

Abstract
The speed, sensitivity, and tolerance of heterogeneity of native mass spectrometry, as well as the kinetic trapping of solution-like states during electrospray, makes mass spectrometry an attractive method to study protein structure. Increasing resolution of ion mobility measurements and mass resolving power and range are leading to the increase of the information content of intact protein measurements, and an expanded role of mass spectrometry in structural biology. Herein, a suite of different length noncovalent (sulfonate to positively charged side chain) crosslinkers was introduced via gas-phase ion/ion chemistry and used to determine distance restraints of kinetically trapped gas-phase structures of native-like cytochrome c ions. Electron capture dissociation allowed for the identification of crosslinked sites. Different length linkers resulted in distinct pairs of side chains being linked, supporting the ability of gas-phase crosslinking to be structurally specific. The gas-phase lengths of the crosslinkers were determined by conformational searches and density functional theory, allowing for the interpretation of the crosslinks as distance restraints. These distance restraints were used to model gas-phase structures with molecular dynamics simulations, revealing a mixture of structures with similar overall shape/size but distinct features, thereby illustrating the kinetic trapping of multiple native-like solution structures in the gas phase.
Keywords: Native mass spectrometry, Ion/Ion reactions, Ion mobility mass spectrometry, Crosslinking, Molecular Dynamics
Graphical Abstract
Introduction
Mass spectrometry (MS) has been used to study intact proteins since the advent of electrospray ionization (ESI) MS. The earliest experiments revealed two important fundamentals of the behavior of protein ions formed by ESI: (1) charge state distributions (CSDs) of proteins reflect their overall structure in solution, with higher CSDs reflecting unfolded structures and lower CSDs reflecting folded structures and (2) noncovalently bound protein complexes and porphyrin groups can be retained upon ionization.1 Native mass spectrometry depends on the retention of important aspects of protein structure during ESI and transfer to the gas phase. Measurements from native MS, where ions are ionized in salty, aqueous conditions at physiological pH, have revealed that specific, noncovalent binding between proteins and ligands or proteins and other proteins can be retained in the gas phase.2 Native MS has become a transformative technique for structural characterization of heterogeneous protein systems, allowing for intact proteins and protein complexes to be measured with speed, sensitivity, and selectivity3, thus expanding our knowledge of biological systems.4
Ion mobility mass spectrometry (IM/MS) measurements have shown that the collision cross sections (CCS) measured for globular proteins and protein complexes correlate strongly to X-ray and NMR structures.5–7 In addition, the number of unfolding domains for proteins and protein complexes in the gas phase is equal to the number of known folding domains in solution.8 Clearly, the overall size/shape and important inter-residue contacts9 can be maintained in the gas phase. Though the protein ions undergo a change in environment from solution to charged, evaporating droplets, to vacuum, it has been suggested that solution structures are “kinetically trapped,” and without significant activation, the equilibrium gas-phase unfolded structures are not observed.10, 11 Kinetic trapping allows for the detection of folding intermediates which are not observable in solution,9, 12 a beautiful example of the versatility of structural MS measurements towards addressing the so-called “protein folding problem.”13
Not all aspects of protein solution structures are maintained upon ionization and transmission into the gas phase. Structures are known to compact in the gas phase. Compaction is the primary hypothesis behind why CCS measurements are systematically smaller than CCS values calculated by trajectory methods14 applied to molecular coordinates directly from high resolution solution structures. For example, molecular dynamics (MD) simulations predict that macromolecular rings will shrink into vacant cavities due to the lack of solvent.15 Charged side chains also collapse onto the protein surface for charge solvation by either carbonyl oxygens or acidic residues.15 Microsolvation studies of lysines in cytochrome c16 and ubiquitin17 with 18-crown-6 ether suggested that the effects of vacuum on charged side chains led to a reduction in CCS for folded proteins and an increase in CCS (i.e., destabilization) for unfolded proteins. These factors are combined with the possibility of pH changes, electrochemical processes, and changes in droplet temperature during electrospray.18, 19
The gas-phase tools used to observe the preservation versus loss of solution-phase structure in native MS look at the averaged, overall structure that emerges from each charge state (e.g., gas-phase IR spectroscopy and IM).20 These measurements do not provide localized structural details. For example, a single CCS can correlate to many possible solution structures.21 Although the native inter-residue contacts have been shown by molecular modeling to be maintained after electrospray,22 these details have been obtained only by molecular modeling approaches to which CCS and IM spectra are fit without additional experimental validation. The experimental measurements themselves do not elucidate these interactions, as CCS measurements, while useful, are a rotationally averaged measurement of the entire conformer/conformer family and cannot inform on localized three-dimensional structural information.
The combination of molecular modeling with experimentally distance restraints, from, for example, NMR,23 single-molecule FRET,24 and chemical crosslinking25 and footprinting26, has led to the successful determination of structures of natively folded proteins. This combination has also been applied to proteins in vacuo by using radical-donor and radical-acceptor pairs in radical-directed dissociation (RDD)27 as well as chemical crosslinking utilizing gas-phase reactions of oppositely charged ions28, 29 (i.e., ion/ion reactions). Previously, our group used the gaseous dianion of the sulfo-EGS (ethylene glycol bis(sulfosuccinimidyl succinate)) from negative mode electrospray ionization to provide distance constraints for the protein ubiquitin upon ion/ion reactions followed by electron capture dissociation.28 Herein, we have expanded our panel of non-covalent crosslinkers ranging in size from 3.5 to ~20 Å to provide distance restraints used in gas-phase molecular dynamics simulations to characterize gas-phase structures of native-like cytochrome c ions. We expect that this development will be an important next step towards characterizing the changes in protein structure from solution to inside the mass spectrometer.
Materials and Methods
Materials and Sample Preparation.
Cytochrome c from equine heart, ammonium acetate, potassium benzene-1,2-disulfonate (1,2-BDSA), 1,3-benzenedisulfonic acid disodium salt (1,3-BDSA), 1,5-napthalenedisulfonic acid, disodium salt hydrate (1,5-NDSA), and pentadecafluoro-1-octanol (PFO) were obtained from Sigma-Aldrich (St. Louis, MO). Ethylene glycol bis(sulfosuccinimidyl succinate) (Sulfo-EGS), bis(sulfosuccinimidyl)suberate (BS3), bis(sulfosuccinimidyl) glutarate-d0 (BS2G) disodium forms and Amberlite IR-120 (H) ion-exchange resin were purchased from ThermoFisher Scientific (Rockford, IL). Methanol, acetonitrile, and ammonium hydroxide were purchased from Fisher Scientific (Fairmont, NJ). Water was obtained from a Milli-Q Millipore A10 (Burlington, MA) water purification system at a resistivity of 18 MΩ or greater. The 7+ charge state of cytochrome c was prepared under aqueous native-like conditions at a final concentration of 10 μM in an aqueous 200 mM ammonium acetate solution, pH 7. The proton transfer reagent, PFO was electrosprayed at 1.25 mM in a 99/1 vol/vol mixture of methanol/ammonium hydroxide and the crosslinking reagents (1,2-BDSA, 1,3-BDSA and 1,5-NDSA) were first prepared at a concentration of 50mM in water and incubated with Amberlite cation exchange resin to facilitate ion exchange of Na+ and K+ and increase the relative concentration of doubly deprotonated form of the reagent. Following the ion exchange step, the stock solutions were diluted to a working concentration of 1mM using an 80/20 vol/vol mixture of acetonitrile/water. The remaining crosslinkers (Sulfo-EGS, BS3 and BS2G) were dissolved in acetonitrile at a final concentration of 1mM.
Ion/ion Reactions and Ion Mobility Mass Spectrometry.
The experiments were performed on a Synapt G2-Si High Definition Mass Spectrometer (Waters Corporation, Wilmslow, U.K.), equipped with an electron transfer dissociation (ETD) glow discharge source, a NanoLockspray source, an external electrospray voltage control module (GAA Custom Electronics LLC, Kennewick, WA) and an ExD cell (e-MSion, Corvallis, OR). Instrumental details for the gas-phase ion/ion reactions30, 31 and electron capture dissociation (ECD) using the ExD cell32 have been described previously. The protein was electrosprayed in positive ion mode using the nanolockspray source via borosilicate tips, pulled using a P-1000 micropipette puller (Sutter Instruments, Novato, CA) while the anionic reagents were infused through the reference sprayer at a flow rate of 1 μL/min. The external voltage control module was used to synchronize the source polarity (+0.90 kV and −0.90 kV) and ion injection times (1s each) to sequentially introduce the anions and cations into the instrument. Cytochrome c charge states and reagent ions (either deprotonated PFO or doubly deprotonated crosslinker) were isolated in the quadrupole and stored in the trap cell with a trap traveling wave height of zero volts to allow for gas-phase ion/ion reactions of specific protein charge states to occur. The ion/ion products were then separated by ion mobility (IM) using nitrogen as the mobility gas at a flow rate of 10 mL/min while the Trap, He and Transfer cell flow rates were set to 10, 10 and 7 mL/min, respectively. This lower pressure regime was used to maintain conditions in the trap cell conducive to stable ion/ion product formation, as previously published.31 A ramping traveling wave velocity of 750 – 2400 m/s over a full IMS cycle and wave height of 40 V were used in the mobility cell. After IM separation, the ions were subjected to ECD fragmentation (using 3.5 eV electrons) in the ExD cell (detailed voltage settings included in Table 1 of supplementary info) and supplemental activation in the transfer cell (Transfer CE of 30V) before mass analysis in the time-of-flight (TOF) mass spectrometer set to ‘Resolution’ mode. The experiments were all acquired in triplicate runs to ensure reproducibility.
