

This paper presents the Domain Auto Finder (DAFi) program and its application to the analysis of single-crystal X-ray diffraction (SC-XRD) data from multiphase mixtures of microcrystalline solids and powders. The DAFi algorithm is designed to quickly find subsets of reflections from individual domains in a whole set of SC-XRD data and neither requires a priori crystallographic information nor is limited by the number of phases or individual domains.
Keywords: single-crystal domain auto finder, DAFi, single-crystal X-ray diffraction, polycrystalline samples, multiphase mixtures
Abstract
This paper presents the Domain Auto Finder (DAFi) program and its application to the analysis of single-crystal X-ray diffraction (SC-XRD) data from multiphase mixtures of microcrystalline solids and powders. Superposition of numerous reflections originating from a large number of single-crystal domains of the same and/or different (especially unknown) phases usually precludes the sorting of reflections coming from individual domains, making their automatic indexing impossible. The DAFi algorithm is designed to quickly find subsets of reflections from individual domains in a whole set of SC-XRD data. Further indexing of all found subsets can be easily performed using widely accessible crystallographic packages. As the algorithm neither requires a priori crystallographic information nor is limited by the number of phases or individual domains, DAFi is powerful software to be used for studies of multiphase polycrystalline and microcrystalline (powder) materials. The algorithm is validated by testing on X-ray diffraction data sets obtained from real samples: a multi-mineral basalt rock at ambient conditions and products of the chemical reaction of yttrium and nitrogen in a laser-heated diamond anvil cell at 50 GPa. The high performance of the DAFi algorithm means it can be used for processing SC-XRD data online during experiments at synchrotron facilities.
1. Introduction
For more than a century, single-crystal X-ray diffraction (SC-XRD) has been a powerful method for determining the structure of crystalline solids. Until very recently it could be applied to single crystals not smaller than dozens of micrometres, but many compounds are only available in a polycrystalline form or as fine powders. State-of-the-art powder X-ray diffraction (XRD) data analysis, including Rietveld refinement in combination with ab initio structure search, can help with structure interpretation if sufficiently large crystals are unavailable and their preparation or growth is infeasible. This concerns investigations of natural objects or drugs, in situ studies of matter under extreme conditions, or processes in solids involving domain formation or reconstructive phase transitions. However, when it comes to multiphase systems with unknown microcrystalline compounds, the problem of structure solution for individual components becomes even more difficult.
In recent decades, the development of third- and fourth-generation synchrotrons, such as the Advanced Photon Source (in Lemont, USA), PETRA III (in Hamburg, Germany) and the ESRF (in Grenoble, France), with the ESRF–EBS (the Extremely Brilliant Source, the ESRF’s facility upgrade over 2015–2022, which increases the brilliance and coherence of the X-ray beams produced by a factor of 100 compared with present-day light sources; https://www.esrf.fr/about/upgrade), has provided users with new opportunities. At the cutting-edge beamlines, such as ID11 at the ESRF, the size of the X-ray beam (0.5 × 0.5 µm FWHM) is commensurate with the size of crystalline domains of polycrystalline samples or fine powder particles, which makes it possible to study each micrometre- to submicrometre-size grain individually by methods of SC-XRD, considering the sphere of confusion of the diffractometer of only a few hundred nanometres. This approach was devised for and first applied to studying products of chemical reactions and phase transformations in laser-heated diamond anvil cells (DACs); this has led to discoveries of many exotic compounds, revealing their crystal structures in situ under high pressure (e.g. Bykova et al., 2016 ▸, 2018 ▸; Laniel, Winkler, Bykova et al., 2020 ▸; Laniel, Winkler, Fedotenko et al., 2020 ▸; Bykov et al., 2020 ▸, 2021 ▸; Aslandukova et al., 2021 ▸; Dubrovinskaia & Dubrovinsky, 2018 ▸; Ceppatelli et al., 2022 ▸).
Still, processing SC-XRD data containing a lot of reflections coming from numerous crystalline grains is a difficult task, especially in the presence of a few different phases in a multicomponent system and/or in the absence of any a priori information about their chemical composition and/or basic crystallographic characteristics, such as the unit-cell parameters. The diffraction data collected from samples under high pressure in a DAC are additionally complicated by undesired but unavoidable reflections from diamond anvils, pressure-transmitting media, gasket materials and other factors. Therefore, the development of software which would allow an automatic separation of the reflections originating from an individual crystalline domain, i.e. a search for the domain in a complex pattern of spots in the reciprocal space, is an urgent task aimed at rationalizing SC-XRD data processing and making it routine for inexperienced users.
To date, several programs have been developed for multigrain indexing. If the unit-cell parameters are known a priori, e.g. from powder XRD data, indexing means finding the orientation matrices of the grains in the sample and sorting the reciprocal-space vectors with regard to the grain of origin. Following the presentation of the program GRAINDEX (Lauridsen et al., 2001 ▸), several alternative approaches have been proposed (Wright, 2006 ▸; Ludwig et al., 2009 ▸; Moscicki et al., 2009 ▸; Schmidt, 2014 ▸). The programs ImageD11 (Wright, 2006 ▸) and GrainSpotter (Schmidt, 2014 ▸) are now incorporated into the FABLE (Fully Automatic BeamLine Experiments) package (Sørensen et al., 2012 ▸). The main limitation of the above-mentioned software is that it is designed to be applied almost exclusively to the analysis of mono-phase materials. Furthermore, the multigrain indexing programs mentioned above all assume that the space group (or at least symmetry) and the unit-cell parameters of phases are known. One straightforward way to generalize the previous approaches is to apply the multigrain indexing algorithms repeatedly, once for each phase (Jimenez-Melero et al., 2011 ▸; Sørensen et al., 2012 ▸), but this still requires the phases to be identified in advance.
