

Abstract
High-dose ascorbate (vitamin C) has shown promising anti-cancer activity. Two redox mechanisms have been proposed: hydrogen peroxide generation by ascorbate itself or glutathione depletion by dehydroascorbate (formed by ascorbate oxidation). Here we show that the metabolic effects and cytotoxicity of high-dose ascorbate in vitro result from hydrogen peroxide independently of dehydroascorbate. These effects were suppressed by selenium through antioxidant selenoenzymes including glutathione peroxidase 1 (GPX1) but not the classic ferroptosis-inhibiting selenoenzyme GPX4. Selenium-mediated protection from ascorbate was powered by NADPH from the pentose phosphate pathway. In vivo, dietary selenium deficiency resulted in significant enhancement of ascorbate activity against glioblastoma xenografts. These data establish selenoproteins as key mediators of cancer redox homeostasis. Cancer sensitivity to free radical-inducing therapies, including ascorbate, may depend on selenium, providing a dietary approach for improving their anticancer efficacy
Keywords: Cancer, ascorbate, metabolism, selenium, ROS
Introduction
The past decade has seen a renewed interest in the use of high-dose ascorbate (vitamin C) as a cancer therapy. Intravenous infusion of pharmacologic ascorbate raises the serum ascorbate concentration into the millimolar range, a concentration at which ascorbate has been shown to kill cancer cells in vitro [1–4]. Pharmacologic ascorbate has been shown to synergize with multiple chemotherapeutic agents in animal models and is well-tolerated in human patients [1,4], motivating ongoing clinical trials. Despite its extensive characterization as a cytotoxic agent in vitro, the mechanism through which ascorbate selectively kills cancer cells remains under debate [2,3].
In its classical role as a vitamin and antioxidant, ascorbate acts as a radical scavenger and reducing agent. The concentration of ascorbate in normal human serum is approximately 50 µM [5]. At higher concentrations, ascorbate can spontaneously oxidize to dehydroascorbate (DHA), particularly in the presence of catalytic metals such as iron that are present in serum and common culture media [6,7]. Inside the cell, DHA reduction consumes glutathione (GSH), depleting intracellular defenses against reactive oxygen species (ROS) (Fig 1a) [2,7]. It has been suggested that cancer cells are especially susceptible to high-dose ascorbate due to enhanced expression of facilitative glucose transporter GLUT1, which also carries DHA [2,7]. However, the role of intracellular DHA as the active cytotoxic agent has not been directly demonstrated, motivating further study on this potential mechanism [2,3,8].
Figure 1: The metabolic phenotype of pharmacologic ascorbate mirrors H2O2 but differs from dehydroascorbate (DHA) or diamide.
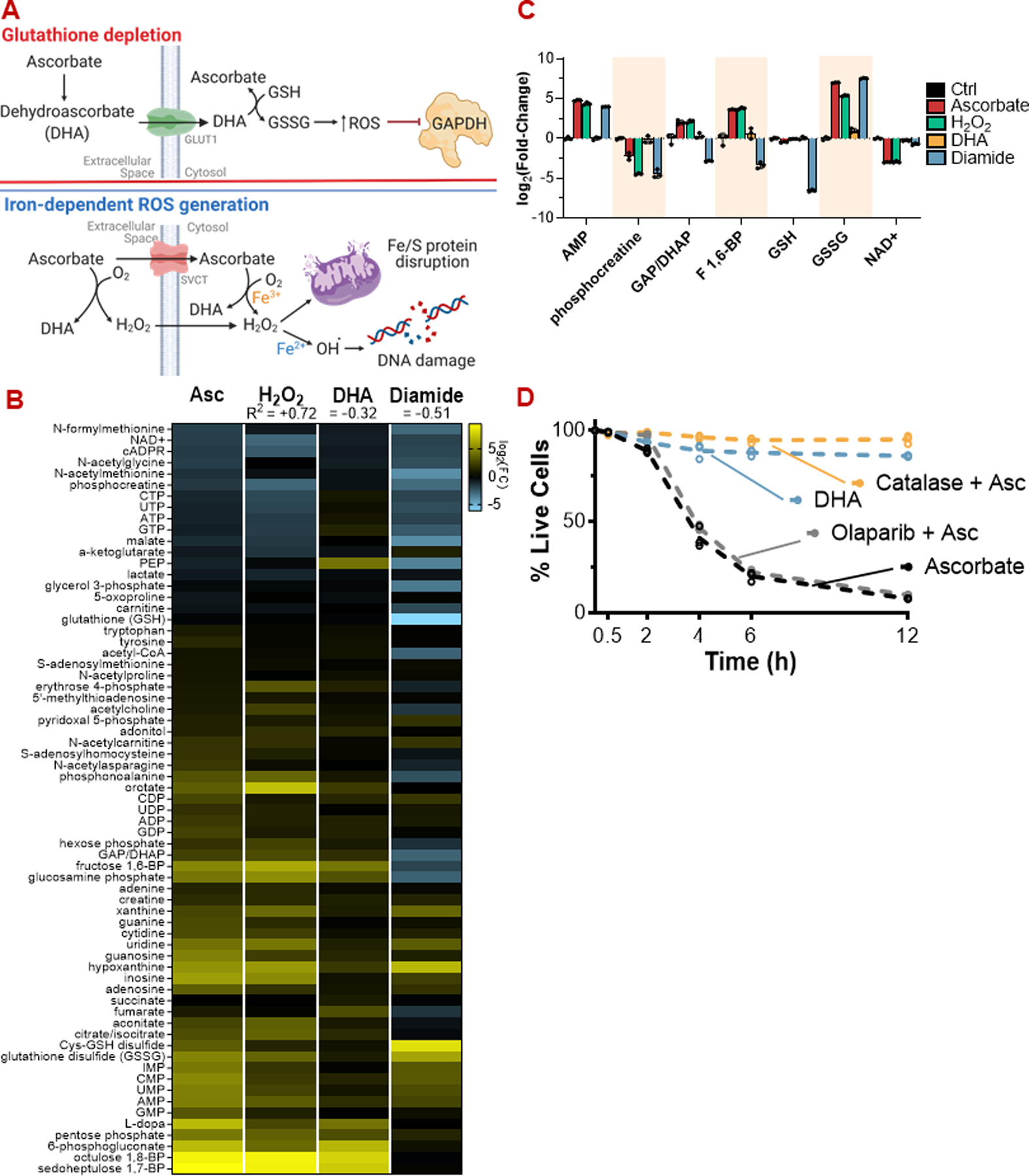
(A) Proposed mechanisms of high-dose ascorbate toxicity. Top: DHA-dependent glutathione depletion. Bottom: Iron-dependent ROS generation.
(B) Metabolic profiles of HCT116 cells challenged with ascorbate (10 mM), H2O2 (200 µM), DHA (10 mM), or diamide (1 mM) for 30 minutes, quantitated by LC-MS.
(C) Bar graphs of selected metabolites from (B) highlighting the overlap in metabolic profile between ascorbate and H2O2 treatment.
(D) Extracellular H2O2 scavenging by catalase (10 U/mL), but not PARP inhibition (10 µM olaparib), protects HCT116 cells from ascorbate (10 mM). DHA (10 mM) itself is markedly less toxic than ascorbate.
ROS: Reactive oxygen species; DHA: Dehydroascorbate; PARP: poly (ADP-ribose) polymerase
For viability assays and metabolomics, n = 3 biological replicates.
Millimolar concentrations of ascorbate can also produce H2O2 by generating the superoxide radical (O2−), a process which occurs in vivo in the extracellular space (Fig 1a) [6]. Indeed, the role of ascorbate as either a pro- or anti-oxidant has been suggested to depend on concentration, with low doses mitigating ROS and high doses generating them [8]. H2O2 generation by ascorbate has been associated with DNA damage and subsequent PARP activation, which can deplete NAD and thereby inhibit glycolysis [2].
Ascorbate cytotoxicity depends on the intracellular labile iron pool (Fig 1a) [3,9]. One explanation for this phenomenon is that ascorbate-generated H2O2 causes toxicity through Fenton chemistry, the iron-dependent conversion of H2O2 into the highly reactive hydroxyl radical. A related possibility is that high-dose ascorbate induces cell death via ferroptosis, an iron-dependent form of necrotic cell death characterized by extensive lipid peroxidation [10,11]. Ascorbate-iron systems have been used to induce lipid peroxidation in microsomes in vitro [12]. Although similarities between ascorbate toxicity and ferroptosis have been noted, the role of lipid peroxidation in ascorbate-induced cell death has not been assessed [10,13].
Several enzymes that fight oxidative stress incorporate the non-canonical amino acid selenocysteine in their active site, and accordingly, their expression and activity depend on an adequate cellular supply of selenium [14,15] Among these proteins is the most well-established suppressor of ferroptosis, the lipid hydroperoxidase GPX4, which reduces lipid hydroperoxides using glutathione as an electron donor. Additional antioxidant selenoproteins include GPX1–3, which function as glutathione-dependent H2O2 detoxifying enzymes and thioredoxin reductases 1 and 2, which reduce oxidized thioredoxin using NADPH
Here we report that ascorbate and DHA exert distinct effects on cellular metabolism in vitro, with the metabolic profile of acute ascorbate administration mimicking that of H2O2. DHA is less cytotoxic than ascorbate itself, and genetic blockage of glutathione-dependent DHA reduction does not rescue cells from ascorbate toxicity. Instead, such toxicity is diminished by H2O2 detoxification, iron chelation, and selenium supplementation. Selenium fortifies cells mainly through GPX1 rather than the ferroptosis-suppressor GPX4. In vivo, dietary selenium depletion enhances the antitumor effect of ascorbate. Collectively, these data suggest that ascorbate kills cancer cells by overwhelming their capacity to clear H2O2 and prevent the iron-catalyzed conversion of H2O2 to the toxic hydroxyl radical. More generally, they establish dietary selenium depletion as a potential means of sensitizing tumors to free radical stress.
