

Abstract
We assessed the activities of 24 different antiviral compounds against smallpox (two strains of variola major and one of variola minor), monkeypox, vaccinia and cowpox viruses by a neutral red uptake assay. To establish assay parameters, we examined viral replication and its inhibition at various times postinfection and at several multiplicities of infection. Drugs were selected to target a range of functions involved in viral replication. Eight compounds (cidofovir, cyclic HPMPC (cHPMPC), HPMPA, ribavirin, tiazofurin, carbocyclic 3-deazaadenosine, 3-deazaneplanocin A and DFBA (1-(2,4-difluorobenzyloxy)adenosine perchlorate)—a derivative of adenosine N1-oxide) inhibited the replication of all three variola strains and the other orthopoxviruses at drug concentrations within a pharmacologically achievable range. Two others (methisazone and bis-POM-PMEA) showed a lesser degree of antiviral effect, while the remainder were inactive. To examine possible naturally occurring drug resistance among a large number of variola isolates obtained from different geographical regions and at different times, we examined the sensitivity of 35 different strains of variola as well as other orthopoxviruses to a subset of three of the most active compounds: cidofovir, cHPMPC, and ribavirin. Preliminary data indicate that nearly all isolates appear to have similar drug sensitivities. These findings are currently being verified and expanded.
Keywords: Orthopox, Cidofovir, Smallpox, Monkeypox, Variola, Antiviral
1. Introduction
Smallpox was officially declared to have been eradicated from the world in 1980 by the 33rd World Health Assembly at the end of a decade-long intensive surveillance and vaccination campaign. Member states of the World Health Organization (WHO) subsequently agreed to either destroy remaining laboratory samples of variola (VAR) major (the more severe form with case fatality rates of 10–30%) and minor (the less severe form with case fatality rates of <5%) viruses, or to consolidate them into two locations: the Centers for Disease Control and Prevention (CDC) in Atlanta, GA, USA and the Russian State Research Center of Virology, Novosibirsk, Russia. These “official” stocks were originally scheduled to be destroyed, but in January 2002, the WHO Smallpox Advisory Group decided to recommend that the existing smallpox stocks be retained until satisfactory antiviral drugs and a safer vaccine are developed because there is still concern that smallpox might be employed as a weapon of terrorism or biowarfare. This fear stems in part from the increasing vulnerability of the entire human race to smallpox infection. Ironically, this vulnerability is a consequence of the successful smallpox eradication program. Because worldwide vaccination was discontinued in the late 1970s, few individuals under 30 years old are now adequately protected against smallpox. Older individuals who were vaccinated decades ago are believed to have little or no residual immunity. In addition, immunodeficient persons, such as those infected with the human immunodeficiency virus (HIV), are especially vulnerable to infection by orthopoxviruses, including the standard smallpox vaccine (vaccinia virus, VAC) and thus cannot be vaccinated (Fenner et al., 1988). Recent data indicate that it may be possible to engineer orthopoxviruses that are able to circumvent protection by the standard vaccine (Jackson et al., 2001). Clearly, the use of VAR as an agent of biowarfare or bioterrorism would have serious public health consequences (Lutwick et al., 2002). Finally, the complete genome of camelpox (CML) has been published (Gubser and Smith, 2002), showing that CML is closely related to VAR and thus may be a suitable starting point for an engineered bioweapon.
In contrast to vaccines, for which the protective effect is delayed, treatment with antiviral drugs of persons already infected with a virulent orthopoxvirus would provide immediate benefit. Because effective antiviral drugs were not available during the smallpox eradication campaign, their role in the control of an outbreak has not been evaluated. Early, mass administration of stockpiled drugs, given as soon as the attack agent is identified, could provide lifesaving therapy and should form part of the overall medical response. Depending upon the recipient’s stage of infection, treatment with an antiviral drug could attenuate illness, reduce infectivity and decrease mortality. In addition, prophylactic drug treatment could protect unvaccinated, front-line workers dealing with the first wave of patients and prevent secondary infection in other case contacts. In contrast to vaccines, antiviral medications could be administered to all exposed individuals, including those with underlying immunodeficiency disorders.
Unlike smallpox, which had no animal reservoir, monkeypox (MPX) cannot be eradicated. There is evidence that routine smallpox vaccination in the 1970s was able to reduce the incidence of illness after known MPX exposure by as much as 90% (Fenner et al., 1988). The high prevalence of HIV infection in central Africa today, with the accompanying increased risk of complications from VAC inoculation, makes the reintroduction of vaccination highly problematic. In the absence of vaccination, antiviral therapy would clearly be of benefit for the treatment of MPX patients and in controlling the spread of infection.
The successful eradication of smallpox as well as the remote location and small numbers of observed cases of MPX mean that there is little commercial market for drugs that specifically target these viruses. VAC is generally not even included in antiviral screening assays performed by the pharmaceutical industry as part of their drug discovery efforts. Recent events have brought the threat of bioterrorism to the forefront of public attention throughout the world. It has now become a priority to develop, license and stockpile sufficient quantities of antiviral drugs effective against virulent orthopoxviruses.
Smallpox vaccination, which uses live VAC, can cause serious and life-threatening complications including disseminated and progressive vaccinia infections. Vaccine complications are another potential target for antiviral therapy. Currently, the only approved treatment for such infections is the administration of vaccinia immune globulin (VIG). The efficacy of VIG treatment for these conditions has never been demonstrated. Treatment of smallpox vaccine adverse reactions with cidofovir was recently approved under an investigational new drug protocol. There has been no published research comparing the relative sensitivity of various orthopoxviruses to antiviral drugs in clinical use, partly because of biosafety considerations. A number of compounds have been shown to inhibit VAC replication, both in vitro and in vivo (reviewed in De Clercq, 2001), but it has never been demonstrated that the pattern of drug sensitivity of VAC is representative of the entire orthopoxvirus spectrum, including the most virulent agents. In particular, no benign virus has been established as a predictor of drug activity against VAR.
Our research therefore focused on three goals: (1) to identify antiviral drugs that inhibit VAR and MPX; (2) to test a diverse group of VAR isolates against some of the most promising antiviral compounds to look for variations in drug sensitivity; and (3) to identify a virus less virulent than VAR, that displays a similar pattern of drug sensitivities and could be used as a surrogate in drug screening. To accomplish these goals, we adapted a neutral red uptake assay to determine the antiviral efficacy against orthopoxviruses. We screened 24 antiviral drugs against a 1975 Bangladesh isolate of VAR major (VAR-BSH), the 1946 Japan Yamada isolate of VAR major (VAR-YAM), and the 1966 Brazil Garcia isolate of VAR minor (VAR-GAR), along with MPX, VAC, and CPX. The VAR-BSH and VAR-YAM viruses were chosen to represent viruses that caused the major form of smallpox as well as diverse years and geographical areas in which they were isolated. The complete genome sequence of VAR-BSH has also been published (Massung et al., 1994). VAR-GAR was chosen as a virus causing the minor form of smallpox. Five drugs that showed strong in vitro efficacy and that had different known modes of action were further tested against 35 additional isolates of VAR.