To our knowledge, there have only been a few proposals for dealing with unknown phases, based on a fast Fourier transform approach (Sørensen et al., 2012 ▸) or on pattern recognition (Sørensen et al., 2012 ▸), or involving a search of reflections and subsequent unit-cell optimization in 3D space (Wejdemann & Poulsen, 2016 ▸). Testing of these programs was performed on data sets artificially created by randomly rotating ‘grains’ with exactly defined unit-cell parameters, and there is no information on how well these programs would work with real data sets where one may need to consider statistical and instrumental errors in the positions of reflections in the reciprocal space, or deal with ‘junk’ reflections characteristic of the XRD data sets originating from high-pressure experiments in DACs. Another important problem is the long program running time; e.g. according to Wejdemann & Poulsen (2016 ▸), indexing of 500 cementite grains takes 5 days.
In this article, we describe our methodological approach to the analysis of XRD data from polycrystalline materials and present the DAFi program which helps to automate the search for individual crystalline domains. A flowchart of the analysis is shown in Fig. 1 ▸. The DAFi program can be applied at that stage of the analysis when the diffraction from individual crystalline domains should be sorted. The algorithm does not need any a priori crystallographic knowledge, and there is no limitation on the phase composition of polycrystalline material and the number of crystalline domains of each phase. The algorithm is implemented with C++ code. Its important advantage is the extremely high speed of data processing. With the number of reflections in the input XRD data set (input peak table) equal to N
reflections, the algorithm has
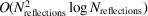 time complexity on a single-core processor, so that a typical computational time is several minutes. Implemented multithreading capability allows a further decrease of the computational time by dividing by the number of processor cores.
time complexity on a single-core processor, so that a typical computational time is several minutes. Implemented multithreading capability allows a further decrease of the computational time by dividing by the number of processor cores.
Figure 1.

A data flowchart for the analysis of single-crystal XRD data from polycrystalline materials. The DAFi program is implemented to sort groups of reflections originating from individual single-crystal domains (see the text for detailed explanations).
While the DAFi program enables the diffraction data of each domain to be separated from those of other domains, the data can be further processed using standard methods of single-crystal X-ray crystallography aimed at structure solution and refinement. The output of the current version of the DAFi program is compatible with the CrysAlisPro software, which performs indexing of each found domain individually with just one click. However, there will not be a problem using the DAFi output file(s) with other standard indexing algorithms implemented in any available crystallographic programs. The algorithm of the DAFi program is described in detail below.
2. Algorithm
2.1. Input and output data
The algorithm requires only a set of coordinates of all reflections in the reciprocal space. Currently, the DAFi program reads these coordinates from the peaktable.tabbin file generated by the CrysAlisPro software after ‘peak hunting’ (Fig. 1 ▸). If the XRD data originate from high-pressure experiments in a DAC, ‘advanced filtering’ (Koemets, 2020 ▸) is applied to eliminate the peaks produced by diamonds and the other diffraction artifacts associated with such a type of XRD raw data. After the ‘DAFi input peak table’ data processing, the DAFi program generates the output file(s), which is the ‘DAFi output peak table’ with the subsets of peaks sorted and numbered in the course of the search (see below for details). This means that the DAFi program updates the initial CrysAlisPro peaktable.tabbin file by marking each reflection with the number of the subset (subset ID) to which it belongs.
2.2. General structure of the algorithm
The ‘peak table’ generated by the CrysAlisPro software presents all diffraction peaks produced by a polycrystalline sample, which are visualized as a set of points in the reciprocal space. The whole set of points is a result of a superposition of numerous ‘subsets’ – the reciprocal-lattice points which belong to individual crystalline domains. Thus, if a subset is identified, then it can be indexed separately using standard crystallographic programs, and the crystal structure of the associated domain can be solved and refined.
Sorting subsets in the whole pattern of points in the reciprocal space is exactly the task of the DAFi program. The advantage of the implemented algorithm is that it selects the subsets purely geometrically, considering only a definition of a lattice (no time-consuming indexing is involved). As any 3D lattice is defined by three lattice vectors, the latter define three directions in a 3D space and the distances between the adjacent lattice points in these three directions. Obviously, a lattice can be recognized if considered as rows of equally distant points aligned in one direction, so the algorithm relies on finding such rows (i.e. a direction vector and a ‘proper’ distance between the adjacent points along the direction vector). This simplifies the search, which is realized iteratively. As soon as one subset of points is found, it is separated from the pool of all points, and only the remaining ones are considered in the next search.
The algorithm consists of two main stages:
(i) Finding a set of possible directions [Fig. 2 ▸(a)] and selecting the ‘best’ ones to consider at the second stage.
Figure 2.

Illustration of the two main stages of the algorithm. (a) First stage: finding a set of possible directions (shown here by arrows) for a given set of reflections (here blue points A through F) and selecting the ‘best’ one(s) to consider at the second stage. Among the ten directions found for the set of points A, B, C, D, E, F, the ‘best’ one (shown by the green arrow) is identified as that corresponding to the largest number of collinear vectors. (b) The second stage: finding the ‘proper’ distance between the reflections (here denoted as ‘d’) in the selected direction. (c) An example of a view of the reciprocal space with the subset of points (orange dots) found in the initial set (blue dots).
(ii) Finding the ‘proper’ distance between the reflections for a given direction [Fig. 2 ▸(b)] and identifying the nodes of the reciprocal lattice [Fig. 2 ▸(c)] generated by the chosen pair (direction, distance).