Data Analysis.
The IM peaks of the 1x crosslinked adduct or the 2x proton transfer product (by PFO) were selected, and the m/z data extracted using MassLynx v4.2 (Waters Corp.). The data was smoothed and centered and a lockmass correction (2134.1621 or 2650.4683 m/z corresponding to z18+ and z23+ fragments respectively) was applied. The processed data was exported as .mgf (Mascot Generic Format) files, and c and z fragments were annotated using LCMS spectator33 (https://omics.pnl.gov/software/lcmsspectator), using a mass error tolerance of 20 ppm34 and a raw intensity threshold of 1000 counts. In addition to an acetyl group at the N-terminus (42.01056 Da) and a Fe-containing heme group (616.17728 Da) covalently bound to Cys14 and Cys17 of cytochrome c, custom modifications were included at the N and C termini to identify the modified ECD fragments. These corresponded to 237.9606 Da (1,2-BDSA and 1,3-BDSA), 287.9762 Da (1,5-NDSA), 485.9886 Da (BS2G), 528.0356 Da (BS3) and 616.0153 Da (sulfo-EGS). The triplicate data was filtered to only include fragments observed in at least two out of three runs and manually verified before visualizing the sequence maps in Prosight Lite.35
Molecular Modeling and CCS Calculations.
The gas-phase optimized geometries of the anionic crosslinkers were used to determine the Nζ – Cζ and Nζ – Nζ distance (i.e., the lengths of restraints to use). First, the linkers containing rings were energy minimized in Avogadro36 using default settings and used as is for geometry optimization with quantum mechanical methods due to the rigidity and planarity of the structures. BS2G, BS3, and sulfo-EGS conformers were searched with the Avogadro systematic rotor search. The twenty lowest energy conformers were used as starting structures for quantum mechanical geometry optimization. Geometries were optimized and zero-point corrected energies were calculated with density functional theory using the Becke, 3-parameter, Lee–Yang–Parr functional combinations37, 38 and the basis set 6–31+G(d) in Gaussian 16.39 Additionally, to account for the electrostatic bond (i.e. ‘salt bridge’ distance) between the sulfonate sulfur and either the ammonium Nζ of Lys or the guanidinium Cζ of Arg, the energy-minimized structure of 1,5-NDSA was modeled with butyl ammonium and ethyl guanidinium respectively, resulting in distances of 3.2 Å and 4.2 Å. The sulfonate sulfur atom was used since all 3 oxygens are the same bond order and partial negative charge. Thus for the Nζ – Nζ distance restraint, 6.4 Å (two Lys ‘salt bridge’ distances) was added to the crosslinker length while the Nζ – Cζ restraint was calculated by adding the crosslinker length and 7.4 Å (sum of Arg and Lys ‘salt bridge’ distances, Figure 1 and Figure 5). Gas-phase structures of cytochrome c were simulated using an existing protocol for gas-phase protein molecular dynamics (MD) that has been validated against experimental ion mobility measurements.15, 40 Briefly, five replicates of MD run for 100 ns in GROMACS 2019.441 using the GROMOS 43a2 force field42 were simulated, incorporating distance restraints with a force constant of 100 kJ/mol Å2.27 Since 1,3-BDSA and 1,5-NDSA are structurally rigid, a restraint tolerance of ±0.1 Å (i.e., the distance where the potential in the restraint is minimized) was used during MD simulations. For the more flexible crosslinkers, the restraint distance at minimal potential was set over a wider range, with 24.7 to 31.5 Å used as the limits for BS2G/ BS3 and 24.7 to 31.5 Å for Sulfo-EGS. Six different distance restraints parameters were used, including applying all four restraints (all restraints), none of the restraints (no restraint) and using three out of four restraints (no 1,3-BDSA, no 1,5-NDSA, no BS2G and no Sulfo-EGS). Charged sites were determined from ECD of cytochrome c 7+ (vide infra) and acidic residues were protonated (neutral) for simulations. Crosslinked distances were measured in Visual Molecular Dynamics (VMD)43 over the progress of the entire 100 ns MD run. Following the conclusion of these calculations, representative final structures were rendered visually using PyMOL Molecular Graphics System, v1.7.2.0 (Schrodinger, LLC, New York, NY) and were used to measure the final crosslinked distances reported in Tables 1 and S2. CCSHe values for representative final MD structures were calculated in IMoS software44, 45 using the Trajectory Method (TM) algorithm with a helium drift gas and default Lennard-Jones parameters and protein charge set to 7. Helium was used as the ion/ion chemistry takes place in the helium-filled trap cell.
Figure 1.
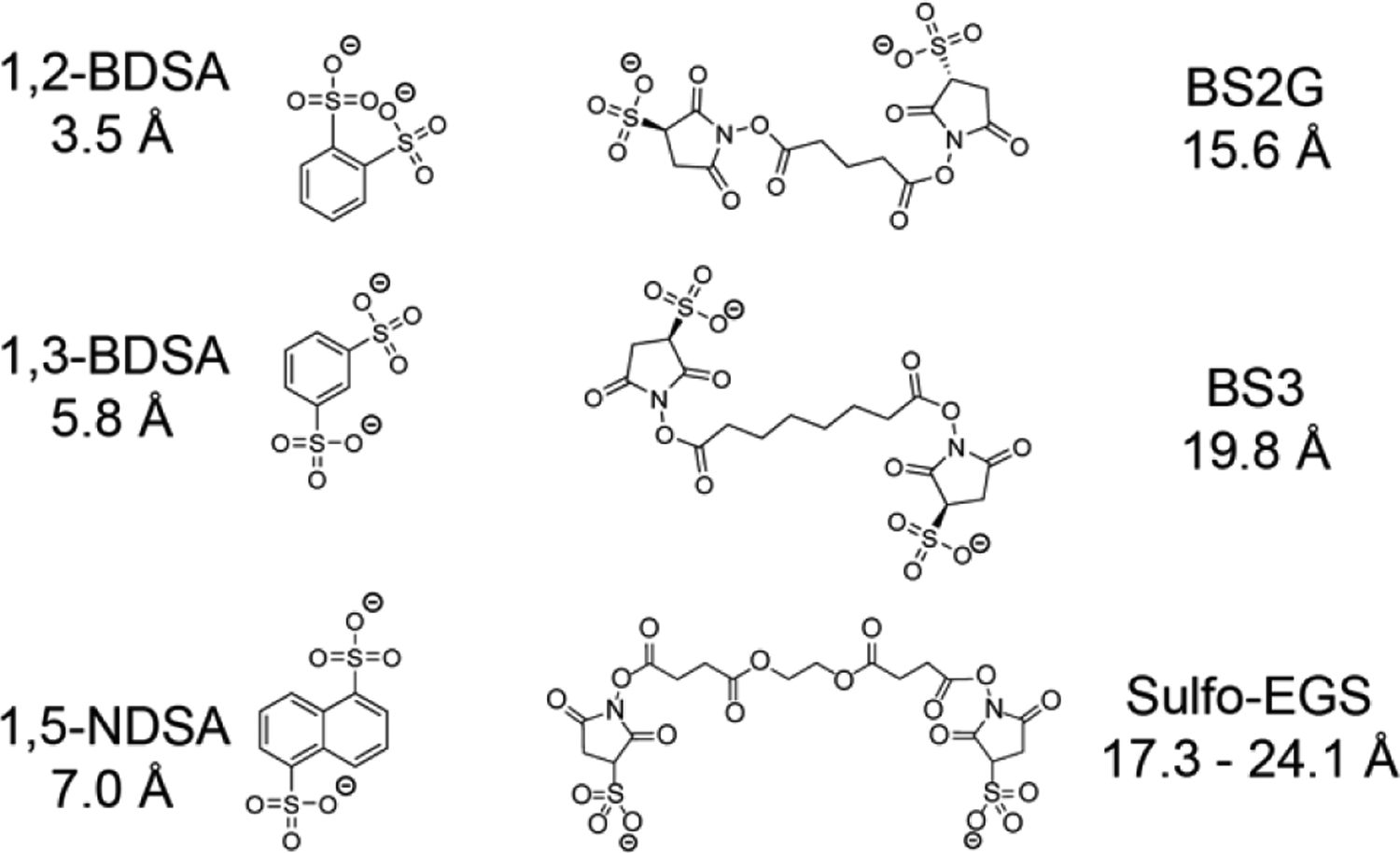
Panel of electrostatic crosslinking reagents with the respective spacer length between the two sulfonate groups calculated from geometry-optimized structures modeled in the gas phase.
Figure 5.
