

Abstract
In a recent article published in Nature, Chen et al. reported that pregestational hyperglycemia rendered offspring vulnerable to glucose intolerance due to insufficient TET3‐mediated 5‐methylcytosine oxidation and DNA demethylation, resulting in hypermethylation and reduction of insulin‐secreting genes.

Since the widespread recognition of the ‘developmental origins of health and disease (DOHaD)’ hypothesis, the concept of ‘fetal origins of adult disease’ has been well recognized and has sparked intensive research interest 1 . The interrelation and environmental insults (nutrition, stress, and chemicals) in early life (fertilization, embryo, and fetus) can contribute to an increased long‐term risk of metabolic disorders. Several lines of evidence has indicated that prolonged gestational hyperglycemia rendered the offspring epigenome susceptible to being perturbed and there being an ensuing amplified risk of metabolic diseases 2 , 3 , 4 . Although gamete‐mediated epigenetic inheritance has been reported, the underlying mechanism of epigenetic inheritance dependent on the maternal germline has never been investigated.
A recent study in Nature has reported that preconception hyperglycemia is likely to render offspring susceptible to metabolic abnormities via maternal germline‐dependent epigenetic inheritance 5 . To distinguish the oocyte‐derived phenotypic transition, the authors performed in vitro fertilization (IVF) from a normal male mouse with hyperglycemic females (HG mice) and embryo transfer (ET) to normal acceptors to exclude the influence of gestational hyperglycemia on the health of progeny. The male F1 generation displayed impaired glucose tolerance, mainly exhibited as defective insulin secretion at 16 weeks of age, and the female F1 generation showed glucose intolerance at 1 year of age. The protective phenotype of sex hormones might account for the delayed onset in females. Upon treatment with a high‐fat diet, glucose intolerance occurred earlier, from 12 weeks of age, both in the male and female F1 generation. However, further intercross and outcross did not generate F2 offspring with glucose intolerance, indicating intergenerational but not transgenerational inheritance of maternal pregestational hyperglycemia.
Given that only oocytes from hyperglycemic mice would receive initial exposure to maternal hyperglycemia and trigger the inherited phenotype, the researchers profiled the transcriptome to investigate potential epigenetic molecules in the oocytes. It was observed that Tet3 gene expression was reduced significantly in oocytes from hyperglycemic mice. Similar decreases were verified in oocytes from mice with type 2 diabetes and diabetic women, and in cultured oocytes stimulated with high glucose in vitro. TET3 is a deoxyribonucleic acid (DNA) demethylation enzyme belonging to the ten‐eleven translocation (TET) family of DNA demethylases [ten‐eleven translocation methylcytosine dioxygenase 1 (TET1) to ten‐eleven translocation methylcytosine dioxygenase 3 (TET3)] that catalyzes the conversion of 5‐methylcytosine (5mC) to 5‐hydroxymethylcytosine (5hmC) during the oocyte stage. The authors next sought to profile abnormally hypermethylated genes via genome‐wide association studies (GWAS) sequencing and it was found that the insulin‐secreting genes ranked top, among which was glucokinase (Gck) in detailed studies. By the combined use of bisulfite sequencing and apolipoprotein B messenger ribonucleic acid editing of the catalytic polypeptide‐like family‐coupled epigenetic sequence (ACE‐Seq), the authors then verified the possibility that hyperglycemia‐induced oocyte TET3 insufficiency led to hypermethylation of the Gck promoter and a decrease in Gck expression that persisted in pancreatic islets from zygote to adulthood. Interestingly, further pyrosequencing analysis revealed that the hypermethylation of a cluster of insulin‐secreting genes was derived explicitly from the paternal allele. These data suggested that TET3 was the key transgenerational factor that epigenetically reprogrammed the insulin‐secreting genes and drove the inheritance of diabetes susceptibility.
To confirm the direct link between TET3 insufficiency and a predisposition of the offspring to diabetes, the authors produced Tet3 knock‐out mice. The offspring that were produced from oocytes in TET3 heterozygotic and homozygotic deficient mice exhibited a similar phenotype to those from oocytes in hyperglycemic mice, suggested by aberrant hypermethylation at the Gck promoter and the subsequent glucose intolerance. In contrast, supplementing exogenous TET3 in oocytes could alleviate abnormal hypermethylation, increase Gck expression, and reverse the glucose intolerance in the resultant offspring.
Taken together, the study by Chen et al. 5 revealed the unequivocal association between pregestational hyperglycemia and the susceptibility of offspring to metabolic disorders. The maternal impact, such as hyperglycemia, causes oocyte TET3 insufficiency, which in turn reprograms and impairs the zygotic paternal methylation. Impaired DNA demethylation leads to dysregulation in insulin‐secreting genes from zygote to adulthood, resulting in glucose intolerance in the offspring. Figure 1 shows the schematic depiction of the mechanism.