Combining these two parts we can find the ‘best’ pair (direction, distance), which corresponds to the biggest group of reflections belonging to one single-crystal domain. In Section 2.3 we describe different approaches to finding a set of possible directions, while in Section 2.4 we present an effective way to find the correct group of reflections for the given direction.
For the convenience of further mathematical description of the algorithm, the terms used below are defined as follows:
A point is a single diffraction reflection in the reciprocal space. The points are denoted as
 and are represented as radius vectors
and are represented as radius vectors
 in a 3D space.
in a 3D space.
A row is a subset of reflections that lie on the same line in the reciprocal space.
A group is a subset of reflections in the reciprocal space belonging to a distinct single-crystal domain.
2.3. First stage of the algorithm
Before the main algorithm, point normalization is applied:
(i) All radius vectors are shifted by the vector
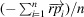 , which shifts the center of the points’ system to the coordinate
, which shifts the center of the points’ system to the coordinate
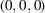 .
.
(ii) The coordinates
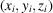 of each radius vector are divided by the maximum absolute value of the corresponding coordinate among all radius vectors (i.e.
of each radius vector are divided by the maximum absolute value of the corresponding coordinate among all radius vectors (i.e.
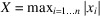 ,
,
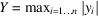 ,
,
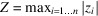 ). After that all radius vectors’ coordinates are transformed into
). After that all radius vectors’ coordinates are transformed into
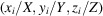 and belong to the range [−1; 1].
and belong to the range [−1; 1].
The shift described in the first step of the normalization procedure aims exclusively to improve the stability of the algorithm during the calculations. Although in practice the shift is very small, the shifting at the very beginning makes the algorithm more stable due to coordinates being transformed into a more uniform distribution. At the same time, the second part of the normalization procedure is important for further calculations [especially for the correct use of allowed absolute and relative errors (epsilon constants) in the second stage].
It is easy to see that the direction vector that determines the group will be equal to the direction vector between some two initial points. So, the most straightforward approach is to create a set of possible directions as a set of all direction vectors between each pair of initial points. However, such a set has a size of
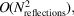 which is too large for the second stage of the algorithm. In Section 2.3.1 we propose a simple way to select only
which is too large for the second stage of the algorithm. In Section 2.3.1 we propose a simple way to select only
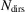 ‘best’ directions out of all
‘best’ directions out of all
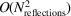 , where
is any integer constant (naive approach), and in Section 2.3.2 we propose an improved version of such a selection (smart approach). Both naive and smart approaches are implemented in DAFi and the user can select which one to use in the configuration file.
, where
is any integer constant (naive approach), and in Section 2.3.2 we propose an improved version of such a selection (smart approach). Both naive and smart approaches are implemented in DAFi and the user can select which one to use in the configuration file.
2.3.1. Naive approach
Ideally, we would like to select directions along which the second part of the algorithm will produce the largest possible group. We do not know in advance which directions are the ‘best’; however, we can see that if a group consists of k
rows with sizes
 , then there are exactly
, then there are exactly
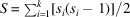 pairs of initial points that produce the same direction vector. This allows us to define the ‘best’ direction as the direction with a maximum number of pairs of initial points that produce it. However, because the initial points are real valued (have non-integer coordinates), all S generated vectors can differ slightly. To compare different real-valued vectors we transform them in two steps.
pairs of initial points that produce the same direction vector. This allows us to define the ‘best’ direction as the direction with a maximum number of pairs of initial points that produce it. However, because the initial points are real valued (have non-integer coordinates), all S generated vectors can differ slightly. To compare different real-valued vectors we transform them in two steps.
Before the first step of transforming a vector
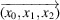 , where
, where
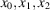 are the coordinates of the real-valued vector that we are transforming, index
are the coordinates of the real-valued vector that we are transforming, index
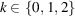 is found such that
is found such that
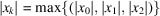 .
.
In the first step we make a transformation after which opposite vectors are considered to be equal:
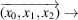
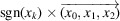 .
.
In the second step we transform the obtained vector to an integer-valued triplet:
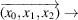
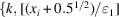 ,
,
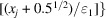 , where i and j are two indices from
, where i and j are two indices from
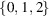 not equal to k,
not equal to k,
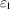 is a constant representing the allowed absolute error, and square brackets denote the integer part of a fractional number.
is a constant representing the allowed absolute error, and square brackets denote the integer part of a fractional number.
To illustrate this, let us consider a numerical example. Suppose that we want to transform a direction vector
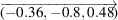 .
.
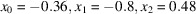 . Note that
. Note that
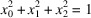 , because we are working with just a direction. Then the three steps of the transformation will be the following:
, because we are working with just a direction. Then the three steps of the transformation will be the following:
(i)
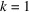 because
because
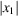 is maximum among
is maximum among
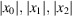 .
.
(ii)
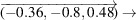
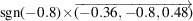 =
=
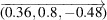 .
.
(iii)
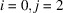 because there are only two indices from
which are not equal to
.
because there are only two indices from
which are not equal to
.
Let us also assume that
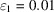 . Then we get the following integer-valued triplet:
. Then we get the following integer-valued triplet:
 |
After such a transformation, each direction is represented as an integer-valued triplet with values in the range
 , where
, where
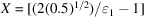 , so we can calculate a distribution of all directions using an array of size
, so we can calculate a distribution of all directions using an array of size
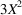 . After distribution calculation, we can find the top
directions in time
. After distribution calculation, we can find the top
directions in time
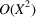 using a standard selection algorithm for finding the kth-order statistic (Blum et al., 1973 ▸). To transform the integer-valued triplet back to the real-valued vector, we can use the following formula:
using a standard selection algorithm for finding the kth-order statistic (Blum et al., 1973 ▸). To transform the integer-valued triplet back to the real-valued vector, we can use the following formula:
where
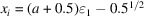 ,
,
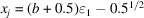 ,
,
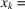
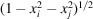 , and
, and
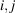 are two indices from
not equal to k.
are two indices from
not equal to k.