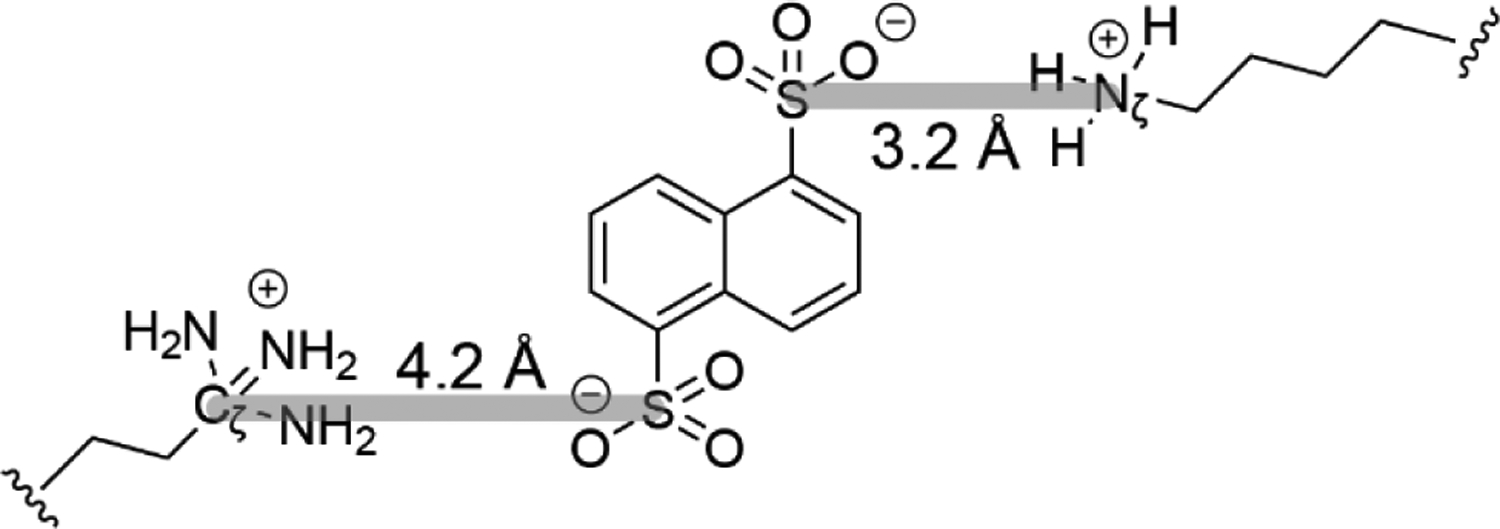
The S – Cζ and S – Nζ gas-phase electrostatic bond distances are measured between sulfonate sulfur and either ethylene guanidinium Cζ (4.2 Å) or butylene ammonium Nζ (3.2 Å) respectively (highlighted in gray).
Table 1.
Summary of Measured Crosslinking Distances and CCSHe Values Calculated for X-ray Structure (PDB ID 1HRC) and MD Models.1
| 1,3-BDSA R38Cζ - K55Nζ ≤ 13.2 Å | 1,5-NDSA R38Cζ - K73Nζ ≤ 14.4 Å | Sulfo-EGS R38Cζ - K88Nζ ≤ 24.7 Å | Sulfo-EGS R38Cζ - K88Nζ ≤ 31.5 Å | BS2G K25Nζ - K73Nζ ≤ 22.0 Å | BS3 K25Nζ - K73Nζ ≤ 26.2 Å | CCSHe ≤ 1247 Å2 | ||
|---|---|---|---|---|---|---|---|---|
| X-ray (1HRC) | 17.4 | 26.2 | 25.6 | 25.6 | 33.4 | 33.4 | 1310 | |
| no BS2G | 1 | 13.4 | 14.6 | 25.2 | 25.2 | 16.5 | 16.5 | 1220 |
| 2 | 13.1 | 14.2 | 25.6 | 25.6 | 19.5 | 19.5 | 1231 | |
| 3 | 13.2 | 14.2 | 24.6 | 24.6 | 13.6 | 13.6 | 1194 | |
| 4 | 13.2 | 14.3 | 24.9 | 24.9 | 18.5 | 18.5 | 1206 | |
| 5 | 13.0 | 14.3 | 24.8 | 24.8 | 21.3 | 21.3 | 1256 | |
Distances highlighted in light blue represent experimental distances measured using X-ray and MD structures that are smaller than the restraint distance set during the MD simulations.
Results and Discussion
Electrostatic Crosslinking Using Various Reagent Length and Flexibility.
Crosslinking reagents were each reacted with cytochrome c 7+ in the gas phase, resulting in an electrostatic adduct formed between the two negatively charged sulfonate groups of the crosslinker and accessible, protonated amino acid residues on cytochrome c. The aromatic disulfonate salts (1,2-BDSA, 1,3-BDSA and 1,5-NDSA) provide a more rigid and shorter range of crosslinking distances while the more linear sulfo-NHS containing compounds (BS2G, BS3 and sulfo-EGS) are able to capture longer distances (Figure 1). Listed distances are measured between the two sulfonate sulfurs after gas-phase geometry optimization by averaging the distances for all structures within 2 kcal/mol of the lowest energy structure (zero-point corrected energies). Of note, only sulfo-EGS was dynamic and adopted multiple structures spanning a range of crosslinking distances within 2 kcal/mol of the lowest energy conformer. The range of distances for BS3 and BS2G were less than an angstrom above or below the listed distance, which is likely within the experimental error of chemical crosslinking.46 Since cytochrome c was crosslinked in the gas phase, the reagent structures were modeled in the absence of solvent to allow for charge solvation by the sulfonate groups to minimize charge repulsion while ignoring any solvent-mediated stabilization owing to charge and hydrogen bonds typically present in solution conditions.
After the formation of a stable electrostatic complex in the trap cell, the reaction products and unmodified protein are separated by ion mobility (IM), with a drift time of 14.1 ms observed for the 1x crosslinked adduct formed using 1,3-BDSA (Figure 2A). By keeping the trap pressure relatively low at ~ 0.038 mbar (= 10mL/min), the ion/ion reaction was tuned to favor the formation of the 1x crosslinked adduct while minimizing additional crosslinks.30 Thus there is still some unmodified protein (drift time = 9.68 ms) observed after the reaction in the trap. The multiple (n) crosslinks species were not considered in our analyses since addition of n number of crosslinks effectively reduces the overall charge state by −2 × n, thereby limiting the number of protonated sites available during electron capture dissociation (ECD) and hindering efficient fragmentation. The presence of excessive crosslinks can also potentially lock the protein structure into an irrelevant conformation that is different from the starting structure. The smaller peak (drift time = 11.8 ms) corresponds to [cytochrome c + 7H + ♦]6+, representing a “dead-end” link where the reagent is attached at only one protonated site using one of the two available sulfonate groups and the other sulfonate group has been protonated (i.e., neutralized) by proton transfer and is thus unreactive. Mobility-separated ion species are fragmented by ECD to identify the sites of modification on cytochrome c 7+. However, the overall sequence coverage was poor with the transfer CE voltage ‘off’ (at 4 V) (Figure S1). To improve sequence coverage and facilitate site annotation, ECD fragments are further subjected to supplemental activation in the transfer cell (transfer CE 30 V), which provides enough energy to disrupt the intramolecular interactions preventing the separation of the c and z fragment ions while minimizing the formation of collision induced dissociation (CID) products, which involve mobilization of protons and the possible loss of the crosslinkers. The energy in the transfer cell was carefully tuned to preserve the electrostatic complex as shown in the extracted mass spectrum of the IM peak (drift time = 14.10 ms), where the [cytochrome c + 7H + ♦]5+ complex (2519.26 m/z) is the major peak relative to the unmodified protein (2471.67 m/z). Efficient ECD is also demonstrated with two subsequent electron capture peaks [M + ♦]4+ and [M + ♦]3+ at 3149.07 and 4198.76 m/z respectively (Figure 2B). The IM and m/z plots for the other two aromatic disulfonate compounds are similar to 1,3-BDSA (Figure S2). Interestingly, for all three sulfo-NHS based crosslinkers, the additional energy in the transfer cell is sufficient to drive the formation of the covalent product [M + *]5+, corresponding to the loss of the two sulfo-NHS groups (Figure S2). However, since these covalent bonds are formed after ECD, the current implementation of the instrument platform prevents fragmentation of these gas-phase covalent crosslinked proteins. Thus, covalent crosslinking was not included in the analyses. Tuning the reactivity of the crosslinking chemistry and the instrument for more facile covalent crosslinking is currently in progress. The gas phase ion/ion reaction between 1-hydroxy-7-aza-benzotriazole (HOAt) ester reagents and nucleophilic side chains on peptides has previously been demonstrated, with the covalent reaction yield of HOAt ester reagent higher compared to that of N-hydroxysuccinimide (NHS) ester reagents since such gas-phase reactions are under kinetic control.47 Using our instrument setup, we have used an HOAt ester reagent (sulfo benzoyl HOAt) to covalently label ubiquitin in the gas phase via ion/ion reaction31 and are currently synthesizing crosslinkers containing HOAt, which will help improve the yield of covalent crosslinked products formed in the gas phase.
Figure 2.