Materials and Methods
Materials
Cell Culture
DMEM (Commercial, Corning 10–017-CV)
FeNO3 (Sigma F8508)
Catalase (Sigma C1345)
Sodium selenite (Sigma S5261)
Pharmacologic Compounds
Sodium ascorbate (Sigma A4034)
Ascorbic acid (Sigma A5960)
Dehydroascorbic acid (Sigma 261556)
H2O2 (VWR BDH7103)
Olaparib (Cayman 10621)
Diamide (Sigma 87751)
Deferoxamine mesylate (DFO; Cayman 14595)
Imidazole ketone erastin (IKE; Cayman 27088)
FIN56 (Cayman 25180)
RSL3 (Cayman 19288)
Auranofin (Cayman 15316)
Ferrostatin (Cayman 17729)
Liproxstatin (Cayman 17730)
Seahorse PMP Reagent (Agilent 102504-100)
MitoB (Cayman 17116)
MitoP (Cayman 17117)
MitoB-d15 (Cayman 17470)
MitoP-d15 (Cayman 19296)
Metabolomics
LCMS water (Fisher W9500)
LCMS MeOH (Fisher A456–500)
Antibodies, Staining, and Blotting
Mouse SEPP1/Selenoprotein P ELISA Kit (LSBio LS-F24254-1)
Anti-GAPDH (Abcam Cat#ab8245, RRID:AB_2107448; 1:10,000)
Anti-yH2AX (Abcam Cat#ab81299, RRID:AB_1640564; 1:10,000)
Anti-GPX1 (Abcam Cat#ab108429, RRID:AB_10865045; 1:2000)
Anti-GPX2 (Abcam Cat#ab140130, RRID:AB_2910235 1:2000)
Anti-GPX3 (Abcam Cat#ab59524, RRID:AB_941788; 1:500)
Anti-GPX4 (Abcam Cat#ab125066, RRID:AB_10973901; 1:1000)
Anti-Txn1 (Abcam Cat#ab133524, RRID:AB_2910242 1:10,000)
Anti-Txn2 (Abcam Cat#ab185544, RRID:AB_2737587; 1:10,000)
Anti-TxnRd1 (Abcam Cat#ab124954, RRID:AB_10975643; 1:5000)
Anti-TxnRd2 (Abcam Cat#ab180493, RRID:AB_2732914; 1:5000)
Anti-Catalase (Abcam Cat#ab76110, RRID:AB_1310035; 1:1000)
Anti-GSTO1 (Abcam Cat#ab129106, RRID:AB_11141057; 1:1000)
Anti-Mouse (Licor IRDye 680RD Donkey anti-Mouse IgG, RRID:AB_10953628; 1:5000)
Anti-Mouse (Licor IRDye 800CW Donkey anti-Mouse IgG, RRID:AB_621847; 1:5000)
Anti-Rabbit (Licor IRDye 800CW Donkey anti-Rabbit IgG, RRID:AB_621848; 1:5000)
Anti-Goat (Licor IRDye 680RD Donkey anti-Goat IgG, RRID:AB_10956736; 1:5000)
Halt Protease and Phosphatase Inhibitor (ThermoFisher 78442)
Propidium iodide (PI, ThermoFisher P3566)
Phen Green FL Diacetate (PGFL, Thermofisher P6763)
Standards and Chemicals
GSH (Sigma G4251)
GSSG (Sigma G4501)
NEM (Thermo Scientific 23030)
4-acetamido-4’-maleimidylstilbene-2,2’-disulfonic acid, disodium salt (AMS, Setareh Biotech 6508)
Enzyme Assays
Thioredoxin Reductase Colorimetric Assay Kit (Cayman 10007892)
Glutathione Peroxidase Assay Kit (Cayman 703102)
Plasmids
PX461 (Addgene #48140, RRID:Addgene_48140)
PX462 (Addgene #48141, RRID:Addgene_48141)
AIO-GFP (Addgene #74119, RRID:Addgene_74119)
AIO-mCherry (Addgene #74120, RRID:Addgene_74120)
Cell Lines
HCT116 (ATCC CCL-228, RRID:CVCL_0291)
HT29 (ATCC HTB-38, RRID:CVCL_0320)
MDA-T85 (ATCC CRL-3354, RRID:CVCL_QW84)
MDA-T120 (ATCC CRL-3355, RRID:CVCL_QW85)
Panc1 (ATCC CRL-1469, RRID:CVCL_0480)
MIA PaCa-2 (ATCC CRM-CRL-1420, RRID:CVCL_0428)
MCF7 (Celeste Nelson Lab, Princeton University; ATCC HTB-22, RRID:CVCL_0031)
MDA-MB-231 (Celeste Nelson Lab, Princeton University; ATCC CRM-HTB-26, RRID:CVCL_0062)
U-87 MG (ECACC 89081402 via Sigma, RRID:CVCL_0022)
U-118 MG (ATCC HTB-15, RRID:CVCL_0633)
Mouse Diet
L-Amino Acid Rodent Diet (Research Diets #A10021Bi)
L-Amino Acid Rodent Diet, No Added Selenium (Research Diets #A14042301i)
Methods
H2O2 Detection and Quantitation
H2O2 concentration was measured using the luminescent ROS-Glo H2O2 Assay (Promega) and cross-validated via platinum reduction using a YSI 2900 Biochemistry Analyzer (Xylem Analytics).
Briefly, 75 µL base media was spiked with 20 µL H2O2 substrate solution and 5 µL base media ± ascorbate or ± H2O2, incubated for 30 minutes in a 37 °C incubator maintained at 5% CO2, then developed with 100 µL ROS-Glo detection solution for 20 minutes at room temperature and the luminescence was read using an HT Synergy Multi-Mode Plate Reader (BioTek).
Alternatively, base media was spiked with ascorbate and incubated for 30 minutes. Aliquots were taken after 30 minutes, placed on ice, and run directly on a YSI 2900 Biochemistry Analyzer (Xylem Analytics) with calibration to a standard H2O2 solution performed every 5 samples. Comparable quantitation was obtained using each method for samples prepared in parallel.
Cell Culture, Ascorbate, and Drug Exposure
Stock cells were maintained in DMEM supplemented with 10% complete (non-dialyzed) fetal bovine serum (cFBS). For experimental purposes, cells were seeded and grown in DMEM supplemented with 10% dialyzed fetal bovine serum (dFBS). Cells were seeded in at a packed-cell volume (PCV) of 0.25 µL/mL and grown to approximately 70–80% confluence. Comparable cell density was confirmed both visually under a microscope and by cell count using a Countess II Hemocytometer. Cells were routinely tested for mycoplasma contamination using the PlasmoTest Mycoplasma Detection Kit (Invitrogen).
One hour prior to ascorbate exposure, blank media was pre-equilibrated for temperature and pH in a 5% CO2 incubator at 37 °C. Growth media was aspirated, equilibrated media was spiked with ascorbate and/or other indicated compounds, and the fresh, spiked media was applied to cells for the time indicated.
Metabolomics
Following incubation, metabolites were extracted from cells on dry ice using 80% methanol ± 1 mM N-ethylmaleimide (NEM) pre-chilled on dry ice (extraction solvent) [16; 17]. NEM was included in the extraction buffer for all experiments shown except where explicitly indicated. Briefly, media was aspirated and 500 – 800 µL extraction solvent was applied to each well and the plate placed on dry ice. Insoluble debris was then scraped from each well and transferred to microcentrifuge tubes, placed on dry ice for 30 minutes to facilitate protein precipitation, and spun down at 17,000 x g for 15 minutes at 4 °C before the supernatant metabolite extract was transferred to a fresh tube and maintained on dry ice. For experiments in which intracellular ascorbate was measured, cells were rinsed thrice with warmed PBS prior to extraction.
Metabolite extracts were either dried under gaseous N2 and reconstituted in LC-MS-grade H2O, or directly injected onto the LC-MS instrument in the 80% MeOH extraction buffer. Metabolite extracts re-dissolved in water were analyzed via reverse-phase ion-pairing chromatography (RPLC) coupled to an Exactive Orbitap mass spectrometer (Thermo) as previously described [18]. Metabolite extracts that were directly injected were analyzed using the same instrument or with a Q-Exactive Orbitrap mass spectrometer (Thermo) with separation by hydrophilic interaction chromatography (HILIC) as previously described [19]. LC-MS data was analyzed using El-MAVEN [20]. In all experiments where NEM was used, the GSH-NEM adduct was used for quantitation.
Viability Measurements, Iron Quantification, and Flow Cytometry
Viability was measured after compound exposure for the indicated periods of time by propidium iodide (PI) fluorescence using an LSRII Flow Cytometer (BD Biosciences). Briefly, media was collected (containing dead/detached cells), adhered cells were trypsinized, and the fractions were combined. Cells were spun down, media aspirated, and cells were resuspended in PBS supplemented with 10% cFBS containing 0.5 µg/mL propidium iodide. The single-cell suspension was then analyzed by flow cytometry and viability determined by the relative proportion of PI-positive and negative cells.
Relative Intracellular Labile Iron Quantitation
Relative labile iron content was measured using Phen Green FL Diacetate (PGFL) [21]. Cell populations at near confluence were treated with saline or DFO (500 µM, 12 hours) to chelate the labile iron pool. Culture media was replaced with HBSS containing 2 µM PGFL for 2 hours. Media was aspirated, then cells were trypsinized and quenched with cold HBSS containing dFBS prior to analysis by flow cytometry. The size of the labile iron pool was determined by calculating the difference between the median fluorescence intensity of control and chelated cells.
Glutathione Analytical Chemistry
Glutathione (GSH) and glutathione disulfide (GSSG) solutions were prepared using ice-cold water or 80% methanol ± 1 mM NEM on ice. Samples were run via LC-MS as described above.
Protein Extraction, Western Blotting, and Protein Quantitation
Proteins were extracted using RIPA buffer supplemented with Halt Protease and Phosphatase Inhibitor Cocktail. Cells were exposed to the indicated compounds and the media was aspirated. Cells were rinsed twice with PBS and protein was extracted with cold RIPA buffer on wet ice. The plates were scraped and transferred to microcentrifuge tubes, then sonicated and spun down at 4 °C. Protein content was determined for normalization using the Pierce BCA Protein Assay.
For blotting of serum proteins, protein was precipitated from the soluble fraction using methanol. Protein was precipitated on dry ice and then spun at 4 °C for 15 minutes before protein was extracted in RIPA buffer as described above.
For redox blotting, proteins were extracted and derivatized with 4-acetamido-4’-maleimidylstilbene-2,2’-disulfonic acid (4-AMS) as described [22] with some modifications. Cells were exposed to the indicated compounds and the media was aspirated. Cells were rinsed twice with HBSS and protein was extracted in ice-cold 10% trichloroacetic acid (TCA) buffer. TCA extracts were incubated on wet ice for 30 minutes, then spun in a cold room. The TCA buffer was aspirated, and the pellet was re-suspended in 80% MeOH on dry ice and cooled for an additional 30 minutes before being spun again in a cold room. The 80% MeOH was aspirated, and the protein pellet was subsequently sonicated in a modified RIPA-AMS buffer (10 mM AMS, 1% SDS) and samples were incubated at room temperature for 3 hours with agitation, then spun down. The protein content of the supernatant was then determined via BCA assay as above.
Discontinuous poly-acrylamide gels were cast at a concentration appropriate to resolve the molecular weight of the target protein and loaded with 20–30 µg protein lysate using a reducing, denaturing loading buffer or a non-reducing, denaturing loading buffer for redox blotting. Protein lysates were heated at 95 °C for 10 minutes, which was necessary to reduce GPX proteins to their monomeric states. Western blots were performed using a wet transfer to a PVDF membrane in Towbin Buffer containing 20% methanol at 4 °C on ice using a cooling block pre-chilled to −80 °C.
