

Abstract
The most common cause of amyotrophic lateral sclerosis and frontotemporal dementia (c9ALS/FTD) is an expanded G4C2 RNA repeat [r(G4C2)exp] in chromosome 9 open reading frame 72 (C9orf72), which elicits pathology through several mechanisms. Here, we developed and characterized a small molecule for targeted degradation of r(G4C2)exp. The compound was able to selectively bind r(G4C2)exp’s structure and to assemble an endogenous nuclease onto the target, provoking removal of the transcript by native RNA quality control mechanisms. In c9ALS patient–derived spinal neurons, the compound selectively degraded the mutant C9orf72 allele with limited off-targets and reduced quantities of toxic dipeptide repeat proteins (DPRs) translated from r(G4C2)exp. In vivo work in a rodent model showed that abundance of both the mutant allele harboring the repeat expansion and DPRs were selectively reduced by this compound. These results demonstrate that targeted small-molecule degradation of r(G4C2)exp is a strategy for mitigating c9ALS/FTD-associated pathologies and studying disease-associated pathways in preclinical models.
One Sentence Summary:
Small molecule recruitment of an endogenous nuclease rescues pathologies associated with c9ALS/FTD in vivo by targeting an RNA repeat expansion.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are progressive neurodegenerative disorders that manifest by motor impairment and cognitive, behavioral, and language deficits. The most common genetic cause of ALS and FTD is a GGGGCC hexanucleotide repeat expansion (HRE; G4C2exp) in intron 1 of chromosome 9 open reading frame 72 (C9orf72), and the associated disease has been designated as c9ALS/FTD (1, 2). Whereas healthy individuals typically carry 2 to 30 G4C2 repeats, individuals with c9ALS/FTD typically harbor hundreds to thousands of repeats (3, 4). Various studies have shown that the RNA transcribed from the expanded repeat, r(G4C2)exp, is a toxic agent that plays a role in the development of c9ALS/FTD pathogenesis (fig. S1A, top). r(G4C2)exp (i) can operate by a gain-of-function mechanism in which the repeat binds and sequesters a subset of proteins affecting gene expression (1), (ii) is aberrantly translated into toxic dipeptide repeat proteins (DPRs) via repeat associated non-AUG (RAN) translation (5–9), and (iii) results in the formation of r(G4C2)exp-containing foci in a subset of central nervous system cells (9, 10). [Note: The C9orf72 antisense strand is also transcribed, producing r(G2C4)exp, which is also RAN-translated and forms foci (8, 9, 11).] Previous studies have shown that r(G4C2)exp-associated toxicity can be mitigated by small molecules that selectively bind r(G4C2)exp (fig. S1A, bottom) (12, 13).
We hypothesized that a small molecule that eliminates the disease-causing agent, r(G4C2)exp, could ameliorate all c9ALS/FTD molecular defects, a substantial advantage over targeting a particular c9ALS/FTD pathway. This strategy could be further benefitted by the fact that r(G4C2)exp is located within an intron, not an open reading frame. More specifically, we envisioned a dual-functioning small molecule that selectively binds the three-dimensional (3D) structure formed by r(G4C2)exp and that recruits an endogenous ribonuclease (RNase) to cleave r(G4C2)exp, or an RNase recruiting chimera (RIBOTAC). A RIBOTAC would effect elimination of the toxic transcript by native RNA quality control pathways. That is, a chemical compound would unnaturally interface a transcript with RNA quality control mechanisms to eliminate a toxic RNA and alleviate disease-associated defects.
To enable such an approach, we first designed and optimized a high-affinity compound that selectively recognizes r(G4C2)exp using structure-activity relationships and biophysical and structural analyses. The optimal compound, as determined through a battery of cellular assays, was then converted into a RIBOTAC by appending it to a small-molecule RNase L recruiter (14). By cleaving r(G4C2)exp, this c9ALS/FTD RIBOTAC ameliorated C9orf72 HRE-associated pathologies in cellular model systems, including patient-derived lymphoblastoid cell lines (LCLs), patient-derived induced pluripotent stem cells (iPSCs), iPSC-derived spinal neurons (iPSNs), and a c9ALS/FTD bacterial artificial chromosome (BAC) transgenic mouse model. Transcriptome-wide studies in iPSNs and in the transgenic mouse confirm that the RIBOTAC is selective for the expanded r(G4C2) repeat in intron 1 of C9orf72.
RESULTS
Rational design of a dimeric compound that binds r(G4C2)exp
The r(G4C2)exp that causes toxicity in c9ALS/FTD folds into two structures that are in equilibrium: a G-quadruplex and a hairpin with a periodic array of 1 × 1 nucleotide G/G internal loops in the stem [(5′CCGGGGCC/3′GGGCCGGG)n] (12, 15). Using our lead identification strategy, Inforna (16, 17), and subsequent optimization, we previously designed a small molecule, 1 (Fig. 1A), that binds these internal loops selectively (12). This chemical probe enabled studies that demonstrated that the hairpin structure, but not the G-quadruplex structure, is RAN translated in vitro and in cells (12). Furthermore, small molecules that recognize this structure can reduce expression of the DPR poly(GP) produced by RAN translation and the number of r(G4C2)exp foci in a cellular model of c9ALS/FTD (12). We have previously shown that a facile route to increase a small molecule’s selectivity and potency, is to design a single molecule that simultaneously binds two or more structural elements within the RNA of interest (18). The molecule reads out not only the presence of the two structural elements but also their juxtaposition, providing a source of increased selectivity and avidity.
Fig. 1.

Small molecules targeting r(G4C2)exp in C9orf72 selectively affect disease-associated pathways.
(A) The monomer compound used in this paper. Compound 1 was previously reported to selectively binding to the G:G loop. (B) Compound 2 was obtained by linker optimization. An amino NH group was introduced into the PEG linker to generate compound 3 to afford higher binding affinity.
We therefore applied this strategy to r(G4C2)exp, guided by a model of 1 bound to r(G4C2) repeats (12). In our model, positions 6 and 9 within 1 were solvent-exposed and thus candidates for functionalization; that is, they could be modified with chemical handles to enable the synthesis of a dimeric compound without affecting molecular recognition. We previously reported functionalization of position 9 with an alkyne (1a) (12). Modification of position 6 with an alkyne moiety afforded 1b, which was synthesized as previously described (see the Supplementary Materials) (12). To confirm that these modifications do not affect molecular recognition, we measured the affinities of 1a and 1b for r(G4C2) repeats by biolayer interferometry (BLI) and compared them to 1 (table S1). Whereas the avidity of 1a [dissociation constant (Kd) = 0.6 ± 0.03 μM] was similar to that of 1 (Kd = 0.3 ± 0.03 μM), 1b’s affinity was ~37-fold weaker with a Kd of 11 ± 0.14 μM (table S1). Thus, position 9 can be functionalized without affecting molecular recognition, in contrast to position 6.
On the basis of these data, we synthesized a panel of dimers that display two copies of the RNA-binding module, conjugated to polyethylene glycol (PEG) linkers of different lengths via position 9 (see the Supplementary Materials). To identify the PEG scaffold with optimal display of 1, that is, the same distance separating the loops in r(G4C2)exp, we measured their potencies for inhibiting the binding of heterogeneous nuclear ribonucleoprotein H1 (hnRNP H1), a known r(G4C2)exp-binding protein, in vitro by a facile time-resolved fluorescence resonance energy transfer (TR-FRET) assay (12). The most potent dimer, 2 (Fig. 1B) had a median inhibitory concentration (IC50) of ~1.8 μM, 10-fold more potent than 1 (IC50 ~19 μM) (12), and a Kd of 50 ± 8 nM, as measured by BLI (table S1). The binding of 2 with related oligonucleotides, d(G4C2)8, r(G2C2)10 (a hairpin with a fully paired stem), and r(G2C4)8, which models the C9orf72 antisense repeat expansion, was much weaker, from ~26- to ~640-fold (fig. S1B and table S1). To further increase the binding affinity of 2 and provide a chemical handle for functionalization, we replaced the oxygen atom within the ethylene glycol linker with an NH group to generate 3 (Fig. 1B). Compound 3 bound ~13-fold more avidly to r(G4C2)8 than 2 with a Kd of 4 ± 0.7 nM (table S1). Moreover, 3 had a slower dissociation rate (koff) compared to 2, providing longer residence time on the target RNA (table S1).