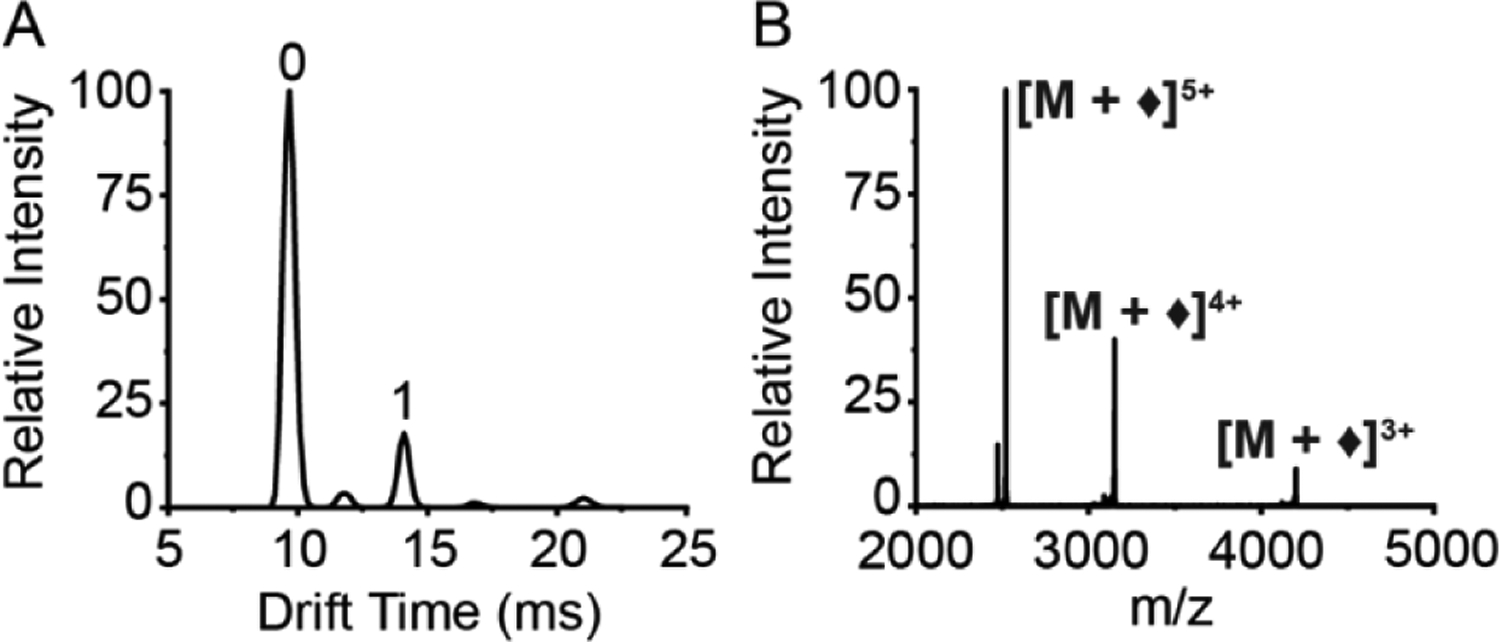
Gas-phase electrostatic reaction between [cytochrome c + 7H]7+ and [1,3-BDSA]2−. (A) Ion mobility spectrum following ion/ion reaction, with numbered labels corresponding to the number of electrostatic attachments. (B) Mass spectrum extracted from ‘1’ electrostatic attachment IM peak (denoted by ♦).
A zoomed-in view of the 3200 – 4200 m/z range shows some annotated c and z fragments shifted by the additional mass of the 1,3-BDSA crosslinker in contrast to unmodified ions resulting from the cytochrome c 5+ precursor obtained from two consecutive proton transfer reactions from cytochrome c 7+ using PFO (Figure 3), which serves as a charge-reduced “control”, having the same net charge as the crosslinked cytochrome c 7+ without the modifications present. Corresponding m/z shifts of the modified ions in red (top 1,3-BDSA spectra) are observed in comparison to the unmodified ions in blue (bottom PFO spectra). Annotated spectra for the other crosslinkers are included in the supplementary material (Figure S3). The unmodified c and z ions generated after the ion/ion reaction with PFO are also observed across the entire protein sequence, thereby demonstrating efficient ECD sequencing of cytochrome c (despite resulting charge prior to ECD equal to +5) (Figures 4, S4). Therefore, changes in the fragmentation patterns between the charge reduced control and the crosslinked 7+ are likely due to the presence of the linker, aiding in the interpretation of the crosslinking ECD fragments results.
Figure 3.
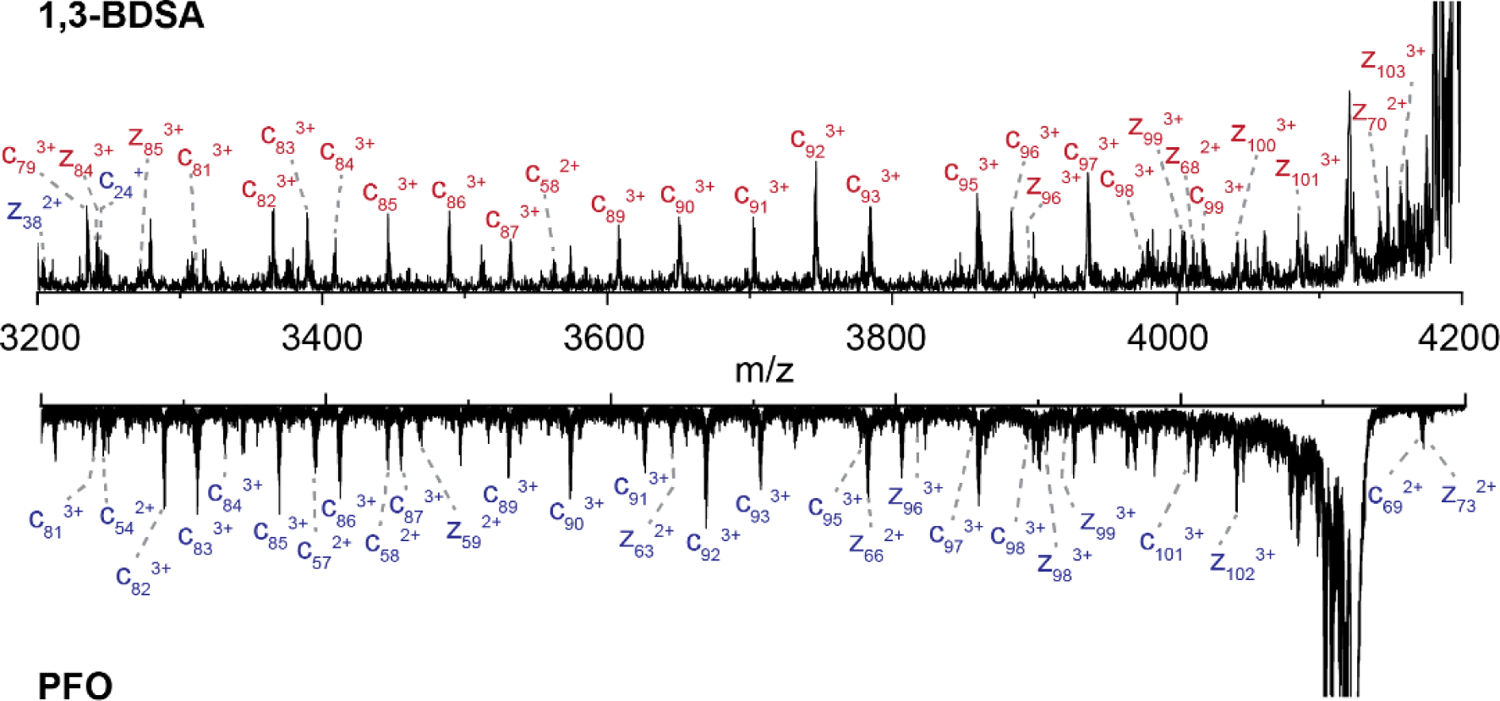
Extracted mass spectra from [M + ♦]5+ precursor ion comparing the c and z ions from crosslinking by 1,3-BDSA (top) and two consecutive proton transfer by PFO (bottom), with modified and unmodified fragments denoted in red and blue, respectively.
Figure 4.

Sequence ladders generated from the ECD fragmentation of the ion/ion PFO proton transfer reaction and crosslinking reaction products of cytochrome c 7+. Modified fragments including the added mass of the reagent are labeled in red while unmodified ions are denoted in blue. Crosslinked regions are highlighted in gray.
The protonated sites are first assigned to identify where electrostatic attachment by the sulfonate groups may occur. To do so, the ECD fragmentation data of unmodified cytochrome c 7+ is analyzed to track where incremental increases in charge occur, as c and z fragments get larger, due to the presence of an ionizing proton. Hence, the protonated sites for unmodified cytochrome c 7+ include Cys14 (Heme), Lys25, Arg38, Lys55, Lys73, Lys88 and Lys99 (Figure S5A). The oxidized form of cytochrome c (FeIII) is present in ESI mass spectrometry, with the heme undergoing electron capture during ECD.48 Other residues are not considered as possible protonated sites since there is not an increase in charge after (C-terminal to the residue for c ions, N-terminal to the residue for z ions) the fragments included the residues. Only the residues that were the closest basic residues to the increase in charge of fragments were considered. The charge remaining the same before and after fragmentation nearby the residues not chosen, but the charge increasing nearby the residues that were chosen, is evidence that the residues we did not consider as protonated were indeed not protonated. This is the same strategy used for identifying protonated sites in the literature.49, 50 Since oxidized heme containing FeIII is already covalently attached to C14 and C17 and in the interior of the protein, it likely cannot participate in gas-phase crosslinking. However, the side chains of the remaining protonated residues are all surface accessible (Figure S5B).