Membranes were blocked using Licor Odyssey or Intercept Blocking Buffer. Antibody solutions were prepared in the same buffer.
Primary antibodies were diluted in blocking buffer, and membranes were rocked overnight in primary antibody solution at 4 °C. Membranes were washed with PBS-T (0.1% Tween20) and incubated with secondary antibody at room temperature for 1 hour. Membranes were washed with PBS-T and PBS, then imaged using a LI-COR Odyssey Fluorescent Imaging System. Images were processed and quantitated using LI-COR Image Studio Software, then pseudocolor LUTs were adjusted using ImageJ with the Bio-Formats Plugin independent of any quantitation.
Selenoprotein P was quantitated by ELISA using the manufacturer’s protocol.
Respirometry and Determination of Complex-Driven Respiration in Permeabilized Cells
80–100,000 HCT116 cells/well were seeded to near confluence on a 96-well Seahorse XFe96 plate and allowed to adhere overnight in a 5% CO2 incubator at 37 °C. Media was aspirated and replaced with DMEM spiked with ascorbate and/or other compounds for 2 hours. Media was aspirated, and cells were rinsed twice with mitochondrial assay solution 1 (MAS-1), then permeabilized with the Seahorse Plasma Membrane Permeabilization Reagent (PMP, a recombinant perfringolysin) to allow mitochondria direct access to exogenous substrates.
Compared to respirometry using isolated mitochondria, the use of permeabilized cells carries the advantage of maintaining mitochondria in their native locale and avoids biasing the mitochondrial pool, especially when mitochondrial integrity may be compromised by drug treatment [23]. Oxygen consumption was measured using the Seahorse XFe96 Analyzer (Agilent) using conditions appropriate to the indicated respiratory complex.
Baseline respiration in MAS-1 buffer was measured for 3 cycles with a 30-second mixing step, a 30-second waiting step, and a 2-minute measurement period. The PMP reagent was injected to a final concentration of 1 nM, and these measurements were repeated for 5 cycles to ensure respiratory decline following permeabilization. Finally, appropriate compounds were injected to the indicated concentration and oxygen consumption rate was measured for 10 cycles as indicated above.
Complex I → III → IV: ADP (4 mM), 3-hydroxybutyrate (10 mM), malate (10 mM) ± rotenone (2 µM)
Complex II → III → IV: ADP (4 mM), succinate (10 mM), rotenone (2 µM), ± antimycin A (2 µM), myxothiazol (2 µM), malonate (20 mM)
Complex IV: ADP (4 mM), ascorbate (10 mM), TMPD (100 µM), antimycin A (2 µM) ± azide (20 mM)
Enzymatic Assays
TxnRd and GPX assays were performed according to the manufacturer’s protocol using 10 and 40 µg protein for each assay, respectively.
Xenograft Studies
Male 6–8 week-old Cd1 mice were purchased from Charles River and housed in the Animal Care Facility at Princeton University. All procedures were approved by the Princeton University Institutional Animal Care and Use Committee and conformed to NIH guidelines. Mice were acclimated to the facility for at least one week before any experimental manipulations were performed. Mice were acclimated to a selenium-free diet for 3 weeks prior to xenograft implantation.
HCT116 and U87 cells were cultured as described above. Cells were trypsinized, then suspended in DMEM (without FBS) and mixed with matrigel. 1e6 cells were injected in 200 µL into the flanks of mice and allowed to grow until palpable (7–14 days) before ascorbate was administered. Ascorbate solutions were prepared from ascorbic acid, pH adjusted, and filter sterilized prior to administration by intraperitoneal (i.p.) injection. Tumor size was measured using calipers and determined according to the formula V = (W2 * L)/2 [24]. A humane endpoint of 2000 mm3 was used.
Statistical Analysis, Data Visualization, and Illustrations
Statistical analysis and data visualization were performed using GraphPad Prism. LD50 values were calculated by interpolating a survival curve fitted using a four-parameter logistic regression. Statistical significance was assessed using a student’s t-test, ANOVA, and Kaplan-Meier survival analysis.
Illustrations were created using Biorender under an institutional license.
Results
Pharmacologic ascorbate damages cells via H2O2, not dehydroascorbate
Pharmacologic ascorbate exposure has been proposed to elicit cytotoxic stress through either H2O2 generation or dehydroascorbate (DHA)-dependent glutathione depletion (Fig 1a) [2,3]. To help distinguish between these possibilities, we performed metabolomics on colorectal cancer HCT116 and glioblastoma U87-MG cells following a short exposure to equimolar ascorbate or DHA (Fig 1b,c; S1a,b). Compared to DHA, ascorbate more broadly impacted the metabolome by elevating glutathione disulfide, depleting NAD, and inducing an energy stress signature of low creatine phosphate and nucleotide triphosphates coupled with high nucleotide monophosphates and diphosphates, nucleosides, and bases (Fig 1b,c; S1a,b). The response to ascorbate was closely mimicked by H2O2, but not by diamide, a thiol-oxidizing agent (Fig 1b,c; S1a). The similarity between the ascorbate and H2O2 responses is consistent with the reported generation of H2O2 by high concentrations of ascorbate [8,25] and suggests that ascorbate impacts metabolism primarily by producing H2O2.
One of the metabolites most strongly depleted by both ascorbate and H2O2 is NAD (Fig 1b,c S1b). Like H2O2, ascorbate treatment has been shown to induce double-stranded DNA (dsDNA) breaks, activating the DNA repair enzyme poly-ADP ribose polymerase (PARP) [2], which consumes NAD as a substrate. Both ascorbate and H2O2 treatment resulted in histone H2AX phosphorylation, a marker of dsDNA breaks, whereas the response to equimolar DHA was less (Fig S1c). Administration of the PARP inhibitor olaparib attenuated the acute metabolic response of HCT116 cells to ascorbate by sparing NAD (Fig S1d). The preservation of NAD mitigated the energy stress signature and prevented accumulation of fructose bisphosphate and dihydroxyacetone phosphate (Fig S1d), which are located upstream of the NAD-dependent glyceraldehyde phosphate dehydrogenase (GAPDH) step in glycolysis. Thus, ascorbate-induced GAPDH inhibition likely arises primarily from PARP-mediated NAD depletion rather than redox changes in GAPDH itself [2,26]. Co-administration of PARP inhibitor also prevented the accumulation of sedoheptulose and octulose bisphosphates, which rise when GAPDH is blocked [27,28].
High concentrations (mM) of ascorbate have been shown to generate H2O2 in vitro [8,25]. We confirmed that, when added to standard cell culture media, a pharmacologic concentration of ascorbate (10 mM) generated high levels of H2O2, whereas generation by DHA was substantially less (Fig S1e) [25]. We also found that ascorbate exhibited a much stronger cytotoxic effect in HCT116 cells compared to an equimolar dose of DHA (Fig 1d) and that adding the H2O2-scavenging enzyme catalase to the media prevented ascorbate-mediated cytotoxicity (Fig 1d). In contrast, PARP inhibition did not prevent ascorbate-mediated cell death (Fig 1d), implicating DNA damage itself, or other steps upstream of the energetic crisis, as the cause of cytotoxicity.
Although ascorbate toxicity was ultimately dependent on extracellular H2O2 generation, we wondered if DHA could still play a role in sensitizing cells to H2O2 by depleting reduced glutathione via the glutathione-dependent DHA reductase GSTO1 (Fig 1a) [29,30]. Cells treated with ascorbate rapidly accumulated intracellular ascorbate and DHA, with the DHA level at approximately 10% that of ascorbate (Fig S2a,b). To explore GSTO1’s role, we engineered clonal knock-out cell lines (HCT116-ΔGSTO1) (Fig S2c). These cells lacked DHA reductase activity (Fig S2d) but were nevertheless indistinguishable from control cells in both viability and metabolic response when challenged by ascorbate or DHA (Fig S2e,f), indicating that glutathione depletion via GSTO1 does not play a prominent role in high-dose ascorbate toxicity.
Abiotic oxidation of glutathione by ascorbate
Ascorbate was previously shown to profoundly decrease the cellular pool of reduced glutathione (GSH) [2]. However, we observed only a minimal change (Fig 1b). In attempting to understand this discrepancy, we noted that, in the previous work, metabolite extracts were dried prior to LC-MS analysis. We hypothesized that the presence of ascorbate in metabolite extracts was causing abiotic GSH consumption during the drying process. Consistent with this, we observed profound GSH loss upon drying of ascorbate-treated samples (Fig 2a,b). This reflects reactivity at the thiol moiety of GSH, as thiol derivatization with N-ethylmaleimide (NEM) prevented the loss of GSH (Fig 2b) [17]. Experiments where cells were extracted in the presence of NEM (which selectively reacts with GSH and not GSSG) (Fig S3a,b) and directly analyzed by LC-MS revealed that pharmacologic ascorbate treatment increases total GSSG, and thus, the GSSG/GSH ratio, but causes only a mild decrease in GSH (Fig 2b,c). As GSSG readily accumulates abiotically, the effect of ascorbate on cellular GSSG was most clearly observed with direct analysis of NEM-derivatized samples (Fig 2c). Thus, while ascorbate causes bona fide thiol redox stress in HCT116 cells, it does not cause substantial GSH depletion.
Figure 2: Glutathione oxidation by pharmacologic ascorbate is an abiotic phenomenon.