This approach is the most straightforward way to select the ‘best’
directions; however, it has drawbacks. The main one is that this approach does not use information about distances between points, which means that even with a large number of points lying on the same line, the second part of the algorithm may still not find the group if these points are located at unequal distances.
2.3.2. Smart approach
Below we present the second approach to select the ‘best’
directions, which does not have the drawbacks mentioned above. We are still going to select the ‘best’
directions from some distribution; however, instead of creating a distribution from all
vectors, we will use only some of the more important of them. Namely, let us iterate over the ‘center’ point
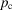 and find all possible rows of size at least 4 that go through the point
and consist of only equidistant points. In order to do this, first of all let us group all other
and find all possible rows of size at least 4 that go through the point
and consist of only equidistant points. In order to do this, first of all let us group all other
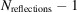 points in rows with respect to our center point
. This can be done by clustering all direction vectors
points in rows with respect to our center point
. This can be done by clustering all direction vectors
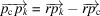
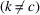 , similarly to the method described in Section 2.3.1. After this, for each row, we can independently find the largest subset of points where each point lies at an equivalent distance from the previous one. To do this, let us find out, for each point
, similarly to the method described in Section 2.3.1. After this, for each row, we can independently find the largest subset of points where each point lies at an equivalent distance from the previous one. To do this, let us find out, for each point
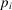 , at which distances d it will be in the same row as a point
. Let us denote by D the distance between points
and
. Then we can say that
is the kth point in a row with 0th point
if the following holds:
, at which distances d it will be in the same row as a point
. Let us denote by D the distance between points
and
. Then we can say that
is the kth point in a row with 0th point
if the following holds:
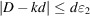 , where
, where
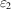 is some small constant that allows a small absolute error. From this inequality we can obtain that valid distances form the following range:
is some small constant that allows a small absolute error. From this inequality we can obtain that valid distances form the following range:
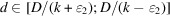 . After finding such ranges for all points
we can find the value of d that belongs to the largest number of ranges using the scanline algorithm (Klee, 1977 ▸). If this value is at least 3, then there exists a row that contains at least 4 points and with high probability belongs to a group. Only in such a case will we use the corresponding direction vector in our distribution. Such an approach takes
. After finding such ranges for all points
we can find the value of d that belongs to the largest number of ranges using the scanline algorithm (Klee, 1977 ▸). If this value is at least 3, then there exists a row that contains at least 4 points and with high probability belongs to a group. Only in such a case will we use the corresponding direction vector in our distribution. Such an approach takes
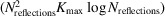 time, where
time, where
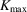 is the maximum point’s relative number on the row under consideration and
is the maximum point’s relative number on the row under consideration and
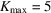 works well in practice.
works well in practice.
The smart approach catches fewer ‘junk’ reflections (Fig. 3 ▸) and, therefore, provides a better distribution of direction vectors to the second stage of the algorithm. However, this approach is a bit slower, because instead of
time, it requires
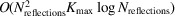 .
.
Figure 3.

Comparison of naive and smart approaches. The naive approach implies consideration of all directions, while the smart one considers only the directions with rows of equidistant points.
2.4. Second stage of the algorithm
Given a direction vector
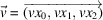 , we have to find the ‘best’ distance d between adjacent points towards a direction
, we have to find the ‘best’ distance d between adjacent points towards a direction
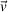 that generates the group of maximum size. Let
that generates the group of maximum size. Let
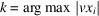 . Then we can project all initial points to a plane
. Then we can project all initial points to a plane
 : radius vector
: radius vector
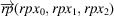 of point
p will be transformed to
of point
p will be transformed to
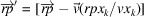 . After such a transformation, all points that belong to the same row in the direction
will be projected to the same point on a plane. This allows all different rows to be obtained by clustering of all projected points. Such clustering can be done in linear time using radix sort (Cormen et al., 2001 ▸) and two linear passes that select equal points in 2 × 2 grid squares. After grouping all points into rows, we can create an array ds of all distances between adjacent points in the same row and choose d as the most frequent number in the array ds. Because all distances are real numbers, we have to use tolerance
. After such a transformation, all points that belong to the same row in the direction
will be projected to the same point on a plane. This allows all different rows to be obtained by clustering of all projected points. Such clustering can be done in linear time using radix sort (Cormen et al., 2001 ▸) and two linear passes that select equal points in 2 × 2 grid squares. After grouping all points into rows, we can create an array ds of all distances between adjacent points in the same row and choose d as the most frequent number in the array ds. Because all distances are real numbers, we have to use tolerance
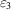 and choose d such that an interval
and choose d such that an interval
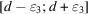 contains the most values from the array ds. Such a d can be found in linear time using the two pointers technique for maintaining a sliding window of size
contains the most values from the array ds. Such a d can be found in linear time using the two pointers technique for maintaining a sliding window of size
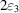 after sorting the array ds.
after sorting the array ds.
After finding the d value, we can find the exact group formed by a pair
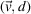 as a union of all largest valid subsets of points for each independent row. In order to find the largest valid subset for a given row, we introduce an auxiliary array ‘shifts’, where
as a union of all largest valid subsets of points for each independent row. In order to find the largest valid subset for a given row, we introduce an auxiliary array ‘shifts’, where
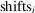 denotes the distance from a point
to the plane
towards the direction
. Since the distance between all adjacent points in a group’s row is equal to d, for a valid subset of points it holds that all remainders
denotes the distance from a point
to the plane
towards the direction
. Since the distance between all adjacent points in a group’s row is equal to d, for a valid subset of points it holds that all remainders
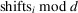 are equal, where mod denotes the modulo operation, i.e.
are equal, where mod denotes the modulo operation, i.e.