Similar to 2, 3 was also selective for r(G4C2)8 over d(G4C2)8 (~225-fold lower affinity), r(G2C2)10 (~200-fold lower affinity), and r(G2C4)8 (~100-fold lower affinity) (fig. S1B). This increase in selectivity also increased its potency in the TR-FRET assay, affording an IC50 of 0.9 ± 0.1 μM (table S1). Complex formation between 3 and r(G4C2)4 was verified by monitoring the imino proton region via 1D nuclear magnetic resonance (1D NMR) spectroscopy, in which two imino proton peaks emerged at 10.5 parts per million, where noncanonically paired G residues appear (fig. S1C). No changes were observed in the 1D NMR spectrum of r(G2C2)4, a hairpin with a fully paired stem, upon addition of 3 (fig. S1D). We then analyzed 2D nuclear Overhauser effect spectra and completed molecular dynamics (MD) simulations to generate a 3D model of 3 bound to the repeats (Fig. 2A and fig. S1E). The 3-r(G4C2)4 repeat complex was primarily stabilized by stacking interactions between each RNA-binding module and the 1 × 1 nucleotide GG internal loop and a closing GC pair of each loop. Methyl groups on the ellipticines fill cavities in the major and minor grooves of the RNA, further stabilizing the complex via van der Waals interactions (Fig. 2A and fig. S1E).
Fig. 2.

Compound 3 recognizes the 3D structure of r(G4C2)exp in cells and inhibits disease-associated RAN translation in a variety of cellular models.
(A) 3D model of 3 bound to a r(G4C2) repeat model construct, as determined by NMR spectroscopy and MD simulations. A space filling model of the complex was used to display the lowest–binding energy state. Compound 3 is highlighted in cyan. (B) Schematic representation of the transcript variants from C9orf72 that were measured by RT-qPCR. (C) Schematic representation of Chem-CLIP method. (D) Fold enrichment of C9orf72 mRNA variants in Chem-CLIP studies completed in c9ALS patient–derived LCLs or iPSCs. Intron 1 harbors the r(G4C2)exp repeat expansion, whereas transcript variants containing exon 1b do not (n = 1 C9orf72 LCL and 1 C9orf72 iPSC line, three replicates per line). (E) Effect of 3 on RAN translation, as assessed in HEK293T cells dually transfected with (G4C2)66-Nano Luciferase (disease) and SV40-Firefly (normalization) (n = 3 replicates). (F) Poly(GP) abundance in protein lysates from patient-derived lymphoblastoid cells treated with 3. Quantification was normalized to vehicle (n = 3 C9orf72 LCL lines, three replicates per line). (G) Poly(GP) abundance in protein lysates from patient-derived iPSCs treated with 3. Quantification was normalized to vehicle (n = 4 C9orf72 iPSC lines, three replicates per line). (H) Percentage of (G4C2)66-No ATG-GFP mRNA transcript present within monosome- and polysome-containing fractions from HEK293T cells treated with vehicle or 200 nM of 3, quantified by RT-qPCR analysis of each fraction (n = 2 independent experiments). Fractions from the polysome gradients labeled as “monosomes” contain 40S, 60S, and 80S ribosomal subunits (fractions 1 to 7); “LMW” indicates low–molecular weight polysomes (fractions 8 to 10); and “HMW” indicates high–molecular weight polysomes (fractions 11 and 12). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, as determined by an unpaired t test with Welch’s correction (D and H), a one-way ANOVA test with multiple comparisons (E), or a repeated measures ANOVA test with Tukey’s multiple comparisons (F and G). Error bars indicate SD.
To validate engagement of r(G4C2)exp by 3 in vitro and in cells, we used a method developed in our laboratory dubbed Chemical Cross-Linking and Isolation by Pull-down (Chem-CLIP) and its competitive (C) variant (C-Chem-CLIP) (fig. S1F) (13, 18–20). These approaches enable target engagement studies in cells by cross-linking a small molecule to its target(s) followed by the subsequent isolation and analysis of the small molecule–RNA covalent adducts. Compound 3 was functionalized to contain a diazirine cross-linking module, which, upon exposure to light, reacts with proximal targets, to afford Chem-CLIP probe 4. A control Chem-CLIP probe lacking RNA-binding modules, 5, was also synthesized (fig. S1G). In vitro, 4 pulled down r(G4C2)8 dose dependently, with ~40% of the RNA pulled down with 500 nM compound (fig. S1H). In contrast, control Chem-CLIP probe 5 pulled down <25% RNA at all concentrations tested (fig. S1H). To confirm that 3 and 4 bind the same site within r(G4C2)8, we completed C-Chem-CLIP studies with a constant concentration of Chem-CLIP probe 4 (500 nM) and increasing concentrations of 3. Addition of 3 reduced the percentage of RNA pulled down in a dose-dependent fashion (fig. S1H); 4 did not pull down other nucleic acids significantly (P > 0.05), including the quadruplex form of r(G4C2) repeats (induced through addition of K+) and base-paired r(G2C2)10, d(G4C2)8, and d(G2C2)10, compared to control compound 5 (fig. S1H). These results indicate that observed pulldown of these other targets by 4 is due to the nonselective reaction of the diazirine.
We next sought to study target engagement in c9ALS patient–derived LCLs and iPSCs harboring G4C2 repeat expansions by C-Chem-CLIP, as measured by quantitative reverse transcription polymerase chain reaction (RT-qPCR). [Note: No evident toxicity (fig. S2A) or effect on cellular proliferation (fig. S2, B and C) was observed upon treatment of patient-derived cells with 3.] When the mutant C9orf72 allele is processed, it is alternatively spliced such that it produces variants that (i) contain exon 1a and intron 1, which harbors r(G4C2)exp, or (ii) lack exon 1a and intron 1 but contain exon 1b (Fig. 2B). Therefore, we used two sets of RT-qPCR primers to determine whether 4 selectively cross-links to variants containing the repeat expansion (Fig. 2C). In both c9ALS patient–derived LCLs and iPSCs, 4 selectively pulled down variants with r(G4C2)exp, enriching the RNA’s abundance by 2.3 ± 0.2– and 1.9 ± 0.4–fold, respectively, whereas no enrichment was observed in variants that do not contain the repeat expansion (Fig. 2D).
To further validate 3’s binding site, we used a method developed in our laboratory named ASO-Bind-Map, which uses competition between antisense oligonucleotides (ASOs) and a small molecule to define the small molecule’s binding site within an RNA target (21, 22). That is, a small molecule that binds and thermodynamically stabilizes the RNA target will prevent binding of the ASO and hence its subsequent degradation by RNase H. We first treated c9ALS patient–derived LCLs with 3 followed by transfection of an oligonucleotide complementary to r(G4C2)exp [5′-mG*mG*mC*C*C*C*G*G*C*C*C*C*G*G*C*C*C*mC*mG*mG, where m indicates a 2′-O-methyl residue and * indicates a locked nucleic acid (LNA) residue]. This ASO is different from previously reported ASOs that target the intron upstream of the repeat [5′-CCCGGCCCCTAGCGCGCGAC, 5′-GCCTTACTCTAGGACCAAGA, or 5′-TACAGGCTGCGGTTGTTTCC (Ionis)] (11, 23–25). As expected, we observed a dose-dependent increase in intron 1 abundance of C9orf72 mRNA, verifying that 3 competes with the complementary ASO for the r(G4C2)exp repeat (fig. S2D). Collectively, each of these target engagement studies shows that 3 binds r(G4C2)exp in patient-derived cells.
Compound 3 inhibits RAN translation in c9ALS/FTD patient–derived cells
After demonstrating target engagement of r(G4C2)exp by 3 in c9ALS patient–derived cells, we next systematically evaluated its effect on c9ALS/FTD biology in various cellular models. To study whether 3 could selectively modulate RAN translation, human embryonic kidney (HEK) 293T cells were co-transfected with (i) a plasmid expressing (G4C2)66-No ATG-Nano-luciferase, which solely undergoes RAN translation (12), and (ii) a plasmid encoding SV40-Firefly luciferase (reporter for canonical translation). Our data showed that 3 inhibited RAN translation of r(G4C2)66 (as measured by reduction of Nano-luciferase activity normalized to firefly luciferase signal; Fig. 2E) without affecting r(G4C2)66 abundance; that is, 3 did not inhibit transcription (fig. S2, E and F). Consistent with its lack of effect on C9orf72 mRNA abundance, 3 did not inhibit topoisomerase activity in vitro, indicating that the compound does not bind DNA, further supporting specific engagement of r(G4C2)exp (fig. S2G).
We next treated c9ALS patient–derived LCLs with 3 at concentrations of 5, 50, and 500 nM for 4 days and then measured poly(GP) abundance by an electrochemical luminescent sandwich immunoassay. We observed a dose-dependent decrease in poly(GP) expression, significant (P < 0.05) at all concentrations tested (Fig. 2F). [Unfortunately, we were unable to assess reduction of other DPRs by electrochemical luminescence immunoassay because of current limitations of antibody sensitivity and protein solubility.] Compound 3 had no effect on the abundance of C9orf72 mRNA variants harboring the HRE, as determined by RT-qPCR with primers specific for intron 1 where r(G4C2)exp resides (fig. S2E). Likewise, 3, at a concentration of 50 nM, significantly (P = 0.0003) reduced the number of r(G4C2)exp-containing nuclear foci by ~60%, as assessed by RNA fluorescence in situ hybridization (FISH) (fig. S2H).
