging and osteoarthritis: Central role of the extracellular matrix. Ageing Res. Rev. 40, 20–30. 10.1016/j.arr.2017.07.004 [DOI] [PubMed] [Google Scholar]
- Regan E., Flannelly J., Bowler R., Tran K., Nicks M., Carbone B. D., et al. (2005). Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis Rheum. 52 (11), 3479–3491. 10.1002/art.21387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S. G., Woo H. A., Kil I. S., Bae S. H. (2012). Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 287 (7), 4403–4410. 10.1074/jbc.R111.283432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F., Hazane-Puch F., Pinosa C., Nguyen M. V., Grange L., Soldini A., et al. (2015). IL-1beta mediates MMP secretion and IL-1beta neosynthesis via upregulation of p22(phox) and NOX4 activity in human articular chondrocytes. Osteoarthr. Cartil. 23 (11), 1972–1980. 10.1016/j.joca.2015.02.167 [DOI] [PubMed] [Google Scholar]
- Ruiz-Romero C., Calamia V., Mateos J., Carreira V., Martínez-Gomariz M., Fernández M., et al. (2009). Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: A decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol. Cell. Proteomics 8 (1), 172–189. 10.1074/mcp.M800292-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Romero C., López-Armada M. J., Blanco F. J. (2006). Mitochondrial proteomic characterization of human normal articular chondrocytes. Osteoarthr. Cartil. 14 (6), 507–518. 10.1016/j.joca.2005.12.004 [DOI] [PubMed] [Google Scholar]
- Rushworth G. F., Megson I. L. (2014). Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 141 (2), 150–159. 10.1016/j.pharmthera.2013.09.006 [DOI] [PubMed] [Google Scholar]
- Salvemini D., Mazzon E., Dugo L., Serraino I., De Sarro A., Caputi A. P., et al. (2001). Amelioration of joint disease in a rat model of collagen-induced arthritis by M40403, a superoxide dismutase mimetic. Arthritis Rheum. 44 (12), 2909–2921. 10.1002/1529-0131(200112)44:12<2909::aid-art479>3.0.co;2-# [DOI] [PubMed] [Google Scholar]
- Sandell L. J. (2012). Etiology of osteoarthritis: Genetics and synovial joint development. Nat. Rev. Rheumatol. 8 (2), 77–89. 10.1038/nrrheum.2011.199 [DOI] [PubMed] [Google Scholar]
- Scanzello C. R., Goldring S. R. (2012). The role of synovitis in osteoarthritis pathogenesis. Bone 51 (2), 249–257. 10.1016/j.bone.2012.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher J. U., Pillinger M. H., Abramson S. B. (2007). Nitric oxide synthases and osteoarthritis. Curr. Rheumatol. Rep. 9 (1), 9–15. 10.1007/s11926-007-0016-z [DOI] [PubMed] [Google Scholar]
- Scott J. L., Gabrielides C., Davidson R. K., Swingler T. E., Clark I. M., Wallis G. A., et al. (2010). Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 69 (8), 1502–1510. 10.1136/ard.2009.119966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shane Anderson A., Loeser R. F., ShAne Anderson A. (2010). Why is osteoarthritis an age-related disease? Best. Pract. Res. Clin. Rheumatol. 24 (1), 15–26. 10.1016/j.berh.2009.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma L. (2021). Osteoarthritis of the knee. N. Engl. J. Med. 384 (1), 51–59. 10.1056/NEJMcp1903768 [DOI] [PubMed] [Google Scholar]
- Stewart H. L., Kawcak C. E. (2018). The importance of subchondral bone in the pathophysiology of osteoarthritis. Front. Vet. Sci. 5, 178. 10.3389/fvets.2018.00178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suantawee T., Tantavisut S., Adisakwattana S., Tanpowpong T., Tanavalee A., Yuktanandana P., et al. (2015). Upregulation of inducible nitric oxide synthase and nitrotyrosine expression in primary knee osteoarthritis. J. Med. Assoc. Thai 98 (1), S91–S97. [PubMed] [Google Scholar]
