

Abstract
Objective
To characterize potential drug safety signals identified from the US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS), from 2008 to 2019, to determine how often these signals resulted in regulatory action by the FDA and whether these actions were corroborated by published research findings or public assessments by the Sentinel Initiative.
Design
Cross sectional study.
Setting
USA.
Population
Safety signals identified from the FAERS and publicly reported by the FDA between 2008 and 2019; and review of the relevant literature published before and after safety signals were reported in 2014-15. Literature searches were performed in November 2019, Sentinel Initiative assessments were searched in December 2021, and data analysis was finalized in December 2021.
Main outcome measures
Safety signals and resulting regulatory actions; number and characteristics of published studies, including corroboration of regulatory action as evidenced by significant associations (or no associations) between the drug related to the signal and the adverse event.
Results
From 2008 to 2019, 603 potential safety signals identified from the FAERS were reported by the FDA (median 48 annually, interquartile range 41-61), of which 413 (68.5%) were resolved as of December 2021 (372 of 399 (93.2%) signals ≥3 years old were resolved). Among the resolved safety signals, 91 (22.0%) led to no regulatory action and 322 (78.0%) resulted in regulatory action, including 319 (77.2%) changes to drug labeling and 59 (14.3%) drug safety communications or other public communications from the FDA. For a subset of 82 potential safety signals reported in 2014-15, a literature search identified 1712 relevant publications; 1201 (70.2%) were case reports or case series. Among these 82 safety signals, 76 (92.7%) were resolved, of which relevant published research was identified for 57 (75.0%) signals and relevant Sentinel Initiative assessments for four (5.3%) signals. Regulatory actions by the FDA were corroborated by at least one relevant published research study for 17 of the 57 (29.8%) resolved safety signals; none of the relevant Sentinel Initiative assessments corroborated FDA regulatory action.
Conclusions
Most potential safety signals identified from the FAERS led to regulatory action by the FDA. Only a third of regulatory actions were corroborated by published research, however, and none by public assessments from the Sentinel Initiative. These findings suggest that either the FDA is taking regulatory actions based on evidence not made publicly available or more comprehensive safety evaluations might be needed when potential safety signals are identified.
Introduction
Post-marketing pharmacovigilance is an essential component for monitoring drug safety, providing early identification of potential adverse events related to drugs through a combination of voluntary and active surveillance efforts.1 2 Although clinical trials conducted before drugs come to market are used to establish drug toxicity and assess potential major adverse events, rare adverse events can be missed, such as those that result from extended periods of use or that are more common in specific subsets of individuals.3 For many years, the primary post-marketing surveillance system in the US has been the Food and Drug Administration (FDA)’s database of spontaneous adverse event reports from drug manufacturers, healthcare staff, or consumers, the FDA Adverse Event Reporting System (FAERS).4 Reporting of adverse events has steadily increased over the past 10 years, from 780 000 in 2011 to >2.3 million in 2021.5 To manage this volume, the FDA uses various data mining techniques to analyze reports and identify potential safety alerts from the FAERS that require further investigation or regulatory changes.4 6
The strength and accuracy of spontaneous adverse event reporting surveillance systems have been extensively debated, however, with reported limitations of disproportionate reporting of serious adverse events and uncertainty in determining causality.7 8 For the FAERS specifically, underreporting, redundancies, selection bias, and incomplete data have been described.9 10 Also, >90% of adverse events in the FAERS are reported by drug manufacturers, most likely because of mandatory reporting requirements, suggesting the possibility of single source bias.11 Reports from non-medical staff can also confuse true associations; for example, many reports identifying adverse cardiovascular events from the use of celecoxib were submitted by legal professionals.12 Finally, stimulated reporting can be an issue with spontaneous reporting systems, whereby media attention and regulatory influences temporarily increase reports of adverse events and, in turn, the probability of false positive results.13
Despite these limitations, spontaneous reporting surveillance systems, such as the FAERS, are the main method of post-marketing pharmacovigilance.5 Potential signals from the FAERS have resulted in numerous safety communications and boxed warnings, each of which has the potential to affect which drug treatments patients use, change prescribing habits of physicians, and even influence decisions about insurance coverage.14 15 In recent years, efforts have increased to look at these widely reported failings and improve the accuracy of passive surveillance systems, such as data mining techniques, examination of the medical literature, and active analysis of ongoing clinical trials or studies.4 Also, in 2008, the FDA launched the Sentinel Initiative, an active surveillance system intended to complement the FAERS to assess the safety of drugs approved by the FDA under real world conditions.16 17 The program uses a distributed data network that has selected claims and electronic health data from multiple health systems in the US that allows the FDA to conduct analyses to assess potential drug safety signals as part of their surveillance efforts. The extent to which these additional measures are used, however, and their influence on potential safety signal assessments is not clear.
In 2007, the FDA Amendments Act required the FDA to report potential safety signals identified from the FAERS in quarterly reports, regardless of whether the investigation of the safety signal had reached a conclusion or whether a regulatory change had occurred.6 Public reporting of these safety signals provides an opportunity to retrospectively examine safety signals that were identified with the FAERS, to better understand this pharmacovigilance system. Therefore, the objective of this study was to characterize recently reported potential safety signals and describe whether these signals were confirmed or supported by the medical literature. Specifically, we examined all potential safety signals reported from 2008 to 2019, determining how often a safety signal led to a regulatory action by the FDA. Then, for a subset of signals reported in 2014-15, we determined whether relevant published research existed that might have informed the regulatory actions and whether the research corroborated the regulatory action (ie, if a significant association described in the published research aligned with the regulatory action), to provide insight into the FDA’s actions for potential safety signals identified from the FAERS. Finally, to better understand whether regulatory information outside of published research was used to corroborate regulatory actions, we also searched assessments publicly reported by the Sentinel Initiative.5 18
Methods
The study was prepared in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for cross sectional studies.