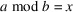 ,
,
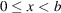 ,
,
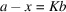 ,
,
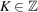 . This allows us to find the largest valid subset as the largest subset of points with equal values of
and pairwise different values of
. This allows us to find the largest valid subset as the largest subset of points with equal values of
and pairwise different values of
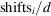 . It can be found in
. It can be found in
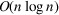 time using the two pointers technique for maintaining the set of all values
in a sliding window, where n is the number of points in the current row. Similarly to Section 2.3.2, the values
time using the two pointers technique for maintaining the set of all values
in a sliding window, where n is the number of points in the current row. Similarly to Section 2.3.2, the values
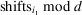 and
and
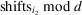 are considered equal iff
are considered equal iff
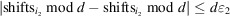 . The program has a configuration file that allows one to flexibly adjust all necessary parameters and in particular values
and
. Smaller values of tolerance will result in a more precise group; however, the found group will contain fewer reflections.
. The program has a configuration file that allows one to flexibly adjust all necessary parameters and in particular values
and
. Smaller values of tolerance will result in a more precise group; however, the found group will contain fewer reflections.
The time complexity of this stage can be estimated as
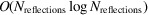 per direction, so the total time complexity for processing all best
directions found in the previous stage is
per direction, so the total time complexity for processing all best
directions found in the previous stage is
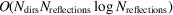 .
.
2.5. Speed optimizations
Without any optimizations, the program finds all groups one by one, so the total time complexity is
There are, however, some implemented optimizations that allow the algorithm to be significantly speeded up:
(i) Both stages of the algorithm allow the use of multithreading (in the first stage several threads uniformly process
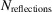 ‘center’ points, and in the second stage several threads uniformly process
different best directions from the first stage).
‘center’ points, and in the second stage several threads uniformly process
different best directions from the first stage).
(ii) The distribution of the ‘best’ directions is calculated only at the beginning of the program and, instead of recalculation from scratch on the following iterations, the distribution is just maintained by subtracting the impact of the removed points from the found group in time
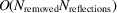 , where
, where
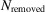 is the number of points in the last group found.
is the number of points in the last group found.
(iii) In fact, the algorithm finds
different groups in one iteration (one for each direction from the first stage), so there is an option to choose not just the largest group, but
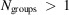 largest groups at once. This is done by firstly selecting the largest group, then the largest group with points not selected in the first group, and so on. Such an option allows the algorithm to be speeded up
largest groups at once. This is done by firstly selecting the largest group, then the largest group with points not selected in the first group, and so on. Such an option allows the algorithm to be speeded up
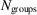 times; however, it may slightly decrease the quality of the search.
times; however, it may slightly decrease the quality of the search.
When combined, such optimizations allow the algorithm to be speeded up to the time complexity
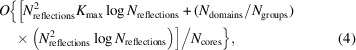 |
where
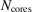 is the number of processor cores and
is the number of groups to be found in one iteration. Assuming that
,
,
is the number of processor cores and
is the number of groups to be found in one iteration. Assuming that
,
,
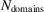 and
are all constants, the total time complexity can be simplified to
.
and
are all constants, the total time complexity can be simplified to
.
3. Examples of application
The testing of the DAFi program was performed on SC-XRD data sets obtained from real polycrystalline samples: (i) a natural basalt rock and (ii) a piece of yttrium (Y) embedded into molecular nitrogen and laser-heated in a DAC. The results of these tests are described below as examples 1 and 2.
Example 1. Study of a sample of basalt rock from the Rauher Kulm mountain/SC-XRD data collected using an in-house diffractometer. Basalt rock is a natural polycrystalline aggregate of several minerals. A sample of basalt was collected by LD and ND at the Rauher Kulm mountain, which is a paleovolcano located in the Upper Palatinate region of the state of Bavaria, 23 km southeast of Bayreuth (Germany). A small isometric dark-gray grain of the rock (of about 40 µm in diameter) with sub-grains barely distinguishable under an optical microscope (×200) was mounted on a goniometer head. A single-crystal XRD data set was collected using a diffractometer equipped with a Bruker D8 platform (the three-axis goniometer), an APEX detector and an Ag Kα Incoatec IµS source (beam size of ∼50 µm FWHM, half-sphere data collection, a collection time of 60 s with a step of 0.3°, 1265 frames). The peak hunting procedure in the CrysAlisPro software found 2928 reflections.
The search for 18 groups of reflections (the number set by the user) in a pool of 2928 reflections took the DAFi program 31 s (Fig. 4 ▸ and Table 1 ▸). Each of the 18 groups found had its own size (the number of reflections included in the group). In the course of further data processing and indexing using CrysAlisPro , some groups were merged, as the CrysAlisPro program recognized them as related to the same single-crystal domain. For example, eight groups (3, 5, 6, 9, 10, 14, 16 and 18) were merged with group 1, whose size increased from 421 (as found by DAFi) to 1312 reflections after indexing (see Table 1 ▸). Further processing in CrysAlisPro revealed crystallographic parameters of the mineral olivine. The olivine crystallite is mosaic, and its nine slightly misaligned domains were recognized by DAFi separately, whereas CrysAlisPro , due to the higher tolerance (0.125 in this particular case), counted the whole crystallite as one domain. Thus, CrysAlisPro revealed the crystallographic data for seven independent single-crystal domains of three different minerals: three domains of phlogopite, three of chromite and one domain of olivine (Table 1 ▸).