In agreement with these observations, 3 also induced a dose-dependent reduction of poly(GP) abundance in iPSCs from patients with c9ALS (Fig. 2G), with no effect on C9orf72 intron 1 transcript abundance (fig. S2E) or wild-type (WT) C9ORF72 protein, as determined by Western blot (fig. S3, A and B). Collectively, these data indicate that only RAN translation, and not canonical translation, was inhibited by treatment with compound 3. Additionally, r(G4C2)exp causes retention of C9orf72 intron 1 (26). Whether the small molecule binds r(G4C2)exp in the context of the entire transcript or the spliced intron, 3 selectively inhibited RAN translation in patient-derived cells (Fig. 2, E to G and fig. S3, A and B) but did not affect overall C9orf72 mRNA or intron 1 abundance (fig. S2, E and F).
Compound 3 inhibits ribosome loading
Our data showing that 3 did not reduce C9orf72 intron 1 or WT C9ORF72 protein abundance in c9ALS patient–derived cells (Fig. 2, E to G, and figs. S2E and S3, A and B) suggested that the compound either inhibits ribosome binding of r(G4C2)exp or induces ribosome stalling on r(G4C2)exp, both of which can be explored by polysome profiling (27, 28). To gain insight into the mechanism of action of 3, we transfected HEK293T cells with a plasmid that encodes r(G4C2)66-No ATG fused to a green fluorescent protein (GFP) marker [r(G4C2)66-No ATG-GFP] and performed polysome profiling. Treatment of HEK293T cells expressing r(G4C2)66 with 3 reduced the amount of the transcript loaded into high–molecular weight (HMW) polysome fractions by 22 ± 2% and increased its abundance in low–molecular weight (LMW) fractions by 17 ± 1% (Fig. 2H and fig. S3, C and D). Recent studies suggested that blocking formation of HMW polysomes leads to inhibition of RAN translation of r(G4C2)exp (29). Further, our data show that 3 had no effect on polysome loading of glyceraldehyde 3-phosphate dehydrogenase (GAPDH; control transcript) under the same experimental conditions (Fig. 2H and fig. S3, C and D). Previous studies have found that small molecules targeting r(CGG)exp similarly block loading of ribosomes onto the repeat mRNA, thus inhibiting RAN translation but not canonical translation (30). Having validated lead compound 3 for selectively reducing C9orf72 HRE-associated pathologies and defining its mechanism of action, we turned our efforts to improve potency and selectivity such that it could be studied in a preclinical in vivo model of c9ALS/FTD.
Design and in vitro validation of a r(G4C2)exp-targeting RIBOTAC
We recently developed a new class of small molecules, dubbed RIBOTACs, that potently and selectively cleave an RNA target by recruiting an endogenous RNase (fig. S4A) (14, 31, 32). Using this approach, we designed a r(G4C2)exp-targeting RIBOTAC using the small-molecule RNase L recruiter ethyl (Z)-5-(3-hydroxy-4-methoxybenzylidene)-4-oxo-2-(phenylamino)-4,5-dihydrothiophene-3-carboxylate (14), which was optimized from a previously reported small molecule (33). RNase L, or latent RNase, is expressed in minute quantities as an inactive monomeric subunit in all cells. It is dimerized and activated by 2′−5′polyadenylate, which is synthesized in response to viral infections (34). In contrast, our RIBOTAC locally recruits RNase L selectively to r(G4C2) expansions, increasing its local effective concentration to trigger cleavage (fig. S4A). Thus, this approach will interface the r(G4C2)exp target with RNA quality control pathways in a programmable manner to allow systematic elimination of the toxic transcript.
In brief, we synthesized three compounds: (i) 6, in which 3’s linker’s amino group was acetylated (Fig. 3A) to study the effects of functionalization; (ii) 7, a r(G4C2)exp-targeting RIBOTAC in which the RNase L–recruiting heterocycle was installed via 3’s linker amino group (Fig. 3B); and (iii) control compound, 8, in which a less efficient RNase L–recruiting compound was conjugated to 3 (Fig. 3B). We first studied the effect of modifying 3 on molecular recognition by evaluating the potency of 6 and 7 (in the absence of RNase L) for inhibiting r(G4C2)8-hnRNP H1 complex formation, as assessed by the TR-FRET assay. As expected, the IC50s of 6 and 7 were similar to each other, 2.3 ± 0.3 and 2.4 ± 0.1 μM, respectively, about 2-fold greater than 3 (0.9 ± 0.1 μM) (table S1). We next assessed the ability of 7 to recruit RNase L and cleave the RNA using a FRET-based assay in which r(G4C2)8 was dually labeled with Cy5 (5′ end) and Cy3 (3′ end) (fig. S4B). The addition of 7 and RNase L dose-dependently decreased the FRET signal, whereas no change in FRET signal was observed upon addition of control RIBOTAC 8 and RNase L (fig. S4C). Collectively, these results suggest that 7 induced r(G4C2)8 cleavage by recruiting and activating RNase L. These findings were confirmed using radioactively labeled r(G4C2)8 and analysis by gel electrophoresis (fig. S4D).
Fig. 3.

A designer small molecule binds to r(G4C2)exp and recruits RNase L to selectively cleave r(G4C2)exp in cells.
(A) Compound 6 is a derivative featuring the RNA-binding modules but lacks the RNase L–recruiting module and was used as a control. (B) Compound 7 was obtained by conjugation of a nuclease RNase L recruiter module to a r(G4C2)exp-targeting dimer. Control compound 8 is a dimer conjugated to a control, less efficient RNase L–recruiting module with altered regioselectivity.
Activity of a r(G4C2)exp-targeting RIBOTAC in a HEK293T cellular model
To assess whether the r(G4C2)exp cleavage observed in vitro translated to cellular activity, we first assessed 7’s ability to inhibit RAN translation in the HEK293T cellular model. HEK293T cells were co-transfected with plasmids encoding (G4C2)66-No ATG-Nano-luciferase (RAN translation) and SV40-Firefly luciferase (control; canonical translation) and treated with 7. Indeed, 7 inhibited RAN translation of r(G4C2)66 by ~65% at a dose of 500 nM, as measured by reduction of Nano-luciferase activity normalized to firefly luciferase activity (Fig. 4A). Furthermore, 7 reduced quantities of the r(G4C2)exp-containing transcript in a dose-dependent fashion, with a 46 ± 2% reduction observed upon treatment with 500 nM compound, as assessed in HEK293T cells transfected with a plasmid encoding (G4C2)66-No ATG-GFP (Fig. 4B). We next used a small interfering RNA (siRNA) to knock down RNase L, eliciting a reduction in 7’s cleavage capacity (Fig. 4B). In contrast, no decrease in 7’s activity was observed upon treatment with a control siRNA (Fig. 4B). [Note: The siRNA directed against RNase L reduced its transcript abundance by ~70%, whereas a scrambled control had no effect (Fig. 4C).] To further validate the mechanism of action, we immunoprecipitated the ternary complex using an anti–RNase L antibody, demonstrating an ~2-fold enrichment of the r(G4C2)exp-containing mRNA in immunoprecipitated fractions, as compared to vehicle-treated samples (Fig. 4, D and E).
Fig. 4.
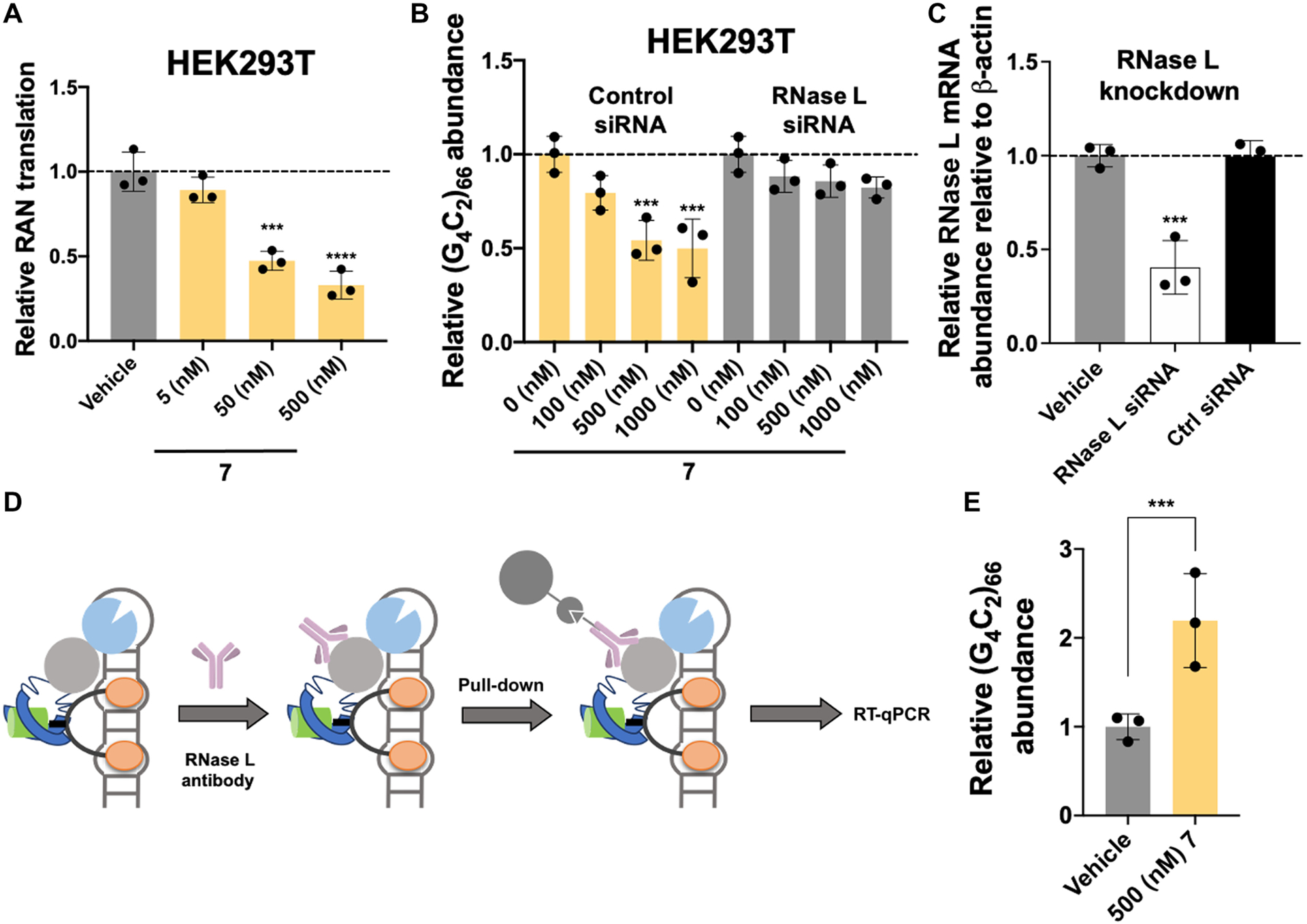
RIBOTAC 7 degrades r(G4C2)exp in a cellular model of c9ALS/FTD by local recruitment of RNase L.