Characterization of FAERS signals and subsequent FDA safety actions
In accordance with Title IX, Section 921 of the FDA Amendments Act of 2007, the FDA screens the FAERS database every two weeks, later amended to “regular screenings” by the 21st Century Cures Act,19 and publishes all aggregated safety signals in a quarterly report, Potential Signals of Serious Risks/New Safety Information Identified from the FDA Adverse Reporting System (FAERS).6 From these quarterly reports, we identified all potential safety signals, including the drug name, adverse events for the signal, and FDA actions taken for the signal, for the period 2008-19. We excluded signals for non-therapeutic products, contrast dyes, medical devices, quasi-drugs, and drug-drug interactions as the adverse event.
Next, signals were classified by their resolution status: signals associated with the regulatory description “FDA is evaluating the need for regulatory action” were classified as unresolved and all others were classified as resolved. Resolved signals were then categorized as requiring or not requiring regulatory action. Regulatory actions included public communications from the FDA, dear healthcare provider letters, changes to drug labeling, including boxed warnings, and withdrawal of drugs. Before 2010, the FDA used different communication methods to alert clinicians about new safety events, such as news releases, healthcare professional sheets, and public health advisories. From 2010, the FDA used drug safety communications as its primary method of notification for post-market adverse event alerts. In this study, safety communications included all previous methods of communication as well as drug safety communications.
Corroboration of reported safety signals
To determine whether published research studies or public assessments by the Sentinel Initiative corroborated or refuted the safety signals identified from the FAERS, we focused on a subset of signals reported in 2014-15. We anticipated that by November 2019, the time of our searches, most signals reported during 2014-15 would be resolved. For all identified signals, two authors (MMD and XS) extracted the therapeutic type (drug or biologic) and adverse event type (cardiovascular v not cardiovascular), with a method developed previously.20
Evidence cited as part of FDA safety actions
Before searching the literature and the Sentinel Initiative website for corroborating evidence, we examined the evidence cited in the regulatory action in response to the subset of safety signals. For each regulatory change to the drug label, we reviewed the evidence cited by the label to determine if the safety signal identified from the FAERS was explicitly cited, and reviewed any other evidence, as well as the context and timing of when the safety signal was identified. For signals that resulted in a drug safety communication, we examined the communication to determine the evidence cited within the data summary section of the drug safety communication to support the relation between the safety signal and the drug.
Corroboration of reported safety signals: literature search for published studies
For each signal reported, we searched for text words and medical subject headings terms for the generic drug or brand name of the drug and its associated adverse event (supplemental table 1). Searches were conducted in Ovid Medline, Ovid Embase, and Web of Science Core Collection, from when the database was established to 1 November 2019. We used recommended search filters for adverse events and searched the reference lists of all of the studies so that other relevant articles not captured in the original search were identified.21 22 Duplicate search results were removed manually by comparing titles and authors of publications.
Literature search eligibility criteria and study selection
Two reviewers (EMC and MMD) independently screened the titles and abstracts of all of the records identified, and one reviewer (MMD) screened the full texts of the identified records for inclusion in the study. Disagreement was resolved through discussion and with a third reviewer (JSR) if necessary. We included published case reports, case series, clinical trials, meta-analyses, intervention studies, cohort studies, case-control studies, and other observational studies that included human patients and reported adverse events from drug therapeutics corresponding to FDA signals. We excluded studies if there was no English language version, if they included animal subjects, or if they were abstracts, opinions or perspectives, consensus statements, reviews without quantitative syntheses, letters, and editorials. We also excluded studies if the adverse event related to the signal was not a study outcome (primary, secondary, or safety endpoint), and we excluded interventional studies if the drug or drugs implicated in the signal were given in all of the study arms. We did not limit inclusion based on the year of the study or the study population.
Data extraction from literature search
For all identified studies, two authors (MMD and XS) extracted general study characteristics: journal and date of online publication, study design, lead sponsor (first sponsor listed if multiple sponsors), and whether any authors reported funding from a pharmaceutical company. Next, studies were divided into one of five groups for extraction of study details: meta-analyses, prospective studies, retrospective non-incidence or prevalence studies (ie, inferential observational research), retrospective incidence or prevalence studies (ie, descriptive observational research), and case series or case reports. Studies were considered incidence or prevalence studies if the primary aim of the study was to describe the incidence or prevalence of the adverse event related to the signal.
For meta-analyses, we extracted the number of studies included and whether the adverse event was reported as significant among the at-risk group, along with the relative effect estimate (ie, odds ratio, hazard ratio, or risk ratio) and the corresponding 95% confidence interval. For prospective studies, we determined whether the study was randomized, double blinded, controlled, or a multicenter study; whether the adverse event was a primary endpoint, secondary endpoint, or safety endpoint; and whether the adverse event associated with the signal was reported as significant among the at-risk group, along with the relative effect estimate and the corresponding 95% confidence interval.
For retrospective non-incidence or prevalence studies, we extracted the study design (case-control, retrospective cohort, or other); whether the study was a multicenter study; and whether the adverse event associated with the signal was reported as significant among the at-risk group, along with the relative effect estimate and the corresponding 95% confidence interval. For retrospective incidence or prevalence studies, we extracted the number of adverse events associated with the signal and the number of patients at risk overall. For case series and case reports, we extracted the number of adverse events associated with the signal and the number of patients at risk overall. All data were extracted independently by two reviewers (MMD and XS). All disagreements were resolved by discussion with a third author (JSR).
Published studies: regulatory action corroboration
We determined whether regulatory actions were corroborated by the published literature. Regulatory actions that led to changes in drug labeling or other safety communications were considered corroborated when at least one study type confirmed a significant relation between the safety signal and the drug in the same direction (ie, increased risk of harm). For signals that did not result in action by the FDA, actions were considered corroborated if no identified studies confirmed a significant relation between the signal and the drug. Case series, case reports, and other descriptive studies, such as retrospective incidence or prevalence studies, cannot provide inferential evidence of associations between a safety signal and a drug, and therefore these studies were not considered to corroborate a regulatory action.