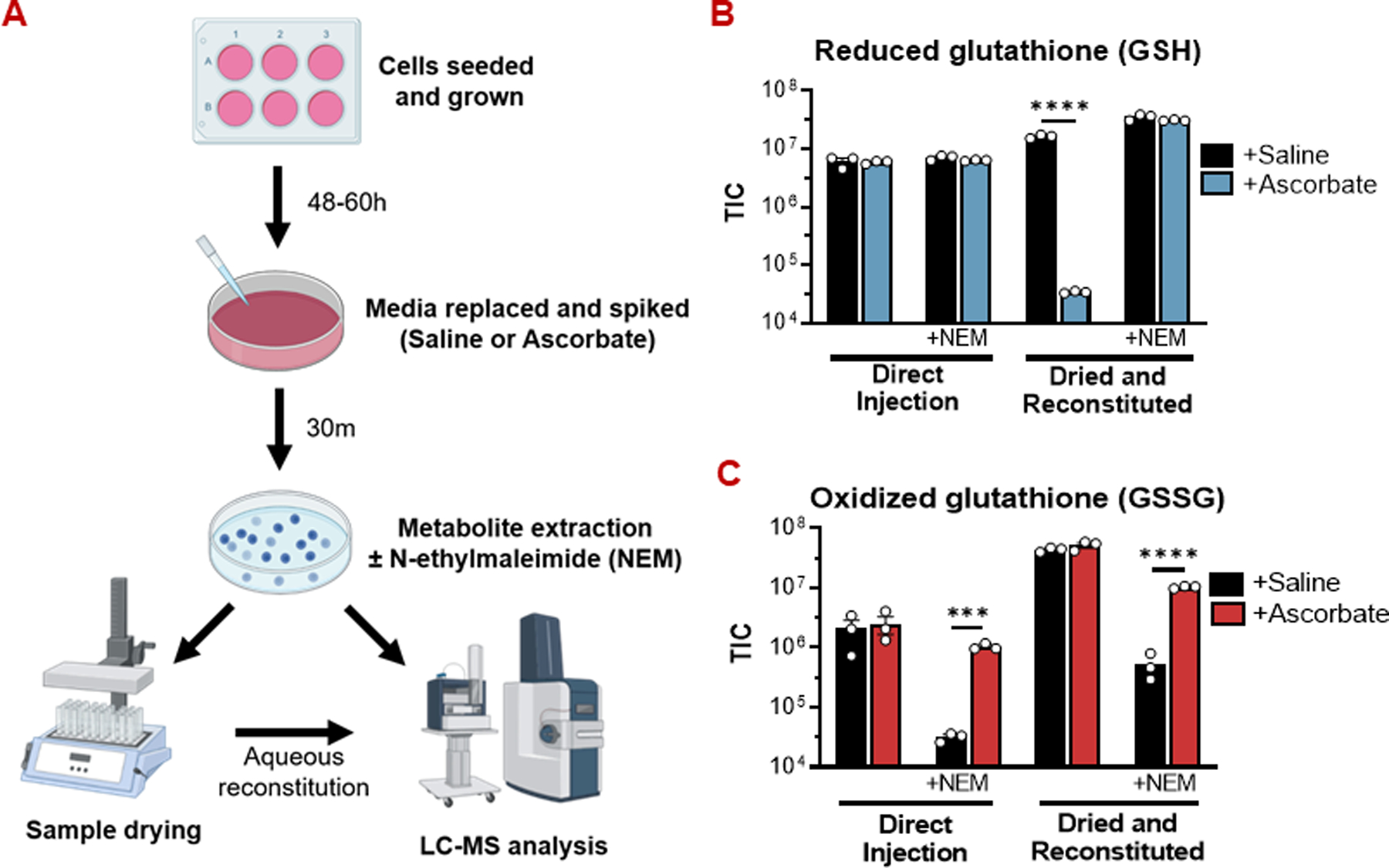
(A) Experimental workflow for preparing metabolite extracts for LC-MS.
(B) Drying metabolite extracts from ascorbate-treated cells in the absence of N-ethylmaleimide (NEM) causes a dramatic reduction in reduced glutathione (GSH) abundance. In experiments where NEM was included in the extraction solvent, the GSH-NEM adduct was measured. Otherwise, free GSH was measured.
(C) Drying metabolite extracts without NEM protection masks the true extent of oxidized glutathione (GSSG) accumulation in ascorbate-treated cells.
For all GSH/GSSG measurements, n = 3 biological replicates.
All data are mean ± SEM. **** p < 0.0001 by unpaired t-test.
Ascorbate toxicity depends on intracellular iron
Generation of H2O2 by ascorbate has been proposed to be an iron-dependent process [3]. We observed that ascorbate generated H2O2 more extensively in cell culture media (DMEM), which contains iron, than in phosphate buffered saline (PBS). Adding iron to PBS, however, did not enhance H2O2 generation by ascorbate nor did the preparation of DMEM without iron, nor iron chelation in DMEM, decrease H2O2 generation (Fig S4a,b). Consistent with these observations, we found that cells exposed to ascorbate in iron-free media exhibited a similar degree of metabolic stress compared to cells exposed in normal media (Fig 3a). Thus, extracellular iron does not appear to be required for ascorbate’s effects. A previous report has shown that supplementing higher concentrations (5–10 µM) of iron into culture media (DMEM concentration approximately 230 nM) has a protective effect against high-dose ascorbate by stimulating iron-catalyzed decomposition of peroxide in culture media, damaging macromolecules in the extracellular space rather than within cells [31]. Consistent with this mechanism, we found that supplementing 10–100 µM iron into culture media prevented DNA damage and subsequent energetic stress to a similar extent as catalase (Fig S4c,d).
Figure 3: Ascorbate toxicity is iron-dependent but does not occur via ferroptosis.
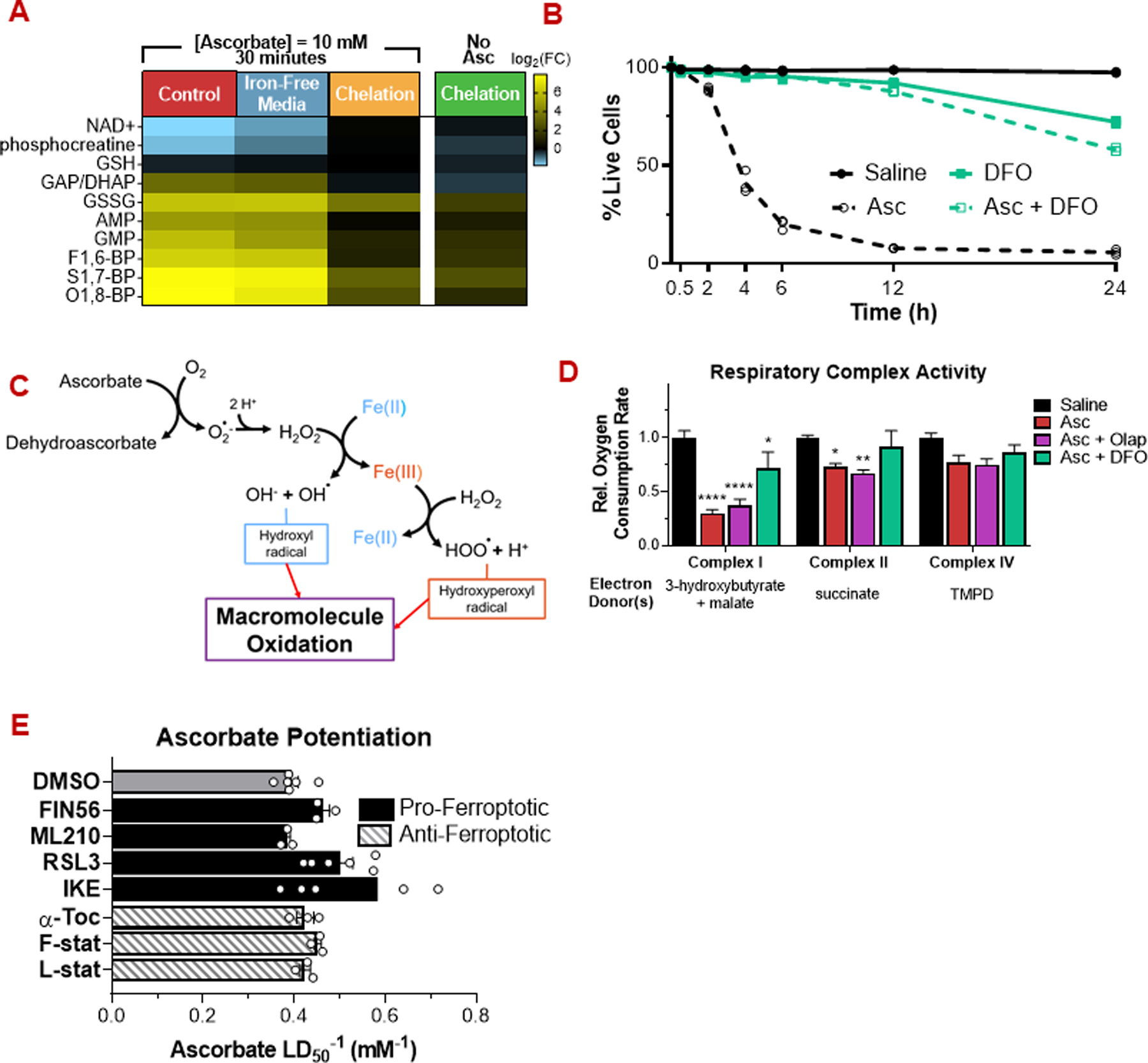
(A) Depletion of the intracellular labile iron pool by chelation (deferoxamine (DFO), 500 µM, 12 h) prevents induction of energetic stress by pharmacologic ascorbate (10 mM, 30 min). In contrast, depletion of the extracellular iron pool by exposure in iron-free media has a minimal effect.
(B) Deferoxamine prevents ascorbate cytotoxicity.
(C) Schematic of ROS generation by ascorbate and iron.
(D) Ascorbate (10 mM, 2h) decreases respiratory chain complex activity, in a manner rescuable by deferoxamine but not PARP inhibition (10 µM olaparib).
(E) Pro-ferroptotic (10 µM) and anti-ferroptotic (1 µM) small molecules have a minimal effect on ascorbate cytotoxicity.
For metabolomics and viability assays, n ≥ 3 biological replicates. For respiratory complex activity assays, n = 5 biological replicates.
All data are mean ± SEM. * p < 0.05; ** p < 0.005; *** p < 0.0005; **** p < 0.0001 by unpared t-test or two-way ANOVA.
In contrast to the lack of requirement of extracellular iron, intracellular iron appears to be critical to ascorbate cytotoxicity. Specifically, we found that pre-loading HCT116 or U87-MG cells with the iron-chelating agent deferoxamine (DFO) protected them from ascorbate’s metabolic and cytotoxic effects (Fig 3a,b; S5a). A possible explanation for the role of iron in ascorbate toxicity is that Fe2+ reduces H2O2 to the more reactive hydroxyl radical (Fig 1a; 3c). Hydroxyl radicals are known to damage enzymes in the respiratory chain [3]. Indeed, the activities of mitochondrial respiratory complexes I and II were significantly decreased following exposure to high-dose ascorbate (Fig 3d). This decline was not driven by a PARP-dependent decrease in NAD+ availability (Fig S1d; 3d). Iron chelation largely rescued respiratory complex activity (Fig 3d) without decreasing mitochondrial H2O2 levels (Fig S5b), suggesting that respiratory complex damage is, in part, mediated by the hydroxyl radical formed by the reaction of H2O2 with free intracellular iron. We examined the relative levels of free intracellular iron in a panel of cancer cell lines (Fig S5c). We did not observed an obvious correlation between free intracellular iron levels and ascorbate sensitivity (Fig S5d), suggesting that additional factors may contribute to the differential sensitivity displayed by cancer cell lines.