2. Materials and methods
2.1. Biological containment
VAR was handled only within the maximum-containment laboratory at the CDC, under Biosafety Level 4 (BSL-4) conditions. Personnel wore positive-pressure protective suits (ILC, Dover, Frederica, DE) equipped with high-efficiency particulate air (HEPA) filters and supplied with umbilical-fed air. All other orthopoxvirus work was done under BSL-3 containment at the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID), Frederick, MD.
2.2. Viruses
VAC Copenhagen, CPX Brighton, and MPX Zaire (V79-1-005-Scab) were obtained from the USAMRIID collection. All VAR strains were obtained from the CDC reference collection.
2.3. Cells
Vero 76 (ATCC CRL 1587), Vero E6 (VERO C1008, ATCC CRL 1586), LLC-MK2 (ATCC CCL 7) and BSC-40 (a derivative of ATCC CCL 26) were propagated in RPMI-1640 (Gibco), 10% fetal bovine serum (FBS), 1% glutamine, 12.5 ng/ml of fungizone and 50 μg/ml of gentamycin at 37 °C in a 5% CO2 atmosphere. The same medium with 2% FBS was used as replacement medium after viral infection of cells.
2.4. Growth and titration of viral stocks
Frozen chicken egg chorionic allentoic membrane (CAM) or scab material from the CDC smallpox repository were homogenized and used by Dr. Inger Damon, Poxvirus section, CDC, to inoculate BSC-40 cells. These seed stocks were then used to inoculate 150 cm2 cell culture flasks of BSC-40 cells at a multiplicity of infection (MOI) of 0.01. Infected cells were incubated for approximately 5 days until a cytopathic effect (CPE) of ≥4 was observed. Virus was harvested by scraping the cells into a small quantity of medium followed by three freeze–thaw cycles and sonication to disrupt intact cells. Cell debris was then pelleted by centrifugation at 3000×g for 10 min. Supernatants representing working viral stocks were stored in liquid nitrogen. Working stocks of all viruses used were titrated by serial 10-fold dilutions and inoculation of 0.1 ml into confluent monolayers of Vero E6 cells in six-well plates, in duplicate, for 2 h. Medium was added to 2 ml and plates were incubated for 3–5 days until plaques could be seen in the microscope. After incubation, 2 ml of crystal violet staining solution (1.3 mg/ml crystal violet, 5% ethanol, 30% formalin) was added to each well. Plates were incubated at room temperature for 20 min, excess stain was removed by gentle washing with water and plaques were counted visually.
2.5. Antiviral compounds
PMEA (adefovir, 9-[2-(phosphonomethoxy)ethyl]adenine), bis-POM-PMEA (adefovir dipivoxil, 9-[2-[[bis[(pivaloyloxy)-methoxy]phosphinyl]methoxy]ethyl]adenine), cidofovir (HPMPC, Vistide®, 1-[(S)-3-hydroxy-2-(phosphonomethoxy)-propyl]cytosine), cyclic cidofovir (1-[(S)-2-hydroxy-2-oxo-1-[4,2-dioxaphosphorinan-5-yl]methyl]cytosine) and HPMPA ((S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine) were provided by Dr. Norbert Bischofberger of Gilead Sciences, Inc., Foster City, CA. Brovavir (sorivudine, BV-araU), Lobucavir (cycovir, cygalovir, [1R(1α,2β,3α)]-9-[2,3-bis(hydroxymethyl)cyclobutyl]guanine), didanosine (ddI, Videx), stavudine (d4T, Zerit), efavirenz (Sustiva) and hydroxyurea (Hydrea) were provided by Bristol–Myers Squibb Pharmaceutical Research Institute, Princeton, NJ. Ribavirin (Virazole) was provided by Dr. Humberto Fernandez, ICN Pharmaceuticals, Costa Mesa, CA. 3-Deazaneplanocin A ((−)-9-[trans-2′-trans-3′-dihydroxy-4′-hydroxymethyl-cyclopent-4′-enyl]-3-deazaadenine, C3-Npc A) was obtained from Dr. John S. Driscoll, National Cancer Institute, Bethesda, MD. Carbocyclic 3-deazaadenosine (3-deazaaristeromycin, C-ca3-Ado) (Montgomery et al., 1982) and DFBA (1-(2,4-difluorobenzyloxy)-adenosine perchlorate) (Kwong et al., 1998) were obtained from Dr. John A. Secrist III, Southern Research Institute, Birmingham, AL. Acyclovir (Zovirax), methisazone (Marboran) and zidovudine (Retrovir, AZT) were provided by Dr. Karen Biron, Glaxo Wellcome, Research Triangle Park, NC. Saquinavir (Fortovase) and ddC (zalcitabine) were obtained from Hoffmann-La Roche Inc., Nutley, NJ. Ritonavir (Norvir) was obtained from Abbott Laboratories, Abbott Park, IL. Nelfinavir (Viracept) was obtained from Agouron Pharmaceuticals Inc., La Jolla, CA. Tiazofurin (2-β-d-ribofuranosylthiazole-4-carboxamide) and d4C (2′,3′-dideoxydidehydrocytidine) were from the USAMRIID repository.
2.6. Neutral red uptake assay
Stocks of antiviral compounds were made by dissolving each compound in DMSO to a concentration of 20 mg/ml. Drugs were then diluted to 400 μg/ml in RPMI-1640, serially diluted three-fold in RPMI-1640, and 50 μl added to 96-well microtiter plates of confluent Vero 76 or LLC-MK2 cells already containing 100 μl of medium. At each drug concentration, three wells were infected with 105 pfu/well (MOI=0.1) of orthopoxvirus in 50 μl of medium, while three were left uninfected for toxicity determination (50 μl of medium added to each well). Plates were examined daily, and were stained once virus-infected, untreated cells showed CPE of ≥4. Fifty microliter neutral red (1.11 mg/ml) was added to the medium to give a final concentration of 0.22 mg/ml, and cells were returned to the incubator for 90 min. The medium was removed, the wells were rinsed twice with buffered saline solution, and retained stain was solubilized by adding 100 μl of developing solution (50% ethanol, 5 mM ammonium phosphate (NH4H2PO4), pH 3.5). Plates were rocked for 30 min at 150 rpm, and the optical density (OD) of the wells at a wavelength of 450 nm was measured on a plate reader. The data were graphed and analyzed by using the four parameter–logit curve fit option of a curve-fitting program (Molecular Devices, Menlo Park, CA) to determine the 50% inhibitory and cytotoxic drug concentrations. Drugs with any detectable activity are shown.