(A) Effect of 7 on RAN translation, as assessed in HEK293T cells dually transfected with (G4C2)66-No ATG-Nano-Luciferase (disease) and SV40-Firefly (normalization) (n = 3 replicates). (B) Effect of 7 on r(G4C2)exp in HEK293T cells transfected with a plasmid encoding (G4C2)66-No ATG-GFP (n = 3 biological replicates) and ablation of this effect upon RNase L knockdown with an RNase L–targeting siRNA (n = 3 replicates). (C) Knockdown of RNase L by siRNA in HEK293T cells (n = 3 replicates). (D) Scheme of an RNase L immunoprecipitation (IP) assay to study ternary complex formation between r(G4C2)66, RNase L, and 7. (E) Results of the RNase L IP assay, used to assess ternary complex formation in cells (n = 3 replicates). ***P < 0.001; ****P < 0.0001, as determined by a one-way ANOVA with multiple comparisons (A and C) or unpaired t test with Welch’s correction (E). Error bars indicate SD.
A r(G4C2)exp-targeting RIBOTAC rescues pathological hallmarks in c9ALS patient–derived LCLs
We next assessed whether 7 could selectively cleave r(G4C2)exp in c9ALS patient–derived LCLs. [Note: No toxicity or effect on cellular proliferation was observed in c9ALS patient–derived cells upon dosage with up to 1000 nM of 7 (fig. S5, A to C)]. Treatment with 7 reduced C9orf72 intron 1 abundance with an IC50 of ~50 nM (Fig. 5A and fig. S5D). Furthermore, C9orf72 intron 1 abundance was dose-dependently restored when patient-derived LCLs were co-treated with increasing concentrations of 6, which binds but does not cleave the repeat expansion, and a constant concentration of 7 (Fig. 5B), confirming that 6 and 7 compete for the same binding site, r(G4C2)exp. In addition, 7 did not affect C9orf72 intron 1 abundance in LCLs from a healthy donor (Fig. 5C). As expected, control compound 8, which cannot effectively recruit RNase L, elicited no changes in intron 1 transcript abundance in both c9ALS disease–affected and healthy LCLs, further confirming specificity for the repeat expansion (Fig. 5C and fig. S5E). The observed decrease in C9orf72 intron 1 abundance due to cleavage of r(G4C2)exp resulted in reduced abundance of poly(GP). In particular, 500 nM of 7 reduced poly(GP) abundance by 71 ± 11% (Fig. 5D and fig. S5F). Thus, nuclease recruitment resulted in a 5-fold increase in potency over the parent compound 3 (Figs. 2F and 5D).
Fig. 5.

The r(G4C2)exp-targeting RIBOTAC 7 selectively cleaves the r(G4C2)exp-containing intron 1 in C9orf72 and alleviates disease-associated defects in patient-derived cells.
(A) Effect of 7 on C9orf72 intron 1, which harbors r(G4C2)exp, in patient-derived lymphoblastoid cells, as determined by RT-qPCR with intron 1–specific primers (n = 3 C9orf72 LCLs, three replicates per line). (B) Effect of co-treating patient-derived lymphoblastoid cells with parent dimer 6 (increasing concentrations) and RIBOTAC 7 (constant concentration), as determined by RT-qPCR using intron 1–specific primers (n = 1 C9orf72 LCL line, three replicates per line). (C) Effect of 7 and 8 in lymphoblastoid cells from healthy donors that do not express the RNA repeat expansion, as determined by quantification of intron 1 by RT-qPCR (n = 1 control LCL, three replicates per line). (D) Effect of 7 on poly(GP) abundance in protein lysate extracted from patient-derived lymphoblastoid cells (n = 3 C9orf72 LCLs, three replicates per line). (E) Effect of 7 on C9orf72 exon 1b, present only in transcripts that do not have r(G4C2)exp, in patient-derived lymphoblastoid cells, as determined by RT-qPCR with exon 1b–specific primers (n = 1 C9orf72 LCL, three replicates per line). (F) Effect of 7 (500 nM) or an ASO complementary to the repeat (100 nM) on human transcripts containing short, nonpathogenic r(G4C2) repeats in a patient-derived LCL (n = 1 C9orf72 LCL, three replicates per line), as determined by RT-qPCR using gene-specific primers (table S2) and compared to vehicle. The relative abundance of each target RNA was calculated relative to GAPDH. The sequence of the ASO is complementary to the repeat expansion itself [5′-mG*mG*mC*C*C*C*G*G*C*C*C*C*G*G*C*C*C*mC*mG*mG, where m indicates a 2′-O-methyl residue and * indicates a locked nucleic acid (LNA) residue]. This ASO is different from a previously reported ASO that targets the intron upstream of the repeat (11, 23–25). (G) Effect of 7 on C9orf72 intron 1, which harbors r(G4C2)exp, in patient-derived iPSCs, as determined by RT-qPCR with intron 1–specific primers (n = 4 C9orf72 iPSC lines, three replicates per line). (H) Effect of 7 on poly(GP) abundance in protein lysate extracted from patient-derived iPSCs (n = 4 C9orf72 iPSC lines, three replicates per line). (I) Effect of 7 on C9orf72 intron 1, which harbors r(G4C2)exp, in patient-derived iPSNs, as determined by RT-qPCR with intron 1–specific primers (n = 2 C9orf72 iPSC lines, three replicates per line). (J) Effect of 7 on poly(GP) abundance in protein lysate extracted from patient-derived iPSN (n = 1 C9orf72 iPSC line, three replicates per line). (K) Intron 1: Exon 2 as determined by RNA-seq analysis of patient-derived iPSNs treated with 7 relative to vehicle (n = 1 C9orf72 iPSC line, three replicates per line). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, as determined by a repeated measures ANOVA test with Tukey’s multiple comparisons (A, D, and G to J), a one-way ANOVA test with multiple comparisons (B, C, E, and F), or an unpaired t test with Welch’s correction (K). Error bars indicate SD.
These data suggest that 7 only cleaves C9orf72 transcripts containing r(G4C2)exp present in intron 1. To determine whether this was indeed the case, we quantified C9orf72 transcripts using RT-qPCR primers selective for exon 1b, which is only present in transcripts lacking r(G4C2)exp (Fig. 2B). In transcripts that lack r(G4C2)exp, no decrease in C9orf72 was observed after treatment with 7 at any concentration (Fig. 5E). To further assess compound selectivity, known genes containing short (that is, nonpathogenic) r(G4C2) repeats were analyzed by RT-qPCR for off-target cleavage. No decrease in abundance was observed for any of the eight transcripts tested upon treatment with 500 nM of 7 (Fig. 5F).