Ferroptosis modulators do not affect ascorbate sensitivity
The dsDNA breaks observed following treatment with H2O2 or ascorbate (Fig S1c; S4c) likely result from the hydroxyl radical formed by the Fenton reaction between H2O2 and Fe2+ (Fig 1a, 3c) [32]. Iron-dependent hydroxyl radical production can cause cell death via lipid peroxidation in a necrotic mode of cell death termed ferroptosis [10; 11]. To assess whether ascorbate kills cells via ferroptosis, we tested it in combination with pro-ferroptotic and anti-ferroptotic small molecules. The lipophilic antioxidants α-tocopherol, ferrostatin, and liproxstatin, which prevent ferroptosis, [32] failed to protect against pharmacologic ascorbate (Fig 3e). Moreover, the pro-ferroptotic compounds FIN56, which promotes degradation of the lipid hydroperoxidase GPX4 and ML210, a GPX4 inhibitor, failed to augment ascorbate’s activity. RSL3, another GPX4 inhibitor, and imidazole ketone erastin (IKE), an inhibitor of cystine uptake and glutathione synthesis, only mildly enhanced ascorbate activity (Fig 3e). Collectively, these observations argue that ascorbate induces an iron-dependent form of cell death distinct from ferroptosis.
Selenium supplementation protects cells from ascorbate
Selenium supplementation has been shown to protect cells against iron-dependent cell death by supporting increased expression of selenoproteins, including GPX4, which defend against oxidative stress [15]. Despite the minimal impact of GPX4 inhibition on ascorbate activity, we wondered if selenium supplementation would protect cells from ascorbate. Cultured cells suffer from chronic selenium deficiency and show low selenoprotein expression under normal culture conditions [14]. We found that cancer cell of multiple origins grown in medium supplemented with sodium selenite displayed a concentration-dependent resistance to ascorbate, with a maximal effect at approximately 30 nM selenite (Fig 4a,b; S6a–c). This resistance appeared to derive from increased capacity to clear intracellular ROS (Fig 4c). In HCT116 cells, selenium supplementation increased cellular TxnRd and GPX activities and GPX expression (Fig 4d; S6d,e). Increased TxnRd expression was not observed (Fig S6d,e), possibly because this protein can be expressed (with decreased catalytic activity) with cysteine, rather than selenocysteine, in its active site [33]. Consistent with an increased ability to detoxify ROS, selenium-supplemented cells suffered less severe oxidative damage in response to ascorbate (Fig S6a,b).
Figure 4: Selenium availability suppresses ascorbate cytotoxicity by enhancing selenoprotein activity.
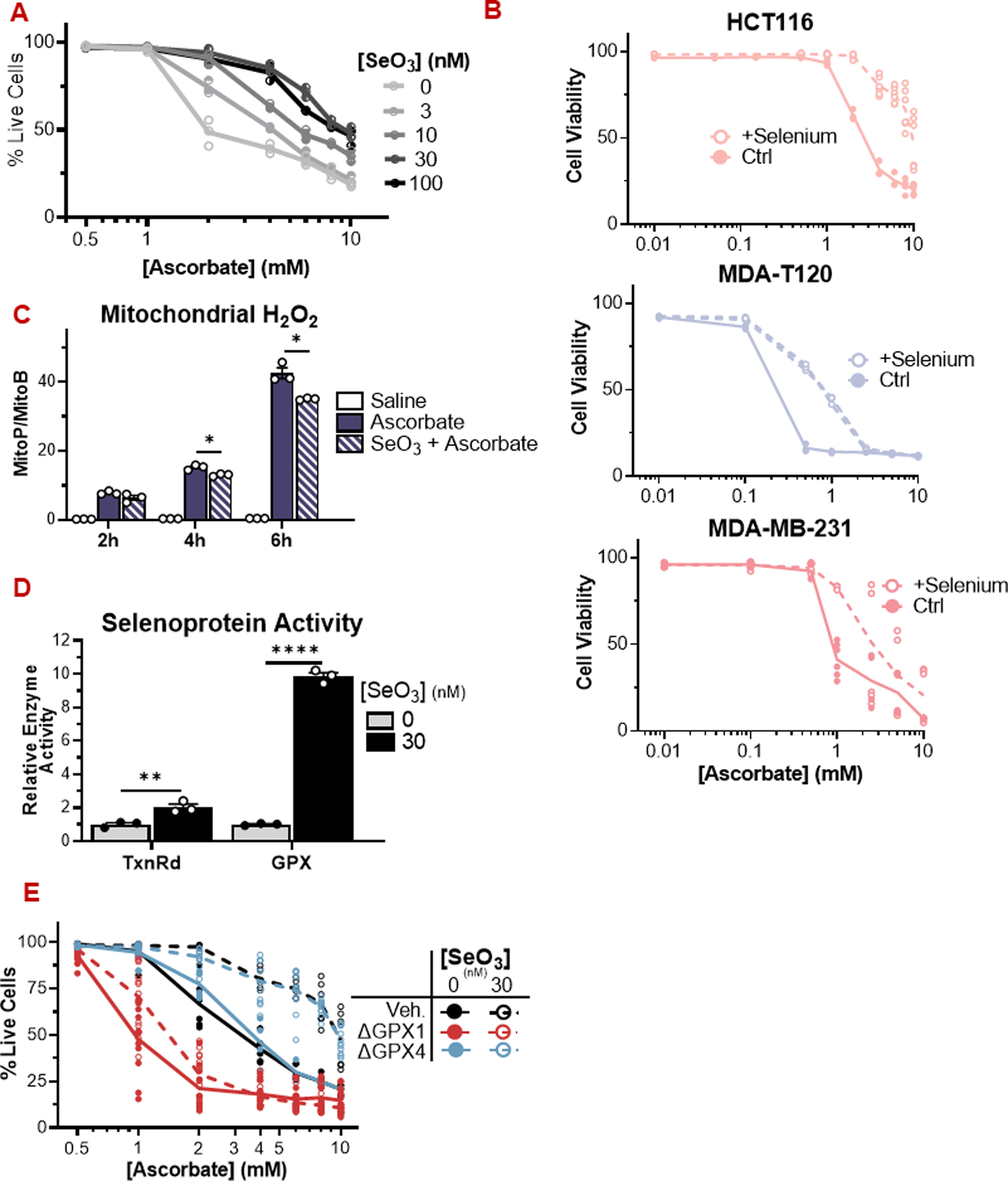
(A) HCT116 cells grown in media supplemented with selenite are resistant to ascorbate (24 h).
(B) HCT116 (colorectal), MDA-T120 (thyroid), and MDA-MB-231 (breast) cancer cells are more resistant to ascorbate when grown in DMEM supplemented with 30 nM sodium selenite.
(C) HCT116 cells grown in selenite-supplemented media display a reduced mitochondrial peroxide load over time compared to control cells exposed to pharmacologic ascorbate (1 mM).
(D) Cells grown in selenite-supplemented media display increased activity of the antioxidant selenozyme sytems TxnRd and GPX. The activity assays measure gross TxnRd and GPX activity, reflecting the combined activities of TxnRd1/2 and multiple GPX isoforms in each assay.
(E) HCT116 cells lacking GPX1, but not GPX4, are sensitized to pharmacologic ascorbate under low and high selenium conditions.
GPX: Glutathione peroxidase; TxnRd: Thioredoxin reductase; SeO3: selenite.
For enzymatic assays, n = 3 biological replicates. For viability assays, n ≥ 3 biological replicates.
All data are mean ± SEM. ^ p < 0.1; * p < 0.05; ** p < 0.005; *** p < 0.0005; **** p < 0.0001 by unpaired t-test or two-way ANOVA.
As selenoproteins are intimately associated with maintaining redox homeostasis, we wondered if the increased activity of one or more of GPX1, GPX4, TxnRd1, and/or TxnRd2 could be responsible for the ascorbate resistance afforded by selenium supplementation. To investigate the role of each selenoprotein in protecting cells from pharmacologic ascorbate, we engineered clonal HCT116 cell lines lacking each of the four selenoproteins (Fig S6e). We also knocked out catalase, which detoxifies H2O2 in a selenium-independent manner (Fig S6e). Under both standard and selenium-supplemented conditions, HCT116-ΔGPX1 cells were dramatically more sensitive to ascorbate than control cells (Fig 4e). HCT116-ΔCatalase, ΔTxnRd1, and ΔTxnRd2 were also sensitized to ascorbate, whereas HCT116-ΔGPX4 cells were not (Fig 4e; S6f). Strikingly, loss of GPX1 eliminated the protective effect of selenium against ascorbate. Thus, in addition to catalase, the selenoproteins TxnRd1, TxnRd2, and especially GPX1 protect cancer cells from ascorbate-induced oxidative stress.
The pentose phosphate pathway supplies NADPH that supports GPX1 and TxnRd1 activity
To further assess the roles of the thioredoxin reductase system in defending against ascorbate toxicity, we employed the pan-TxnRd inhibitor auranofin [34]. Auranofin co-administration sensitized cells to ascorbate toxicity (Fig 5a) Similar to auranofin, the TxnRd1 inhibitor TRi-1 also synergized with ascorbate (Fig 5a) [35]. Redox analysis of cytosolic/nuclear and mitochondrial thioredoxins (Txn1 and Txn2, respectively) showed that Txn2, but not Txn1, was rapidly oxidized following exposure to pharmacologic ascorbate (Fig S7a). We wondered whether HCT116 cells maintained cytosolic Txn1 in the reduced state due to a robust cytosolic antioxidant metabolism.
Figure 5: The pentose phosphate pathway defends against ascorbate via selenoproteins.