Corroboration of reported safety signals: Sentinel Initiative assessments
We also determined whether regulatory actions were corroborated by assessments reported by the Sentinel Initiative (search conducted 13-24 December 2021). For each signal reported, one author (MMD) searched for the generic drug or brand name of the drug and its associated adverse event, and examined all publicly available assessments examining the relation between the safety signal and the drug. We extracted results from all reported assessments. A 10% random sample of the signals searched was cross referenced with search results obtained by another author (JSR), with complete inter-rater agreement.
Statistical analysis
Descriptive data are reported as number (percentage), median (interquartile range), and mean (standard deviation). The proportion of regulatory actions with and without corroborated findings within the published literature are reported with descriptive statistics. All statistical analyses were conducted in R 3.6.0 (R foundation for Statistical Computing, Vienna, Austria), JMP statistical software version 15.0.0 (SAS Institute), Python statistical software version 3.8 (Python), and Excel spreadsheet software version 16.16.27 (Microsoft).
Patient and public involvement
Improving data access and transparency are core concepts that inspired the development of the FAERS interactive dashboard. FAERS spurs submission of more detailed and complete reports from consumers, healthcare providers, and other members of the public. Complete and detailed reports are immensely helpful to the agency when identifying safety signals and choosing particular products for further scrutiny. In this way, patients and members of the public contributed to the data used in our reporting although they were not directly involved in producing this research with us. We used publicly available data and information from the FDA website, the Sentinel Initiative website, and published journal articles. No patients were involved in setting the research question or the outcome measures, nor were they involved in developing plans for design or implementation of the study. No patients were asked to advise on interpretation or writing up of results. Unfortunately, we could not determine what proportion of the potential safety signals reported in the quarterly reports aggregated by the FDA from the FAERS were initiated by patients.
Results
Potential safety signals and subsequent FDA actions
From 2008 to 2019, 603 potential safety signals were reported in the quarterly reports compiled by the FDA from the FAERS (median 48 annually, interquartile range 41-61). Of these 603 signals, 413 (68.5%) were resolved (as of December 2021, fig 1), including 372 of 399 (93.2%) signals ≥3 years old; 120 of 399 (29.1%) signals applied to multiple drugs in one class. Among the resolved potential safety signals, 91 (22.0%) required no regulatory action and 322 (78.0%) resulted in regulatory action, including 319 (77.2%) changes in drug labeling and 59 (14.3%) drug safety communications or other public communications from the FDA (table 1).
Fig 1.
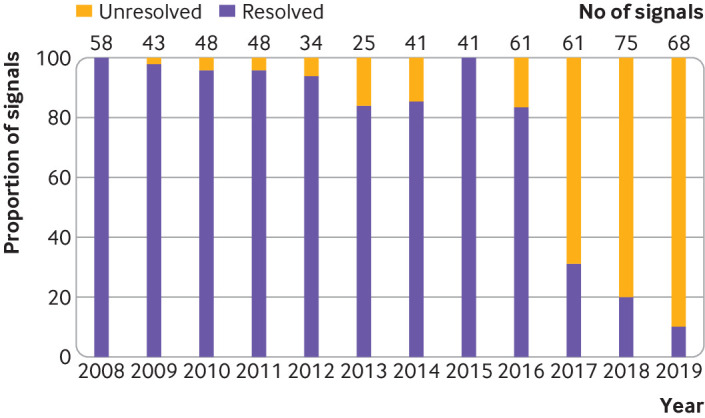
Proportion of potential safety signals identified from the US Food and Drug Administration (FDA) Adverse Event Reporting System by the FDA that were resolved, 2008-19. Numbers above each column are total number of potential safety signals identified by the FDA in each year
Table 1.
Potential safety signals identified from the US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) publicly reported by the FDA between 2008 and 2019
| Year | No of signals | No of signals with resolution | No of signals requiring no regulatory action | No of signals requiring regulatory action: drug label change | No of signals requiring regulatory action: early warning communication | No of signals requiring regulatory action: drug safety communications | |
|---|---|---|---|---|---|---|---|
| 2008 | 58 | 58 | 18 | 38 | 17 | 1 | |
| 2009 | 43 | 42 | 10 | 32 | 15 | 4 | |
| 2010 | 48 | 46 | 7 | 38 | 15 | 11 | |
| 2011 | 48 | 46 | 2 | 43 | 14 | 11 | |
| 2012 | 34 | 32 | 7 | 25 | 6 | 7 | |
| 2013 | 25 | 21 | 4 | 17 | 7 | 5 | |
| 2014 | 41 | 35 | 4 | 31 | 18 | 3 | |
| 2015 | 41 | 41 | 12 | 29 | 17 | 10 | |
| 2016 | 61 | 51 | 12 | 41 | 3 | 4 | |
| 2017 | 61 | 19 | 6 | 12 | 1 | 0 | |
| 2018 | 75 | 15 | 8 | 7 | 0 | 1 | |
| 2019 | 68 | 7 | 1 | 6 | 0 | 2 | |
| Total | 603 | 413 | 91 | 319 | 113 | 59 |
Eighty two potential safety signals identified from the FAERS were reported by the FDA in 2014-15, of which 66 (80.5%) were for drugs and 16 (19.5%) were for biologics. Seven (8.5%) were signals associated with cardiovascular adverse events and 75 (91.5%) were other adverse event types (table 2). Among these 82 safety signals, 76 (92.7%) were resolved and 60 (73.2%) resulted in regulatory action, including 13 (15.9%) drug safety communications, 35 (42.6%) early warning communications from the FDA, and 60 (73.2%) changes in drug labeling.