A r(G4C2)exp-targeting RIBOTAC rescues pathological hallmarks in c9ALS patient–derived iPSCs
We next confirmed our observations from LCLs in c9ALS patient–derived iPSCs. After treatment with 50 or 500 nM of 7, r(G4C2)exp-containing intron 1 abundance was reduced by 45 ± 11 and 64 ± 10%, respectively (Fig. 5G). The observed decrease in intron 1 abundance and the mature transcript, as assessed using RT-qPCR primers that amplify the exon 2–3 junction, was observed whether normalized to GAPDH, 18S, or β-actin housekeeping genes (fig. S5, G and H). Treatment with 7 had no effect on C9orf72 intron 1 quantities in iPSC lines from healthy donors (fig. S6, A to C). In agreement with reduced C9orf72 intron 1 abundance, we also observed a significant (P < 0.05) reduction in the abundance of poly(GP) (Fig. 5H). The reduction of RAN-translated poly(GP) was selective given that canonically translated WT C9ORF72 protein abundance was unchanged by treatment with 7, as determined by Western blotting (relative to β-actin and compared to vehicle-treated samples; fig. S7A). Nuclease recruitment resulted in a significant (P < 0.05) increase in potency and a prolonged duration of activity for reducing poly(GP) abundance, as compared to parent compound 3 (fig. S7, B and C).
To quantify the selectivity of 7 in c9ALS patient–derived iPSCs, we performed full RNA sequencing (RNA-seq) analysis after treatment with 50 nM of 7 for 4 days. Abundance of C9orf72 transcripts containing the r(G4C2)exp-harboring intron 1 was reduced, with no significant (P > 0.05) effect on other transcripts containing short, nonpathogenic r(G4C2) repeats (fig. S8, A to C). Furthermore, no changes in the transcriptome were observed in iPSCs derived from healthy donors (fig. S8D) nor was there an effect on C9orf72 expression (fig. S8, E and F).
A r(G4C2)exp-targeting RIBOTAC rescues pathological hallmarks in c9ALS patient–derived iPSNs
We extended our studies of 7 into c9ALS iPSC–derived spinal neurons, which recapitulate the genetic, transcriptional, and biochemical signatures of brain tissue from patients with c9ALS (24, 35). Upon treatment of patient-derived iPSNs with 7, a dose-dependent reduction in intron 1 abundance was observed, with significant (P < 0.05) reduction observed at a concentration as low as 50 nM (Fig. 5I). As expected, no decrease in intron 1 abundance was observed in neurons derived from two different healthy individuals that do not express the HRE (fig. S8G). Protein analysis via the sandwich electrochemical luminescence immunoassay also revealed a dose-dependent decrease in poly(GP) abundance upon treatment with 7, with a 33 ± 4% reduction observed for iPSNs treated with 50 nM of compound (Fig. 5J and fig. S5F).
To quantify the selectivity of 7 in patient-derived iPSNs, we performed full RNA-seq analysis after treatment with 50 nM of 7 for 2.5 weeks. Abundance of C9orf72 transcripts containing the r(G4C2)exp-harboring intron 1 was reduced (Fig. 5K), with no statistically significant (P > 0.05) effects on other transcripts containing short, nonpathogenic r(G4C2) repeats (fig. S8H), in agreement with RT-qPCR analysis of 7-treated patient-derived lymphoblastoid cells (Fig. 5F).
Previous studies have established a link between C9orf72-mediated ALS/FTD and defects in the nuclear pore complex (NPC) (36, 37). In particular, a recent study identified several nuclear pore proteins to be reduced in C9orf72 neurons, reversible upon ASO treatment (37). Using superresolution structured illumination microscopy, we found that treatment of patient-derived iPSNs with 50 nM 7 reversed the loss of Nup98, a component of the central channel of the NPC involved in bidirectional transport previously reported in c9ALS iPSNs (38), mirroring the abundance found in healthy control iPSNs (fig. S9, A and B).
A r(G4C2)exp-targeting RIBOTAC ameliorates c9ALS/FTD pathology in vivo
Last, we assessed the therapeutic potential of 7 in a C9orf72 BAC mouse model (+/+PWR500), which expresses 500 r(G4C2) repeats (39). These mice exhibit several pathological hallmarks associated with c9ALS/FTD including formation of RNA foci and increased abundance of DPRs (39). We focused on these pathological hallmarks, rather than other phenotypic defects, because of their robustness in this mouse model.
Mice were treated with RIBOTAC 7 (33 nmol) by a single intracerebroventricular (ICV) injection for 3 weeks (table S6). After treatment, abundance of the r(G4C2)exp-containing intron 1 mRNA was measured by RT-qPCR, revealing a reduction of 44 ± 22% compared to vehicle-treated controls (Fig. 6A). The abundance of transcripts containing exon 1b, which do not contain the repeat expansion, was not altered (Fig. 6B). We measured the amount of all C9orf72 transcripts after treatment with 7 (using RT-qPCR primers for the exon 2–exon 3 junction common to all transcripts; Fig. 2A). C9orf72 abundance was reduced by 7 to an extent that reflects the percentage of transcripts that contain intron 1 and proportional to the observed cleavage of intron 1 (fig. S10A). Furthermore, no effect was observed on transcripts with short, nonpathogenic r(G4C2) repeats, as determined by RNA-seq analysis (fig. S10B). Collectively, these data suggest that cleavage by 7 is specific for the repeat expansion.
Fig. 6.

The r(G4C2)exp-targeting RIBOTAC 7 alleviates c9ALS/FTD–associated toxicity in a preclinical mouse mode of disease.
(A) Effect of 33 nmol of 7 on r(G4C2)exp-containing intron 1 in the +/+PWR500 C9orf72 BAC mouse model, as determined by RT-qPCR analysis of total RNA isolated from treated and untreated mice with intron 1–specific primers (n = 6; each data point represents a sample from a different mouse). (B) Effect of 7 on C9orf72 exon 1b, found only in transcripts lacking r(G4C2)exp, in +/+PWR500 mice, as determined by RT-qPCR analysis of total RNA isolated from treated and untreated mice with exon 1b–specific primers (n = 6; each data point represents a sample from a different mouse). N.S., not significant. (C) Representative RNA-FISH images of r(G4C2)exp-containing foci from the cortex of +/+PWR500 mice treated with 7 compared with vehicle mice. Scale bars, 20 μm. Arrows point to r(G4C2)exp foci. DAPI, 4′,6-diamidino-2-phenylindole. (D) Quantification of the effect of 7 on r(G4C2)exp-containing foci in +/+PWR500, as determined by RNA-FISH (n = 3). (E) Effect of 7 on poly(GP) abundance in brain tissue harvested from +/+PWR500 mice, as determined by an electrochemical luminescence immunoassay. Relative poly(GP) abundance was normalized to total protein and vehicle-treated mice (n = 6; each data point represents a sample from a different mouse). (F) Effect of 7 on the abundance of control protein β-actin abundance in brain tissue harvested from +/+PWR500 mice, as determined by an electrochemical luminescence immunoassay. Relative β-actin abundance was normalized to total protein and vehicle treated mice (n = 6; each data point represents a sample from a different mouse). (G) Representative histological images of cortex from +/+PWR500 mice treated with 7 compared with vehicle, visualizing poly(GP), poly(GA), or TDP-43 by immunohistochemistry. Staining highlights cellular nuclei and respective aggregates or inclusions indicated by arrows. Scale bars, 200 μm. Arrows point to poly(GP) aggregates (left), poly(GA) aggregates (middle), and TDP-43 inclusions (right). (H) Quantification of poly(GP) aggregates from histological analysis, including images in (G) [n = 4 mice; all cells (~200) were counted per section; three sections counted per mouse]. (I) Quantification of poly(GA) aggregates from histological analysis, including images in (G) [n = 4 mice; all cells (~200) were counted per section; three sections counted per mouse]. (J) Quantification of TDP-43 inclusions from histological analysis, including images in (G) [n = 4 mice; all cells (~200) were counted per section; three sections counted per mouse]. **P < 0.01; ***P < 0.001; ****P < 0.0001, as determined by an unpaired t test with Welch’s correction (A, B, D to F, and H to J). Error bars indicate SD.
As expected on the basis of the reduction of r(G4C2)exp abundance, the number of r(G4C2)exp-containing nuclear foci were also decreased in the brains of +/+PWR500 mice treated with 7 compared to vehicle-treated mice, as determined by FISH (Fig. 6, C and D; fig. S10C shows lack of foci by FISH staining in WT mice, whether treated or untreated). No effect was observed on the abundance of antisense r(G2C4)exp foci in treated mice compared to vehicle, demonstrating that compound 7 is selective for the sense transcript (fig. S10, D and E) (39). Poly(GP) translated from the expanded repeat was also significantly (P < 0.0001) reduced in +/+PWR500 mice treated with 7, by 74 ± 24% (Fig. 6E). No decrease of β-actin protein expression was observed in treated +/+PWR500 mice or treated WT mice, as determined by an electrochemical luminescence immunoassay (Fig. 6F and fig. S10F). These data indicate a selective reduction of RAN translation, at least for poly(GP). Rescue of c9ALS/FTD pathological hallmarks was observed as early as 1 week (the shortest time analyzed after treatment) and persisted to at least 6 weeks (the longest time period evaluated after treatment), both to similar extents as observed 3 weeks after administration of 7 (fig. S11).