3. Results
3.1. Viral growth and plaque morphology
As other investigators have noted, the various orthopoxviruses differed in their growth rate and plaque morphology (Mathew, 1970, Sheek et al., 1975). We found that VAC, probably because of its extensive adaptation to cell culture, grew rapidly to produce 2 mm plaques on monolayers of Vero 76, Vero E6 or BSC-40 cells within 3 days after infection. MPX and CPX took 4–5 days to produce plaques of similar size. All three of these viruses showed a mixed population of plaques, with 10–20% being much smaller than the majority. VAR produced only very small plaques after 5–6 days and showed low to moderate syncytia formation at high MOIs, depending on the strain examined.
3.2. Neutral red uptake assay
We tested several different MOIs and length of infection to attempt to establish standard assay parameters using the sensitivity of VAR-BSH to cidofovir on Vero cells (Table 1 ). We found that a higher MOI and a longer incubation time led to higher apparent IC50. On Day 5 postinfection, the IC50 values appeared to reach their maximum and remain relatively stable on Day 6. Similarly, the IC50 obtained at an MOI of 0.1 on Days 5 and 6 appeared similar to those for an MOI of 1 on the same days. Similar results were seen with cHPMPC and ribavirin (data not shown). We therefore set an MOI of 0.1 and an incubation period of 5 days as standard assay conditions. Finally, we have compared the IC50 values obtained using a variety of assays including neutral red uptake, plaque reduction and reporter gene activity and find good agreement between all three methods (manuscript in preparation).
Table 1.
Effects of MOI and period of infection on apparent IC50 values for cidofovir inhibition of VAR-BSH on Vero cells
| MOI | Day of postinfection |
|||
| Day 3 | Day 4 | Day 5 | Day 6 | |
| 0.01 | 2.1 | 2.9 | 10.9 | 10.3 |
| 0.1 | 5.2 | 8.0 | 8.1 | 8.2 |
| 1.0 | 6.1 | 11.8 | 13.8 | 14.1 |
3.3. Antiviral drug activity
In order to determine which viruses to use for drug screening, we tested 35 VAR isolates for sensitivity to three different antiviral drugs (cidofovir, cHPMPC, and ribavirin). Data were graphed as shown in Fig. 1 and are summarized in Table 2 . Antiviral activity is presented in terms of the 50% inhibitory drug concentration (IC50). Cidofovir had a mean IC50 value 12±1 cHPMPC showed an IC50 value of 17±2 μg/ml for Vero cells. Ribavirin was observed to have a value of 50±8. It should be noted that the relatively high observed IC50 values for ribavirin against V77-1605 and V70-222 are obtained from a single experiment and are most likely statistical anomalies. Additional repeats of these experiments are planned. VAR strains were isolated from geographically separated areas and at different times as shown in Fig. 2 . We concluded that the Garcia, Yamada and Bangladesh isolates represented a reasonable sampling of the available strains and would be appropriate for further testing against the larger panel of drugs.
Fig. 1.
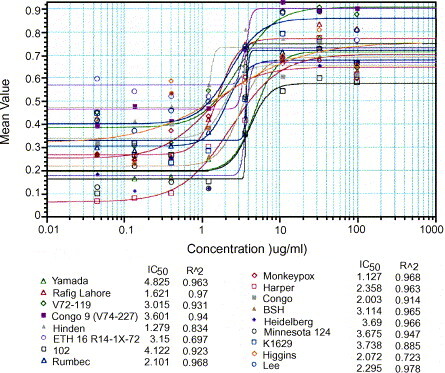
Representative data from a single experiment for cidofovir against various orthopoxviruses on Vero cells. Data shown are for a single replicate set of assays. IC50 values (μg/ml) for each strain are shown below the graph, as is the R2 fit of the points to the curve.
Table 2.
IC50 values (μg/ml) for cidofovir (CDV), cHPMPC and ribavirin against all VAR isolates tested on Vero cells
| VAR isolate | CDV | cHPMPC | Ribavirin | VAR isolate | CDV | cHPMPC | Ribavirin |
| Butlera | 6 ± 4 (3) | 6 ± 4 (3) | 28 ± 1 (3) | Kali-Muthu | 11 ± 0 (3) | ND | ND |
| Garciaa | 7 ± 3 (5) | 4 ± 1 (2) | 39 ± 12 (3) | Jee | 10 ± 6 (3) | 7 (1) | 47 ± 2 (2) |
| Minn124a | 5 ± 1 (3) | 9 ± 1 (2) | 32 (1) | Rafig Lahore | 12 ± 9 (4) | 18 ± 11 (3) | 30 ± 5 (3) |
| V68-59a | 13 ± 3 (4) | 20 (1) | 41 (1) | Rumbec | 10 ± 2 (5) | 16 (1) | ND |
| 102 | 23 ± 6 (5) | 38 (1) | 110 (1) | Shahzaman | 11 ± 1 (4) | 14 (1) | 15 (1) |
| 7124 | 20 ± 7 (5) | 26 ± 10 (3) | 36 ± 14 (3) | Somalia | 7 ± 2 (3) | 8 ± 2 (3) | 84 ± 45 (3) |
| 7125 | 12 ± 3 (4) | 24 ± 8 (4) | 37 ± 19 (3) | V70-46 | 7 ± 3 (4) | 13 ± 4 (3) | 45 ± 7 (3) |
| Bangladesh | 17 ± 4 (5) | 17 ± 6 (5) | 30 ± 8 (4) | V70-222 | 12 ± 5 (4) | 23 (1) | 221 (1) |
| Eth17 | 11 ± 8 (4) | 17 ± 9 (3) | 61 ± 25 (3) | V72-119 | 10 ± 5 (4) | 18 ± 3 ( 3) | 35 ± 18 (3) |
| Harper | 28 ± 13 (3) | ND | 40 (1) | V73-175 | 12 ± 2 (4) | 16 (1) | 17 (1) |
| Heidelberg | 15 ± 6 (3) | 14 ± 5 (3) | 43 ± 23 (3) | V73-225 | 5 ± 2 (3) | 9 (1) | 26 ± 6 (2) |
| Higgins | 14 ± 6 (5) | 17 ± 9 (3) | 48 ± 5 (3) | V74-227 | 10 ± 2 (7) | 9 ± 4 (3) | 54 ± 20 (3) |
| Hinden | 10 ± 3 (5) | 19 (1) | 33 (1) | V77-1605 | 19 ± 2 (4) | 22 (1) | 453 (1) |
| Horn | 11 ± 5 (4) | 12 ± 7 (3) | 28 ± 2 (3) | Yamada | 13 ± 9 (3) | 31 ± 23 (2) | 40 ± 6 (2) |
| Iran 2602 | 8 ± 2 (4) | 7 (1) | 11 (1) | VAR mean | 12 ± 1 | 17 ± 2 | 50 ± 8 |
| Juba | 14 ± 3 (3) | ND | ND | MPX | 11 ± 2 (6) | 21 ± 4 (6) | 11 ± 2 (6) |
| K1629 | 17 ± 7 (7) | 25 ± 17 (3) | 24 ± 8 (3) | CPX | 13 ± 3 (6) | 52 ± 11 (6) | >300 (6) |
MPX and CPX values for these compounds are shown for comparison. Numbers shown are mean values±S.E. of the mean for the indicated orthopoxviruses. Numbers in parentheses indicate the number of replicates (N). ND: not determined.