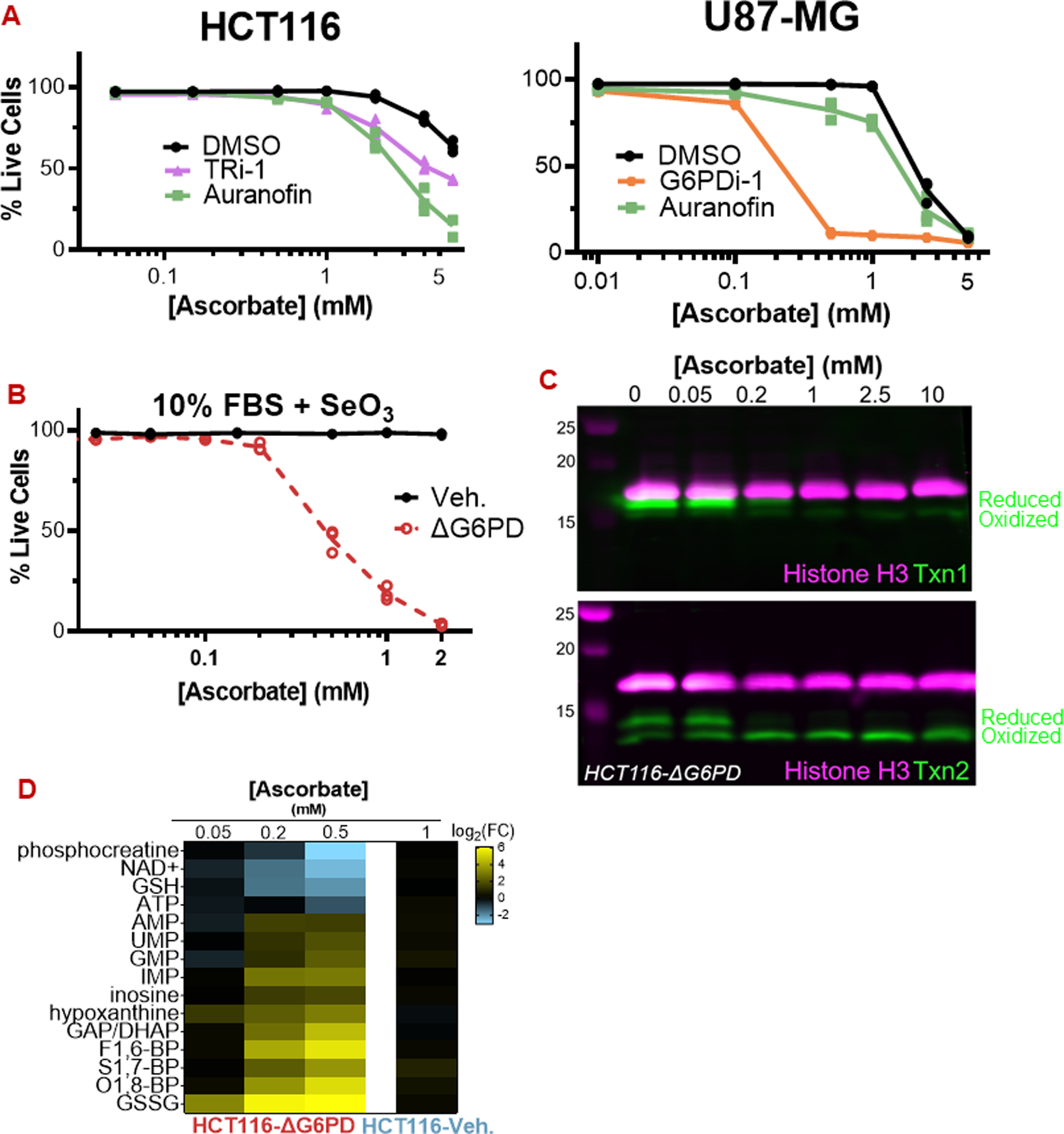
(A) Inhibition of TxnRd1 by TRi-1 (10 µM) and auranofin (5 µM) sensitize HCT116 and U87-MG cells to ascorbate. G6PDi-1 (50 µM) also sensitized U87-MG cells.
(B) HCT116-ΔG6PD cells are highly sensitive to ascorbate even when grown with 30 nM selenite.
(C) Ascorbate (30 min) causes degradation of cytosolic Txn1 and oxidation of mitochondrial Txn2 in HCT116-ΔG6PD.
(D) Ascorbate (30 min) induces energetic stress and GSH depletion at sub-millimolar concentrations in HCT116-ΔG6PD cells.
For all data, n = 3 biological replicates. Representative immunoblots are shown.
All data are mean ± SEM.
Thioredoxin reductase enzymes require NADPH as an electron donor to reduce the oxidized sulfhydryl residues in thioredoxin proteins. Moreover, NADPH is critical for maintaining reduced glutathione, the electron donor for the GPX enzymes. To assess the importance of NADPH in preventing ascorbate toxicity, we employed HCT116-ΔG6PD cells, which cannot generate NADPH from the pentose phosphate pathway [36]. ΔG6PD cells were exquisitely sensitive to ascorbate under both low and high-selenium conditions (Fig 5b; S7b). Similarly, the small-molecule G6PD inhibitor G6PDi-1 sensitized HCT116 and U87-MG cells to ascorbate (Fig 5a; S7c) [37]. Ascorbate depleted the reduced (active) form of Txn1 selectively in ΔG6PD cells and Txn2 in both control and ΔG6PD cells, though the KO cells were more sensitive (Fig 5c; S7a). Thus, selenoprotein activity cannot protect cells from ascorbate when NADPH supply is compromised.
Indeed, we found that the metabolome of HCT116-ΔG6PD cells exhibited signs of oxidative stress at just 50 µM ascorbate, while control cells remained insensitive at 1 mM, a 20-fold difference in concentration (Fig 5d). GSH was depleted by more than 4-fold in ∆G6PD cells challenged with 0.5 mM ascorbate, in contrast to control cells that maintained more than 85% of their GSH pool when challenged with 10 mM ascorbate (Fig 5d; 1b,c). Consistent with TxnRd1 being functionally incapacitated by lack of G6PD-derived NADPH, G6PD-deficient cells were not further sensitized to ascorbate by the co-administration of auranofin or TRi-1 (Fig S7b). Collectively, these data demonstrate that ascorbate-derived H2O2, in combination with intracellular iron, generates hydroxyl radicals, and that cells control the resulting damage using selenoproteins powered by NADPH derived from the pentose phosphate pathway.
Dietary selenium restriction and pharmacologic ascorbate decrease U87-MG tumor growth in vivo
As selenium strongly suppresses ascorbate toxicity in vitro, we wondered if restricting dietary selenium could sensitize tumors to pharmacologic ascorbate in vivo. To assess this possibility, we fed Cd1-nude mice to a selenium-deficient diet. After three weeks, mice exhibited a dramatic reduction in the levels of the circulating selenoproteins GPX3 and selenoprotein P (SELP) (Fig 6a), which account for 60–90% of circulating selenium [38,39]. Body weight remained unchanged (Fig S8a).
Figure 6: Dietary selenium restriction enhances ascorbate activity against U87-MG glioblastoma tumors.
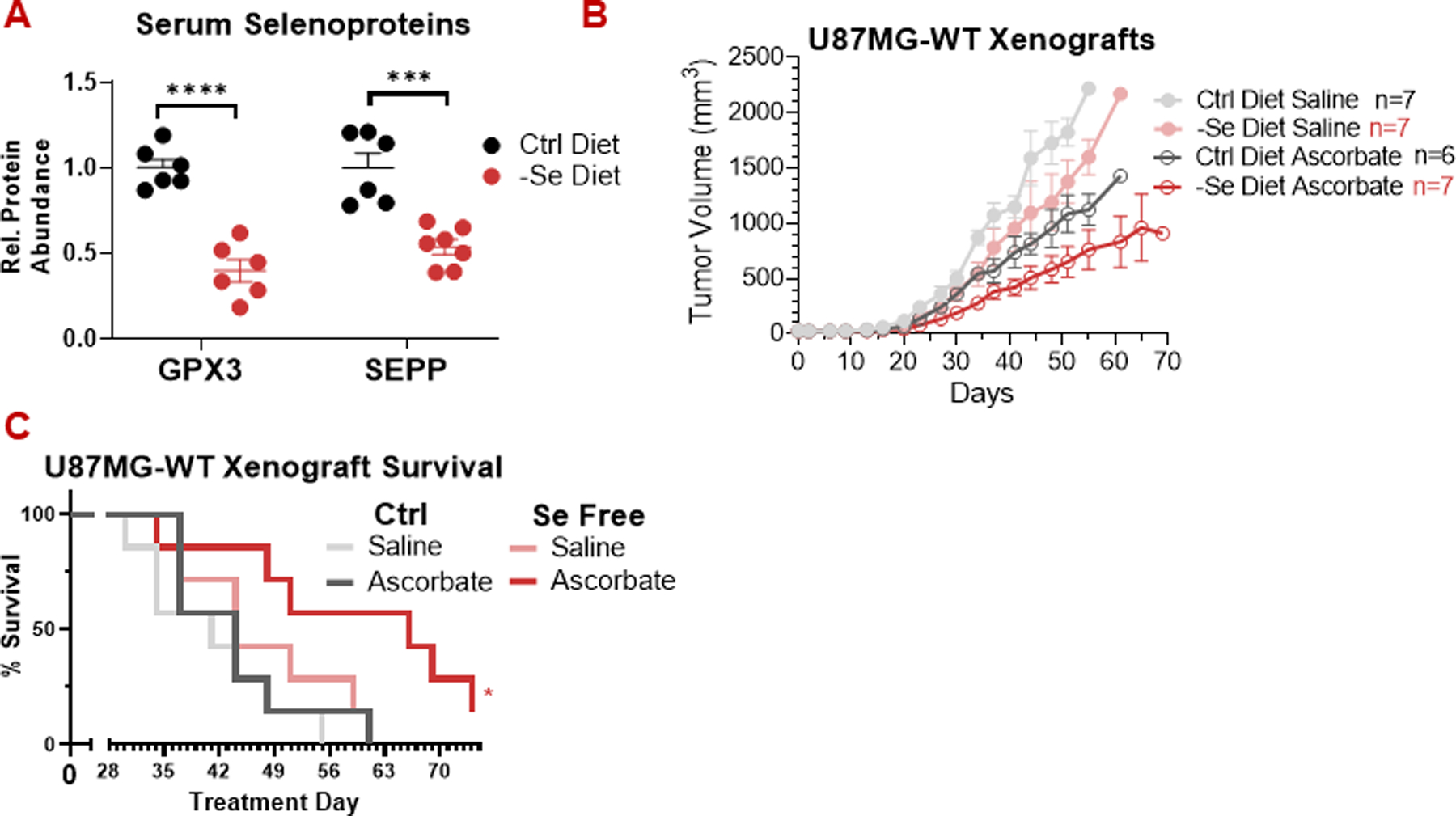
(A) Maintaining mice on a selenium-free diet for 3-weeks prior to xenograft implantation decreased serum glutathione peroxidase 3 (GPX3) and selenoprotein P (SEPP) levels.
(B) Pharmacologic ascorbate (4 g/kg daily) slowed the growth of U87-MG xenografts in both control and selenium-free diets. Selenium restriction alone did not significantly decrease tumor size, but it did lead to stronger response to ascorbate.
(C) In mice harboring U87-MG xenografts, the combination of dietary selenium restriction and pharmacologic ascorbate (4 g/kg daily) enhanced survival.
All data, n ≥ 6 mice.
All data are mean ± SEM. * p < 0.05; ** p < 0.005; *** p < 0.0005; **** p < 0.0001 by unpaired t-test or Kaplan-Meier survival analysis.
We implanted mice on selenium-replete (control) and deficient diets with HCT116 and U87-MG xenografts and monitored tumor growth in response to treatment with high-dose ascorbate, which has previously been shown to reduce tumor growth as a monotherapy in HCT116 xenografts and extend lifespan in combination with irradiation in U87-MG xenografts [2,3]. Irrespective of dietary selenium status, we observed no effect of ascorbate on HCT116 tumors (Fig S8b,c). In U87-MG tumors, we did observe growth suppression by ascorbate in both the control and selenium-deficient diet conditions (Fig 6b). Ascorbate was more effective at suppressing tumor growth in the context of the selenium-deficient diet. Importantly, we observed significant survival benefits selectively in the mice treated with a combination of selenium-deficient diet and pharmacologic ascorbate (Fig 6c).