Figure 4.

Reciprocal space representing SC-XRD data from a sample of basalt: (a) all reflections; (b) reflections of group 1 as found by the DAFi program; (c) reflections of group 1 extended by CrysAlisPro software.
Table 1. Results of the DAFi run on the data set collected from a sample of basalt.
| Lattice parameters | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group No. | Size of group found by DAFi | Size of group after indexing in CrysAlisPro † | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Minerals |
| 1 | 421 | 1312 | 4.7881 (7) | 6.0329 (10) | 10.3079 (2) | 90 | 90 | 90 | Olivine |
| 2 | 197 | 494 | 5.2989 (17) | 8.911 (3) | 9.755 (3) | 90 | 105.47 (3) | 90 | Phlogopite |
| 3 | 171 | Merged with group 1 | |||||||
| 4 | 137 | 372 | 8.425 (3) | 8.425 (3) | 8.425 (3) | 90 | 90 | 90 | Chromite |
| 5 | 107 | Merged with group 1 | |||||||
| 6 | 57 | Merged with group 1 | |||||||
| 7 | 82 | Merged with group 2 | |||||||
| 8 | 57 | Merged with group 3 | |||||||
| 9 | 38 | Merged with group 1 | |||||||
| 10 | 41 | Merged with group 1 | |||||||
| 11 | 25 | 140 | 8.425 (3) | 8.425 (3) | 8.425 (3) | 90 | 90 | 90 | Chromite |
| 12 | 28 | Merged with group 3 | |||||||
| 13 | 15 | 98 | 5.317 (8) | 8.907 (5) | 9.723 (9) | 90 | 105.35 (12) | 90 | Phlogopite |
| 14 | 16 | Merged with group 1 | |||||||
| 15 | 21 | 86 | 5.286 (9) | 8.970 (17) | 9.752 (19) | 90 | 106.0 (2) | 90 | Phlogopite |
| 16 | 20 | Merged with group 1 | |||||||
| 17 | 20 | 85 | 8.398 (6) | 8.398 (6) | 8.398 (6) | 90 | 90 | 90 | Chromite |
| 18 | 26 | Merged with group 1 | |||||||
Indexing performed with a tolerance of 0.125.
Example 2. Study of products of the reaction of yttrium and nitrogen in a double-sided laser-heated DAC at 50 GPa. A piece of yttrium was placed in the sample chamber of a BX90-type large X-ray aperture DAC (Kantor et al., 2012 ▸) equipped with Boehler–Almax-type diamonds with 250 µm culets. Molecular nitrogen was then loaded into the DAC using a high-pressure gas loading system. The sample was compressed to ∼50 GPa and laser-heated (λ = 1064 nm) to 2000 (200) K using the double-sided laser heating system operating at the P02.2 beamline at the PETRA III synchrotron. A single-crystal data set was collected at the same P02.2 beamline (λ = 0.2908 Å, beam size 1.8 × 2 µm FWHM, acquisition time 4 s, angular ω step 0.5o, 132 frames). See Aslandukov et al. (2021 ▸) for more experimental details.
The peak hunting procedure in CrysAlisPro found 68 846 reflections. Since this high-pressure experiment was conducted in a DAC, a lot of undesired reflections from diamonds, the pressure-transmitting medium, the material of the gasket and other artifacts were present in the data set. Therefore, a procedure of ‘advanced filtering’ was applied to remove such reflections before the execution of the DAFi program. To realize such a ‘clean-up’, a special script was written by E. Koemets and M. Bykov, and then incorporated into the CrysAlisPro software. After the filtering, 44 312 reflections were left out of 68 846. The size of the DAFi input data set (‘DAFi input peak table’) was still huge. In such cases, it is more reasonable to search for several strongly diffracting domains of different phases than for all single-crystal domains. A search for ten groups in 44 312 reflections took the DAFi program 5 min 25 s.
The results of the search are shown in Fig. 5 ▸ and Table 2 ▸. It appeared that all ten groups of reflections belong to the same phase. Each group was indexed independently in CrysAlisPro [see Table 2 ▸ and Figs. 5 ▸(b) and 5 ▸(c) as an example], and the crystal structure of the phase (identified as Y5N14) was solved and refined for each of its single-crystal domains (Aslandukov et al., 2021 ▸) (e.g. for domain 6, the integration led to R int = 6.47%; based on 597 independent reflections, the structure of Y5N14 was solved and refined to R 1 = 4.88%). Note that the unusual stoichiometry of the Y5N14 phase was not known initially and was determined as a result of the crystal structure solution and refinement using the standard crystallographic software OLEX2 (Dolomanov et al., 2009 ▸), considering that the elements present in the system were known. In the example of Y5N14, only a piece of yttrium and nitrogen were loaded into the DAC, thus limiting the set of possible elements (Y, N) in the new compound. Other possible elements (for example, C from the diamond anvils, Re from the gasket or other impurities in the initial sample) would have to be taken into consideration in the case of unsatisfactory structure refinement (which was not the case for Y5N14).
Figure 5.

Reciprocal space representing SC-XRD data from a sample of Y+N2 in a DAC: (a) all reflections (cyan reflections are those filtered after applying ‘advanced filtering’); (b) reflections of group 1 belonging to the first Y5N14 domain found by the DAFi program; (c) reflections of group 1 belonging to the Y5N14 domain extended by CrysAlisPro ; (d) reflections of ten groups (1 through 10) belonging to ten Y5N14 domains marked by ten different colors.