VAR minor strain.
Fig. 2.
Locations and dates of isolation of VAR strains used in this study. Dates are shown in parentheses and locations of original isolation are indicated by the arrows.
Twenty-four compounds targeting a variety of virus-specific functions were tested for inhibitory activity against three strains of VAR and three other orthopoxviruses by means of the neutral red uptake assay. Table 3 shows mean IC50s for active compounds tested in this study, and grouped by their molecular target, if known (De Clercq, 1993a). Drugs deemed inactive at the highest concentrations tested (100 μg/ml) are omitted from Table 3. Drug toxicity was assessed by the extent to which confluent monolayers of uninfected drug-treated cells actively took up neutral red dye. The most potent antiviral agents were those that produced a 50% inhibitory effect at the lowest concentrations, i.e. had the smallest IC50. The least toxic were those which had a high 50% cytotoxic concentration (TC50). The most clinically beneficial drugs were those that had a high ratio of TC50 to IC50 (the “therapeutic index” (TI)) (Table 4 ).
Table 3.
Inhibition of orthopoxvirus growth by antiviral compounds
| Drug target | VAR-BSH |
VAR-YAM |
VAR-GAR |
MPX |
CPX |
VAC |
Cytotoxicity |
|||||||||||||||||||||
| Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | |||||||||||||||
| DNA polymerase | ||||||||||||||||||||||||||||
| bis-POM-PMEA | 84.2 | >100 | >100 | >100 | 90.0 | 85.0 | >100 | >100 | 75.5 | 86.0 | 44.0 | 66.2 | >100 | >100 | ||||||||||||||
| HPMPA | 12.6 | 6.1 | 11.4 | 6.8 | 6.6 | 4.6 | 10.9 | 9.2 | 8.4 | 7.7 | 8.1 | 8.0 | >100 | >100 | ||||||||||||||
| Cidofovir | 9.5 | 3.2 | 12.1 | 2.7 | 4.5 | 2.5 | 6.7 | 3.7 | 7.5 | 1.2 | 8.1 | 4.3 | >100 | >100 | ||||||||||||||
| cHPMPC | 25.3 | 20.3 | 28.2 | 6.1 | 7.9 | 5.9 | 12.3 | 3.4 | 26.9 | >100 | 18.1 | 4.4 | 80.0 | >100 | ||||||||||||||
| IMP dehydrogenase | ||||||||||||||||||||||||||||
| Ribavirin | 18.4 | 2.1 | 15.8 | 3.4 | 17.0 | 3.6 | 5.9 | 4.1 | 30.6 | 10.6 | >100 | 23.9 | >100 | >100 | ||||||||||||||
| Tiazofurin | 22.6 | 5.5 | 21.4 | 3.9 | 19.8 | 6.1 | 20.0 | 5.2 | 34.6 | 11.0 | >100 | 33.3 | 90.0 | >100 | ||||||||||||||
| SAH hydrolase | ||||||||||||||||||||||||||||
| C-ca3-Ado | 1.2 | 1.6 | 5.3 | 1.4 | 1.6 | 4.4 | 0.4 | 0.8 | >100 | 25.2 | 1.2 | 3.4 | >100 | >100 | ||||||||||||||
| C3-Npc A | 0.03 | 0.47 | 0.04 | 0.24 | 0.04 | 0.45 | 0.01 | 0.05 | >100 | >100 | 0.02 | 0.05 | >100 | >100 | ||||||||||||||
| Other | ||||||||||||||||||||||||||||
| DFBA | 0.16 | >100 | 0.14 | >100 | 0.14 | >100 | 0.95 | >100 | 0.71 | >100 | 1.18 | >100 | 75.0 | >100 | ||||||||||||||
| Methisazone | 34.6 | 41.2 | 39.0 | 43.3 | 21.6 | 44.1 | 66.2 | 80.4 | 49.2 | 45.2 | 45.8 | 52.0 | >100 | >100 | ||||||||||||||
Inhibition of orthopoxvirus replication in Vero and cells by a series of antiviral compounds, as measured by neutral red uptake assay. Values are IC50 for each virus or TC50 for cytotoxicity (μg/ml).
Table 4.
Therapeutic indices (TI) of active compounds, expressed as the ratio of IC50 to TC50
| Drug | VAR-BSH |
VAR-YAM |
VAR-GAR |
MPX |
CPX |
VAC |
||||||
| Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | Vero | LLC-MK2 | |
| bis-POM-PMEA | 1.2 | 1.0 | 1.0 | 1.0 | 1.1 | 1.2 | 1.0 | 1.0 | 1.3 | 1.2 | 2.3 | 1.5 |
| HPMPA | 7.9 | 16.4 | 8.8 | 14.7 | 15.2 | 21.7 | 9.2 | 10.9 | 11.9 | 13.0 | 12.3 | 12.5 |
| Cidofovir | 10.5 | 31.3 | 8.3 | 37.0 | 22.2 | 40.0 | 14.9 | 27.0 | 13.3 | 83.3 | 12.3 | 23.3 |
| cHPMPC | 3.2 | 4.9 | 2.8 | 16.4 | 10.1 | 16.9 | 6.5 | 29.4 | 3.0 | 1.0 | 4.4 | 22.7 |
| Ribavirin | 5.4 | 47.6 | 6.3 | 29.4 | 5.9 | 27.8 | 16.9 | 24.4 | 3.3 | 9.4 | 1.0 | 4.2 |
| Tiazofurin | 4.0 | 18.2 | 4.2 | 25.6 | 4.5 | 16.4 | 4.5 | 19.2 | 2.6 | 9.1 | 1.0 | 3.0 |
| C-ca3-Ado | 83.3 | 62.5 | 18.9 | 71.4 | 62.5 | 22.7 | 250 | 125 | 1.0 | 4.0 | 83.3 | 29.4 |
| C3-Npc A | 3333 | 213 | 2500 | 417 | 2500 | 222 | 10000 | 2000 | 1.0 | 1.0 | 5000 | 2000 |
| DFBA | 469 | 1.0 | 536 | 1.0 | 536 | 1.0 | 1250 | 1.0 | 78.9 | 1.0 | 63.6 | 1.0 |
| Methisazone | 3.0 | 2.4 | 2.6 | 2.3 | 4.6 | 2.3 | 1.5 | 1.2 | 2.0 | 2.2 | 2.2 | 1.9 |
Note: For those drugs which have a cytotoxicity >100 μg/ml, the minimum TI is shown and may be larger.