Discussion
While numerous studies have demonstrated that high-dose ascorbate is cytotoxic to cancer cells in vitro, the mechanism underlying this toxicity has remained unclear [1–3]. Two distinct models have arisen based on different features of ascorbate chemistry and cancer cell metabolism [2,3]. Our results support a model in which ascorbate exerts cytotoxicity through extracellular H2O2 generation and independent of the oxidized metabolite DHA (Fig 7).
Figure 7: Iron-dependent, selenium-repressible ascorbate toxicity.
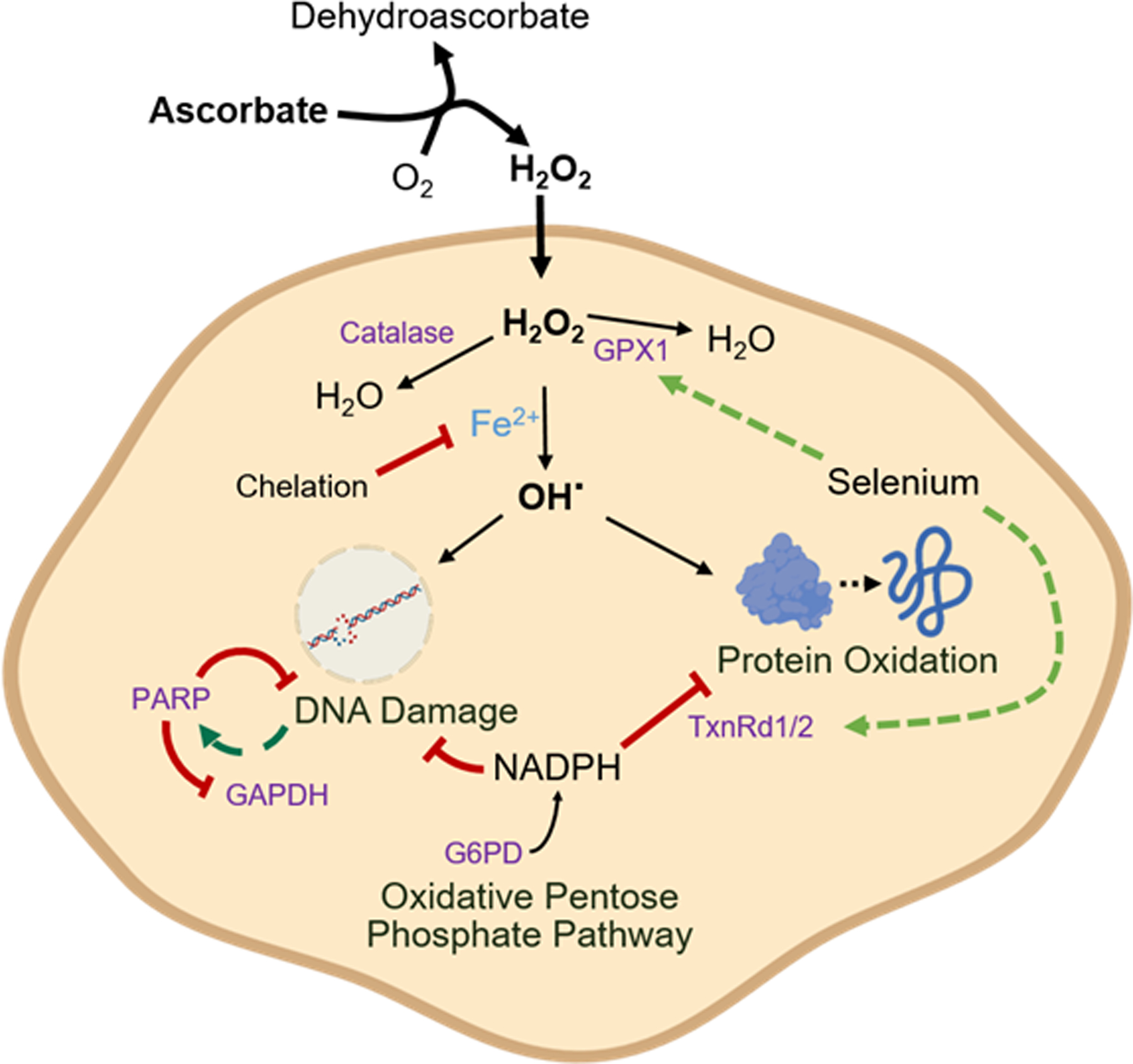
Ascorbate generates H2O2 in the extracellular space, which crosses the plasma membrane. Inside the cell, H2O2 reacts with free iron (Fe2+), generating the reactive hydroxyl radical (OH·). H2O2 is detoxified by catalase and GPX1, and selenium availability drives GPX1 expression. The hydroxyl radical causes both protein oxidation, which is repaired by the thioredoxin reductase (TxnRd) system, and DNA damage, which activates PARP, consumes NAD, and thereby inhibits GAPDH. The cellular defense against these oxidative injuries ultimately relies on NADPH generated in the pentose phosphate pathway by the activity of glucose 6-phosphate dehydrogenase (G6PD).
This toxicity depends on free intracellular iron, which acts in opposition to selenium bioavailability. Intracellular iron sensitizes cells to H2O2, presumably by promoting hydroxyl radical generation. Selenium protects cells by supporting the expression of antioxidant defense selenoproteins including GPX1 and thioredoxin reductase 1 and 2. Cancer cells become further sensitized to the pro-oxidant effects of pharmacologic ascorbate when the activity of any of these proteins is lost and especially when the pentose phosphate pathway – which provides NADPH to power these proteins – is blocked (Fig 7).
Although it has been proposed that cancer cell sensitivity may derive from elevated expression of the ascorbate transporter SVCT2 [40], our data suggest that in vitro ascorbate toxicity comes from extracellular H2O2 generation, not intracellular ascorbate accumulation (Fig 1a–d; S1a–e; S2a,b). H2O2 was a metabolic phenocopy for high-dose ascorbate (Fig 1b,c; S1a), high-dose ascorbate generates H2O2 under standard culture conditions, (Fig S1e) and ascorbate’s cytotoxicity is blocked by extracellular catalase (Fig 1d).
Similarly, it has been proposed that cancer cell sensitivity depends on GLUT1-mediated uptake of dehydroascorbate, which is reduced to ascorbate by glutathione in a reaction catalyzed by GSTO1, potentially depleting the intracellular reduced glutathione pool. However, we found that ΔGSTO1 cells were not protected from either ascorbate or DHA, that ascorbate does not deplete glutathione markedly, and that ascorbate is the stronger cytotoxic agent (Fig 1b–d; S2a–f; 2a–c).
Another hypothesis regarding the sensitivity of cancer cells to ascorbate involves an increased intracellular labile iron pool in cancer cells, which promotes the generation of hydroxyl radicals from Fenton chemistry [3,41]. We confirm that sensitivity to ascorbate requires iron, and that toxic free radical damage accumulates in both DNA (leading to PARP activation) and Fe/S cluster proteins in the mitochondrial respiratory chain (Fig 3a–d). Based on environmental, pharmacological, and genetic modulators of ascorbate toxicity, the cytotoxic pathway runs through extracellular and intracellular H2O2, hydroxyl radicals, and thioredoxin-repairable protein oxidation (Fig 7). Lipid peroxidation and canonical ferroptosis inducers do not appear to be on the critical path (Fig 3e; 4e).
Selenium antagonizes the effects of ascorbate and intracellular iron by promoting expression of the glutathione-dependent H2O2-clearing enzyme GPX1 and thioredoxin reductase 1 and 2 (Fig 4a–e; S6a–f). The importance of these systems is easily underappreciated in cell culture due to low selenium availability. Maintenance of adequate reduced NADPH and glutathione to power these selenoproteins depends on the pentose phosphate pathway and its gating enzyme G6PD, as exemplified by the oxidation of Txn1 by ascorbate in ΔG6PD cells. (Fig 5a–d; S7a–c, 7)
Intravenously administered ascorbate has consistently been shown to be well-tolerated in cancer patients and healthy individuals [4], with the notable exception of G6PD-deficient individuals in whom intravenous ascorbate administration causes hemolysis [42]. Pending the advent of selective G6PD inhibitors with suitable pharmacologic properties, the tolerability of acute pharmacological G6PD inhibition with ascorbate remains to be determined. However, our data suggest that limiting dietary selenium may be sufficient to sensitize tumors to ascorbate and other pro-oxidant therapies.
Overall, we observed less anticancer activity from ascorbate than prior studies in the same model [2; 9]. This may reflect bias towards publication of positive results [43], which impacts anticancer research more generally [44].
Mineral bioavailability is a key determinant for how oxidative stressors affect cells [45], and essential minerals have been linked to cancer in a variety of contexts. Iron has been linked to increased cancer risk, and iron metabolism is dysregulated in many types of cancer cells [41]. High serum selenium levels have been suggested to prevent some cancers [46]. Similarly, magnesium has been associated with decreased cancer risk [47] with its deficiency inhibiting primary tumor growth but promoting metastasis [48]. Manganese is indispensable for immune sensing of tumors and enhances the adaptive immune response [49]. Here, we add to these results by showing that dietary selenium deficiency sensitizes tumors to ascorbate (and perhaps, more generally, to oxidative damage).
Collectively, our results imply that high-dose ascorbate acts through a free radical mechanism. Tumor cells may be preferentially sensitive due to increased labile iron, enhanced endogenous oxidative stress, or higher sensitivity to DNA damage. A key, and until now, underappreciated, determinant of oxidative defense status is selenium bioavailability. To our knowledge, this is the first study to assess dietary selenium depletion as part of an anticancer treatment. Whether selenium or selenoprotein manipulations can be applied therapeutically against cancer merits further investigation.
Limitations
Our studies of ascorbate’s mode of action were largely conducted in vitro. Our in vivo evidence for tumor sensitization to ascorbate by selenium deficiency is limited to a single xenograft model. In this model, the combination of pharmacologic ascorbate and dietary selenium deprivation shows therapeutic benefits, but we did not assess whether the combination is synergistic or merely additive, nor whether its benefits occur via the mechanism observed in vitro by enhancing tumor oxidative stress. Further studies of ascorbate’s mechanism of action in vivo and of the interaction between dietary selenium and pharmacologic ascorbate are merited.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Significance.
Selenium restriction augments ascorbate efficacy and extends lifespan in a mouse xenograft model of glioblastoma, suggesting that targeting selenium-mediated antioxidant defenses merits clinical evaluation in combination with ascorbate and other pro-oxidant therapies.
