

Abstract
Background
The impact of the new “borderline” hemodynamic class for pulmonary hypertension (PH) (mean pulmonary artery pressure [mPAP], 21–24 mm Hg and pulmonary vascular resistance, [PVR], ≥3 wood units, [WU]) in chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD) is unclear.
Objectives
The aim of this study was to assess the effect of borderline PH (BLPH) on survival in COPD and ILD patients.
Method
Survival was analyzed from retrospective data from 317 patients in 12 centers (Italy, Spain, UK) comparing four hemodynamic groups: the absence of PH (NoPH; mPAP <21 mm Hg or 21–24 mm Hg and PVR <3 WU), BLPH (mPAP 21–24 mm Hg and PVR ≥3 WU), mild-moderate PH (MPH; mPAP 25–35 mm Hg and cardiac index [CI] ≥2 L/min/m2), and severe PH (SPH; mPAP ≥35 mm Hg or mPAP ≥25 mm Hg and CI <2 L/min/m2).
Results
BLPH affected 14% of patients; hemodynamic severity did not predict survival when COPD and ILD patients were analyzed together. However, survival in the ILD cohort for any PH level was worse than in NoPH (3-year survival: NoPH 58%, BLPH 32%, MPH 28%, SPH 33%, p = 0.002). In the COPD cohort, only SPH had reduced survival compared to the other groups (3-year survival: NoPH 82%, BLPH 86%, MPH 87%, SPH 57%, p = 0.005). The mortality risk correlated significantly with mPAP in ILD (hazard ratio [HR]: 2.776, 95% CI: 2.057–3.748, p < 0.001) and notably less in COPD patients (HR: 1.015, 95% CI: 1.003–1.027, p = 0.0146).
Conclusions
In ILD, any level of PH portends worse survival, while in COPD, only SPH presents a worse outcome.
Keywords: Borderline pulmonary hypertension, Chronic lung disease, Chronic obstructive pulmonary disease, Interstitial lung disease, Survival
Introduction
Pulmonary hypertension (PH) is a common complication of chronic lung disease (CLD) and is associated with higher morbidity [1] and mortality [2, 3, 4]. The vast majority of patients with CLD presents with mild-moderate PH (MPH), but a small group presents with severe PH (SPH); several studies have found that patients with SPH in the context of chronic obstructive pulmonary disease (COPD) have a different phenotype, characterized by mild airflow obstruction but severe hypoxemia, with an increased mortality [5]. In idiopathic pulmonary fibrosis (IPF), on the other hand, the presence of PH greatly increases the mortality risk, while its severity does not seem to affect the outcome to the same degree [6]. Severity of PH therefore seems to have a different effect in patients with different underlying CLD.
The threshold for the diagnosis of PH has been defined since the World Health Organization 1973 Geneva Symposium at a mean pulmonary artery pressure (mPAP) of ≥25 mm Hg [7], this being the case also for PH associated with CLD; however, this pressure threshold has recently been put into question for all groups of PH during the proceedings of the 6th World Symposium on Pulmonary Hypertension [8]. The proposed change was based on studies reporting data from large patient cohorts [9, 10], which found that a lower level of mPAP previously called “borderline PH” (BLPH), defined by various values of mPAP with a lower limit between 17 and 24 mm Hg, could adversely influence prognosis and exercise capacity [11]. A recent study on data collected in the United Network for Organ Sharing (UNOS) [12] database evaluated how the new definition changes prevalence of PH in COPD and IPF and found that, despite both old and new definitions of pre-capillary PH are able to discern outcomes in these two diseases, there was a trend toward a better discrimination of the new definition of PH in IPF patients. Until now, however, the burden of this hemodynamic state (defined in the 6th World Symposium on Pulmonary Hypertension proceedings as mPAP of 21–24 mm Hg and pulmonary vascular resistance [PVR] of ≥3 wood units [WU]) on patient survival, as considered separately, has not been evaluated specifically in group 3 PH.
The objective of our study was to assess the prevalence and impact on survival of BLPH and compare them to those of patients without PH and with moderate PH and SPH, according to the hemodynamic classification of PH in CLD [13] in a multicenter, international cohort of interstitial lung disease (ILD) and COPD patients.
Materials and Methods
We retrospectively included data from patients with a diagnosis of COPD and ILD (both according to the European Respiratory Society guidelines) who had been evaluated at an expert PH center between January 2000 and March 2020 with a baseline right heart catheterization as part of their clinical evaluation; all patients were evaluated during stable respiratory disease and under optimal oxygen therapy, and none received pulmonary vasodilators at the time of hemodynamic assessment. The indications for right heart catheterization were referral for lung transplant workup, suspicion of SPH and/or inclusion in a clinical trial. Patients' data were derived from: (1) 9 Spanish hospitals participating in the Spanish Registry of Pulmonary Hypertension in Respiratory Disease (Registro Español de Hipertensión asociada a Enfermedad Respiratoria [REHAR]; full list of hospitals in online suppl. materials; for all online suppl. material, see www.karger.com/doi/10.1159/000524263) (2) the IRCCS ISMETT in Palermo (Italy); (3) the National Pulmonary Hypertension Service at Royal Brompton Hospital in London (UK); and (4) the IRCCS San Matteo Foundation in Pavia (Italy). Exclusion criteria were a diagnosis of combined pulmonary fibrosis-emphysema, lung transplant or post-capillary PH: thus, all included patients met the criteria for pre-capillary hemodynamic profile. The patients were divided into four hemodynamic subgroups as follows: absence of PH (NoPH; mPAP, <21 mm Hg or mPAP 21–24 mm Hg and PVR <3 WU), “borderline” PH (BLPH, mPAP 21–24 mm Hg and PVR ≥3 WU), MPH (mPAP 25–35 mm Hg and cardiac index [CI] ≥2.0 L/min/m2) and SPH (mPAP ≥35 mm Hg or mPAP ≥25 mm Hg and CI <2.0 L/min/m2). Due to the characteristics of the REHAR registry, where a diagnosis of PH was necessary for enrolment, patients with BLPH were included only following the change in hemodynamic definition at the Nice 2018 proceedings [13] (April 2019). We analyzed all the variables according to underlying diagnosis and within each diagnostic group according to the hemodynamic subgroup.