3.3.1. DNA polymerase inhibitors
Several nucleoside phosphonates significantly inhibited all orthopoxviruses tested (Table 3). Cidofovir (HPMPC) and HPMPA were especially active against the panel of viruses (having mean IC50 values of 5.5±0.95 and 8.4±0.67 μg/ml and TIs of 18.2 and 11.9, respectively). Cyclic HPMPC was moderately effective, inhibiting all viruses tested with a mean IC50 value of 21.6±7.6 μg/ml. Neither PMEA nor bis-POM-PMEA was effective at inhibiting the viruses tested, though bis-POM-PMEA showed some moderate activity against VAC (IC50=55.1 μg/ml). No other test compounds of this group (acyclovir, brovavir, lobucavir, didanosine, ddC, or d4C) showed any activity.
3.3.2. Inosine mononphosphate (IMP) dehydrogenase inhibitors
Two IMP dehydrogenase inhibitors, ribavirin and tiazofurin, inhibited the replication of all the orthopoxviruses tested. VAR and MPX were more sensitive than the other viruses to both drugs. However, drug sensitivities varied considerably, depending on the virus and cell line used. Ribavirin had mean IC50 values of 31.3±14.1 μg/ml on Vero cells and 8.0±3.4 μg/ml on LLC-MK2 cells while tiazofurin had mean IC50 values of 36.4±12.9 on Vero cells and 10.8±4.6 on LLC-MK2 cells. It was noteworthy to learn the relative insensitivity of VAC to both of these drugs. We also found that both drugs had three- to nine-fold greater activity in LLC-MK2 cells as compared to Vero 76, with the exception of MPX sensitivity to ribavirin. Despite the apparent low in vitro efficacy of ribavirin against CPX, it can protect CPX-infected mice (Smee et al., 2000a).
3.3.3. S-Adenosylhomocysteine (SAH) hydrolase inhibitors
The SAH hydrolase inhibitors, carbocyclic-3-deazaadenosine and 3-deazaneplanocin A, were highly active against VAR and the other viruses tested, with the notable exception of CPX which for unknown reasons was almost completely insensitive to these drugs. Their IC50 values were the lowest of any class of compounds, while their toxic levels were sufficiently elevated to give a high TI (Table 4). This suggests that SAH hydrolase inhibitors might exert a potent antiviral effect in vivo without producing significant side effects. In support of this conclusion and despite the low in vitro activity, these same drugs are at least partially effective in treating mice infected with CPX virus (R. Baker and M. Bray, unpublished data).
3.3.4. Reverse transcriptase (RT) and protease inhibitors
As expected, the RT inhibitors (stavudine, efavirenz, and zidovudine) were inactive against orthopoxviruses, as these viruses do not possess RT activity. In addition, all HIV protease inhibitors tested (saquinavir, ritonavir, and nelfinavir) were inactive.
3.3.5. Other compounds
Methisazone (Marboran) showed only minimal activity against the test viruses (mean IC50=469±4.3 μg/ml). Hydroxyurea was inactive. DFBA showed high activity against all viruses tested (mean IC50=0.55±0.19 μg/ml) as well as high TIs, but only for Vero cells—this compound showed no activity on LLC-MK2 cells against any test virus.
4. Discussion
4.1. Neutral red uptake assay
To determine antiviral efficacy in this study, we used a neutral red uptake assay to measure cell viability. We attempted to speed the process of antiviral drug discovery by developing simple, rapid screening assays for the evaluation of new compounds against orthopoxvirus infections. Drug screens based on cellular uptake of neutral red dye as the index of cell viability have been in use for many years, and are employed with many different viruses and cell lines (Finter, 1969). Healthy, uninfected cells will take up neutral red dye through pinocytosis, while cells infected with a cytopathic virus such as the orthopoxviruses will not. The unabsorbed dye can then be washed away, and the absorbed dye solubilized by an ethanol solution. Thus, the amount of dye taken into cells is inversely proportional to the amount of viral growth. We currently use such assays for measuring antiviral activity of compounds against Ebola and Marburg viruses. In this report we have attempted to standardize the assay conditions for orthopoxviruses, and to examine the effects of variations on these parameters. We conclude that an MOI of 0.1 and a 5-day period of infection to be the standard conditions used for this study, as these are the minimum time and MOI necessary to obtain consistent IC50 values.
The neutral red uptake assay is more efficient than the plaque reduction method, since eight sequential dilutions of drug could be tested in triplicate for both antiviral activity and cytotoxicity on a single 96-well plate. Many combinations of drugs and viruses could thus be studied in a single batch, and the data for each plate could be acquired and analyzed within minutes using a computer-linked reader. In addition, this method alleviated the ambiguity of variable plaque size and the potential subjectivity of plaque counting by a human operator. It should be noted that cytotoxicity in this assay is measured in confluent cell monolayers. Thus, compounds exhibiting little or no toxicity in this assay may still show considerable toxicity when measured in a cell proliferation assay.
4.2. DNA polymerase inhibitors
We identified a number of antiviral compounds with differing mechanisms of action that strongly inhibited the replication of VAR and three other orthopoxviruses. We had expected that certain DNA polymerase inhibitors would be effective as some of them had already been reported to inhibit VAC (reviewed in De Clercq, 2001). There are two major classes of nucleoside analogs that interfere with viral DNA polymerase activity. The first group requires an initial phosphorylation by a viral enzyme to become a substrate for cellular kinases (De Clercq, 1993a). The drug acyclovir, as well as others, are members of this group. These compounds are taken up by all cells, but are phosphorylated only by the thymidine kinase (TK) of herpes simplex viruses or by the nucleoside kinase of CMV. Subsequent conversion of the compound to its di- and triphosphate form by host cell enzymes leads to competition for the active site of the viral DNA polymerase, incorporation of the drug into viral DNA, and chain disruption or termination. As expected, we found that three nucleoside analogs in this category (acyclovir, brovavir and lobucavir) (De Clercq, 1993b) failed to inhibit orthopoxvirus replication, indicating that either the orthopoxvirus TK (product of the BSH VAR L2R gene (GenBank accession no. L22579)) does not recognize them as a substrate for phosphorylation or that the phosphorylated drugs are not a substrate for the viral polymerase.