Table 2. Results of the DAFi run on the data set collected from a sample of Y+N2 in a DAC at 50 GPa.
| Lattice parameters | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group No. | Size of group found by DAFi | Size of group after indexing in CrysAlisPro † | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Phase |
| 1 | 617 | 1286 | 8.4595 (3) | 8.4595 (3) | 4.7032 (5) | 90 | 90 | 90 | Y5N14 |
| 2 | 590 | 1044 | 8.4788 (6) | 8.4788 (6) | 4.6883 (3) | 90 | 90 | 90 | Y5N14 |
| 3 | 499 | 954 | 8.4527 (8) | 8.4527 (8) | 4.7034 (11) | 90 | 90 | 90 | Y5N14 |
| 4 | 485 | 997 | 8.4459 (5) | 8.4459 (5) | 4.7182 (7) | 90 | 90 | 90 | Y5N14 |
| 5 | 531 | 1199 | 8.4538 (4) | 8.4538 (4) | 4.711 (2) | 90 | 90 | 90 | Y5N14 |
| 6 | 483 | 1095 | 8.4737 (5) | 8.4737 (5) | 4.6922 (4) | 90 | 90 | 90 | Y5N14 |
| 7 | 478 | 1011 | 8.4710 (5) | 8.4710 (5) | 4.6977 (4) | 90 | 90 | 90 | Y5N14 |
| 8 | 375 | 984 | 8.4690 (5) | 8.4690 (5) | 4.7047 (19) | 90 | 90 | 90 | Y5N14 |
| 9 | 361 | 823 | 8.4606 (5) | 8.4606 (5) | 4.7065 (3) | 90 | 90 | 90 | Y5N14 |
| 10 | 308 | 935 | 8.4677 (9) | 8.4677 (9) | 4.699 (3) | 90 | 90 | 90 | Y5N14 |
Indexing performed with a tolerance of 0.05.
The DAFi program could have been run to find more domains. However, this would have made sense only if there were other phases in the sample. In this particular case, a quick check of the powder diffraction pattern generated for the whole data set showed no extra reflections apart from the found phase; therefore there was no reason to continue the search.
4. Summary
Existing indexing algorithms for single-crystal data analysis implemented in available crystallographic programs have no proven record of application to SC-XRD data processing from a multiphase mixture of microcrystalline samples. Superposition of numerous reflections originating from a large number of single-crystal domains of the same and/or different (especially unknown) phases precludes the sorting of reflections coming from individual domains, making their automatic indexing impossible. The DAFi algorithm presented in this work is designed for a quick search for subsets of reflections from individual domains in a whole set of SC-XRD data from a seemingly polycrystalline sample. Further indexing of all found subsets can be easily performed in one click using widely accessible crystallographic packages such as CrysAlisPro . The fact that the algorithm presented above neither requires a priori crystallographic information nor is limited by the number of the various phases and their individual domains makes DAFi a powerful software tool to be used for studies of multiphase polycrystalline and microcrystalline (powder) materials. It has been shown to be especially valuable for the analysis of single-crystal diffraction data from products of chemical reactions being realized in laser-heated DACs. Such data are always very complex due to (i) the presence of undesired reflections from diamond anvils and gaskets, and other technical and diffraction artifacts (e.g. ‘bad’ or ‘saturated’ detector pixels, or reflections from the body of the DAC itself), and (ii) the limited opening angle of DACs, which shadows a part of the Ewald sphere. To our knowledge, there are no existing software tools capable of finding the domains of unknown phases in such a complicated XRD data set as in example 2. The DAFi program tackles the task within a few minutes and finds several strongly diffracting domains, so that their XRD patterns can be indexed, the data integrated, and the crystal structures solved and refined. The high performance of the proposed algorithm allows the use of this program for online processing of the XRD data directly during experiments at synchrotron facilities.
The DAFi program is not designed to be effective with non-merohedral twins, where a large fraction of reflections are overlapped, while some of them overlap only partially or do not overlap. Once DAFi finds the reflection group, the program removes it from consideration for the next iterations. If reflections do not overlap, DAFi finds two separate reflection groups which can be processed afterwards by the user. If reflections overlap partially, DAFi finds two separate reflection groups; however, the first group would contain all reflections of the first crystal in a twin, while the second group would contain only non-overlapped reflections of the second one. If a large number of reflections overlap, the second group most likely will not be found.
The current version of the DAFi program does not find all reflections belonging to a particular single-crystal domain, as the algorithm searches for rows of at least three reflections along a certain direction, so that single reflections or those which are only two in a row are overlooked. Also, several groups of reflections can be found to belong to the same domain, as in the 3D reciprocal space the algorithm searches for rows of reflections in only one direction. These technical peculiarities of the program are not crucial for further data processing, as the input and output format of the DAFi program are compatible with the CrysAlisPro software. Moreover, the input and output formats of the DAFi program could be adapted to users’ needs and made to be compatible with other crystallographic software.
5. Distribution
The DAFi program and its documentation can be downloaded from https://github.com/AsMaNick/Domain-Auto-Finder/.
Acknowledgments
The authors thank D. Laniel for testing the program, reporting bugs and giving useful hints for further improvement. Open access funding enabled and organized by Projekt DEAL.
Funding Statement
ND and LD thank the Federal Ministry of Education and Research, Germany (BMBF, grant No. 05K19WC1), and the Deutsche Forschungsgemeinschaft (DFG projects DU 954-11/1, DU 393-9/2, DU 393-13/1) for financial support. ND also thanks the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009 00971).
References
- Aslandukov, A., Aslandukova, A., Laniel, D., Koemets, I., Fedotenko, T., Yuan, L., Steinle-Neumann, G., Glazyrin, K., Hanfland, M., Dubrovinsky, L. & Dubrovinskaia, N. (2021). J. Phys. Chem. C, 125, 18077–18084.