Statistics
Qualitative variables are described by frequency distributions, while quantitative variables are expressed by medians ± interquartile ranges. To compare the four hemodynamic groups, the χ2 test and Fisher's exact test were used for categorical variables, while the 2-samples T test or Wilcoxon-Mann-Whitney test were used for continuous variables when appropriate. Cox models were applied to obtain survival curves with respect to the hemodynamic groups and the diagnoses. All statistical analyses were performed using SAS software, version 9.4 (SAS Institute Inc, Cary, NC, USA). p values <0.05 were considered statistically significant.
Results
The records from 525 patients with COPD and ILD who had undergone right-heart catheterization in 9 centers from Spain, 2 centers in Italy, and 1 center in the UK were available for analysis. As detailed in the online supplementary Figure 1, of the initial 525 patients, 317 patients were retained in the final analysis (REHAR registry n = 104; ISMETT n = 126; IRCCS S. Matteo n = 42; Royal Brompton Hospital n = 45). The cohorts grouped by hemodynamic class and underlying disease are shown in Figure 1.
Fig. 1.

Summary of cases. Overall study population categorized by hemodynamic class and underlying respiratory condition. BLPH, borderline pulmonary hypertension group; COPD, chronic obstructive pulmonary disease; ILD, interstitial lung disease; MPH, mild-moderate pulmonary hypertension group; NoPH, group of patients without pulmonary hypertension; SPH, severe pulmonary hypertension group.
Description of the Cohort
Anthropometric, clinical, and functional characteristics of the study subjects, classified according to underlying respiratory diagnosis, are shown in Table 1. Patients with ILD had a higher BMI than patients with COPD, as well as fewer accumulated pack/years of cigarette smoking, different spirometric profiles as expected but no differences in DLCO, severely reduced in both groups, or partial oxygen pressure in arterial blood. Interestingly, despite no difference in the 6-min-walking-distance, patients with ILD had lower PVR and mPAP and more preserved right-heart function. We found no differences between ILD and COPD in left ventricular ejection fraction, which was preserved in all cases or in the rate of comorbidities (dyslipidemia, diabetes mellitus, and coronary artery disease). Within the ILD cohort, 126 patients (74%) were diagnosed with IPF, 26 patients were classified as nonspecific interstitial pneumonia (15%), 13 patients suffered from hypersensitivity pneumonitis (8%), while the other 5 subjects (3%) suffered from NSIP in the context of connective-tissue disease, Langerhans cells histiocytosis, pleuropulmonary fibroelastosis, fibrosing organizing pneumonia, and one unclassifiable idiopathic interstitial pneumonia. Thus, 91% of our sample represented idiopathic interstitial pneumonias.
Table 1.
Characteristic of the study subjects, stratified according to underlying disease
| All patients (n = 317) | COPD (n = 147) | ILD (n = 170) | |
|---|---|---|---|
| Age, years | 62 (57–68) | 63 (57–70) | 62 (56–66) |
| Male gender, n (%) | 213 (67) | 106 (72) | 107 (63) |
| Body mass index, kg/m2 | 26 (23–29) | 25 (22–28) | 27 (25–29)* |
| Smoking status | |||
| Current smokers, n (%) | 12 (4) | 12 (8) | 0 (0)* |
| Ex-smokers, n (%) | 162 (51) | 98 (67) | 65 (38)* |
| Pack-years smoking | 30 (10–48) | 40 (30–70) | 10 (0–30)* |
| FEV1, % pred | 52 (33–66) | 34 (22–63) | 55 (46–67)* |
| FVC, % pred | 58 (45–74) | 69 (50–86) | 53 (42–64)* |
| FEV1/FVC, % | 65 (42–89) | 44 (33–59) | 89 (83–107)* |
| TLC, % pred | 87 (52–108) | 106 (91–119) | 50 (41–60)* |
| DLCO, % pred | 24 (17–33) | 26 (17–39) | 23 (17–32) |
| PaO2, mm Hg | 62 (52–72) | 61 (51–72) | 62 (53–73) |
| 6MWD, meters | 297 (184–376) | 279 (206–374) | 301 (167–383) |
| Hemodynamic group, N-B-M-S | 81-45-97-94 | 22-16-45-64 | 59-29-52-30 |
| sPAP, mm Hg | 40 (33–60) | 46 (35–68) | 36 (32–48)* |
| dPAP, mm Hg | 18 (14–25) | 21 (16–28) | 15 (12–21)* |
| mPAP, mm Hg | 27 (22–36) | 32 (24–42) | 24 (21–32)* |
| Cardiac output, L/min | 4.60 (3.90–5.30) | 4.40 (3.60–5.10) | 4.80 (4.13–5.50)* |
| CI, L/min/m | 2.60 (2.20–3.00) | 2.50 (2.00–3.00) | 2.70 (2.30–3.00)* |
| PAWP, mm Hg | 9 (7–12) | 10 (8–11) | 9 (6–12) |
| PVR, dyn·s·cm·2 | 4.2 (2.7–6.3) | 4.9 (3.4–8.0) | 3.6 (2.4–5.0)* |
| RAP, mm Hg | 5 (3–8) | 6 (4–9) | 4 (2–7)* |
| LVEF, % | 60 (60-60) | 60 (60–65) | 60 (60-60) |
| Dyslipidemia, n (%) | 32 (10) | 18 (12) | 14 (8) |
| Diabetes mellitus, n (%) | 38 (12) | 20 (14) | 18 (11) |
| Coronary artery disease, n (%) | 42 (13) | 20 (14) | 22 (13) |
| Pulmonary vasodilators, n (%) | 51 (17) | 38 (26) | 13 (9%)*,# |
Results expressed in median (percentile 25–percentile 75). COPD, chronic obstructive pulmonary disease; ILD, interstitial lung disease; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; TLC, total lung capacity; RV, residual volume; DLCO, diffusing capacity for carbon monoxide; PaO2, arterial partial oxygen pressure; 6MWD, 6-min walking distance test; hemodynamic group N, No pulmonary hypertension; B, borderline pulmonary hypertension; M, mild-moderate pulmonary hypertension, S, severe pulmonary hypertension; sPAP, systolic pulmonary artery pressure; dPAP, diastolic pulmonary artery pressure; mPAP, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; LVEF, left ventricular ejection fraction.
p < 0.05 compared to COPD.