The second group of DNA polymerase inhibitors consists of nucleoside analogs containing a phosphonyl group, which serves as a substrate for phosphorylation by cellular enzymes. These compounds can potentially be taken up and phosphorylated in all cells; their therapeutic usefulness and safety are based on the much higher affinity of their diphosphate form for the viral than the cellular DNA polymerase. Cidofovir diphosphate, for example, inhibits the DNA polymerase of CMV at a 10-fold lower concentration than that needed to inhibit human DNA polymerase α to a comparable extent (Hitchcock et al., 1996). The intracellular form of the compound, cidofovir 5′-diphosphate, forms an adduct with choline, from which it slowly dissociates, giving the drug a very long intracellular half-life (t 1/2=17–65 h) (Hitchcock et al., 1996). Cidofovir has potent in vitro and in vivo activity against a broad spectrum of herpesvirus infections. De Clercq et al. (1987) demonstrated that cidofovir, HPMPA and other phosphonylmethoxyalkyl- and phosphonylethoxyalkyl-nucleotide analogs also inhibit VAC replication in vitro. We have now extended the known range of antiviral activity of this class of agents, by showing that cidofovir and other nucleoside phosphonates were active in vitro against a broad panel of orthopoxviruses. Work in this and other laboratories has shown cidofovir to be active in vivo against poxviruses as well, in both the CPX and VAC mouse models (Bray et al., 2000a, Neyts and De Clercq, 1993, Smee et al., 2001, Smee et al., 2000b). Cidofovir is in clinical use in the United States for the treatment of CMV retinitis in AIDS patients (Khare and Sharland, 2001, Salmon-Ceron, 2001) and the pharmacokinetics of cidofovir treatment are well established (reviewed in Cundy, 1999). Effective treatment is possible with infrequent dosing. A typical schedule for treatment of a chronic viral infection consists of weekly or biweekly i.v. injections to maintain therapeutic tissue levels but this would not be required for an acute orthopoxvirus infection. The requirement for i.v. administration is a definite disadvantage of cidofovir. Various prodrugs which have been chemically modified to improve their oral bioavailability would be easier to administer and are currently under investigation.
As shown in Table 3, HPMPA was active against the orthopoxviruses tested and has activity in several other systems as well (De Clercq, 2001). HPMPA is closely related to cidofovir and functions to inhibit viral replication through the same mechanism. Because cidofovir is already licensed for use in the United States and thus is much more likely to actually be used for the treatment of poxvirus infections, HPMPA is not likely to be put into actual clinical use in the United States. Both PMEA (adefovir) and bis-POM-PMEA (adefovir dipivoxil) have proven useful in the inhibition of retroviruses (Noble and Goa, 1999) and hepatitis B virus, though they did not appear to have significant activity against poxviruses. While cHPMPC has shown activity against CMV in immunosuppressed patients and now at least some activity against poxviruses, development of this drug has been put on hold by the manufacturer (Hoffman and Skiest, 2000).
4.3. IMP dehydrogenase inhibitors
Ribavirin and tiazofurin inhibited all of the orthopoxviruses tested, and were especially active against VAR. These drugs inhibit inosine monophosphate dehydrogenase, the rate limiting enzyme in GMP biosynthesis resulting in depleted intracellular pools of guanosine, interference with transcription of viral messenger RNA and with 5′ cap formation (Jordan et al., 1999). There is also evidence that ribavirin inhibits the helicase activity associated with viral RNA polymerase (Rankin et al., 1989). In hepatitis C and other RNA virus replication, ribavirin is incorporated into the viral RNA during replication, and causes an increase in deleterious mutations (Crotty et al., 2000, Maag et al., 2001). Both ribavirin and tiazofurin have broad-spectrum antiviral activity (Kirsi et al., 1983), but only ribavirin is licensed for clinical use. The drug is currently approved in the United States only for the aerosol treatment of respiratory syncytial virus infection, and orally for the treatment of hepatitis C in combination with interferon. Ribavirin is being used to treat other viral infections in humans including Lassa fever and hantaviruses under investigational protocols. There is little clinical experience in using ribavirin for the treatment of DNA virus infections. Ribavirin inhibited VAC replication in vitro (Katz et al., 1976, Kirsi et al., 1984) and was active in murine tailpox models, including infections by recombinant VAC (De Clercq et al., 1976, Tignor et al., 1992). Combination chemotherapy with ribavirin and acyclovir improves the outcome of HSV infection in experimental animals (Shishkov and Pancheva, 1990, Pancheva, 1991), while combination therapy of ribavirin and cidofovir increases the survival of mice infected with CPX over either drug alone (Smee et al., 2000a). In the only reported case of ribavirin treatment of a human orthopoxvirus infection (a case of progressive vaccinia), the drug was used in combination with VIC, making it difficult to judge its effect alone (Kesson et al., 1997). We conclude that ribavirin might be useful in the treatment of VAR or MPX infections, especially in combination with other drugs.
4.4. SAH hydrolase inhibitors
We also found that two adenosine analogs (C-ca3-Ado and C3-Npc A) which have been shown to inhibit a host cell enzyme, SAH hydrolase, strongly inhibited orthopoxviral replication. SAH is a product of cellular methylation reactions, which use S-adenosylmethionine (SAM) as the methyl group donor. Inhibition of SAH hydrolase results in the intracellular accumulation of SAH, blocking cellular and viral transmethylation reactions through feedback inhibition. Viral transmethylases tend to be much more readily inhibited than their cellular counterparts by rising SAR levels (De Clercq, 2001). The use of SAH hydrolase inhibitors as antiviral drugs is therefore a reasonable strategy. These compounds have broad-spectrum antiviral activity and they are active against VAC in vitro (De Clercq, 1987, De Clercq and Montgomery, 1983). Interestingly, these compounds show little or no detectable activity against CPX in vitro. Despite the dramatic difference in activity in cell culture for these drugs against VAC and CPX, both C-ca3-Ado and C3-Npc A have a modest protective effect on either VAC or CPX-infected mice in vivo (R. Baker and M. Bray, unpublished data). These drugs have also shown strong protective activity in a lethal murine model of Ebola virus infection (Huggins et al., 1999, Bray et al., 2000b, Bray et al., 2002). Despite extensive laboratory investigation however, SAH hydrolase inhibitors have not been tested in humans, chiefly because of concerns about possible drug toxicity. These compounds may prove to be uniquely beneficial for treating severe, rapidly progressive viral infections, such as smallpox or Ebola hemorrhagic fever, in which the risk of death outweighs the risk of drug toxicity.
4.5. Other compounds
Adenosine N1-oxide (ANO) inhibits VAC virus replication in vitro by blocking the translation of viral mRNAs (Kane and Shuman, 1995) through an as yet undetermined mechanism. While we saw little activity of ANO against the orthopoxviruses used in this study (data not shown), the ANO derivative DFBA showed significant activity against all viruses tested, but only in Vero cells. Adding certain nucleotides to LLC-MK2 cell cultures causes proliferation through an unknown mechanism (Lemmens et al., 1996), indicating that these cells employ altered nucleotide uptake, salvage or response pathways than Vero cells. These different pathways may be responsible for the differences in DFBA poxvirus inhibitory activity that we observed and may warrant further investigation. Other derivatives of ANO have been tested for activity against these same orthopoxviruses (R. Baker, manuscript in preparation).
The first goal of this investigation was to identify antiviral drugs that have activity against VAR and MPX. This objective has unequivocally been met. All three VAR isolates, as well as the Zaire isolate of MPX, were highly sensitive to eight different antiviral compounds. Only three of these (cidofovir, ribavirin and cHPMPC) are approved for clinical use or are in advanced clinical development for other indications.