Table 2.
Characteristics of potential safety signals identified from the US Food and Drug Administration (FDA) Adverse Event Reporting System publicly reported by the FDA, 2014-15
| No (%) of signals | |
| Total No of signals | 82 (100) |
| Associated product type: | |
| Drug | 66 (80.5) |
| Biologic | 16 (19.5) |
| Therapeutic area: | |
| Autoimmune/musculoskeletal | 3 (3.7) |
| Cancer | 10 (12.2) |
| Cardiovascular, diabetes, or lipids | 19 (23.2) |
| Dermatology | 1 (1.2) |
| Infectious disease | 13 (15.9) |
| Neurology | 3 (3.7) |
| Other | 28 (34.1) |
| Psychiatry | 5 (6.1) |
| Associated adverse event type: | |
| Cardiovascular adverse event | 7 (8.5) |
| Non-cardiovascular adverse event | 75 (91.5) |
Evidence cited as part of FDA safety actions
Sixty safety signals reported in 2014-15 were resolved and resulted in regulatory action. All 60 led to changes in drug labeling but none stated that the evidence supporting the change was explicitly from the FAERS. Fifty two (86.7%) signals stated that the adverse event had occurred in patients taking the drug, whereas six (10%) also stated that the adverse events were reported in clinical trials that supported the original approval. Eight (13.3%) changes in drug labeling did not include an explicit statement of any evidence supporting the safety action.
Of the 13 drug safety communications from the safety signals reported in 2014-15, 11 (84.6%) explicitly cited the FAERS as evidence for the association between the drug and the adverse event, including one that also cited case reports from the literature and two that also cited case reports submitted by the sponsoring pharmaceutical company. Of the two (15.4%) drug safety communications that did not explicitly cite the FAERS as evidence, one cited one case report from the literature and another stated that the adverse event had occurred in patients taking the drug (but did not state where this evidence had been reported).
Safety signal literature search
In total, 8465 studies were identified for screening by the corresponding searches for each of the 82 potential safety signals reported in 2014-15 (supplemental figure 1). After screening of the titles and abstracts, 3700 full text records were manually assessed for eligibility; 1712 publications, published 1964-2019, were found to be eligible. At least one relevant publication was identified for 61 (74.4%) signals but no relevant publications were identified for 21 (25.6%) signals. The median number of publications identified for each safety signal was 3.5 (interquartile range 0.75-10.5) and the safety signals associated with the largest number of publications were signals examining the risk of angiogenesis inhibitors or bisphosphonates for osteonecrosis of the jaw (n=510) and the risk of tumor necrosis factor α inhibitors for psychiatric or nervous system disorders (n=314) (fig 2). Of the 1712 eligible publications, 1201 (70.2%) were case series or case reports, 188 (11.0%) were prospective studies, 159 (9.3%) were retrospective non-incidence or prevalence studies (ie, inferential observational research), 105 (6.1%) were retrospective incidence or prevalence studies (ie, descriptive observational research), and 59 (3.4%) were meta-analyses.
Fig 2.
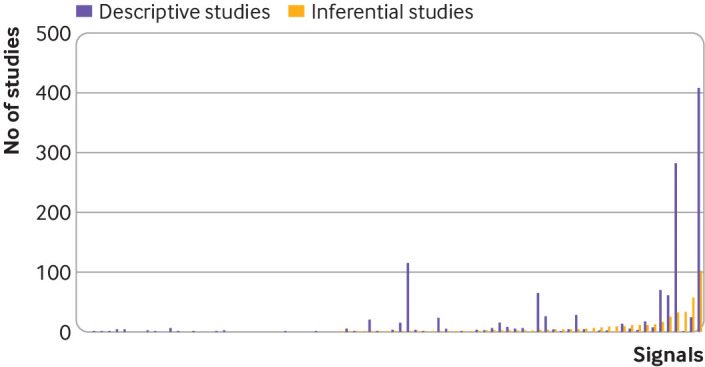
Number of published descriptive and inferential studies identified for each potential safety signal identified from the US Food and Drug Administration Adverse Event Reporting System in 2014-15. Each column pair represents one signal. Inferential studies=meta-analyses, prospective studies, and retrospective non-incidence or prevalence studies. Descriptive studies=retrospective incidence or prevalence studies and case series or case reports. Column pairs are ordered from left to right according to ascending number of inferential studies
The 59 meta-analyses were published between 2001 and 2019. The lead sponsor was a pharmaceutical company in nine (15.3%) studies, a government or public organization in 15 (25.4%), and a non-profit organization in seven (11.9%); seven (11.9%) had at least one author who was employed by a pharmaceutical company. The median number of studies included in each meta-analysis was 11 (interquartile range 6-39). Nineteen (32.2%) reported a significant association between the drug related to the signal and the adverse event.
The 188 relevant prospective studies were published between 1992 and 2019. The lead sponsor was a pharmaceutical company in 100 (53.2%) studies, a government or public organization in 21 (11.2%), and a non-profit organization in 10 (5.3%); 66 (35.1%) had at least one author who was employed by a pharmaceutical company. Of these 188 prospective studies, 129 (68.6%) were controlled, 117 (62.2%) were randomized, 73 (38.8%) were double blinded, and 114 (60.6%) were multicenter studies. Fifty three (28.2%) studies reported a significant association between the drug related to the signal and the adverse event.
The 159 relevant retrospective non-incidence or prevalence studies were published between 1981 and 2019. The lead sponsor was a pharmaceutical company in 23 (14.5%) studies, a government or public organization in 37 (23.3%), and a non-profit organization in 19 (11.9%); 25 (15.7%) had at least one author who was employed by a pharmaceutical company. Of these 159 retrospective non-incidence or prevalence studies, 107 (67.3%) were retrospective cohort studies and 30 (18.9%) were case-control studies. Sixty five (41.0%) studies reported a significant association between the drug related to the signal and the adverse event.