Data available for 150 out of 170 patients.
Overall, PH was present in 236 (74%) of the cohort and BLPH in 45 patients (14%). We then looked at the two cohorts separately and stratified them for hemodynamic severity. In COPD (Table 2), we found that patients with SPH were older compared to the MPH group and had accumulated a higher pack/year count, compared to both NoPH and MPH groups; they also suffered much more frequently of diabetes and coronary artery disease. Moreover, patients with SPH had a more preserved lung function profile, with the exception of DLCO which was severely reduced in all four groups, and worse hypoxemia. Again, despite the expected differences in hemodynamic severity which defined the four groups, we found no differences in the 6-min walking distance. In the ILD cohort (Table 3), the four hemodynamic groups were very homogenous with the exception of older patients in the MPH group compared to the NoPH group and of the expected differences in hemodynamic severity; once more, no difference was found in the 6-min-walking-distance.
Table 2.
Characteristic of the COPD cohort, stratified according to hemodynamic severity
| All patients (n = 147) | NoPH (n = 22) | BLPH (n = 16) | MPH (n = 45) | SPH (n = 64) | |
|---|---|---|---|---|---|
| Age, years | 63 (57–70) | 62 (55–65) | 62 (57–72) | 60 (55–66) | 68 (60–74)† |
| Male gender, n (%) | 106 (72) | 12 (55) | 10 (63) | 34 (76) | 50 (78) |
| Body mass index, kg/m2 | 25 (22–28) | 21 (21–25) | 24 (22–25) | 26 (22–29) | 25 (22–29)* |
| Smoking status | |||||
| Current smokers, n (%) | 12 (10) | 0 (0) | 0 (0) | 8 (18) | 4 (6) |
| Ex-smokers, n (%) | 98 (83) | 15 (68) | 14 (88) | 24 (53) | 45 (70) |
| Pack-years smoking | 40 (30–70) | 35 (29–40) | 27 (13–35) | 60 (40–80)# | 80 (65–105)*, # |
| FEV1, % pred | 34 (22–63) | 25 (20–37) | 32 (22–36) | 29 (19–40) | 62 (39–74)*,#, † |
| FVC, % pred | 69 (50–86) | 60 (45–72) | 69 (54–82) | 55 (46–78) | 82 (60–91)*,† |
| FEV1/FVC, % | 44 (33–59) | 35 (31–47) | 32 (27–44) | 38 (31–50) | 59 (50–62)*,#, † |
| TLC, % pred | 106 (91–119) | 100 (83–116) | 112 (97–128) | 116 (93–137) | 100 (89–109)† |
| DLCO, % pred | 26 (17–39) | 24 (15–30) | 22 (10–41) | 30 (22–45) | 25 (16–29) |
| PaO2, mm Hg | 61 (51–72) | 70 (64–77) | 70 (60–77) | 65 (57–76) | 51 (46–61)*,#,† |
| 6MWD, meters | 279 (206–374) | 240 (168–359) | 337 (240–377) | 334 (240–410) | 276 (177–357) |
| sPAP, mm Hg | 46 (35–68) | 30 (26–32) | 33 (31–35) | 40 (37–47)*, # | 71 (61–81)*,#,† |
| dPAP, mm Hg | 21 (16–28) | 15 (12–15) | 15 (12–16) | 19 (17–21)*,# | 29 (25–34)*,#,† |
| mPAP, mm Hg | 32 (24–42) | 21 (18–22) | 23 (22–24) | 28 (26–32)*,# | 43 (38–52)*,#,† |
| Cardiac output, L/min | 4.40 (3.60–5.10) | 5.15 (4.50–5.85) | 4.05 (3.83–4.58)* | 4.50 (4.00–5.25) | 3.85 (3.03–4.60)*,† |
| CI, L/min/m | 2.50 (2.00–3.00) | 3.05 (2.80–3.55) | 2.30 (2.15–2.68)* | 2.60 (2.30–3.00) | 2.15 (1.70–2.70)*,† |
| PAWP, mm Hg | 10 (8–11) | 10 (9–11) | 9 (7–11) | 9 (7–12) | 10 (7–12) |
| PVR, dyn·s·cm·2 | 4.9 (3.4–8.0) | 1.8 (1.5–2.3) | 3.4 (3.0–3.7) | 4.3 (3.6–5.0)* | 8.3 (6.3–11.6)*,#,† |
| RAP, mm Hg | 6 (4–9) | 5 (5–7) | 5 (4–6) | 6 (3–8) | 7 (5–11) |
| LVEF, % | 60 (60–65) | 61 (60–65) | 60 (53–60) | 60 (53–61) | 60 (60-60) |
| Dyslipidemia, n (%) | 18 (19) | 2 (9) | 0 (0) | 3 (7) | 13 (20) |
| Diabetes mellitus, n (%) | 20 (21) | 1 (5) | 0 (0) | 3 (7) | 16 (25)*,#,† |
| Coronary artery disease, n (%) | 20 (21) | 1 (6) | 2 (20) | 3 (10) | 14 (32)*,#,† |
| Pulmonary vasodilators, n (%) | 38 (26) | 0 (0) | 0 (0) | 2 (4) | 36 (56)*,#,† |
Results expressed in median (percentile 25–percentile 75). NoPH, group of patients without pulmonary hypertension; BLPH, borderline pulmonary hypertension group; PH, pulmonary hypertension group; SPH, severe hypertension group; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; TLC, total lung capacity; RV, residual volume; DLCO, diffusing capacity for carbon monoxide; PaO2, arterial partial oxygen pressure; 6MWD, 6-min walking distance test; sPAP, systolic pulmonary artery pressure; dPAP, diastolic pulmonary artery pressure; mPAP, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; LVEF, left ventricular ejection fraction.
p < 0.05 compared to the NoPH group.
p < 0.05 compared to the BLPH group.
p < 0.05 compared to the MPH group.
Table 3.