4.6. Drug sensitivity of multiple VAR isolates
The second goal of this project was to identify an orthopoxvirus other than VAR which shares its pattern of drug sensitivities to be used as a surrogate for antiviral drug evaluation should the remaining laboratory stocks of VAR in Atlanta and Novosibirsk be destroyed. This surrogate should also have the lowest human virulence possible to allow for testing of antivirals in labs that are not equipped with high-level containment facilities. This goal has been only partially met. Several viruses displayed sensitivities similar to VAR for drugs that target the viral DNA polymerase, reflecting the near-identity of the enzyme among the orthopoxviruses (R. Baker, S. Ibrahim and M. Frace, unpublished data). Any of the less virulent viruses could therefore serve as surrogates for screening new members of this class of compounds. For other enzyme targets, the molecular mode of action of a new drug must be well established and the structures of the target enzymes of VAR and its surrogate must be known to be identical. Unfortunately, this level of understanding in most cases does not yet exist. More variation in sensitivity to drugs with other mechanisms of action, such as the ANO derivative DFBA, was seen among the test viruses. Of greatest importance was the greater sensitivity of VAR to several antiviral agents. For example, VAR was more sensitive to ribavirin than either CPX or VAC. If a surrogate virus, rather than VAR itself, had been used in drug testing there may have been little reason to consider ribavirin as a potential therapeutic agent for smallpox. We must therefore conclude that none of the three other orthopoxviruses in the panel can truly qualify as a surrogate for VAR in antiviral drug screening. Thus, far, drug-sensitivity data generated for over 800 compounds (R. Baker, unpublished data) indicate that CPX is the virus that has sensitivities most similar to those of VAR.
An important and as yet unresolved question involves resistance to antiviral drugs. This resistance could either be naturally occurring differences between the various isolates of VAR or could potentially be engineered into the virus. An attack with a drug-resistant virus would seriously complicate treatment options and efficacy. Several varieties of cidofovir-resistant orthopoxviruses have already been derived in our lab (Smee et al., 2002). Thus, the third goal of this study was to evaluate some of the most promising antiviral compounds against a diverse group of VAR isolates to assess the degree of naturally occurring variations in drug sensitivity. We tested three compounds (cidofovir, cHPMPC and ribavirin) against 35 strains of VAR that were isolated at different time periods and from different regions of the globe. In these preliminary studies most VAR isolates tested displayed similar drug sensitivities, and while these data indicate some isolates of VAR may have altered drug sensitivities, we believe that with further investigation most if not all of these differences will disappear. A more detailed analysis of these findings and the potential differences in drug sensitivities of VAR isolates is currently underway. This should allow us to determine the potential for naturally occurring or deliberately created drug-resistant mutants. These findings will have a significant impact on future antiviral drug development.
In conclusion, we have identified several antiviral agents with promise for treating human VAR and MPX infection. Two drugs, cidofovir and ribavirin, are already in clinical use. Their continued availability seems assured. Others are in development, and at least some are likely to receive FDA approval. In the meantime, in the event of a VAR or MPX infection, our data can provide an interim guide for therapeutic decision-making.
Acknowledgements
The authors acknowledge the outstanding technical work of Rafael Herrera, Debbie Kefauver, and Joshua Shamblin. We also thank Dr. Jeanette Simpson for useful discussion and critical review of this manuscript. This work was funded in part by a fellowship from the National Academy of Sciences to Dr. Robert O. Baker.
Footnotes
The views, opinions, and/or findings contained herein are those of the authors and should not be construed as an official Department of the Army position, policy, or decision unless so designated by other documentation.
References
- Bray M., Martinez M., Smee D.F., Kefauver D., Thompson E., Huggins J.W. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J. Infect. Dis. 2000;181:10–19. doi: 10.1086/315190. [DOI] [PubMed] [Google Scholar]
- Bray M., Driscoll J., Huggins J.W. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-l-homocysteine hydrolase inhibitor. Antiviral Res. 2000;45:135–147. doi: 10.1016/s0166-3542(00)00066-8. [DOI] [PubMed] [Google Scholar]
- Bray M., Raymond J.L., Geisbert T., Baker R.O. 3-Deazaneplanicin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antiviral Res. 2002;55:151–159. doi: 10.1016/s0166-3542(02)00018-9. [DOI] [PubMed] [Google Scholar]
- Crotty S., Maag D., Arnold J.J., Zhong W., Lau J.Y., Hong Z., Andino R., Cameron C.E. Arnold, The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Cundy K.C. Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir and adefovir. Clin. Pharmacokinet. 1999;36:127–143. doi: 10.2165/00003088-199936020-00004. [DOI] [PubMed] [Google Scholar]
- De Clercq E. S-Adenosylhomocysteine hydrolase inhibitors as broad-spectrum antiviral agents. Biochem. Pharmacol. 1987;36:2567–2575. doi: 10.1016/0006-2952(87)90533-8. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Antiviral agents: characteristic activity spectrum depending on the molecular target with which they interact. Adv. Virus. Res. 1993;42:1–55. doi: 10.1016/s0065-3527(08)60082-2. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Antivirals for the treatment of herpesvirus infections. J. Antimicrob. Chemother. 1993;32(Suppl. A):121–132. doi: 10.1093/jac/32.suppl_a.121. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Vaccinia virus inhibitors as a paradigm for the chemotherapy of poxvirus infections. Clin. Microbiol. Rev. 2001;14:382–397. doi: 10.1128/CMR.14.2.382-397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E., Montgomery J.A. Broad-spectrum antiviral activity of the carbocyclic analog of 3-deazaadenosine. Antiviral Res. 1983;3:17–24. doi: 10.1016/0166-3542(83)90011-6. [DOI] [PubMed] [Google Scholar]
- De Clercq E., Luczak M., Shugar D., Torrence P.F., Waters J.A., Witkop B. Effect of cytosine, arabinoside, iododeoxyuridine, ethyldeoxyuridine thiocyanatodeoxyuridine, and ribavirin on tail lesion formation in mice infected with vaccinia virus. Proc. Soc. Exp. Biol. Med. 1976;151:487–490. doi: 10.3181/00379727-151-39241. [DOI] [PubMed] [Google Scholar]
- De Clercq E., Sakuma T., Baba M., Paulwels S.R., Balzarini I., Rosenberg I., Holy A. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines. Antiviral Res. 1987;8:261–272. doi: 10.1016/s0166-3542(87)80004-9. [DOI] [PubMed] [Google Scholar]
- Fenner, F., Henderson, D.A., Arita. I., Jezek, Z., Ladnyi, I.D., 1988. Smallpox and Its Eradication. World Health Organization, Geneva.