Acknowledgements
The authors thank Dr. Melanie McReynolds from the Pennsylvania State University for her advice on NAD+ metabolism and PARP inhibitors and Rishabh Sharan for his assistance writing code to facilitate data analysis. This work is supported by grant R01 CA163591 (J.D.R.), by a Stand Up To Cancer-Cancer Research UK-Lustgarten Foundation Pancreatic Cancer Dream Team Research Grant (Grant Number: SU2C-AACR-DT20-16) and LUSTGARTEN 2015–002 (J.D.R.), and by Ludwig Cancer Research. Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. The authors also thank Christine Decoste and Katherine Rittenbach at the Princeton Flow Cytometry Resource Facility. The Princeton University Flow Cytometry Resource Facility is supported, in part, with funding from NCI-CCSG P30CA072720-5921. C.S.R.J is supported by the NIGMS of the National Institutes of Health under grant T32GM007388.
Funding
This work is supported by grant R01 CA163591 (J.D.R.), by SU2C AACR-DT20-16 and LUSTGARTEN 2015–002 (J.D.R.), and by Ludwig Cancer Research. The Princeton University Flow Cytometry Resource Facility is supported, in part, with funding from NCI-CCSG P30CA072720-5921. C.S.R.J is supported by the NIGMS of the National Institutes of Health under grant T32GM007388.
Footnotes
Competing Interests
J.D.R. is a paid adviser and/or stockholder in Colorado Research Partners, Kadmon Pharmaceuticals, L.E.A.F. Pharmaceuticals, Rafael Pharmaceuticals and its subsidiaries, Empress, and Agios Pharmaceuticals; a paid consultant of Pfizer; a founder, director, and stockholder of Farber Partners and Serien Therpeutics. C.S.R.J. declares no conflicts of interest.
References
- 1.Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, Levine M, et al. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic Biol Med 2011; 50: 1610–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Nature 2015; 350: 1391–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoenfeld JD, Sibenaller ZA, Mapuskar KR, Wagner BA, Cramer-Morales KL, Furqan M, et al. O2 and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 2017; 31: 487–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polireddy K, Dong R, Reed G, Yu J, Chen P, Williamson S, et al. High dose parenteral ascorbate inhibited pancreatic cancer growth and metastasis: mechanisms and a phase I/IIa study. Sci Rep 2017; 7:17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ching SYL, Prins AW, and Beilby JP. Stability of ascorbic acid in serum and plasma prior to analysis. Ann Clin Biochem 2002; 39: 518–20 [DOI] [PubMed] [Google Scholar]
- 6.Du J, Cullen JJ, and Buettner GR. Ascorbic acid: chemistry, biology and treatment of cancer. Biochim Biophys Acta 2012; 1826: 443–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohndiek SE, Ketunnen MI, Hu D, Kennedy BWC, Boren J, Gallagher FA, et al. Hyperpolarized [1–13C]-ascorbic acid and dehydroascorbic acid: vitamin C as a probe for imaging redox status in vivo. J Am Chem Soc 2011; 133: 11795–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakraborty A and Jana NR Vitamin C-conjugated nanoparticle protects cells from oxidative stress at low doses but induces oxidative stress and cell death at high doses. ACS Appl Mater Interfaces 2017; 9: 41807–17 [DOI] [PubMed] [Google Scholar]
- 9.Di Tano M, Raucci F, Vernieri C, Caffa I, Buono R, Fanti M, et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat Commun 2020; 11:2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149: 1060–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imai H, Matsouka M, Kumagai T, Sakamoto T, and Koumura T Lipid peroxidation-dependent cell death regulated by GPX4 and ferrptosis. Curr Top Microbiol Immunol 2017; 403: 143–70 [DOI] [PubMed] [Google Scholar]
- 12.Rosa D, Catala A Fatty acid profiles and non-enzymatic lipid peroxidation of microsomes and mitochondria from bovine liver, kidney, lung and heart. Arch Physiol Biochem 1998; 106: 33–7 [DOI] [PubMed] [Google Scholar]
- 13.Yu H, Yang C, Jian L, Guo S, Chen R, Li K, et al. Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol Rep 2019; 42; 826–38 [DOI] [PubMed] [Google Scholar]
- 14.Karlenius TC, Shah F, Yu WC, Hawkes HJK, Tinggi U, Clarke FM, et al. The selenium content of cell culture serum influences redox-regulated gene expression. Biotechniques 2011; 50: 295–301 [DOI] [PubMed] [Google Scholar]
- 15.Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 2019; 177: 1262–79 [DOI] [PubMed] [Google Scholar]
- 16.Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol 2008; 26: 1179–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu W, Su X, Klein MS, Lewis IA, Fiehn O, Rabinowitz JD Metabolite measurement: pitfalls to avoid and practices to follow. Annu Rev Biochem 2017; 86: 277–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G The small intestine converts dietary fructose into glucose and organic acids. Cell Metab 2018; 27: 351–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Xing X, Chen L, Yang L, Su X, Rabiwtz H, et al. Peak annotation and verification engine for untargeted LC-MS metabolomics. Anal Chem 2019; 91: 1838–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agrawal S, Kumar S, Sehgal R, George S, Gupta R, Poddar S, et al. El-MAVEN: a fast, robust, and user-friendly mass spectrometry data processing engine for metabolomics. Methods Mol Biol 2019; 1978: 301–21 [DOI] [PubMed] [Google Scholar]
- 21.Petrat F, Rauen U, and de Groot H Determination of the chaltable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK. Hepatology 1999; 29: 1171–9 [DOI] [PubMed] [Google Scholar]
- 22.Go YM and Jones DP Thioredoxin redox western analysis. Curr Protoc Toxicol 2009; 17:17.12 [DOI] [PubMed] [Google Scholar]
- 23.Salabei JK, Gibb AA, and Hill BG Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc 2014; 9: 421–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faustino-Rocha A, Poliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG, et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim (NY) 2013; 42: 217–24 [DOI] [PubMed] [Google Scholar]
- 25.Clément MV, Ramalingam J, Long LH, and Halliwell B. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxid Redox Signal 2011; 3: 157–63 [DOI] [PubMed] [Google Scholar]
- 26.Du X, Matsumara T, Edelstein D, Rossetti L, Zsengellér Z, Szabo C, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 2003; 112: 1049–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng ML, Lin JF, Huang CY, Li GJ, Shih LM, Chiu DTY, et al. Sedoheptulose-1,7-bisphosphate accumulation and metabolic anomalies in hepatoma cells exposed to oxidative stress. Oxid Med Cell Longev 2019; 2019:5913635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clasquin MF, Melamud E, Singer A, Gooding JR, Xu X, Dong A, et al. Riboneogenesis in yeast. Cell 2011; 145: 969–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmuck EM, Board PG, Whitbread AK, Tetlow N, Cavanaugh JA, Blackburn AC, et al. Characterization of the monomethylarsonate reductase and dehydroascorbate reductase activities of Omega class glutathione transferase variants: implications for arsenic metabolism and the age-at-onset of Alzheimer’s and Parkinson’s diseases. Pharmacogenet Genomics 2005; 15: 493–501 [DOI] [PubMed] [Google Scholar]
- 30.Zhou H, Brock J, Liu D, Board PG, and Oakley AJ. Structural insights into the dehydroascorbate reductase activity of human omega-class glutathione transferases. J Mol Biol 2012; 420: 190–203 [DOI] [PubMed] [Google Scholar]
- 31.Mojic M, Pristov JB, Maksimovic-Ivanic D, Jones DR, Stanic M, Mijatovic S, et al. Extracellular iron diminishes anticancer effects of vitamin C: an in vitro study. Sci Rep 2014: 4:5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zilka O, Shah S, Li B, Angeli JPF, Briesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci 2017; 3: 232–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu J, Zhong L, Lonn ME, Burk RF, Hill KE, Holmgren A Penultimate selenocysteine residue replaced by cysteine in thioredoxin reductase from selenium-deficient rat liver. FASEB J 2009; 23: 2394–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rigobello MP, Scutari G, Folda A, and Bindoli A Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochem Pharmacol 2004; 67: 689–96 [DOI] [PubMed] [Google Scholar]
- 35.Stafford WC, Peng X, Olofsson MH, Zhang X, Luci DK, Lu L, et al. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci Transl Med 2018; 10:eaaf7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab 2019; 1: 404–15 [PMC free article] [PubMed] [Google Scholar]
- 37.Ghergurovich JM, Garcia-Canaveras JC, Wang J, Schmidt E, Zhang Z, TeSlaa T, et al. A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat Chem Biol 2020; 16: 731–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deagen JT, Butler JA, Zachara BA, and Whanger PD Determination of the distribution of selenium between glutathione peroxidase, selenoprotein P, and albumin in plasma. Anal Biochem 1993; 208: 176–81 [DOI] [PubMed] [Google Scholar]
- 39.Hill KE, Xia Y, Akesson B, Boeglin ME, Burk RF Selenoprotein P concentration in plasma is an index of selenium status in selenium-deficient and selenium-supplement Chinese subjects. J Nutr 1996; 126: 138–45 [DOI] [PubMed] [Google Scholar]
- 40.Cho S, Chae JS, Shin H, Shin Y, Song H, Kim Y, et al. Hormetic dose response to L-ascorbic acid as an anti-cancer drug in colorectal cancer cell lines according to SVCT-2 expression. Nat Sci Rep 2018; 8:11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torti SV and Torti FM Iron and cancer: more ore to be mined. Nat Rev Cancer 2013; 13; 342–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rees DC, Kelsey H, and Richards JD Acute haemolysis induced by high dose ascorbic acid in glucose-6-phosphate dehydrogenase deficiency. BMJ 1993; 306: 841–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boffetta P, McLaughlin JK, La Vecchia C, Tarone RE, Lipworth L, Blot WJ False-positive results in cancer epidemiology: a please for epistemological modestry. J Natl Cancer Inst 2008; 100: 988–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Errington TM, Denis A, Allison AB, Araiza R, Aza-Blanc P, Bower LR, et al. Experiments from unfinished registered reports in the reproducibility project: cancer biology. eLife 2021; 10:e73430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeo JE and Kang SK Selenium effectively inhibits ROS-mediated apoptotic neural precursor cell death in vitro and in vivo in traumatic brain injury. Biochim Biophys Acta 2007; 1772: 1199–210 [DOI] [PubMed] [Google Scholar]
- 46.Cai X, Wang C, Yu W, Fan W, Wang S, Shen N, et al. Selenium exposure and cancer risk: an updated meta-analysis and meta-regression. Sci Rep 2016; 6:19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu X, Jin F, Hao Y, Zhu Z, Li H, Tang T, et al. Nonlinear association between magnesium intake and the risk of colorectal cancer. Eur J Gastroenterol Hepatol 2013; 25: 309–18 [DOI] [PubMed] [Google Scholar]
- 48.Nasulewicz A, Wietrzyk J, Wolf FI, Dzimira S, Madej J, Maier JAM, et al. Magnesium deficiency inhibits primary tumor growth but favors metastasis in mice. Biochim Biophys Acta 2004; 1739: 26–32 [DOI] [PubMed] [Google Scholar]
- 49.Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses via cGAS_STING and improves the efficacy of clinical immunotherapy. Cell Res 2020; 30: 966–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.