Characteristic of the ILD cohort, stratified according to hemodynamic severity
| All patients (n = 170) | NoPH (n = 59) | BLPH (n = 29) | MPH (n = 52) | SPH (n = 30) | |
|---|---|---|---|---|---|
| Age, years | 62 (56–66) | 60 (52–63) | 62 (58–65) | 64 (60–70)* | 62 (57–70) |
| Male gender, n (%) | 107 (63) | 34 (58) | 21 (72) | 35 (67) | 17 (57) |
| Body mass index, kg/m2 | 27 (25–29) | 27 (25–29) | 26 (24–28) | 28 (25–29) | 27 (25–28) |
| Smoking status | |||||
| Current smokers, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Ex-smokers, n (%) | 64 (66) | 30 (51) | 12 (41) | 14 (27) | 9 (30) |
| Pack-years smoking | 10 (0–30) | 15 (4–30) | 3 (0–10) | 12 (0–53) | 35 (8–78) |
| FEV1, % pred | 55 (46–67) | 55 (45–68) | 58 (46–64) | 54 (47–67) | 57 (46–67) |
| FVC, % pred | 53 (42–64) | 51 (40–65) | 54 (42–59) | 54 (41–64) | 55 (47–70) |
| FEV1/FVC, % | 89 (83–107) | 96 (84–114) | 87 (79–103) | 89 (84–99) | 90 (76–99) |
| TLC, % pred | 50 (41–60) | 49 (41–58) | 53 (41–60) | 51 (44–65) | 50 (43–77) |
| DLCO, % pred | 23 (17–32) | 24 (16–33) | 23 (18–38) | 22 (17–26) | 26 (18–32) |
| PaO2, mm Hg | 62 (53–73) | 69 (59–73) | 63 (56–74) | 62 (48–77) | 56 (48–66) |
| 6MWD, meters | 301 (167–383) | 358 (198–438) | 306 (143–365) | 285 (179–379) | 278 (149–316) |
| sPAP, mm Hg | 36 (32–48) | 30 (26–33) | 36 (34–37) | 43 (39–49)*,# | 66 (59–82) *,#,† |
| dPAP, mm Hg | 15 (12–21) | 12 (10–14) | 15 (13–16) | 18 (16–21)* | 27 (25–37) *,#,† |
| mPAP, mm Hg | 24 (21–32) | 19 (16–22) | 23 (22–24) | 29 (27–32)*,# | 41 (37–48) *,#,† |
| Cardiac output, L/min | 4.80 (4.13–5.50) | 5.25 (4.50–6.13) | 4.90 (4.20–5.40) | 4.65 (4.13–5.28)* | 4.10 (3.30–4.95)* |
| CI, L/min/m | 2.70 (2.30–3.00) | 2.80 (2.45–3.35) | 2.65 (2.25–3.03) | 2.70 (2.43–2.90) | 2.00 (1.75–2.85)* |
| PAWP, mm Hg | 9 (6–12) | 10 (6–12) | 9 (6–11) | 8 (6–11) | 13 (9–14)#,† |
| PVR, dyn·s·cm·2 | 3.6 (2.4–5.0) | 1.8 (1.4–2.4) | 3.5 (2.8–4.2)* | 4.7 (3.8–5.3)* | 7.7 (6.0–10.1)*,#,† |
| RAP, mm Hg | 4 (2–7) | 3 (2–5) | 3 (2–5) | 4 (3–7) | 10 (5–13)*,#,† |
| LVEF, % | 60 (60–60) | 60 (60–62) | 60 (59–61) | 60 (59–60) | 60 (60-60) |
| Dyslipidemia, n (%) | 14 (15) | 2 (3) | 4 (14) | 7 (13) | 1 (3) |
| Diabetes mellitus, n (%) | 18 (20) | 6 (10) | 5 (17) | 5 (10) | 2 (7) |
| Coronary artery disease, n (%) | 22 (24) | 8 (14) | 3 (10) | 9 (17) | 2 (7) |
| Pulmonary vasodilators, n (%)§ | 13 (9) | 0 (0) | 0 (0) | 2 (4) | 11 (37)*, #, † |
Results expressed in median (percentile 25–percentile 75). NoPH, group of patients without pulmonary hypertension; BLPH, borderline pulmonary hypertension group; PH, pulmonary hypertension group; SPH, severe hypertension group; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; TLC, total lung capacity; RV, residual volume; DLCO, diffusing capacity for carbon monoxide; PaO2, arterial partial oxygen pressure; 6MWD, 6-min walking distance test; sPAP, systolic pulmonary artery pressure; dPAP, diastolic pulmonary artery pressure; mPAP, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; LVEF, left ventricular ejection fraction.
p < 0.05 compared to the NoPH group.
p < 0.05 compared to the BLPH group.
p < 0.05 compared to the MPH group.
Data available for 150 out of 170 patients.
Data on vasodilator treatment were available for 297 patients (Table 1), of which 51 (17%) has received treatment; 47 of them suffered from SPH. However, only 50% of the whole SPH cohort for which we had treatment data had been treated. Breaking down treatment according to underlying pulmonary diagnosis, 26% of COPD (Table 2) and only 9% of ILD patients (Table 3) had undergone vasodilator treatment.
Survival Analysis
Survival in ILD patients was significantly worse compared to COPD patients: the overall 3-year-survival for the two cohorts was 40% and 73%, respectively (p < 0.001).
When comparing the outcomes of the four hemodynamic groups in the whole cohort of patients enrolled in the study, we did not observe any difference in mortality (Fig. 2a); 3-year-survival was 64% in the NoPH group, 51% in the BLPH group, 55% in the MPH group, and 50% in the SPH group (p = 0.369). The median follow-up time for these subgroups was of 22, 20, 21, and 22 months, respectively (25th−75th percentiles: 9–44, 9–40, 12–43, and 10–40 months), with a median follow-up time for the whole cohort of 21 months.
Fig. 2.

Survival for the whole cohort (a), the COPD (b), and the ILD cohorts (c). Kaplan-Meier analysis with log-rank to determine the probability of all-cause mortality according to different thresholds of mPAP in the whole cohort, the COPD, and the ILD cohort. Each cohort is divided into four subgroups: NoPH (mPAP <21 mm Hg or mPAP 21–24 mm Hg with PVR <3 WU), BLPH (mPAP 21–24 mm Hg with PVR ≥3 WU), MPH (mPAP ≥25 mm Hg and CI, ≥2.0 L/min/m2), and SPH (mPAP ≥35 mm Hg or mPAP ≥25 mm Hg and CI <2.0 L/min/m2).