- Aslandukova, A., Aslandukov, A., Yuan, L., Laniel, D., Khandarkhaeva, S., Fedotenko, T., Steinle-Neumann, G., Glazyrin, K., Dubrovinskaia, N. & Dubrovinsky, L. (2021). Phys. Rev. Lett. 127, 135501. [DOI] [PubMed]
- Blum, M., Floyd, R. W., Pratt, V., Rivest, R. L. & Tarjan, R. E. (1973). J. Comput. Syst. Sci. 7, 448–461.
- Bykov, M., Chariton, S., Bykova, E., Khandarkhaeva, S., Fedotenko, T., Ponomareva, A. V., Tidholm, J., Tasnádi, F., Abrikosov, I. A., Sedmak, P., Prakapenka, V., Hanfland, M., Liermann, H. P., Mahmood, M., Goncharov, A. F., Dubrovinskaia, N. & Dubrovinsky, L. (2020). Angew. Chem. Int. Ed. 59, 10321–10326. [DOI] [PMC free article] [PubMed]
- Bykov, M., Fedotenko, T., Chariton, S., Laniel, D., Glazyrin, K., Hanfland, M., Smith, J. S., Prakapenka, V. B., Mahmood, M. F., Goncharov, A. F., Ponomareva, A. V., Tasnádi, F., Abrikosov, A. I., Bin Masood, T., Hotz, I., Rudenko, A. N., Katsnelson, M. I., Dubrovinskaia, N., Dubrovinsky, L. & Abrikosov, I. A. (2021). Phys. Rev. Lett. 126, 175501. [DOI] [PubMed]
- Bykova, E., Bykov, M., Černok, A., Tidholm, J., Simak, S. I., Hellman, O., Belov, M. P., Abrikosov, I. A., Liermann, H.-P., Hanfland, M., Prakapenka, V. B., Prescher, C., Dubrovinskaia, N. & Dubrovinsky, L. (2018). Nat. Commun. 9, 4789. [DOI] [PMC free article] [PubMed]
- Bykova, E., Dubrovinsky, L., Dubrovinskaia, N., Bykov, M., McCammon, C., Ovsyannikov, S. V., Liermann, H. P., Kupenko, I., Chumakov, A. I., Rüffer, R., Hanfland, M. & Prakapenka, V. (2016). Nat. Commun. 7, 10661. [DOI] [PMC free article] [PubMed]
- Ceppatelli, M., Scelta, D., Serrano–Ruiz, M., Dziubek, K., Morana, M., Svitlyk, V., Garbarino, G., Poręba, T., Mezouar, M., Peruzzini, M. & Bini, R. (2022). Angew. Chem. 134, e20211419. [DOI] [PMC free article] [PubMed]
- Cormen, T. H., Leiserson, C. E., Rivest, R. L. & Stein, C. (2001). Introduction to Algorithms, Section 8.3, pp. 170–173. Cambridge: MIT Press/McGraw-Hill.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Dubrovinskaia, N. & Dubrovinsky, L. (2018). Phys. Scr. 93, 062501.
- Jimenez-Melero, E., van Dijk, N. H., Zhao, L., Sietsma, J., Wright, J. P. & van der Zwaag, S. (2011). Mater. Sci. Eng. A, 528, 6407–6416.
- Kantor, I., Prakapenka, V., Kantor, A., Dera, P., Kurnosov, A., Sinogeikin, S., Dubrovinskaia, N. & Dubrovinsky, L. (2012). Rev. Sci. Instrum. 83, 125102. [DOI] [PubMed]
- Klee, V. (1977). Am. Math. Mon. 84, 284–285.
- Koemets, E. (2020). Dissertation, University of Bayreuth, Germany.
- Laniel, D., Winkler, B., Bykova, E., Fedotenko, T., Chariton, S., Milman, V., Bykov, M., Prakapenka, V., Dubrovinsky, L. & Dubrovinskaia, N. (2020). Phys. Rev. B, 102, 134109. [DOI] [PubMed]
- Laniel, D., Winkler, B., Fedotenko, T., Pakhomova, A., Chariton, S., Milman, V., Prakapenka, V., Dubrovinsky, L. & Dubrovinskaia, N. (2020). Phys. Rev. Lett. 124, 216001. [DOI] [PubMed]
- Lauridsen, E. M., Schmidt, S., Suter, R. M. & Poulsen, H. F. (2001). J. Appl. Cryst. 34, 744–750.
- Ludwig, W., Reischig, P., King, A., Herbig, M., Lauridsen, E. M., Johnson, G., Marrow, T. J. & Buffière, J. Y. (2009). Rev. Sci. Instrum. 80, 033905. [DOI] [PubMed]
- Moscicki, M., Kenesei, P., Wright, J., Pinto, H., Lippmann, T., Borbély, A. & Pyzalla, A. R. (2009). Mater. Sci. Eng. A, 524, 64–68.
- Schmidt, S. (2014). J. Appl. Cryst. 47, 276–284.
- Sørensen, H. O., Schmidt, S., Wright, J. P., Vaughan, G. B. M., Techert, S., Garman, E. F., Oddershede, J., Davaasambuu, J., Paithankar, K. S., Gundlach, C. & Poulsen, H. F. (2012). Z. Kristallogr. 227, 63–78.
- Wejdemann, C. & Poulsen, H. F. (2016). J. Appl. Cryst. 49, 616–621. [DOI] [PMC free article] [PubMed]
- Wright, J. P. (2006). ImageD11, https://github.com/FABLE-3DXRD/ImageD11/.