Following cytochrome c crosslinking with the various disulfonate-containing compounds, the resulting c and z ions were analyzed and used in conjunction with the charge sites for cytochrome c 7+ to determine the site(s) of electrostatic attachment. All the ECD experiments were run in triplicates, with the three datasets combined and filtered to only include reproducible ions (observed in n ≥ 2). Both unmodified and crosslinked c and z ions were then manually validated and mapped onto the protein sequence to identify modified residues. Unmodified fragments (labeled in blue) are annotated without including any mass shift while modified fragments (shown in red) are mapped after including the mass increase due to the reagent (Figure 4). Both unmodified and modified fragments are helpful when determining the position of the crosslinker. The smallest modified c and z ions ([c + ♦] and [z + ♦]) that are observed, bracket the protein region being crosslinked since those require the added mass of the reagent to be assigned, indicating that a crosslinker must be present N-terminal to the smallest [c + ♦] ion and C-terminal to the smallest [z + ♦] ion. The amino acid sequence spanned by the crosslinker is also held together by the reagent, thereby resulting in an apparent silencing of fragmentation between the two crosslinked residues. Furthermore, the largest c and z unmodified fragments indicate that no crosslinking occurs N-terminal to the largest unmodified c ion and C-terminal to the largest unmodified z ion. Combining the location of the ionizing protons, the smallest modified c and z ions, the “silenced” regions, and the location of the largest unmodified c and z ions allows us to assign the location of the crosslinks. Notably, comparing the ECD pattern of the unmodified fragments of the crosslinked complex to that of the 2x proton transfer product using PFO shows that ECD sequence coverage is excellent in the absence of the crosslinker, especially in the Lys25 – Lys88 region where crosslinking is occurring (Figure 4). This provides additional confirmation that the gaps in sequencing are due to the presence of the crosslinker.
To minimize the possibility of ‘scrambling’ of the crosslinking positions, the instrument conditions are tuned to mitigate the loss of the electrostatic crosslinker after the ion/ion reaction occurs in the trap cell. Also, the use of ECD fragmentation to sequence the crosslinked protein helps prevent ‘scrambling’ that could occur using slow heating fragmentation methods such as CID. We do not observe multiple residue pairs crosslinked by the same reagent, suggesting that ‘scrambling’ is not occurring. For the cytochrome c 7+ and 1,2-BDSA adduct, the largest unmodified ions are c49 and z46 while the smallest modified fragments are [c58 + ♦] and [z57 + ♦], suggesting that the crosslinker is present in this Tyr48 – Thr58 region. However, the only protonated site for electrostatic attachment is Lys55 (highlighted in gray), implying that 1,2-BDSA is only bound at that one site. This is not surprising considering that the sulfonate groups on 1,2-BDSA are next to each other (spacer length = 3.9 Å) and are likely unable to bridge the distance between Lys55 and its closest accessible protonated site. In addition, the positions of the sulfonates in 1,2-BDSA (i.e., ortho) likely plays a role in the lack of observed links due to geometric constraints. The remaining reagents are all able to crosslink a pair of residues, with different spacer lengths providing a useful range of crosslinking distances that can be detected. In the case of 1,3-BDSA (spacer length = 5.8 Å), the reagent crosslinks Arg38 and Lys55 since the largest unmodified c and z ions are c38 and z55 while the smallest modified fragments are [c58 + ♦] and [z68 + ♦], with almost complete silencing of sequence coverage between those fragments. Arg38 is a protonated site, crosslinked by 1,3-BDSA and the presence of the unmodified c38 fragment at that Arg residue could be due to the neighboring Lys39, which could play a role in solvating the charge; however, this does not result in a significant change in the models due to the proximity of the two residues (vide infra). With a larger spacer length of 7.0 Å, 1,5-NDSA forms a Arg38 – Lys73 crosslink, given the largest unmodified ions are c37 and z18 while the smallest modified fragments are [c78 + ♦] and [z83 + ♦]. The region between Arg38 and Lys73 shows no sequence coverage, but for the charge reduced only (via PFO) version, there is significant fragmentation in this region, providing additional evidence of the crosslink’s location. Reaction with BS2G results in c11 and z16 as the largest unmodified ions and smallest modified ones as [c85 + ♦] and [z99 + ♦]. The largest unmodified ions observed for BS3 are c8 and z11 while [c78 + ♦] and [z84 + ♦] are the smallest modified fragments. Despite BS2G and BS3 having different spacer lengths of 15.6 Å and 19.8 Å respectively, they both react at Lys25 and Lys73. Finally, with its largest unmodified ions being c37 and z16 and smallest modified fragments being [c92 + ♦] and [z99 + ♦], sulfo-EGS binds at Arg38 and Lys88, indicating that these residues are within 17.3 Å and 24.1 Å apart at the ends of the side chains. Using a panel of crosslinkers with distinct size and flexibility allows the capture of multiple residue pairs used to map the structure of cytochrome c 7+ and, it provides insights into the proximity and orientation of the protonated sites in the gas phase.
Molecular Dynamics Models Generated Using Experimental Gas-phase Crosslinking Data as Distance Restraints.
The various modified sites identified experimentally (Arg38 – Lys55, Arg38 – Lys73, Arg38 – Lys88 and Lys25 – Lys73) restrain distances between crosslinked residues and guide MD simulations of cytochrome c in the gas phase. The spacer length of the crosslinker (sulfonate sulfur to sulfonate sulfur) predicted using the DFT geometry optimized gas-phase structures (Figure 1) is added to the ‘salt bridge’ (i.e. gas-phase electrostatic bond) distance to obtain the Arg Cζ – Lys Nζ and Lys Nζ – Lys Nζ distances used as restraints for the crosslinked pairs. Figure 5 illustrates how the gas-phase electrostatic bond distance was calculated for the interaction from the sulfonate sulfur (S) to the guanidinium carbon (Cζ) of Arg (4.2 Å) and that between the sulfonate sulfur (S) and nitrogen (Nζ) of Lys (3.2 Å). Thus, for the Arg Cζ – Lys Nζ restraint, a total noncovalent bond distance of 7.4 Å was added to the reagent spacer length while 6.4 Å was used in the case of Lys Nζ – Lys Nζ. This approach accounts for the flexibility of the side chains and is an improved approximation for the crosslinked (i.e., restraint) distance. Instead of the full Lys and Arg residues, the shorter butyl ammonium and ethyl guanidinium were used as models for the protonated side chains to simplify the complex process of modeling the electrostatic interaction between the crosslinker and the protonated side chain. The through distance to the Cα of the modified amino acid was not considered when estimating the salt bridge distance between the reactive sulfonate group and labeled residue. Therefore, using the entire Lys and Arg amino acids would not accurately reflect that since the Cα is part of the protein backbone and not necessarily as flexible as the end atoms of the reactive side chains. Analyses of large chemical crosslinking datasets have also shown that using the Nζ – Nζ straight line distances accurately recapitulated condensed phase structures, especially when compared to the more traditional Cα – Cα distance restraint.51, 52
Five replicate gas-phase MD runs of 100 ns restrained by the experimentally determined cross linking distances were simulated using the X-ray structure of cytochrome c (PDB ID: 1HRC) as a starting structure. For a more comprehensive study, several restraint parameters were run, including all four restraints, none of the restraints and three out of four restraints. In addition, the restraint distance was adjusted to account for the flexibility of the crosslinkers. For the more rigid reagents 1,3-BDSA and 1,5-NDSA, restraint tolerance of ±0.1 Å (i.e., the distance where the potential in the restraint is minimized) was used during MD simulations. the more flexible crosslinkers, the restraint distance at minimal potential was set over a wider range, with 24.7 to 31.5 Å used as the limits for BS2G/ BS3 and 24.7 to 31.5 Å for Sulfo-EGS. Therefore, the rigid crosslinks were penalized more strongly for diverting from expected distances than the flexible linkers, which reflects their inability/ability to span a range of distances. In the future, it would also be of interest to further look into the effect of reagent flexibility by using additional crosslinkers such as using anthracene-1,5-disulfonic acid, where three phenyl rings are attached. The percentages of observed states with measured crosslinked distances below restraint limits during MD runs with different restraint parameters are shown in Table S1. The R38Cζ – K73Nζ restraint (1,5-NDSA crosslink) is important during the progress of the MD run since omission of the 1,5-NDSA restraint (no restraints and no NDSA parameters) results in no structures converging to be within the set restraint distance of 14.4 Å. In contrast, the measured K25Nζ – K73Nζ distance (BS2G and BS3 crosslink) is consistently over the restraint limit range of 22.0 – 26.2 Å unless the restraint is omitted (no restraints and no BS2G parameters), where more than 90% of the observed states are below the restraint limit. The measured R38Cζ – K73Nζ distance is the most tolerant of all the various restraint parameters, where the measured distance being below the restraint distance of 24.7 – 31.5 Å is always observed over the course of the 100 ns MD run. The progress of the measured crosslinked distance over the course of the 100 ns MD simulation for the different restraint parameters was also plotted (Figures S6 to S9). Overall, larger variations in the measured distance are observed in the absence of all four restraints as well as the omission of the restraint corresponding to the reagent crosslinking a specific residue pair, such as not using the 1,3-BDSA restraint of 13.2 Å and measuring the Arg38 – Lys55 pair crosslinked experimentally by 1,3-BDSA (Figure S6). Interestingly, including the BS2G/BS3 restraint range of 22.0 – 26.2 Å results in measured Lys25 – Lys73 distances that are consistently larger (> 36 Å) than the set restraint range and do not converge below the restraint distance upon completion of the MD run (Figure S9), likely due to additional flexibility in these crosslinks than what was incorporated in the model.