When we further analyzed COPD and ILD cohorts separately, we found that in the COPD cohort, BLPH (n = 16) stratified closely to NoPH (n = 22) and MPH (n = 45) in the survival analysis, presenting a significantly better prognosis compared to SPH (n = 64; Fig. 2b); the 3-year survival for the hemodynamic groups within the COPD cohort was 82%, 86%, 87%, and 57%, respectively (p = 0.005). However, in the ILD cohort, BLPH (n = 29), MPH (n = 52) and SPH (n = 30) had significantly worse prognosis than the NoPH group (n = 59; Fig. 2c); the 3-year survival was 58%, 32%, 28%, and 33%, respectively (p = 0.002).
The hazard ratio (HR) analysis confirmed our results: the HR was not increased when the overall cohort was considered (HR: 1.002, 95% CI: 0.990–1.014, p = 0.774). However, when analyzing the COPD and ILD cohort separately (Fig. 3), the risk for each increment of mPAP by 1 mm Hg was significant in both the COPD cohort (HR: 1.015, 95% CI: 1.003–1.027, p = 0.0146) and the ILD cohort (HR: 2.776, 95% CI: 2.057–3.748, p < 0.001), in which the mortality risk was notably increased.
Fig. 3.

Association between mPAP and HR stratified by diagnosis. No statistically significant increased risk is observed in the overall cohort for each increase of mPAP by 1 mm Hg: HR 1.002, 95% CI: 0.990–1.014, p = 0.774; instead, in the COPD cohort, the risk is slightly increased (HR: 1.015, 95% CI: 1.003–1.027, p = 0.0146) and it is greatly increased in the ILD cohort: HR 2.776, 95% CI: 2.057–3.748, p < 0.001). The kernel density estimate represents the relative density of patients across mPAP values. mPAP, mean pulmonary artery pressure; COPD, chronic obstructive pulmonary disease; ILD, interstitial lung disease; HR, hazard ratio; CI, confidence interval.
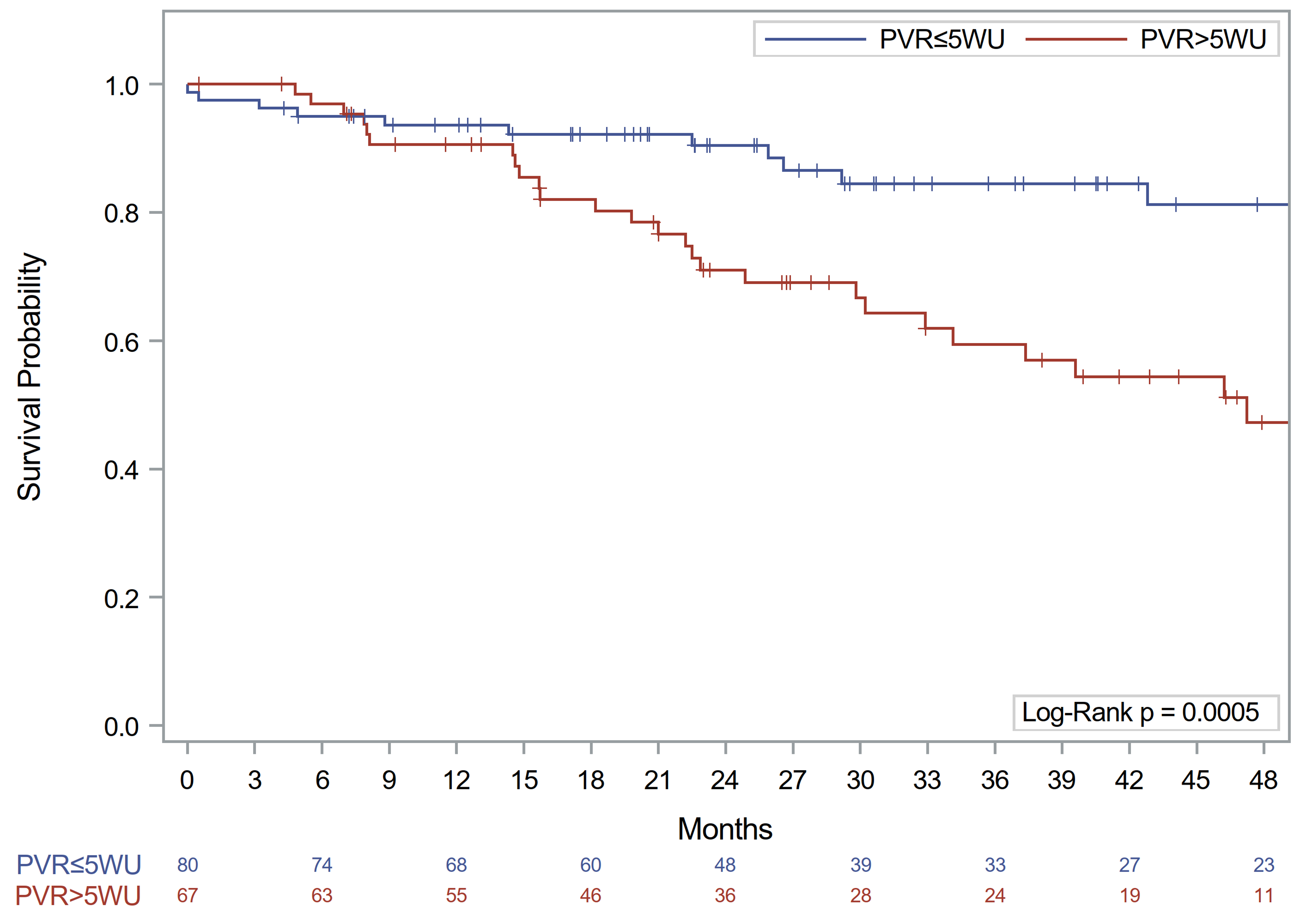
On the other hand, stratification according to a PVR cutoff of 5 WU as suggested by Zeder et al. [14] did not show different results in survival for the overall cohort or when considering the ILD patients alone; however, when this cutoff was applied to COPD patients, those with PVR >5 WU had a markedly worse prognosis (3-year survival: 59% vs. 84% in patients with PVR ≤5 WU, p = 0.001) (online suppl. Fig. 2). When we analyzed the HR of having PVR >5 WU, COPD patients presented an increased risk (HR: 2.774, 95% CI: 1.264–6.089, p = 0.011), while ILD patients did not (HR: 1.204, 95% CI: 0.693–2.092, p = 0.510).