- Teixeira G., Szyndralewiez C., Molango S., Carnesecchi S., Heitz F., Wiesel P., et al. (2017). Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 174 (12), 1647–1669. 10.1111/bph.13532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traber M. G., Atkinson J. (2007). Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 43 (1), 4–15. 10.1016/j.freeradbiomed.2007.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D., Lu W., Ogasawara M. A., Nilsa R. D., Huang P. (2008). Redox regulation of cell survival. Antioxid. Redox Signal. 10 (8), 1343–1374. 10.1089/ars.2007.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552 (2), 335–344. 10.1113/jphysiol.2003.049478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman K., Khurana S., Tai T. C. (2013). Oxidative stress in aging--matters of the heart and mind. Int. J. Mol. Sci. 14 (9), 17897–17925. 10.3390/ijms140917897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Yen F. S., Zhu X. G., Timson R. C., Weber R., Xing C., et al. (2021). SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599 (7883), 136–140. 10.1038/s41586-021-04025-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel A., Cikryt P. (1980). The level and half-life of glutathione in human plasma. FEBS Lett. 120 (2), 209–211. 10.1016/0014-5793(80)80299-7 [DOI] [PubMed] [Google Scholar]
- Whiteman M., Armstrong J. S., Cheung N. S., Siau J. L., Rose P., Schantz J. T., et al. (2004). Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. Faseb J. 18 (12), 1395–1397. 10.1096/fj.03-1096fje [DOI] [PubMed] [Google Scholar]
- Xu R., Tao A., Bai Y., Deng Y., Chen G. (2016). Effectiveness of N-acetylcysteine for the prevention of contrast-induced nephropathy: A systematic review and meta-analysis of randomized controlled trials. J. Am. Heart Assoc. 5 (9), e003968. 10.1161/jaha.116.003968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita Y., Fahey J. W., Dinkova-Kostova A. T., Kensler T. W. (2019). Broccoli or sulforaphane: Is it the source or dose that matters? Molecules 24 (19), E3593. 10.3390/molecules24193593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada E. F., Dos Santos Stein C., Moresco R. N., Bobinski F., Palandi J., Fernandes P. F., et al. (2022). Photobiomodulation and Sida tuberculata combination declines the inflammation's markers in knee-induced osteoarthritis. Lasers Med. Sci. 37 (1), 193–204. 10.1007/s10103-020-03207-8 [DOI] [PubMed] [Google Scholar]
- Yamada E. F., Olin L. C., Pontel C. L., da Rosa H. S., Folmer V., da Silva M. D. (2020). Sida tuberculata reduces oxidative stress and pain caused by the knee osteoarthritis. J. Ethnopharmacol. 248, 112277. 10.1016/j.jep.2019.112277 [DOI] [PubMed] [Google Scholar]
- Yamazaki K., Fukuda K., Matsukawa M., Hara F., Matsushita T., Yamamoto N., et al. (2003). Cyclic tensile stretch loaded on bovine chondrocytes causes depolymerization of hyaluronan: Involvement of reactive oxygen species. Arthritis Rheum. 48 (11), 3151–3158. 10.1002/art.11305 [DOI] [PubMed] [Google Scholar]
- Yang B., Huang H., He Q., Lu W., Zheng L., Cui L. (2021). Tert-butylhydroquinone prevents oxidative stress-mediated apoptosis and extracellular matrix degradation in rat chondrocytes. Evid. Based. Complement. Altern. Med. 2021, 1905995. 10.1155/2021/1905995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H., Xu J. K., Zheng N. Y., Wang J. L., Mok S. W., Lee Y. W., et al. (2019). Intra-articular injection of magnesium chloride attenuates osteoarthritis progression in rats. Osteoarthr. Cartil. 27 (12), 1811–1821. 10.1016/j.joca.2019.08.007 [DOI] [PubMed] [Google Scholar]
- Yao H., Xu J., Wang J., Zhang Y., Zheng N., Yue J., et al. (2021). Combination of magnesium ions and vitamin C alleviates synovitis and osteophyte formation in osteoarthritis of mice. Bioact. Mater. 6 (5), 1341–1352. 10.1016/j.bioactmat.2020.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Khan S., Liu Y., Wu G., Yong V. W., Xue M. (2022). Oxidative stress following intracerebral hemorrhage: From molecular mechanisms to therapeutic targets. Front. Immunol. 13, 847246. 10.3389/fimmu.2022.847246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Makosa D., Miller B., Griffin T. M. (2020). Glutathione as a mediator of cartilage oxidative stress resistance and resilience during aging and osteoarthritis. Connect. Tissue Res. 61 (1), 34–47. 10.1080/03008207.2019.1665035 [DOI] [PMC free article] [PubMed] [Google Scholar]