The final structures observed at the end of the MD runs were used as representative structures to measure the crosslinked distance when different restraint parameters were tested. Out of all the different restraint parameters (Table S2), the final structures obtained when using three of the restraints (1,3-BDSA, 1,5-NDSA and Sulfo-EGS, i.e. no BS2G) provided the best models. Table 1 summarizes the measured distances between the crosslinked pairs as well as the collision cross-section values (CCSHe) calculated using the Trajectory Method algorithm and default Lennard-Jones parameters in IMoS. Calculations were run in helium since the ion/ion reaction takes place in the trap cell with helium as the drift gas. All of the distances for the X-ray structure are larger than the calculated restraint distance as is the computed CCSHe value of 1310 Å2 compared to 1247 Å2 published previously for native cytochrome c 7+ using a drift tube ion mobility instrument with He as the drift gas by Shelimov et al.53 Figure 6A also shows the X-ray structure of cytochrome c and with the side chains oriented outwards and none of the measured distances below the calculated restraint distances. These data correlate with the compaction of solution structure upon transfer to the gas phase, a phenomenon that has been thoroughly studied.54, 55
Figure 6.
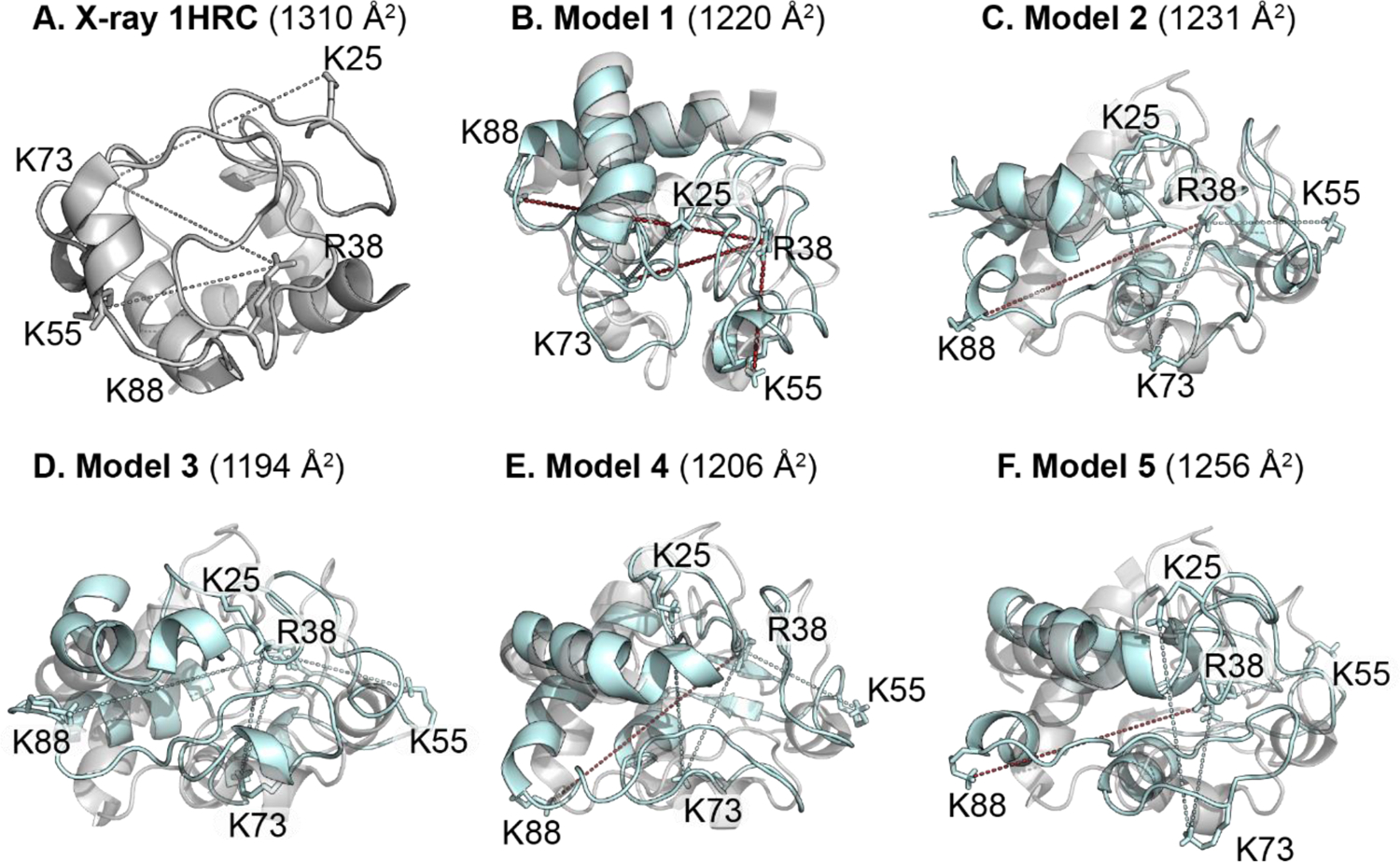
Modified side chains displayed using cytochrome c (A) x-ray structure only and (B – F) MD models 1 – 5 (cyan) aligned to the x-ray structure (gray). For MD models 1 through 5, dotted lines represent the Cζ – Nζ or Nζ – Nζ distances measured for the different crosslinked residue pairs, with measured distances within the calculated restraint distance in cyan and those larger than the restraint distance in red.
The calculated CCSHe for all the MD models are within 4.2 % of the experimental value (1247 Å2) obtained by Shelimov et al. While the CCSHe values provide a coarser approach to study protein structure in the gas phase, the experimental crosslinking distances are useful in gaining residue-level structural details. Only MD model 3 satisfies the restraint distances for all four crosslinking pairs when considering the lower end of the restraint range for Sulfo-EGS (24.7 Å). Models 2, 4 and 5 also satisfy all restraint distances when considering the upper bound of the Sulfo-EGS (31.5 Å). The various models suggest a degree of dynamicity of the side chain residues in the gas phase and the presence of multiple co-existing structures that are being captured using gas-phase crosslinking. For model 1, the Arg38 – Lys55 (1,3-BDSA) and Arg38 – Lys73 (1,5-NDSA) distances are larger compared to the restraint limits by 0.2 Å. Structures similar to model 1 are the least sampled by our crosslinkers since the percentages of observed states with measured distances below the restraint cut-off are the lowest in contrast to models 2 to 5 (Table S1). Sulfo-EGS was the longest reagent used and while only one model contains a R38Cζ – K73Nζ distance below the lower end of the restraint range, all the models are well below the maximum 31.5 Å cut-off. Thus, using crosslinkers longer than Sulfo-EGS might not provide helpful restraints for a protein of the size of cytochrome c.
Alignment of individual gas-phase MD model with the x-ray structure of cytochrome c provides a visual assessment of how dynamic the crosslinked amino acid residues are (Figure 6). Overall, the secondary structure features overlap well between the condensed phase (gray) and gas-phase (cyan) structures, such as α-helices spanning regions such as Thr49 – Lys55, Asn69 – Ile75 and Lys87 – Glu108 (C-terminus), albeit with slight shifts in relative positions. The modified residues Lys55, Lys73 and Lys88 are part of the α-helices while Lys25 and Arg38 are in random coil regions. The decreased overlap for the random coil regions demonstrates compaction in the gas-phase as well as the trapping of kinetically frozen solution structures during the electrospray process. Thus, despite marginal changes in the calculated CCSHe values, our restrained MD models show that cytochrome c undergoes rearrangements at the residue level, which are uniquely probed by gas-phase crosslinking. Moreover, the use of a multipass traveling wave cyclic IM (cIM) device to achieve higher resolution separation has led to the detection of new IM features within a broad IM peak distribution for cytochrome c 7+, likely due to various, highly similar conformers present within that broadened IM peak.56 Thus, our gas-phase crosslinking study using a Synapt G2-Si instrument provides evidence for multiple co-existing conformers in the gas-phase when combined with MD calculations, recapitulating the published results obtained for cytochrome c 7+ after multiple passes in a cIM. However, we note that a fuller exploration of the solution phase conformational landscape through longer simulations or replica exchange methods57, 58 is necessary for gas-phase de novo structural determination.