Discussion
In a cohort of 317 patients with CLD, 74% of which had PH, and BLPH was detected in 45 (14%); BLPH was associated with worse survival in ILD but not in COPD patients. Notably, in the COPD cohort only those in the SPH group had very poor prognosis and clustered separately in terms of survival compared to other hemodynamic strata. Conversely, ILD patients with any level of PH from mPAP 21–24 mm Hg and PVR ≥3 WU upward demonstrated reduced survival compared to patients without PH, and the increased risk of mortality associated with mPAP increments was much higher in ILD than in COPD. Finally, the hemodynamic stratification was not able to predict survival when COPD and ILD were considered together.
Our results confirm the data from the ASPIRE Registry in COPD, which reported that patients with SPH had a significantly worse prognosis than patients with MPH [15]. The 3-year-survival in COPD patients with values of mPAP between 20 and 40 mm Hg was found to be 80% in several studies [4, 16] and not significantly lower than that observed for patients without PH (mPAP <20); this is consistent with our findings, which show that BLPH has no impact on COPD prognosis. We observed worse 3-year survival in PH patients with ILD compared to COPD despite more severe pulmonary hemodynamic compromise in the latter group; the same difference in survival according to underlying diagnosis was reported by the ASPIRE Registry [3] (3-year-survival: COPD 41%; ILD: 16%) and the Giessen Registry (3-year-survival: COPD 66%; ILD 40%) [4]. Two further studies [17, 18] have shown that both COPD and IPF patients with PH, even when treated with pulmonary vasodilators, have significantly worse prognosis compared to patients with idiopathic pulmonary arterial hypertension.
The relevance of BLPH in the survival of ILD patients from our cohort is consistent with preliminary data on the subject. In ILD, others have studied the relevance of BLPH, focusing on the impact of chronic hypercapnia [19] or on exercise capacity and the risk of exacerbation [20]. In the latter study, it was observed that in ILD, mPAP ≥21 mm Hg was associated with lower survival. Previous work in IPF patients described a worse prognosis both in the mPAP 21–24 mm Hg and in the mPAP ≥25 mm Hg groups as compared with mPAP <21 mm Hg [21] and in patients with mPAP of ≥17 mm Hg, compared to those with mPAP <17 mm Hg [22].
The significance of PVR in PH associated with lung disease has recently been in the spotlight: a PVR value greater than 5 WU was suggested [14, 23] as a reliable cutoff to demarcate a worse prognosis. When we applied this threshold to the COPD and ILD cohorts, we confirmed that COPD patients with PVR >5 WU had a significantly worse prognosis; however, this stratification was not predictive of survival in ILD. Olsson et al. [23] showed that a higher cutoff value of PVR >8 WU provided the best discrimination of survival in ILD patients in the COMPERA registry, which enrolls patients with a diagnosis of PH (mPAP ≥25 mm Hg until 2019) and undergoing vasodilator treatment. Our cohort included a significant subgroup of patients without PH as the control group, absent in the COMPERA analysis; furthermore, the great majority of patients in our study were not treated with pulmonary vasodilators. These differences may account for the discrepancy in results; however, it is possible that, had our ILD cohort been larger, we might have detected a similar significance.
In our cohort, the hemodynamic assessment was driven by diverse indications (suspicion of SPH, inclusion in a clinical trial, lung transplantation workup) and performed in 12 European referral centers, all involved in both end-stage lung disease and PH management. This approach reflects a “real-world” scenario; furthermore, it allowed us to recruit patients without PH as a control group, adding robustness to our findings. Moreover, we carefully excluded all post-capillary forms of PH because they might have been more appropriately categorized under PH due to left-heart disease or combined pre- and post-capillary PH, as well as rare and definite entities such as combined pulmonary fibrosis and emphysema. Crucially, we also excluded patients who underwent lung transplantation, given that those patients would have exited the survival analysis not by the natural course of their condition but through therapeutic intervention.
Recent studies have shown that the development of PH in COPD and ILD differs in its pathophysiology [5, 24], natural history [25, 26], pulmonary artery gene expression [27], and treatment strategies; concerning the latter, pulmonary vasodilators should generally be avoided in COPD [28] except possibly in SPH [29], while they have been shown to provide increased exercise tolerance [30] in ILD patients, regardless of hemodynamic severity. In our study, only 17% of patients, 13 of which presented with ILD (9% of the ILD cohort), received vasodilator treatment, a testimony to the scarcity of clinical trials available to CLD-PH patients. Of note, less thanonly half of the patients with severe hemodynamic compromise were treated with vasodilators.
Taken together, our findings suggest that different hemodynamic parameters may have different relevance in COPD and ILD. In COPD, we observe that the Nice 2018 hemodynamic severity classification in CLD [13] (a multifactorial stratification accounting for mPAP, PVR, and mPAP combined with CI, expressing vascular resistance) is correlated with mortality, which is significantly higher for the SPH group; furthermore, increments in mPAP considered as an isolated parameter are only marginally correlated with an increased risk, while stratifying patients according to PVR >5 WU is predictive of worse outcomes, as previously observed [14], and of significantly increased risk for COPD patients in the HR analysis. Conversely, in ILD, the Nice 2018 hemodynamic severity classification shows significantly increased mortality for any level of PH above BLPH; additionally, increments in mPAP are correlated with a significantly increased risk while a PVR >5 WU cutoff is not predictive of worse survival and is not correlated with an increased risk.
Recognizing that there is no single hemodynamic parameter that can predict survival in such complex diseases, we nonetheless speculate that in COPD, patients' PVR is a better descriptor of prognosis, while mPAP is likely less relevant; however, mPAP might be more useful to evaluate prognosis in ILD, whereas the role of PVR may be important only when very SPH occurs and therefore not applicable to the majority of cases. Supporting this hypothesis, COPD and ILD patients with BLPH in our cohort had similar PVR median values (3.4 and 3.5 WU), both below the cutoff of 5, but had divergent prognoses. The Nice 2018 hemodynamic severity classification, taking into account a combination of these parameters, proves to be a strong tool for predicting the outcome in both COPD and ILD patients. Finally, it is possible that other hemodynamic parameters, such as pulmonary vascular compliance as well as respiratory function aspects, might affect the overall survival in such complex and heterogeneous diseases as COPD and ILD. Given the disparities in genetic background, molecular and pathologic mechanisms, and clinical features observed in the two entities, it is therefore not surprising that mild elevations in mPAP and PVR confer different prognostic importance, as evidenced by our findings.