The 105 relevant retrospective incidence or prevalence studies were published between 1977 and 2019. These studies reported an incidence of 4914 observations of the adverse event associated with the signal among 463 116 individuals exposed to the drug, and a total prevalence of 13.8% (190 observations of the adverse event associated with the signal among 1378 individuals).
The 1201 relevant case series or case reports were published between 1964 and 2019. These case series and case reports included 8809 observations of the adverse event associated with the signal among 49 816 individuals who used the drug.
Published research for each safety signal identified
For the 82 potential safety signals reported in 2014-15, the median number of relevant publications was 2.5 (interquartile range 0.75-10): 0 (0-0) meta-analyses, 0 (0-1) prospective studies, 0 (0-1) retrospective non-incidence or prevalence studies, 0 (0-0) retrospective incidence or prevalence studies, and 1 (0-5) case series or case reports. The median number of relevant publications published after the safety signal was identified was 1 (interquartile 0-4): 0 (0-0) meta-analyses, 0 (0-1) prospective studies, 0 (0-1) retrospective non-incidence or prevalence studies, 0 (0-0) retrospective incidence or prevalence studies, and 0 (0-2) case series or case reports.
Regulatory action corroboration by published literature
Of the 82 potential safety signals reported in 2014-15, 76 were resolved, with at least one relevant research publication identified for 57 (75.0%) signals and no relevant published research study for 19 (25.0%) signals. Of the 57 resolved signals with at least one relevant published research study, FDA regulatory actions (or no regulatory action) were corroborated for 17 (29.8%) signals by at least one study showing a significant association between the drug related to the signal and the adverse event (in the case of subsequent FDA regulatory action) or no association (in the case of no subsequent FDA regulatory action) (fig 3).
Fig 3.

Relevant published studies identified for potential safety signals identified from the US Food and Drug Administration (FDA) Adverse Event Reporting System in 2014-15, grouped by study type and corroboration of the FDA’s regulatory action. Shaded bar denotes years 2014-15 when the potential safety signals were made public. Studies that corroborated the FDA’s regulatory action, studies that did not corroborate the FDA’s regulatory action, and descriptive studies that could not corroborate (not applicable) the FDA’s regulatory action are indicated
Thirteen (22.8%) signals were resolved, with at least one relevant published study, that were not followed by FDA regulatory action. Studies showing no significant association between the drug related to the signal and the adverse event were found for 12 (92.3%) signals whereas a study demonstrating a significant association between the drug related to the signal and adverse event was found for 1 (7.7%) signal.
Forty four (77.2%) signals were resolved, with at least one relevant published study, that were followed by FDA regulatory action. A study demonstrating a significant association between the drug related to the signal and the adverse event was found for five (11.4%) signals and no studies showing a significant association between the drug related to the signal and the adverse event were found for 39 (88.6%) signals. Among the two, 17, and 44 resolved signals with at least one relevant published study that were followed by at least a drug safety communication, early warning communication, or change in drug labeling, respectively, a study demonstrating a significant association between the drug related to the signal and the adverse event was found for one (50%), four (23.5%), and five (11.4%) signals, respectively.
Sentinel Initiative assessments
For the 82 safety signals, we identified four (5.3%) with at least one relevant public assessment by the Sentinel Initiative. Of these, all were assessments that quantified the incidence rate of the adverse event related to the signal. No assessments were identified that examined significant associations between the drug related to the signal and the adverse event, which would be needed to corroborate FDA regulatory action.
Discussion
Principal findings
We characterized all safety signals identified from the FAERS that were published by the FDA from 2008 to 2019, evaluated how often a safety signal resulted in regulatory action by the FDA and, for a subset of signals reported in 2014-15, determined whether relevant published research or public Sentinel Initiative assessments existed that might have corroborated or refuted the FDA’s regulatory actions. Among potential safety signals identified from the FAERS by the FDA from 2008 to 2019, about 70% were resolved, including more than 90% of those ≥3 years old, and nearly 80% led to regulatory action, most often changes to drug labeling. Of the subset of signals reported in 2014-15, at least one relevant study was found in the literature for about 75% of the signals but most of these studies were case reports or case series. Moreover, only a third of regulatory actions were corroborated by studies published in the literature. Also, at least one relevant public Sentinel Initiative assessment was found for only 5% of safety signals but none corroborated the regulatory actions by the FDA.
Comparison with other studies
Our study suggests that although most of the potential safety signals identified from the FAERS were resolved, about 30% were unresolved. These unresolved signals were mainly from quarterly reports made public over the past three years. These findings are similar to those of Beninger and Murray who found that 61% of potential safety signals identified from the FAERS were resolved and that most unresolved signals were <3 years old.23 The FDA Amendments Act requires the FDA to publicly report potential safety signals, but no strict deadline is indicated by which safety signal evaluations must be completed.24 To ensure that the process for determining appropriate regulatory action is timely and transparent, encouraging prompt evaluation and resolution of all reported signals while also minimizing uncertainty for patients and clinicians in care decisions, a timeframe for evaluating signals identified from the FAERS, along with annual public reporting of progress, might be necessary.