Finally, we recognize that our models are dependent on our choice of a starting structure, which is the crystal structure of cytochrome c. If we performed a full gas-phase conformational search, our results may have been different, showing evidence for non-native structures. However, rigorous modeling of the energetics of ions in a Synapt G2-Si, the same instrument that we used for this study, showed that protein ion temperatures without applying collision induced unfolding were on the order of much less than 400 K (e.g., for native-like myoglobin 8+ ions, temperatures were near 330 K),59 which would not support an unfolding and refolding mechanism, also consistent with MD of the transition of folded proteins from the droplet to the gas phase.60 Although instrument conditions are not exactly the same for our ion/ion reactions, we do tune the lens elements prior to the trap cell (i.e., to the crosslinking reaction) to minimize collisional activation. In doing so, we consistently observe native like arrival time distributions for small proteins such as ubiquitin and cytochrome c.30, 61 Since the gas phase lacks solvent, and thus refolding back into the native structure can be considered highly unlikely,11 approaches such as gas phase simulated annealing MD performed at much higher temperatures might be expected to give artifactual results for well-folded proteins. Indeed, increasing the temperature (i.e., through collisional activation) of compact cytochrome c structures in the gas phase does produce extended states that compact down to the original CCS values, with the compact structures displaying a “permanent” increase in CCS.62, 63 With these considerations, the most accurate modeling of gaseous structures of well-folded proteins would need to keep temperatures low enough that the gas phase rearrangement barriers are not reached. Additionally, using the crystal structure of folded proteins, followed by relaxation in the gas phase, accurately reproduces experimental observables.15, 40
Conclusions
We have demonstrated, for the first time, the use of an ensemble of different length gas-phase noncovalent crosslinkers via ion/ion reactions to provide insight into the nature of gas-phase structures of a cooperatively folded protein. Crosslinking was performed prior to ion mobility, allowing for ion/ion reactions products to be separated and identifications made based on ECD with mild supplemental activation. We only used c and z ions to determine noncovalent linking identifications to prevent misidentifications due to mobilization of the sulfonate-lysine bonds, though previous work has shown this bond has covalent-like strength in the gas phase.61 The most abundant native-like ion from cytochrome c, the 7+ charge state, yielded different identifications of crosslinks with increasing linker length, evidence that the crosslinks are sensitive to 3-D structure. When used to restrain gas-phase molecular dynamics simulations, the crosslink identifications predicted structures that gave calculated CCSHe in reasonable agreement with experimental values, giving evidence that cytochrome c native-like ions remain compact in the gas phase. This work, along with mobility measurements and infrared spectroscopy20, clearly supports the kinetic trapping of solution-like structures during gentle transfer to the gas phase.
Interestingly, and importantly, the combination of ion/ion reaction-derived distance restraints and molecular dynamics did not converge on a single structure that satisfied all criteria. This correlates with observations based on tandem IM measurements from the Clemmer Laboratory, where they observed that the arrival time distribution of ubiquitin ions was composed of many non-interconverting structures,64 which, again, was recapitulated with cyclic tandem ion mobility experiments with cytochrome c.56 Instead, we predict structures that are similar in CCS, but are not identical. Our structures are not exhaustive; there are several individual conformations that we did not determine that are still supported by our measurements. Thus, in the future, a more thorough conformational search is warranted. However, our modeled structures do show preservation of the overall protein fold, where the “breathing” motion of less ordered regions and compaction of side chains to form new hydrogen bonds/salt bridges account for the differences in structure. Therefore, in the future, we envision the application of this combined experimental and MD method to follow changes in solution structure due to unfolding conditions, ligand and metal binding, etc., and determine their effects on gas-phase structure and whether non-native structures are retained in the gas phase.
Supplementary Material
Acknowledgements
Portions of this work were funded by the National Institutes of Health under R21GM134408 and the Indiana University-Purdue University Indianapolis School of Science. We would like to acknowledge Dr. Amber Rolland and Professor James Prell from the University of Oregon for assistance with the implementation of gas phase molecular dynamics.
Footnotes
Supporting Information
ECD sequence coverage of crosslinked cytochrome c without supplemental activation, IM and m/z spectra of the rest of the crosslinking ion/ion reactions, Annotated m/z spectra (3200 – 4200 m/z range) of the crosslinking ion/ion reactions, ECD sequence coverage of charge reduced cytochrome c via ion/ion reaction with PFO, ECD sequence coverage of unmodified cytochrome c 7+ showing the assignment of ionizing protons via increase in fragment ion charge state and protonation sites mapped on the X-ray structure, Progression of the distance measured between R38Cζ and K55Nζ, R38Cζ and K73Nζ, R38Cζ and K88Nζ and K25Nζ and K73Nζ using different restraint parameters during MD runs, Percentages of observed states with measured crosslinked distances below restraint limits during MD runs with different restraint parameters, Measured distances for the final MD structures using various restraints parameters.
References
- (1).Hamdy OM; Julian RR Reflections on charge state distributions, protein structure, and the mystical mechanism of electrospray ionization. J. Am. Soc. Mass Spectrom 2012, 23, 1–6. [DOI] [PubMed] [Google Scholar]
- (2).Loo JA Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev 1997, 16, 1–23. [DOI] [PubMed] [Google Scholar]
- (3).McLafferty FW Tandem mass spectrometry. Science 1981, 214, 280–287. [DOI] [PubMed] [Google Scholar]
- (4).Leney AC; Heck AJR Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom 2017, 28, 5–13. [DOI] [PubMed] [Google Scholar]
- (5).Butler KE; Takinami Y; Rainczuk A; Baker ES; Roberts BR Utilizing Ion Mobility-Mass Spectrometry to Investigate the Unfolding Pathway of Cu/Zn Superoxide Dismutase. Front. Chem 2021, 9, 614595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ruotolo BT; Benesch JL; Sandercock AM; Hyung SJ; Robinson CV Ion mobility-mass spectrometry analysis of large protein complexes. Nat. Protoc 2008, 3, 1139–1152. [DOI] [PubMed] [Google Scholar]
- (7).Ruotolo BT; Robinson CV Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol 2006, 10, 402–408. [DOI] [PubMed] [Google Scholar]
- (8).Zhong Y; Han L; Ruotolo BT Collisional and Coulombic unfolding of gas-phase proteins: high correlation to their domain structures in solution. Angew. Chem 2014, 53, 9209–9212. [DOI] [PubMed] [Google Scholar]
- (9).Wagner ND; Russell DH Defining Noncovalent Ubiquitin Homodimer Interfacial Interactions through Comparisons with Covalently Linked Diubiquitin. J. Am. Chem. Soc 2016, 138, 16588–16591. [DOI] [PubMed] [Google Scholar]
- (10).Wyttenbach T; Bowers MT Structural stability from solution to the gas phase: native solution structure of ubiquitin survives analysis in a solvent-free ion mobility-mass spectrometry environment. J. Phys. Chem. B 2011, 115, 12266–12275. [DOI] [PubMed] [Google Scholar]
- (11).Clemmer DE; Russell DH; Williams ER Characterizing the Conformationome: Toward a Structural Understanding of the Proteome. Acc. Chem. Res 2017, 50, 556–560. [DOI] [PubMed] [Google Scholar]
- (12).El-Baba TJ; Woodall DW; Raab SA; Fuller DR; Laganowsky A; Russell DH; Clemmer DE Melting Proteins: Evidence for Multiple Stable Structures upon Thermal Denaturation of Native Ubiquitin from Ion Mobility Spectrometry-Mass Spectrometry Measurements. J. Am. Chem. Soc 2017, 139, 6306–6309. [DOI] [PubMed] [Google Scholar]
- (13).Dill KA; MacCallum JL The protein-folding problem, 50 years on. Science. 2012, 338, 1042–1046. [DOI] [PubMed] [Google Scholar]
- (14).Mesleh MF; Hunter JM; Shvartsburg AA; Schatz GC; Jarrold MF Structural information from ion mobility measurements: Effects of the long-range potential. J Phys. 1996, 100, 16082–16086. [Google Scholar]
- (15).Rolland AD; Prell JS Computational Insights into Compaction of Gas-Phase Protein and Protein Complex Ions in Native Ion Mobility-Mass Spectrometry. Trends Anal. Chem 2019, 116, 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Warnke S; von Helden G; Pagel K Protein structure in the gas phase: the influence of side-chain microsolvation. J. Am. Chem. Soc 2013, 135, 1177–1180. [DOI] [PubMed] [Google Scholar]
- (17).Goth M; Lermyte F; Schmitt XJ; Warnke S; von Helden G; Sobott F; Pagel K Gas-phase microsolvation of ubiquitin: investigation of crown ether complexation sites using ion mobility-mass spectrometry. Analyst. 2016, 141, 5502–5510. [DOI] [PubMed] [Google Scholar]