Limitations
This study has a few limitations. First, we present retrospective data where BLPH was found in a minority of patients affected by CLD who underwent right heart catheterization; this is affected by the new definition of PH, as the REHAR registry subgroup, which represent a third of the cohort, did not enroll patients with mPAP <25 mm Hg before April 2019. Our findings should be therefore confirmed in larger, prospective cohorts. However, in the context of group 3 PH, where right heart catheterization is not common or routinely indicated, our cohort is still one of the largest in the literature not derived from automated computerized patient records, where the quality of data is sometimes burdened by potential inconsistencies and errors. Second, we cannot rule out an effect of vasodilator treatment on the outcome of patients; however, only 17% of patients overall had received vasodilators and in no hemodynamic group was the majority of patients treated. Finally, the ILD cohort was somewhat heterogeneous, but 74% of this cohort was represented by IPF, and only a small number of patients (9%) had a diagnosis that was not ascribable to idiopathic interstitial pneumonias.
Conclusions
The impact of BLPH on survival is different according to the underlying respiratory condition: in ILD, any level of PH is associated with reduced survival, while in COPD only SPH is associated with worse outcome.
Statement of Ethics
This study was conducted in accordance with the World Medical Association Declaration of Helsinki. The REHAR Registry and the present retrospective study were approved by the Institutional Review Board (IRB) of Hospital del Mar (2016-7080-I); patients enrolled in the registry signed written informed consent to the use of their data for research purposes, in compliance with the requirements of the IRB that approved the establishment of registry. Patients at the San Matteo Foundation have been enrolled within the scope of the regional registry of PH approved by the local IRB (44784/2011). ISMETT (n. IRRB 23/19) and Royal Brompton (2019-05a) research groups also received approval of the present study by their respective IRBs.
Conflict of Interest Statement
Dr. Piccari reports grants, personal fees, and nonfinancial support from Janssen and nonfinancial support from Menarini, outside the submitted work. Dr. Wort reports grants and personal fees from Actelion UK. He has also received personal fees from MSD, Janssen, and GSK, outside the submitted work. Dr. Price reports personal fees from Janssen, outside the submitted work. Dr. Meloni, Dr. Rizzo, Dr. Martino, Dr. Salvaterra, Dr. Scelsi, Dr. López-Meseguer, Dr. Blanco, Dr. Callari, Dr. Pérez González, Dr. Tuzzolino, Dr. McCabe, Dr. Rodríguez-Chiaradía, and Dr. Vitulo, have nothing to disclose.
Funding Sources
Dr. Piccari has received a PVRI Fellowship (grant no. RGLP003) for the SUPPORT project, which partly supported the present work; the PVRI was not involved in the design or the completion of this study. No other funding was obtained for this study.
Author Contributions
L.P., S.J.W., and P.V. conceived and designed the study and supervised the work; L.P., S.J.W., F.M., L.C.P., L.M., E.S., L.S., M.L.M., I.B., A.C., V.P.G., C.M., D.A.R.C., and P.V. collected data; M.R. and F.T. performed the statistical analysis; L.P., S.J.W., F.M., and P.V. interpreted the results; L.P. prepared the manuscript; L.P., S.J.W., F.M., and P.V. revised the manuscript. All the authors reviewed and approved the final version of the manuscript.
Data Availability Statement
Data collected for this study contain sensitive information on the human subjects that make up the cohort and will be available upon reasonable request from the corresponding author.
Supplementary Material
Supplementary data
{kind=link}
Supplementary data
Supplementary data
Acknowledgments
The present study was part of the SUPPORT project, partly funded by the Pulmonary Vascular Research Institute (PVRI), to which the authors are indebted. The authors wish to thank the REHAR investigators who provided data for this study: a full list of investigators from centers providing data can be found in the online supplementary material. Finally, the authors also wish to thank the patients who selflessly gave access to their medical record for this study.
Funding Statement
Dr. Piccari has received a PVRI Fellowship (grant no. RGLP003) for the SUPPORT project, which partly supported the present work; the PVRI was not involved in the design or the completion of this study. No other funding was obtained for this study.
References
- 1.Kessler R, Faller M, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999 Jan;159((1)):158–64. doi: 10.1164/ajrccm.159.1.9803117. [DOI] [PubMed] [Google Scholar]
- 2.Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, et al. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest. 1995 May;107((5)):1193–8. doi: 10.1378/chest.107.5.1193. [DOI] [PubMed] [Google Scholar]
- 3.Hurdman J, Condliffe R, Elliot CA, Davies C, Hill C, Wild JM, et al. ASPIRE registry: assessing the spectrum of pulmonary hypertension identified at a REferral centre. Eur Respir J. 2012 Apr;39((4)):945–55. doi: 10.1183/09031936.00078411. [DOI] [PubMed] [Google Scholar]
- 4.Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, et al. The giessen pulmonary hypertension registry: survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017 Sep;36((9)):957–67. doi: 10.1016/j.healun.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 5.Blanco I, Piccari L, Barberà JA. Pulmonary vasculature in COPD: the silent component. Respirology. 2016 Aug;21((6)):984–94. doi: 10.1111/resp.12772. [DOI] [PubMed] [Google Scholar]
- 6.Hayes D, Jr, Black SM, Tobias JD, Kirkby S, Mansour HM, Whitson BA. Influence of pulmonary hypertension on patients with idiopathic pulmonary fibrosis awaiting lung transplantation. Ann Thorac Surg. 2016 Jan;101((1)):246–52. doi: 10.1016/j.athoracsur.2015.06.024. [DOI] [PubMed] [Google Scholar]
- 7.Hatano S, Strasser T. Primary pulmonary hypertension: report on a WHO meeting, 1973. Geneva: World Health Organisation; 1973. [consulted 2021 Nov 2] Available from http://www.wsphassociation.org/wp-content/uploads/2019/03/Programma-WSPH-Geneva-1973.pdf. [Google Scholar]