We found that most safety signals identified from the FAERS were resolved, and that among these, 80% led to regulatory action, most commonly changes to drug labeling. Few of these regulatory actions that resulted in a change in drug labeling or a drug safety communication, however, cited evidence other than the safety signal identified from the FAERS. We found that for about a quarter of these regulatory actions, no published evidence or Sentinel Initiative assessments existed to corroborate the risk (or lack of risk) to safety between the drug and the associated adverse event. Also, when published studies in the medical literature were identified, most were case reports or case series, which are often small, lack generalizability, and generate anecdotal evidence comparable with the quality of the evidence available from the FAERS. These findings are consistent with research conducted by FDA staff, who characterized the sources of data triggering and supporting the identification of new safety risks of drugs approved by the FDA communicated through safety related labeling changes, and determined that 70% were prompted by the FAERS safety reports.25
Policy implications
Are published research and public assessments by the Sentinel Initiative necessary to update a product label in response to adverse event reports identified through the FAERS? First, the supporting evidence will vary depending on the severity of the adverse event, how often patients are expected to experience it, and how commonly the drug is used. For example, many potential safety signals are rare adverse events and designing a study that would offer inferential evidence would not be feasible. Second, FDA staff have access to the full details for every adverse event report and can follow-up with the reporting source to get more information, evaluate the likelihood and mechanism of action, and assess the potential for causality. Third, other information sources, including clinical trial data submitted to the agency and decisions made by international regulators, can be scrutinized. The FDA explicitly states that other forms of evidence, beyond the published literature and public assessments by the Sentinel Initiative, can be used to inform the FDA’s regulatory actions.26 These additional information sources or the agency’s reasoning, however, are not routinely made public or cited in changes to drug labeling or in drug safety communications. This lack of transparency has implications globally, because state regulators frequently disagree with one another on whether to issue drug safety warnings.27 28 In the context of more than 2.3 million adverse events reported to the FDA through the FAERS in 2021 alone,5 understanding how the agency makes decisions to change drug labels and based on what evidence is vital.
Our findings suggest that the FDA has a low evidentiary threshold to change drug labeling to include new adverse events after identification from the FAERS. This practice is codified by law, which requires only “reasonable evidence of a causal association with a drug; a causal relationship need not have been definitely established” to add a new adverse reaction to the warnings and precautions section of a product label, and similarly explains that “only those adverse events for which there is some basis to believe there is a causal relationship between the drug and the occurrence of the adverse event” to add a new adverse reaction to the adverse reactions section of a product label. Although this practice could help increase awareness among patients and physicians of possibly rare but serious adverse events, many adverse events derived from low quality evidence could be reported on drug labels. Similar reliance on low quality evidence has been reported for safety communications for drugs and medical devices.29 30 31 Knowing what evidence was used to support regulatory actions to resolve safety signals, as well as what other evidence might have corroborated those actions, could inform and improve clinical decision making. The public and those who prescribe drugs, who might be asked to change the drug treatments they use or prescribe, would benefit from understanding the strength of the evidence on which regulatory safety actions are based.
Our findings highlight the need for robust and larger scale post-market safety studies to strengthen the quality of evidence and to evaluate rare safety events, particularly because many regulatory safety actions are taken only from signals identified from the FAERS. Well designed post-market safety studies might be particularly crucial for drugs approved under the Accelerated Approval program by the FDA, where pivotal trials tend to be smaller and to evaluate surrogate markers as primary endpoints,32 33 as well as for biologics, both of which have been found to be associated with higher rates of post-market safety actions by the FDA, such as withdrawals, black box warnings, and safety communications.34 Although conducting clinical trials in response to every potential safety signal would not be feasible, recent efforts over the past decade to promote sharing of clinical trial data suggest that collecting these data at the drug or class level might offer opportunities for safety analyses examining rare adverse events.35 36
We found that few safety signals were evaluated by the Sentinel Initiative, suggesting that this active surveillance system, or others such as the Observational Health Data Sciences and Informatics network with similar resources, are being under used for post-market safety evaluations. Other efforts to advance evaluation of safety signals with real world data sources could also be considered. These assessments and evaluations must be made public, however, including specifying the safety outcome of interest in advance, to ensure their rigor and reliability.37 The FDA has indicated that it intends to further strengthen the Sentinel Initiative through user fee funds, increase public availability of the system, and enhance its ability to conduct analyses of specific safety issues.38 Further research will be crucial to determine the effectiveness of these initiatives in increasing detection and improving evaluation of potential safety signals identified from the FAERS.
Limitations of this study
Our study has important limitations. First, the more comprehensive in-depth corroboration analysis of potential safety signals from the FAERS was performed on a subset of safety signals reported in 2014-15, which might not be applicable to safety signals reported more recently. No known change to FDA policy in evaluating potential safety signals identified from the FAERS has been reported, however, with the exception of possibly increasing reliance on the Sentinel Initiative for safety assessments. Second, our search of assessments by the Sentinel Initiative was conducted in December 2021, whereas our search for relevant published literature was completed in 2019. Although more studies might have been published since our search was completed, we do not expect substantive changes to our overall findings, particularly because few relevant research studies were found that were published after the potential safety signal was identified from the FAERS. Third, we did not evaluate regulatory actions taken in other countries in response to these safety signals, which might have informed the FDA’s regulatory actions, nor could we consider unpublished studies or other data accessible to the agency but not publicly available. Fourth, when determining whether the evidence reported in the medical literature corroborated a regulatory action taken by the FDA, we did not consider the quality of the published study beyond the study design. For some safety signals, however, case studies or other descriptive evidence might be considered sufficient to corroborate regulatory actions. Finally, although our analysis focused on all changes to drug labeling, we would expect that the implications for patient drug use and clinician prescribing would differ between a black box warning being added to the product label versus an addition to the adverse reaction section.
Conclusions
Among potential safety signals identified from the FAERS that were reported by the FDA from 2008 to 2019, about 70% were resolved, of which nearly 80% led to regulatory action, most often changes to drug labeling. Among a subset of potential safety signals reported in 2014-15, at least one relevant study was found in the literature for about two thirds of the signals; however, most were case reports or case series. Although only a third of regulatory actions were corroborated by studies published in the literature, none was corroborated by public assessments by the Sentinel Initiative. These findings suggest that either the FDA is taking regulatory actions based on evidence not made publicly available or that more comprehensive safety evaluations might be needed when potential safety signals are identified. The findings also highlight the continued need for rigorous post-market safety studies to strengthen the quality of evidence available at the time of regulatory action, as well as the importance of ongoing efforts to leverage real world data sources to evaluate and resolve signals identified from the FAERS and support FDA regulatory decisions.