We additionally performed immunohistochemical (IHC) analysis of sagittal brain sections for inclusions immunopositive for poly(GP), poly(GA), or transactivation response DNA-binding protein 43 (TDP-43), known hallmarks of a subset of cells in c9ALS/FTD (Fig. 6, G to J) (40). A significant (P < 0.01) decrease in poly(GP) and poly(GA) aggregates was observed in the cortex of treated mice as compared to mice that received only vehicle (Fig. 6, G to J), the former consistent with reduction of poly(GP) abundance measured in brain lysates (Fig. 6F). [Poly(GP) and poly(GA) aggregates were not detected by immunohistochemistry in WT mice, as expected (fig. S12).] TDP-43 inclusions were also decreased throughout the cortex of treated mice relative to vehicle (Fig. 6, G and J). Collectively, these results demonstrate that RIBOTAC 7 can rescue c9ALS/FTD pathology in mice.
DISCUSSION
As the biology underpinning the pathology of microsatellite diseases has unfolded, it has become clear that many mechanisms contribute to toxicity, including the binding, sequestration, and loss of function of RNA-binding proteins and the production of toxic proteins translated from the expanded repeats (26, 41–45). The complexity of these mechanisms themselves, as well as their interplay, suggests that eliminating repeat expansion RNA from disease-affected cells could be broadly therapeutically beneficial. The state-of-the-art method to effect elimination of an RNA is its sequence-based recognition by ASOs that recruit endogenous RNase H (46). The ease of design of ASOs makes them attractive therapeutic modalities, and they have been developed for various repeat expansion disorders, including c9ALS/FTD (24, 47, 48). The highly optimized C9orf72-targeting ASO potently ameliorates c9ALS/FTD-associated pathologies in patient-derived iPSCs and iPSNs and in mouse models, demonstrating its therapeutic potential (9, 11, 23, 24). Both the potential and challenges of ASO treatment for repeat expansion disorders are exemplified by the oligonucleotide developed by Roche/Ionis Pharmaceuticals for Huntington’s disease [r(CAG)exp]. The ASO reduced the abundance of the repeat expansion in phase 1 clinical trials but was halted in phase 3 trials because of worsening outcome (49). Rather than targeting the repeats themselves, the most selective ASOs target unique sequences within the transcript in which they are harbored, because repeat-targeting ASOs also down-regulate transcripts with short repeats that do not cause disease. Thus, an ASO would need to be developed against each microsatellite disorder, even if they are caused by the same repeat expansions.
Small molecules offer an important complement to ASOs, recognizing RNA 3D folds rather than sequence and providing avenues for optimization using traditional medicinal chemistry approaches (50, 51). Still in its infancy compared to protein targeting, the number of bioactive small molecules with RNA-centric modes of action is relatively few, by and large binding the RNA target at protein-binding sites, whether enzymatic processing sites as in the case of microRNA precursors, toxic structures in repeat expansions that sequester RNA-binding proteins and the ribosome, or regulatory structures bound by various cellular machineries. The promise of small molecules that modulate RNA (dys)function is exemplified by the discovery and lead optimization of the small molecule risdiplam, a molecular glue that binds an RNA-protein manifold (52). Identified from a phenotypic screen, the U.S. Food and Drug Administration–approved drug directs alternative splicing of a pre-mRNA for the treatment of spinal muscular atrophy (52).
Small molecules have been developed to bind two different structures adopted by r(G4C2)exp, a G-quadruplex or a hairpin with an array of 1 × 1 nucleotide internal loops. For example, one of the first small molecules shown to bind the r(G4C2)exp G-quadruplex structure and inhibit protein binding in vitro was the porphyrin TmPyP4 (53). G-quadruplex–targeting ligands are also bioactive against the c9ALS/FTD repeat expansion (54). A small-molecule library comprising antiparasitic compounds and known G-quadruplex binders was screened for thermally stabilizing r(G4C2) repeats in vitro. Three related dinicotinimidamides were found that rescued c9ALS/FTD pathology in patient-derived iPSC-cortical and iPSC-motor neurons. The most potent, 6,6′-([2,2′-biselenophene]-5,5′-diyl)dinicotinimidamide, was studied in a Drosophila model where the small molecule reduced the amount of DPRs in adult flies and larvae, increasing survival of the latter to the pupal stage (54). Our laboratory has designed various small molecules including two substituted ellipticines and a substituted benzimidazole that bind the hairpin structure formed by r(G4C2)exp in various cellular models, including transdifferentiated neurons from patient-derived fibroblasts (12, 13, 55). In other studies, a peptide was developed that binds r(G4C2)exp in a transfected human neuroblastoma cell line and rescues retinal degeneration in a Drosophila model (56).
Here, we describe the design of the small-molecule functional equivalent of an ASO that selectively targets the unique structure of r(G4C2)exp, rather than its sequence, for degradation by recruiting the endogenous nuclease RNase L. That is, the RIBOTAC’s mode of action is the selective recognition and cleavage of the mutant C9orf72 allele, because the abundance of WT C9orf72 transcripts or other transcripts containing short, nonpathogenic r(G4C2) repeats was unchanged. Various studies have shown that short RNA repeats form different structures than disease-causing expanded repeats (57–60). As demonstrated here, small molecules can exploit these differences, leading to discrimination not only between mutant and WT alleles but also between the mutant allele and other transcripts harboring short repeats. Elimination of the toxic repeat expansion by the RIBOTAC rescued c9ALS/FTD-associated pathological hallmarks in c9ALS patient–derived cells and a mouse model. The ALS-targeting RIBOTAC did not alleviate disease pathology associated with antisense r(G2C4)exp transcript, because it forms a different structure. Dual-targeting of both sense and antisense transcripts could be challenging. The observed lack of an effect on the antisense transcript could be a limitation of the compounds developed herein.
Nonetheless, it should be noted that the binding small molecule and RIBOTAC studied here are lead molecules, and much translational work remains to advance them toward therapeutic modalities, both from a medicinal chemistry and physicochemical property perspective. Although overactivation of the immune system by RIBOTAC molecules has not been observed in cells or rodent models, it certainly warrants careful consideration in man. Likewise, RNase L expression is likely to be cell and tissue dependent and could limit target or disease scope. Such limitation could be overcome by recruiting other cellular RNases or enzymes that act on RNA.
Collectively, although RNA is a challenging target, the complementarity of the ASO and small-molecule fields is triangulating its ligandability and druggability to study its function and to deactivate dysfunction for therapeutic benefit. Expanding small-molecule mode of action to RNase recruitment to cleave repeat expansions provides another avenue to explore for the treatment of microsatellite disorders. The ability to design compounds that target RNA structures from sequence could enable the development of many more compounds that can specifically affect RNA biology to study function and provide lead medicines.
MATERIALS AND METHODS
Study design
In this study, to probe small-molecule targeting of the expanded r(G4C2) repeat associated with ALS/FTD, we aimed to design and evaluate small molecules that selectively bind the structure of the disease-causing RNA. One structure formed by r(G4C2)exp is a hairpin with a periodic array of internal loops. We first designed a dimeric small molecule that engages two internal loops simultaneously to increase both affinity and specificity. The dimer was evaluated in various cellular models, including patient-derived cells, for inhibiting disease-associated pathologies and directly engaging the target RNA. Small-molecule mode of action for inhibiting aberrant translation was then assessed by polysome profiling. Next, we designed the small molecule to direct an endogenous nuclease to cleave r(G4C2)exp in cells and in vivo. The RIBOTAC’s mode of action was assessed by RT-qPCR, a competitive experiment with a noncleaving, binding compound, siRNA knockdown of the endogenous RNase, and immunoprecipitation of the ternary complex. Selective cleavage of the r(G4C2)exp-containing intron 1 was also evaluated by RNA-seq. Inhibition of pathological hallmarks of c9ALS/FTD was demonstrated in patient-derived LCLs, iPSCs, and differentiated spinal neurons, including reduction of poly(GP) abundance and nuclear foci in patient-derived cells. In vivo, the effect of a single ICV injection of 33 nmol to the right hemisphere on the abundance of the RNA repeat expansion was analyzed by RT-qPCR and on poly(GP) abundance by a sandwich immunoassay 1, 3, and 6 weeks after injection. IHC and FISH assays were also used to assess c9ALS/FTD pathology. Experimental protocols were completed as approved by the Scripps Florida Institutional Animal Care and Use Committee, and all relevant ethical regulations were followed. FVB/NJ-Tg(C9orf72)500Lpwr/J “PWR500” mice were acquired from the Jackson Laboratory and bred for study at The Scripps Research Institute.
Study size calculations
Power calculations were not completed for these studies.
Treatment of outliers
Data were treated as outliers if they lied outside 1.5 times the inner quartile range. This criterion was only applied to in vivo poly(GP) measurements; in these cases, the samples were also excluded from other analyses (RT-qPCR analysis and Δweight measurements). RNA samples were not analyzed by RT-qPCR if OD260 (optical density at 260 nm)/OD280 (optical density at 280 nm) was <1.8, indicative of poor quality.
Randomization
Mice were randomly assigned to treatment groups, such that the groups were age- and gender-matched. For cellular experiments, plate wells were randomly assigned to treatment groups.