- 8.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019 Jan 24;53((1)):1801913. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maron BA, Hess E, Maddox TM, Opotowsky AR, Tedford RJ, Lahm T, et al. Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the veterans affairs clinical assessment, reporting, and tracking program. Circulation. 2016 Mar 29;133((13)):1240–8. doi: 10.1161/CIRCULATIONAHA.115.020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Assad TR, Maron BA, Robbins IM, Xu M, Huang S, Harrell FE, et al. Prognostic effect and longitudinal hemodynamic assessment of borderline pulmonary hypertension. JAMA Cardiol. 2017 Dec 1;2((12)):1361–8. doi: 10.1001/jamacardio.2017.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliveira RKF, Faria-Urbina M, Maron BA, Santos M, Waxman AB, Systrom DM. Functional impact of exercise pulmonary hypertension in patients with borderline resting pulmonary arterial pressure. Pulm Circ. 2017 Jul-Sep;7((3)):654–65. doi: 10.1177/2045893217709025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathan SD, Barnett SD, King CS, Provencher S, Barbera JA, Pastre J, et al. Impact of the new definition for pulmonary hypertension in patients with lung disease: an analysis of the united network for organ sharing database. Pulm Circ. 2021 Mar 30;11((2)):2045894021999960. doi: 10.1177/2045894021999960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019 Jan 24;53((1)):1801914. doi: 10.1183/13993003.01914-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeder K, Avian A, Bachmaier G, Douschan P, Foris V, Sassmann T, et al. Elevated pulmonary vascular resistance predicts mortality in COPD patients. Eur Respir J. 2021 Aug 26;58((2)):2100944. doi: 10.1183/13993003.00944-2021. [DOI] [PubMed] [Google Scholar]
- 15.Hurdman J, Condliffe R, Elliot CA, Swift A, Rajaram S, Davies C, et al. Pulmonary hypertension in COPD: results from the ASPIRE registry. Eur Respir J. 2013 Jun;41((6)):1292–301. doi: 10.1183/09031936.00079512. [DOI] [PubMed] [Google Scholar]
- 16.Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducoloné A, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005 Jul 15;172((2)):189–94. doi: 10.1164/rccm.200401-006OC. [DOI] [PubMed] [Google Scholar]
- 17.Hoeper MM, Behr J, Held M, Grunig E, Vizza CD, Vonk-Noordegraaf A, et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS One. 2015 Dec 2;10((12)):e0141911. doi: 10.1371/journal.pone.0141911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vizza CD, Hoeper MM, Huscher D, Pittrow D, Benjamin N, Olsson KM, et al. Pulmonary hypertension in patients with COPD: results from the comparative, prospective registry of newly initiated therapies for pulmonary hypertension (COMPERA) Chest. 2021 Aug;160((2)):678–89. doi: 10.1016/j.chest.2021.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Zuoyou L, Shiota S, Morio Y, Sugiyama A, Sekiya M, Iwakami SI, et al. Borderline pulmonary hypertension associated with chronic hypercapnia in chronic pulmonary disease. Respir Physiol Neurobiol. 2019 Apr;262:20–5. doi: 10.1016/j.resp.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Nemoto K, Oh-Ishi S, Akiyama T, Yabuuchi Y, Goto H, Nonaka M, et al. Borderline pulmonary hypertension is associated with exercise intolerance and increased risk for acute exacerbation in patients with interstitial lung disease. BMC Pulm Med. 2019 Sep 2;19((1)):167. doi: 10.1186/s12890-019-0932-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, et al. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85((6)):456–63. doi: 10.1159/000345221. [DOI] [PubMed] [Google Scholar]
- 22.Hamada K, Nagai S, Tanaka S, Handa T, Shigematsu M, Nagao T, et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007 Mar;131((3)):650–656. doi: 10.1378/chest.06-1466. [DOI] [PubMed] [Google Scholar]
- 23.Olsson KM, Hoeper MM, Pausch C, Grünig E, Huscher D, Pittrow D, et al. Pulmonary vascular resistance predicts mortality in patients with pulmonary hypertension associated with interstitial lung disease: results from the COMPERA registry. Eur Respir J. 2021 Aug 26;58((2)):2101483. doi: 10.1183/13993003.01483-2021. [DOI] [PubMed] [Google Scholar]
- 24.Shino MY, Lynch JP, 3rd, Saggar R, Abtin F, Belperio JA, Saggar R. Pulmonary hypertension complicating interstitial lung disease and COPD. Semin Respir Crit Care Med. 2013 Oct;34((5)):600–19. doi: 10.1055/s-0033-1356548. [DOI] [PubMed] [Google Scholar]
- 25.Elwing J, Panos RJ. Pulmonary hypertension associated with COPD. Int J Chron Obstruct Pulmon Dis. 2008;3((1)):55–70. doi: 10.2147/copd.s1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teramachi R, Taniguchi H, Kondoh Y, Ando M, Kimura T, Kataoka K, et al. Progression of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis with mild to moderate restriction. Respirology. 2017 Jul;22((5)):986–90. doi: 10.1111/resp.12986. [DOI] [PubMed] [Google Scholar]
- 27.Hoffmann J, Wilhelm J, Marsh LM, Ghanim B, Klepetko W, Kovacs G, et al. Distinct differences in gene expression patterns in pulmonary arteries of patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis with pulmonary hypertension. Am J Respir Crit Care Med. 2014 Jul 1;190((1)):98–111. doi: 10.1164/rccm.201401-0037OC. [DOI] [PubMed] [Google Scholar]
- 28.Blanco I, Gimeno E, Munoz PA, Pizarro S, Gistau C, Rodriguez-Roisin R, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med. 2010 Feb 1;181((3)):270–8. doi: 10.1164/rccm.200907-0988OC. [DOI] [PubMed] [Google Scholar]
- 29.Vitulo P, Stanziola A, Confalonieri M, Libertucci D, Oggionni T, Rottoli P, et al. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: a randomized controlled multicentre clinical trial. J Heart Lung Transplant. 2017 Feb;36((2)):166–74. doi: 10.1016/j.healun.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021 Jan 28;384((4)):325–334. doi: 10.1056/NEJMoa2008470. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data
Supplementary data
Data Availability Statement
Data collected for this study contain sensitive information on the human subjects that make up the cohort and will be available upon reasonable request from the corresponding author.