- (18).Konermann L; Silva EA; Sogbein OF Electrochemically induced pH changes resulting in protein unfolding in the ion source of an electrospray mass spectrometer. Anal. Chem 2001, 73, 4836–4844. [DOI] [PubMed] [Google Scholar]
- (19).Soleilhac A; Dagany X; Dugourd P; Girod M; Antoine R Correlating droplet size with temperature changes in electrospray source by optical methods. Anal. Chem 2015, 87, 8210–8217. [DOI] [PubMed] [Google Scholar]
- (20).Seo J; Hoffmann W; Warnke S; Bowers MT; Pagel K; von Helden G Retention of Native Protein Structures in the Absence of Solvent: A Coupled Ion Mobility and Spectroscopic Study. Angew Chem Int Ed Engl 2016, 55, 14173–14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bakhtiari M; Konermann L Protein Ions Generated by Native Electrospray Ionization: Comparison of Gas Phase, Solution, and Crystal Structures. J. Phys. Chem. B 2019, 123, 1784–1796. [DOI] [PubMed] [Google Scholar]
- (22).Bleiholder C; Liu FC Structure Relaxation Approximation (SRA) for Elucidation of Protein Structures from Ion Mobility Measurements. J. Phys. Chem. B 2019, 123, 2756–2769. [DOI] [PubMed] [Google Scholar]
- (23).Cavalli A; Salvatella X; Dobson CM; Vendruscolo M Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 9615–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Uphoff S; Holden SJ; Le Reste L; Periz J; van de Linde S; Heilemann M; Kapanidis AN Monitoring multiple distances within a single molecule using switchable FRET. Nat. Methods 2010, 7, 831–836. [DOI] [PubMed] [Google Scholar]
- (25).Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Sci. Adv 2017, 3, e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khaje NA; Eletsky A; Biehn SE; Mobley CK; Rogals MJ; Kim Y; Mishra SK; Doerksen RJ; Lindert S; Prestegard JH; Sharp JS Validated determination of NRG1 Ig-like domain structure by mass spectrometry coupled with computational modeling. Commun. Biol 2022, 5, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ly T; Julian RR Elucidating the tertiary structure of protein ions in vacuo with site specific photoinitiated radical reactions. J. Am. Chem. Soc 2010, 132, 8602–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cheung See Kit M; Carvalho VV; Vilseck JZ; Webb IK Gas-Phase Ion/Ion Chemistry for Structurally Sensitive Probes of Gaseous Protein Ion Structure: Electrostatic and Electrostatic to Covalent Cross-Linking. Int. J. Mass Spectrom 2021, 463, 116549–116559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Webb IK; Mentinova M; McGee WM; McLuckey SA Gas-phase intramolecular protein crosslinking via ion/ion reactions: ubiquitin and a homobifunctional sulfo-NHS ester. J. Am. Soc. Mass Spectrom 2013, 24, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Webb IK; Morrison LJ; Brown J Dueling electrospray implemented on a traveling-wave ion mobility/time-of-flight mass spectrometer: Towards a gas-phase workbench for structural biology. Int. J. Mass Spectrom 2019, 444, 116177–116185. [Google Scholar]
- (31).Carvalho VV; See Kit MC; Webb IK Ion Mobility and Gas-Phase Covalent Labeling Study of the Structure and Reactivity of Gaseous Ubiquitin Ions Electrosprayed from Aqueous and Denaturing Solutions. J. Am. Soc. Mass Spectrom 2020, 31, 1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Williams JP; Morrison LJ; Brown JM; Beckman JS; Voinov VG; Lermyte F Top-Down Characterization of Denatured Proteins and Native Protein Complexes Using Electron Capture Dissociation Implemented within a Modified Ion Mobility-Mass Spectrometer. Anal. Chem 2020, 92, 3674–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Park J; Piehowski PD; Wilkins C; Zhou M; Mendoza J; Fujimoto GM; Gibbons BC; Shaw JB; Shen Y; Shukla AK; et al. Informed-Proteomics: open-source software package for top-down proteomics. Nat. Methods 2017, 14, 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Donnelly DP; Rawlins CM; DeHart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B; et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fellers RT; Greer JB; Early BP; Yu X; LeDuc RD; Kelleher NL; Thomas PM ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics. 2015, 15, 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lee C; Yang W; Parr RG Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]
- (38).Becke A Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648. [Google Scholar]
- (39).Gaussian 16, Revision C.01, Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- (40).Rolland AD; Biberic LS; Prell JS Investigation of Charge-State-Dependent Compaction of Protein Ions with Native Ion Mobility–Mass Spectrometry and Theory. J. Am. Soc. Mass Spectrom 2022, 33, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015, 1–2, 19–25. [Google Scholar]
- (42).Schuler LD; Van Gunsteren WF On the choice of dihedral angle potential energy functions for n-alkanes. Mol. Simul 2000, 25, 301–319. [Google Scholar]
- (43).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (44).Larriba C; Hogan CJ Jr Free molecular collision cross section calculation methods for nanoparticles and complex ions with energy accommodation. J. Comput. Phys 2013, 251, 344–363. [Google Scholar]
- (45).Larriba C; Hogan CJ Jr Ion mobilities in diatomic gases: measurement versus prediction with non-specular scattering models. J. Phys. Chem. A 2013, 117, 3887–3901. [DOI] [PubMed] [Google Scholar]
- (46).Merkley ED; Rysavy S; Kahraman A; Hafen RP; Daggett V; Adkins JN Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 2014, 23, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bu J; Peng Z; Zhao F; McLuckey SA Enhanced Reactivity in Nucleophilic Acyl Substitution Ion/Ion Reactions Using Triazole-Ester Reagents. J. Am. Soc. Mass Spectrom 2017, 28, 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Breuker K; McLafferty FW Native electron capture dissociation for the structural characterization of noncovalent interactions in native cytochrome c. Angew. Chem 2003, 42, 4900–4904. [DOI] [PubMed] [Google Scholar]
- (49).Morrison LJ; Brodbelt JS Charge site assignment in native proteins by ultraviolet photodissociation (UVPD) mass spectrometry. Analyst. 2016, 141, 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kjeldsen F; Savitski MM; Adams CM; Zubarev RA Determination of the location of positive charges in gas-phase polypeptide polycations by tandem mass spectrometry. Int. J. Mass Spectrom 2006, 252, 204–212. [Google Scholar]
- (51).Gong Z; Ye S-X; Tang C Tightening the Crosslinking Distance Restraints for Better Resolution of Protein Structure and Dynamics. Structure. 2020, 28, 1160–1167. [DOI] [PubMed] [Google Scholar]
- (52).Degiacomi MT; Schmidt C; Baldwin AJ; Benesch JLP Accommodating Protein Dynamics in the Modeling of Chemical Crosslinks. Structure. 2017, 25, 1751–1757. [DOI] [PubMed] [Google Scholar]
- (53).Shelimov KB; Jarrold MF Conformations, unfolding, and refolding of apomyoglobin in vacuum: An activation barrier for gas-phase protein folding. J. Am. Chem. Soc 1997, 119, 2987–2994. [Google Scholar]
- (54).Hall Z; Politis A; Bush MF; Smith LJ; Robinson CV Charge-state dependent compaction and dissociation of protein complexes: insights from ion mobility and molecular dynamics. J. Am. Chem. Soc 2012, 134, 3429–3438. [DOI] [PubMed] [Google Scholar]
- (55).Pacholarz KJ; Porrini M; Garlish RA; Burnley RJ; Taylor RJ; Henry AJ; Barran PE Dynamics of Intact Immunoglobulin G Explored by Drift-Tube Ion-Mobility Mass Spectrometry and Molecular Modeling. Angew. Chem 2014, 53, 7765–7769. [DOI] [PubMed] [Google Scholar]
- (56).Eldrid C; Ujma J; Kalfas S; Tomczyk N; Giles K; Morris M; Thalassinos K Gas Phase Stability of Protein Ions in a Cyclic Ion Mobility Spectrometry Traveling Wave Device. Anal. Chem 2019, 91, 7554–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sugita Y; Okamoto Y Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- (58).Rhee YM; Pande VS Multiplexed-Replica Exchange Molecular Dynamics Method for Protein Folding Simulation. Biophys. J 2003, 84, 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Donor MT; Shepherd SO; Prell JS Rapid Determination of Activation Energies for Gas-Phase Protein Unfolding and Dissociation in a Q-IM-ToF Mass Spectrometer. J. Am. Soc. Mass Spectrom 2020, 31, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).McAllister RG; Metwally H; Sun Y; Konermann L Release of Native-like Gaseous Proteins from Electrospray Droplets via the Charged Residue Mechanism: Insights from Molecular Dynamics Simulations. J. Am. Chem. Soc 2015, 137, 12667–12676. [DOI] [PubMed] [Google Scholar]
- (61).Cheung See Kit M; Shepherd SO; Prell JS; Webb IK Experimental Determination of Activation Energies for Covalent Bond Formation via Ion/Ion Reactions and Competing Processes. J. Am. Soc. Mass Spectrom 2021, 32, 2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Eldrid C; Ben-Younis A; Ujma J; Britt H; Cragnolini T; Kalfas S; Cooper-Shepherd D; Tomczyk N; Giles K; Morris M; et al. Cyclic Ion Mobility–Collision Activation Experiments Elucidate Protein Behavior in the Gas Phase. J. Am. Soc. Mass Spectrom 2021, 32, 1545–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Borotto NB; Osho KE; Richards TK; Graham KA Collision-Induced Unfolding of Native-like Protein Ions Within a Trapped Ion Mobility Spectrometry Device. J. Am. Soc. Mass Spectrom 2022, 33, 83–89. [DOI] [PubMed] [Google Scholar]
- (64).Koeniger SL; Merenbloom SI; Clemmer DE Evidence for many resolvable structures within conformation types of electrosprayed ubiquitin ions. J. Phys. Chem. B 2006, 110, 7017–7021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.