Blinding
IHC staining and analyses of in vivo studies were completed blinded. All other studies were not blinded.
Replication
The number of replicates for each experiment is indicated in the figure legends and the test used to calculate statistical significance.
Quantification and statistical analysis
All quantification and statistical analyses (completed in GraphPad Prism version 8) were completed as described in the figure legends. In brief, the statistical analysis for all experiments completed in c9ALS/FTD patient–derived cells accounted for repeated measurements of the same patient cell line using a repeated measures one- or two-way analysis of variance (ANOVA). Tukey’s multiple comparison test was used to compare multiple samples as indicated in the figure legends. For studies completed in vitro or in cells wherein a single dataset or cell line was analyzed, statistical significance was determined using a one-way ANOVA or an unpaired t test with Welch’s correction (assuming unequal SD), as indicated in each figure legend. For all panels where statistical significance is indicated, *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. Bar graphs display individual data points and report the data as the means ± SD. All compound-treated samples were normalized to vehicle unless otherwise noted. The dimethyl sulfoxide (DMSO) concentration for vehicle-treated samples was always ≤0.1% for cellular assays and was 1% DMSO and 99% H2O for in vivo studies.
Supplementary Material
Removing disease-causing expansion.
Expansion in G4C2 RNA repeat in chromosome 9 open reading frame 72 (C9orf72) is one of the most common genetic causes of amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD). To develop better therapeutics, Bush et al. developed and characterized a small molecule with dual function, able to bind the G4C2 expansion and to recruit an endonuclease to remove the expansion from the native RNA. The small molecule rescued typical cellular hallmarks of ALS/FTD in vitro in several patient-derived models and ameliorated ALS/FTD pathology in a mouse model. Removing pathological GC expansion using this dual-function small molecule could be an effective approach for treating C9orf72-mediated ALS/FTD.
Funding
This study was funded by the NIH (P01 NS099114 to M.D.D., L.P., and J.D.R.; DP1 NS096898 and R35 NS116846 to M.D.D.; and R35 NS097273 to L.P.), Target ALS (to M.D.D.), the Nelson Family Fund (to M.D.D.), the First Family Fund (to M.D.D.), the Deutsche Forschungsgemeinschaft (DFG) for the DFG Postdoctoral Fellowship, and the ALS Association (ALSA) for the Milton Safenowitz Postdoctoral Fellowship (to A.U.).
Footnotes
Competing interests: M.D.D. is a founder of Expansion Therapeutics. M.D.D. is a consultant for Expansion Therapeutics, and E.T.W. was a consultant for Expansion Therapeutics during the course of these studies. A patent disclosure has been filed on aspects of this work.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All RNA-seq data were deposited in Mendeley Data (DOI: 10.17632/332kvzz8xk.1).
References
- 1.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R, Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M; The ITALSGEN Consortium, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ, A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, Campbell T, Uphill J, Borg A, Fratta P, Orrell RW, Malaspina A, Rowe J, Brown J, Hodges J, Sidle K, Polke JM, Houlden H, Schott JM, Fox NC, Rossor MN, Tabrizi SJ, Isaacs AM, Hardy J, Warren JD, Collinge J, Mead S, Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet 92, 345–353 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, Brown PH, Baker MC, Finch NA, Bauer PO, Serrano G, Beach TG, Josephs KA, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Boylan KB, Petrucelli L, Dickson DW, Rademakers R, Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): A cross-sectional cohort study. Lancet Neurol. 12, 978–988 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul III JW, Rademakers R, Boylan KB, Dickson DW, Petrucelli L, Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, Van Broeckhoven C, Kremmer E, Kretzschmar HA, Haass C, Edbauer D, Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D, The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP, RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U.S.A 110, E4968–E4977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, Cosio DM, van Blitterswijk M, Lee WC, Rademakers R, Boylan KB, Dickson DW, Petrucelli L, Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, Castanedes-Casey M, Kurti A, Stankowski JN, Disney MD, Rothstein JD, Dickson DW, Fryer JD, Zhang YJ, Petrucelli L, Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener 14, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J, Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U.S.A 110, E4530–E4539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang ZF, Ursu A, Childs-Disney JL, Guertler R, Yang WY, Bernat V, Rzuczek SG, Fuerst R, Zhang YJ, Gendron TF, Yildirim I, Dwyer BG, Rice JE, Petrucelli L, Disney MD, The hairpin form of r(G4C2)exp in c9ALS/FTD is repeat-associated non-ATG translated and a target for bioactive small molecules. Cell Chem. Biol 26, 179–190.e112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, Johnston A, Overstreet K, Oh SY, Todd PK, Berry JD, Cudkowicz ME, Boeve BF, Dickson D, Floeter MK, Traynor BJ, Morelli C, Ratti A, Silani V, Rademakers R, Brown RH, Rothstein JD, Boylan KB, Petrucelli L, Disney MD, Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 83, 1043–1050 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costales MG, Aikawa H, Li Y, Childs-Disney JL, Abegg D, Hoch DG, Velagapudi SP, Nakai Y, Khan T, Wang KW, Yildirim I, Adibekian A, Wang ET, Disney MD, Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. U.S.A 117, 2406–2411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy K, Zamiri B, Stanley SY, Macgregor RB Jr., Pearson CE, The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem 288, 9860–9866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Disney MD, Winkelsas AM, Velagapudi SP, Southern M, Fallahi M, Childs-Disney JL, Inforna 2.0: A platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol 11, 1720–1728 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velagapudi SP, Gallo SM, Disney MD, Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol 10, 291–297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Disney MD, Targeting RNA with small molecules to capture opportunities at the Intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 141, 6776–6790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guan L, Disney MD, Covalent small-molecule-RNA complex formation enables cellular profiling of small-molecule-RNA interactions. Angew. Chem. Int. Ed. Engl 52, 10010–10013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, Disney MD, Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol 13, 188–193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Childs-Disney JL, Tran T, Vummidi BR, Velagapudi SP, Haniff HS, Matsumoto Y, Crynen G, Southern MR, Biswas A, Wang Z-F, Tellinghuisen TL, Disney MD, A massively parallel selection of small molecule-RNA motif binding partners informs design of an antiviral from sequence. Chem 4, 2384–2404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang P, Park H-J, Zhang J, Junnb E, Andrews RJ, Velagapudi SP, Abegg D, Vishnu K, Costales MG, Childs-Disney JL, Adibekian A, Moss WN, Mouradian MM, Disney MD, Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. U.S.A 117, 1457–1467 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, Chun S, Sun S, Ling SC, Myers B, Engelhardt J, Katz M, Baughn M, Platoshyn O, Marsala M, Watt A, Heyser CJ, Ard MC, De Muynck L, Daughrity LM, Swing DA, Tessarollo L, Jung CJ, Delpoux A, Utzschneider DT, Hedrick SM, de Jong PJ, Edbauer D, Van Damme P, Petrucelli L, Shaw CE, Bennett CF, Da Cruz S, Ravits J, Rigo F, Cleveland DW, Lagier-Tourenne C, Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD, RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang YJ, Prudencio M, Carlomagno Y, Daughrity LM, Jansen-West K, Perkerson EA, O’Raw A, Cook C, Pregent L, Belzil V, van Blitterswijk M, Tabassian LJ, Lee CW, Yue M, Tong J, Song Y, Castanedes-Casey M, Rousseau L, Phillips V, Dickson DW, Rademakers R, Fryer JD, Rush BK, Pedraza O, Caputo AM, Desaro P, Palmucci C, Robertson A, Heckman MG, Diehl NN, Wiggs E, Tierney M, Braun L, Farren J, Lacomis D, Ladha S, Fournier CN, McCluskey LF, Elman LB, Toledo JB, McBride JD, Tiloca C, Morelli C, Poletti B, Solca F, Prelle A, Wuu J, Jockel-Balsarotti J, Rigo F, Ambrose C, Datta A, Yang W, Raitcheva D, Antognetti G, McCampbell A, Van Swieten JC, Miller BL, Boxer AL, Brown RH, Bowser R, Miller TM, Trojanowski JQ, Grossman M, Berry JD, Hu WT, Ratti A, Traynor BJ, Disney MD, Benatar M, Silani V, Glass JD, Floeter MK, Rothstein JD, Boylan KB, Petrucelli L, Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med 9, eaai7866 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sznajder ŁJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, Oliveira R, Nutter CA, Bhatt K, Sobczak K, Ashizawa T, Thornton CA, Ranum LPW, Swanson MS, Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. U.S.A 115, 4234–4239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johannes G, Carter MS, Eisen MB, Brown PO, Sarnow P, Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc. Natl. Acad. Sci. U.S.A 96, 13118–13123 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zong Q, Schummer M, Hood L, Morris DR, Messenger RNA translation state: The second dimension of high-throughput expression screening. Proc. Natl. Acad. Sci. U.S.A 96, 10632–10636 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamada SB, Gendron TF, Niccoli T, Genuth NR, Grosely R, Shi Y, Glaria I, Kramer NJ, Nakayama L, Fang S, Dinger TJI, Thoeng A, Rocha G, Barna M, Puglisi JD, Partridge L, Ichida JK, Isaacs AM, Petrucelli L, Gitler AD, RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat. Neurosci 22, 1383–1388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang WY, He F, Strack RL, Oh SY, Frazer M, Jaffrey SR, Todd PK, Disney MD, Small molecule recognition and tools to study modulation of r(CGG)exp in fragile X-associated tremor ataxia syndrome. ACS Chem. Biol 11, 2456–2465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costales MG, Matsumoto Y, Velagapudi SP, Disney MD, Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc 140, 6741–6744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costales MG, Suresh B, Vishnu K, Disney MD, Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol 26, 1180–1186.