Figure 1.
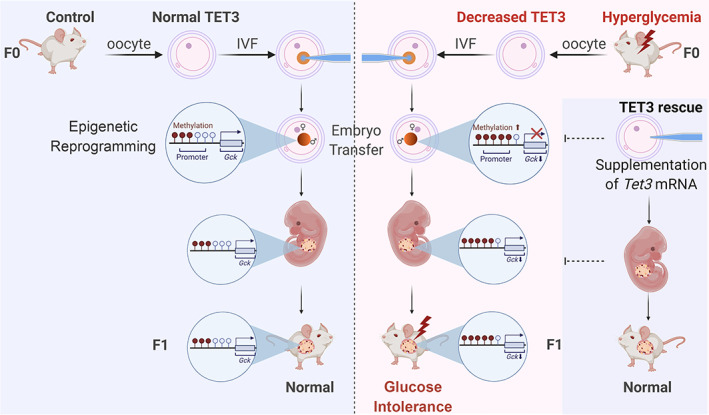
Schematic depiction of the mechanism of maternal epigenetic inheritance of glucose intolerance induced by pregestational hyperglycemia mediated oocyte TET3 insufficiency and impaired DNA demethylation. IVF, in vitro fertilization; mRNA, messenger ribonucleic acid; TET3, ten‐eleven translocation methylcytosine dioxygenase 3; DNA, deoxyribonucleic acid. Brown solid lollipops suggest DNA methylation. [Colour figure can be viewed at wileyonlinelibrary.com]
Multiple human cohorts and experimental animal models have suggested that parental early‐life exposure could potentially influence the phenotype in subsequent generations 4 . Parental health pre‐pregnancy, during pregnancy, and post‐pregnancy are able to affect future health. The mechanisms of transmission across generations occur through maternal or paternal inheritance, or both. Unique maternal factors mainly cover maternal metabolic disorders, an abnormal vaginal or gut microbiome, altered reproductive organs, and mitochondrial‐related inheritance. Epigenetic modification of gene transcription is an intensive candidate to regulate environment‐induced metabolic diseases, causing epigenetic regulation, a strongly dynamic progress throughout the whole lifespan.
The research article 5 suggested and verified the concept of pregestational hyperglycemia sensitizing the progeny to metabolic diseases in humans and mice, and further exposed the critical epigenetic regulator TET3 for the first time. The IVF‐ET and pronucleus track mainly avoids the influence of gestation and maternal metabolism, highlighting the effect of pre‐pregnancy and ensuring full‐chain traceability. Similar findings in humans might enhance extrapolation from bench studies to the bedside. These inspiring results in rescue experiments with a TET3 supplement might inspire the possibility of reprogramming stem cells for prevention and treatment. However, there are still some questions that merit further investigation. For example, what is the upstream regulatory mechanism whereby hyperglycemia regulates TET3 expression? Will dietary change or lifestyle modifications be beneficial for women with pregestational hyperglycemia via regulatory effects on TET3 expression and activity? What are the potential effects of other adverse factors, such as hyperlipidemia and hyperhomocyteinemia on offspring predisposition to metabolic disorders via TET3‐dependent epigenetic mechanisms? Further investigation of TET3‐mediated mechanisms in prospective women and fetus cohorts is definitely needed.
In summary, the study 5 unveiled that pregestational hyperglycemia predisposes offspring to metabolic disorders via an insufficiency of oocyte TET3 and impaired DNA demethylation in insulin‐secreting genes. This study offered revolutionary insights into the intergenerational or transgenerational inheritance of environmental metabolic diseases and acquired phenotypes, and into the early prevention of chronic diseases at origin. The findings further provide a novel perspective and call for special attention to be paid to regular screening and early prevention in women of reproductive age.
DISCLOSURE
The authors declare no conflict of interest.
Approval of the research protocol: N/A.
Informed consent: N/A.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
REFERENCES
- 1. Hales CN, Barker DJ, Clark PM, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991; 303: 1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rando OJ, Simmons RA. I'm eating for two: parental dietary effects on offspring metabolism. Cell 2015; 161: 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaeger K, Saben JL, Moley KH. Transmission of metabolic dysfunction across generations. Physiology (Bethesda) 2017; 32: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sales VM, Ferguson‐Smith AC, Patti ME. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab 2017; 25: 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen B, Du YR, Zhu H, et al. Maternal inheritance of glucose intolerance via oocyte TET3 insufficiency. Nature 2022; 605: 761–766. [DOI] [PubMed] [Google Scholar]