What is already known on this topic
Post-marketing pharmacovigilance is essential for monitoring drug safety
The US Food and Drug Administration (FDA) receives more than two million adverse event reports annually, and reviews all potential safety signals to determine if regulatory action is needed
What this study adds
Most safety signals identified from post-marketing pharmacovigilance adverse event reporting systems were resolved after several years, and 80% led to FDA regulatory action
Less than a third of regulatory actions were corroborated by published research findings or public assessments by the Sentinel Initiative, suggesting that either the FDA is taking regulatory actions based on evidence not made publicly available or that more comprehensive safety evaluations might be needed when potential safety signals are identified
Web extra.
Extra material supplied by authors
Supplementary information:
Contributors: All authors acquired, analyzed, and interpreted the data and critically revised the manuscript for important intellectual content. MMD, EMC, and JSR conceived and designed the study. MMD conducted the statistical analysis and drafted the manuscript. JSR provided supervision. JSR is the study guarantor. MMD and JSR had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Funding: None.
Competing interests: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/disclosure-of-interest/ and declare: no support from any organization for the submitted work; MMD reports receiving grants from the Yale-Mayo Clinic Center of Excellence in Regulatory Science and Innovation (CERSI) Scholars outside the submitted work; RR reports research support through Yale University from Arnold Ventures and the Stavros Niarchos Foundation and was an employee of the Veterans Health Administration, but the views expressed in this article are those of the authors and do not necessarily reflect those of the US Department of Veteran Affairs or the US government; XS is supported by the China Scholarship Council; JDW is supported by the FDA, Johnson and Johnson through Yale University, Arnold Ventures, and the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under award 1K01AA028258; JDW serves as a consultant for Hagens Berman Sobol Shapiro LLP and Dugan Law Firm APLC.111; JSR is the US outreach and associate research editor at The BMJ and currently receives research support through Yale University from Johnson and Johnson to develop methods of clinical trial data sharing, from the Medical Device Innovation Consortium as part of the National Evaluation System for Health Technology (NEST), from the Food and Drug Administration for the Yale-Mayo Clinic Center of Excellence in Regulatory Science and Innovation (CERSI) program (U01FD005938), from the Agency for Healthcare Research and Quality (R01HS022882), from the National Heart, Lung and Blood Institute of the National Institutes of Health (NIH) (R01HS025164, R01HL144644), and from Arnold Ventures; JSR is an expert witness at the request of Relator’s attorneys, the Greene Law Firm, in a qui tam suit alleging violations of the False Claims Act and Anti-Kickback Statute against Biogen Inc; no other relationships or activities that could appear to have influenced the submitted work.
The senior author (the manuscript’s guarantor) affirms that the manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant registered) have been explained.
Dissemination to participants and related patient and public communities: Results from the study will be disseminated through press release and social media (Twitter) and will be directly sent to the FDA’s Office of Surveillance and Epidemiology within the Center for Drug Evaluation and Research to inform the agency’s pharmacovigilance efforts.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Ethical approval
Institutional review board approval or informed consent was not required because the study was based on publicly available information and involved no patient records, in accordance with 45 CFR §46.
Data availability statement
The dataset will be made available via a publicly accessible repository on publication.
References
- 1. Bouvy JC, De Bruin ML, Koopmanschap MA. Epidemiology of adverse drug reactions in Europe: a review of recent observational studies. Drug Saf 2015;38:437-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279:1200-5. [DOI] [PubMed] [Google Scholar]
- 3. Singh S, Loke YK. Drug safety assessment in clinical trials: methodological challenges and opportunities. Trials 2012;13:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duggirala HJ, Tonning JM, Smith E, et al. Use of data mining at the Food and Drug Administration. J Am Med Inform Assoc 2016;23:428-34. [DOI] [PubMed] [Google Scholar]
- 5.US Food and Drug Administration Center for Drug Evaluation and Research. Office of Surveillance and Epidemiology 2021 Annual Report. https://www.fda.gov/media/157388/download. Published 2022. Accessed July 29, 2022.
- 6.US Food and Drug Administration. Potential Signals of Serious Risks/New Safety Information Identified from the FDA Adverse Event Reporting System (FAERS). https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/potential-signals-serious-risksnew-safety-information-identified-fda-adverse-event-reporting-system. Accessed July 29, 2022.
- 7. Hazell L, Shakir SA. Under-reporting of adverse drug reactions: a systematic review. Drug Saf 2006;29:385-96. [DOI] [PubMed] [Google Scholar]
- 8. Heeley E, Riley J, Layton D, Wilton LV, Shakir SA. Prescription-event monitoring and reporting of adverse drug reactions. Lancet 2001;358:1872-3. [DOI] [PubMed] [Google Scholar]
- 9. McConeghy KW, Bress A, Qato DM, Wing C, Nutescu EA. Evaluation of dabigatran bleeding adverse reaction reports in the FDA adverse event reporting system during the first year of approval. Pharmacotherapy 2014;34:561-9. [DOI] [PubMed] [Google Scholar]
- 10. Alatawi YM, Hansen RA. Empirical estimation of under-reporting in the U.S. Food and Drug Administration Adverse Event Reporting System (FAERS). Expert Opin Drug Saf 2017;16:761-7. [DOI] [PubMed] [Google Scholar]
- 11.US Food and Drug Administration. Reports Received and Reports Entered into FAERS by Year. https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/reports-received-and-reports-entered-faers-year Published 2015. Accessed July 29, 2022.