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thakur CS, Jha BK, Dong B, Das Gupta J, Silverman KM, Mao H, Sawai H, Nakamura AO, Banerjee AK, Gudkov A, Silverman RH, Small-molecule activators of RNase L with broad-spectrum antiviral activity. Proc. Natl. Acad. Sci. U.S.A 104, 9585–9590 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, Korennykh A, Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343, 1244–1248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH, Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med 5, 208ra149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP, GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coyne AN, Zaepfel BL, Hayes L, Fitchman B, Salzberg Y, Luo EC, Bowen K, Trost H, Aigner S, Rigo F, Yeo GW, Harel A, Svendsen CN, Sareen D, Rothstein JD, G4C2 repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron 107, 1124–1140.e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HJ, Taylor JP, Lost in transportation: Nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96, 285–297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LPW, C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90, 521–534 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Mackenzie IRA, Frick P, Neumann M, The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 127, 347–357 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Cleary JD, Ranum LP, Repeat associated non-ATG (RAN) translation: New starts in microsatellite expansion disorders. Curr. Opin. Genet. Dev 26, 6–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Echeverria GV, Cooper TA, RNA-binding proteins in microsatellite expansion disorders: Mediators of RNA toxicity. Brain Res. 1462, 100–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohan A, Goodwin M, Swanson MS, RNA-protein interactions in unstable microsatellite diseases. Brain Res. 1584, 3–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nalavade R, Griesche N, Ryan DP, Hildebrand S, Krauss S, Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 4, e752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ranum LPW, Cooper TA, RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci 29, 259–277 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Stephenson ML, Zamecnik PC, Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. U.S.A 75, 285–288 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA, Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K, Wu J, Bezprozvanny I, Corey DR, Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol 27, 478–484 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kingwell K, Double setback for ASO trials in Huntington disease. Nat. Rev. Drug Discov 20, 412–413 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Fedorova O, Jagdmann GE Jr., Adams RL, Yuan L, Van Zandt MC, Pyle AM, Small molecules that target group II introns are potent antifungal agents. Nat. Chem. Biol 14, 1073–1078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen JL, Zhang P, Abe M, Aikawa H, Zhang L, Frank AJ, Zembryski T, Hubbs C, Park H, Withka J, Steppan C, Rogers L, Cabral S, Pettersson M, Wager TT, Fountain MA, Rumbaugh G, Childs-Disney JL, Disney MD, Design, optimization, and study of small molecules that target Tau pre-mRNA and affect splicing. J. Am. Chem. Soc 142, 8706–8727 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, Denk N, Feng Z, Green L, Guerard M, Jablonski P, Jacobsen B, Khwaja O, Kletzl H, Ko C-P, Kustermann S, Marquet A, Metzger F, Mueller B, Naryshkin NA, Paushkin SV, Pinard E, Poirier A, Reutlinger M, Weetall M, Zeller A, Zhao X, Mueller L, Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem 61, 6501–6517 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Zamiri B, Reddy K, Macgregor RB Jr., Pearson CE, TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J. Biol. Chem 289, 4653–4659 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, Woodling NS, Niccoli T, Gilbert-Jaramillo J, Abdelkarim S, Clayton EL, Clarke M, Konrad MT, Nicoll AJ, Mitchell JS, Calvo A, Chio A, Houlden H, Polke JM, Ismail MA, Stephens CE, Vo T, Farahat AA, Wilson WD, Boykin DW, Zetterberg H, Partridge L, Wray S, Parkinson G, Neidle S, Patani R, Fratta P, Isaacs AM, G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol. Med 10, 22–31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ursu A, Wang KW, Bush JA, Choudhary S, Chen JL, Baisden JT, Zhang YJ, Gendron TF, Petrucelli L, Yildirim I, Disney MD, Structural features of small molecules targeting the RNA repeat expansion that causes genetically defined ALS/FTD. ACS Chem. Biol 15, 3112–3123 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Q, An Y, Chen ZS, Koon AC, Lau KF, Ngo JCK, Chan HYE, A peptidylic inhibitor for neutralizing r(GGGGCC)exp-associated neurodegeneration in C9ALS-FTD. Mol. Ther. Nucleic Acids 16, 172–185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Angelbello AJ, Rzuczek SG, McKee KK, Chen JL, Olafson H, Cameron MD, Moss WN, Wang ET, Disney MD, Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U.S.A 116, 7799–7804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ, RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 31, 5469–5482 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Michlewski G, Krzyzosiak WJ, Molecular architecture of CAG repeats in human disease related transcripts. J. Mol. Biol 340, 665–679 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Busan S, Weeks KM, Role of context in RNA structure: Flanking sequences reconfigure CAG motif folding in huntingtin exon 1 transcripts. Biochemistry 52, 8219–8225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH, Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U.S.A 101, 7287–7292 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mathews DH, Sabina J, Zuker M, Turner DH, Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol 288, 911–940 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Hwang TL, Shaka AJ, Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson 112, 275–279 (1995). [Google Scholar]
- 64.Grzesiek S, Bax A, The importance of not saturating water in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc 115, 12593–12594 (1993). [Google Scholar]
- 65.Piotto M, Saudek V, Sklenář V, Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665 (1992). [DOI] [PubMed] [Google Scholar]
- 66.Goddard TD, Kneller DG (University of California, San Francisco, 2004). [Google Scholar]
- 67.Angelbello AJ, Disney MD, Bleomycin can cleave an oncogenic noncoding RNA. Chembiochem 19, 43–47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang WY, Wilson HD, Velagapudi SP, Disney MD, Inhibition of non-ATG translational events in cells via covalent small molecules targeting RNA. J. Am. Chem. Soc 137, 5336–5345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, Sarkar P, Schatz GC, Disney MD, Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem. Biol 9, 538–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Childs-Disney JL, Stepniak-Konieczna E, Tuan T, Yildirim I, Park H, Chen CZ, Hoskins J, Southall N, Marugan JJ, Patnaik S, Zheng W, Austin CP, Schatz GC, Sobczak K, Thornton CA, Disney MD, Induction and reversal of myotonic dystrophy type 1 pre-mRNA splicing defects by small molecules. Nat. Commun 4, 2044 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, Development and testing of a general amber force field. J. Comput. Chem 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Bayly CI, Cieplak P, Cornell WD, Kollman PA, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem 97, 10269–10280 (1993). [Google Scholar]
- 73.Cornell WD, Cieplak P, Bayly CI, Kollman PA, Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc 115, 9620–9631 (1993). [Google Scholar]
- 74.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, J. Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ, Gaussian 09 (Gaussian Inc., 2016). [Google Scholar]
- 75.Sangster MJL, Atwood RM, Interionic potentials for alkali halides. II. Completely crystal independent specification of Born-Mayer potentials. J. Phys. C Solid State Phys 11, 1541–1555 (1978). [Google Scholar]
- 76.Cheatham III TE, Miller JL, Fox T, Darden TA, Kollman PA, Molecular dynamics simulations on solvated biomolecular systems: The particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc 117, 4193–4194 (1995). [Google Scholar]
- 77.Bednářová E, Hybelbauerová S, Jindřich J, Optimized methods for preparation of 6I-(ω-sulfanyl-alkylene-sulfanyl)-β-cyclodextrin derivatives. Beilstein J. Org. Chem 12, 349–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shan M, Bujotzek A, Abendroth F, Wellner A, Gust R, Seitz O, Weber M, Haag R, Conformational analysis of bivalent estrogen receptor ligands: From intramolecular to intermolecular binding. Chembiochem 12, 2587–2598 (2011). [DOI] [PubMed] [Google Scholar]
- 79.Cao Y, Yang J, Development of a folate receptor (FR)-targeted indenoisoquinoline using a pH-sensitive N-ethoxybenzylimidazole (NEBI) bifunctional cross-linker. Bioconjug. Chem 25, 873–878 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dračínský M, Sejbal J, Rygerová B, Stiborová M, An efficient modification of ellipticine synthesis and preparation of 13-hydroxyellipticine. Tetrahedron Lett. 48, 6893–6895 (2007). [Google Scholar]
- 81.Mandl CP, König B, Synthesis of mono protected 1,10-diaza-18-crown-6. Synth. Commun 34, 3573–3578 (2004). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All RNA-seq data were deposited in Mendeley Data (DOI: 10.17632/332kvzz8xk.1).