- 12. Maciejewski M, Lounkine E, Whitebread S, et al. Reverse translation of adverse event reports paves the way for de-risking preclinical off-targets. Elife 2017;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McAdams M, Staffa J, Dal Pan G. Estimating the extent of reporting to FDA: a case study of statin-associated rhabdomyolysis. Pharmacoepidemiol Drug Saf 2008;17:229-39. [DOI] [PubMed] [Google Scholar]
- 14. Dhruva SS, Karaca-Mandic P, Shah ND, Shaw DL, Ross JS. Association between FDA black box warnings and Medicare formulary coverage changes. Am J Manag Care 2017;23:e310-5. [PubMed] [Google Scholar]
- 15. Kesselheim AS, Donneyong M, Dal Pan GJ, et al. Changes in prescribing and healthcare resource utilization after FDA Drug Safety Communications involving zolpidem-containing medications. Pharmacoepidemiol Drug Saf 2017;26:712-21. [DOI] [PubMed] [Google Scholar]
- 16. Psaty BM, Breckenridge AM. Mini-Sentinel and regulatory science--big data rendered fit and functional. N Engl J Med 2014;370:2165-7. [DOI] [PubMed] [Google Scholar]
- 17. Platt R, Brown JS, Robb M, et al. The FDA Sentinel Initiative - An Evolving National Resource. N Engl J Med 2018;379:2091-3. [DOI] [PubMed] [Google Scholar]
- 18.FDA Sentinel Analyses from ARIA and Other Sentinel Data Sources. https://www.sentinelinitiative.org/studies/drugs/individual-drug-analyses#fda-sentinel-queries-from-aria-and-other-sentinel-data-sources. Accessed July 29, 2022.
- 19.21st Century Cures Act. In: Congress t, ed2016.
- 20. Solotke MT, Dhruva SS, Downing NS, Shah ND, Ross JS. New and incremental FDA black box warnings from 2008 to 2015. Expert Opin Drug Saf 2018;17:117-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Golder S, Loke YK. Sensitivity and precision of adverse effects search filters in MEDLINE and EMBASE: a case study of fractures with thiazolidinediones. Health Info Libr J 2012;29:28-38. [DOI] [PubMed] [Google Scholar]
- 22.Health Information Research Unit. Search Filters for MEDLINE in Ovid Syntax and the PubMed translation. McMaster University. https://hiru.mcmaster.ca/hiru/HIRU_Hedges_MEDLINE_Strategies.aspx. Accessed July 29, 2022.
- 23. Beninger P, Murray M. Review of FDA Amendments Act Section 921 Experience in Posting Data-mining Results from the FAERS Database. Clin Ther 2021;43:380-95. [DOI] [PubMed] [Google Scholar]
- 24.Zinderman SOPP. 8420: FDAAA Section 921: Posting of Potential Signals of Serious Risk. In: Center for Biologics Evaluation and Research; 2019:11. [Google Scholar]
- 25. Croteau D, Pinnow E, Wu E, Muñoz M, Bulatao I, Dal Pan G. Sources of Evidence Triggering and Supporting Safety-Related Labeling Changes: A 10-Year Longitudinal Assessment of 22 New Molecular Entities Approved in 2008 by the US Food and Drug Administration. Drug Saf 2022;45:169-80. [DOI] [PubMed] [Google Scholar]
- 26.Center for Drug Evaluation and Research (CDER). Best Practices in Drug and Biological Product Postmarket Safety Surveillance for FDA Staff. In:2019. [Google Scholar]
- 27. Zeitoun JD, Lefèvre JH, Downing N, Bergeron H, Ross JS. Inconsistencies among European Union pharmaceutical regulator safety communications: a cross-country comparison. PLoS One 2014;9:e109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perry LT, Bhasale A, Fabbri A, et al. Comparative Analysis of Medicines Safety Advisories Released by Australia, Canada, the United States, and the United Kingdom. JAMA Intern Med 2019;179:982-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tau N, Shochat T, Gafter-Gvili A, Tibau A, Amir E, Shepshelovich D. Association Between Data Sources and US Food and Drug Administration Drug Safety Communications. JAMA Intern Med 2019;179:1590-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tau N, Shepshelovich D. Assessment of Data Sources That Support US Food and Drug Administration Medical Devices Safety Communications. JAMA Intern Med 2020;180:1420-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rathi VK, Krumholz HM, Masoudi FA, Ross JS. Characteristics of Clinical Studies Conducted Over the Total Product Life Cycle of High-Risk Therapeutic Medical Devices Receiving FDA Premarket Approval in 2010 and 2011. JAMA 2015;314:604-12. [DOI] [PubMed] [Google Scholar]
- 32. Zhang AD, Puthumana J, Downing NS, Shah ND, Krumholz HM, Ross JS. Assessment of Clinical Trials Supporting US Food and Drug Administration Approval of Novel Therapeutic Agents, 1995-2017. JAMA Netw Open 2020;3:e203284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005-2012. JAMA 2014;311:368-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Downing NS, Shah ND, Aminawung JA, et al. Postmarket Safety Events Among Novel Therapeutics Approved by the US Food and Drug Administration Between 2001 and 2010. JAMA 2017;317:1854-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallach JD, Wang K, Zhang AD, et al. Updating insights into rosiglitazone and cardiovascular risk through shared data: individual patient and summary level meta-analyses. BMJ 2020;368:l7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ross JS, Waldstreicher J, Bamford S, et al. Overview and experience of the YODA Project with clinical trial data sharing after 5 years. Sci Data 2018;5:180268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang SV, Pinheiro S, Hua W, et al. STaRT-RWE: structured template for planning and reporting on the implementation of real world evidence studies. BMJ 2021;372:m4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.US Food and Drug Administration. PDUFA Reauthorization performance goals and procedures fiscaly years 2023 through 2027. https://www.fda.gov/media/151712/download. Published 2021. Accessed July 29, 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information:
Data Availability Statement
The dataset will be made available via a publicly accessible repository on publication.