- Finter N. Dye uptake methods for assessing viral cytopathogenicity and their application to interferon assays. J. Gen. Virol. 1969;5:419–427. [Google Scholar]
- Gubser C., Smith G.L. The sequence of camelpox virus shows it is most closely related to variola virus, the cause of smallpox. J. Gen. Virol. 2002;83:855–872. doi: 10.1099/0022-1317-83-4-855. [DOI] [PubMed] [Google Scholar]
- Hitchcock M.J.M., Jaffe H.S., Martin J.C., Stagg R.J. Cidofovir a new agent with potent anti-herpesvirus activity. Antiviral Chem. Chemother. 1996;7:115–127. [Google Scholar]
- Hoffman V.F., Skiest D.J. Therapeutic developments in cytomegalovirus retinitis. Expert Opin. Invest. Drugs. 2000;9:207–220. doi: 10.1517/13543784.9.2.207. [DOI] [PubMed] [Google Scholar]
- Huggins J.W., Zhang Z.X., Bray M. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model. J. Infect. Dis. 1999;179(Suppl. 1):S240–S247. doi: 10.1086/514316. [DOI] [PubMed] [Google Scholar]
- Jackson R.J., Ramsay A.J., Christensen C.D., Beaton S., Hall D.F., Ramshaw I.A. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 2001;75:1205–1210. doi: 10.1128/JVI.75.3.1205-1210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan I., Briese T., Averett D.R., Lipkin W.I. Inhibition of Boma disease virus replication by ribavirin. J. Virol. 1999;73:7903–7906. doi: 10.1128/jvi.73.9.7903-7906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane E.M., Shuman S. Adenosine N1-oxide inhibits vaccinia virus replication by blocking translation of viral early mRNAs. J. Virol. 1995;69:6352–6358. doi: 10.1128/jvi.69.10.6352-6358.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E., Margalith E., Winer B. Inhibition of vaccinia virus growth by the nucleoside analogue 1-beta-d-ribofuranosyl-1,2,4-triazole-3-carboxarnide (virazole, ribavirin) J. Gen. Virol. 1976;32:327–330. doi: 10.1099/0022-1317-32-2-327. [DOI] [PubMed] [Google Scholar]
- Kesson A.M., Ferguson J.K., Rawlinson W.D., Cunningham A.L. Progressive vaccinia treated with ribavirin and immune globulin. Clin. Infect. Dis. 1997;25:911–914. doi: 10.1086/515534. [DOI] [PubMed] [Google Scholar]
- Khare M.D., Sharland M. Cytomegalovirus treatment options in immunocompromised patients. Expert Opin. Pharmacother. 2001;2:1247–1257. doi: 10.1517/14656566.2.8.1247. [DOI] [PubMed] [Google Scholar]
- Kirsi J.J., North J.A., McKernan P.A., Murray B.K., Canonico P.G., Huggins J.W., Srivastava P.C., Robins R.K. Broad-spectrum antiviral activity of 2-beta-d ribofuranosyl selenazole 4-carboxamide, a new antiviral agent. Antimicrob. Agents Chemother. 1983;24:353–361. doi: 10.1128/aac.24.3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsi J.J., Mckernan P.A., Burns N.J., North J.A., Murray B.K., Robins R.K. Broad-spectrum synergistic antiviral activity of selenazofurin. Antimicrob. Agents Chemother. 1984;26:466–475. doi: 10.1128/aac.26.4.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong C.D., Krauffi C.A., Shortnacy-Fowler A.T., Arnett G., Hollingshead M.G., Shannon W.M., Montgomery J.A., III, Secrist J.A. Synthesis and antiviral evaluation of analogs of adenosine-N1-oxide and 1-(benzyloxy)adenosine. Nucleosides Nucleotides. 1998;17:1409–1443. doi: 10.1080/07328319808003478. [DOI] [PubMed] [Google Scholar]
- Lemmens R., Vanduffel L., Teuchy H., Culic O. Regulation of proliferation of LLC-MK2 cells by nucleosides and nucleotides: the role of ecto-enzymes. Biochem. J. 1996;316(Pt. 2):551–557. doi: 10.1042/bj3160551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutwick L.I., Goozner B., Bourke E. Bioterrorism: a primer for 2002. J. Assoc. Acad. Minor. Phys. 2002;13:9–13. [PubMed] [Google Scholar]
- Maag D., Castro C., Hong Z., Cameron C.E. Hepatitis C Virus RNA-dependent RNA polymerase (NS5B) as a mediator of the antiviral activity of ribavirin. J. Biol. Chem. 2001;276:46094–46098. doi: 10.1074/jbc.C100349200. [DOI] [PubMed] [Google Scholar]
- Massung R.F., Liu L.I., Qi J., Knight J.C., Yuran T.E., Kerlavage A.R., Parsons J.M., Venter J.C., Esposito J.J. Analysis of the complete genome of smallpox variola major virus strain Bangladesh—1975. Virology. 1994;201:215–240. doi: 10.1006/viro.1994.1288. [DOI] [PubMed] [Google Scholar]
- Mathew T. Comparative studies on buffalo pox, vaccinia and cow pox viruses in hamster kidney cell cultures. Acta Virol. 1970;14:513. [PubMed] [Google Scholar]
- Montgomery J.A., Clayton S.J., Thomas H.J., Shannon W.M., Arnett G., Bodner A.J., Kion I.K., Cantoni G.L., Chiang P.K. Carbocyclic analogue of 3-deazaadenosine: a novel antiviral agent using S-adenosylhomocysteine hydrolase as a pharmacological target. J. Med. Chem. 1982;25:626–629. doi: 10.1021/jm00348a004. [DOI] [PubMed] [Google Scholar]
- Neyts J., De Clercq E. Efficacy of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine for the treatment of lethal vaccinia virus infections in severe combined immune deficiency (SCID) mice. J. Med. Virol. 1993;41:242–246. doi: 10.1002/jmv.1890410312. [DOI] [PubMed] [Google Scholar]
- Noble S., Goa K.L. Adefovir dipivoxil. Drugs. 1999;58:479–489. doi: 10.2165/00003495-199958030-00010. [DOI] [PubMed] [Google Scholar]
- Pancheva S.N. Potentiating effect of ribavirin on the anti-herpes activity of acyclovir. Antiviral Res. 1991;16:151–161. doi: 10.1016/0166-3542(91)90021-i. [DOI] [PubMed] [Google Scholar]
- Rankin I.T., Jr., Eppes S.B., Antczak J.B., Joiklik W.K. Studies on the mechanism of the antiviral activity of ribavirin against reovirus. Virology. 1989;168:147–158. doi: 10.1016/0042-6822(89)90413-3. [DOI] [PubMed] [Google Scholar]
- Salmon-Ceron D. Cytomegalovirus infection: the point in 2001. HIV Med. 2001;2:255–259. doi: 10.1046/j.1468-1293.2001.00082.x. [DOI] [PubMed] [Google Scholar]
- Sheek M.R., Chapman A.L., Wenner H.A. Human and primate poxviruses: I. Growth characteristics of cytolytic and tumor variants. Arch. Virol. 1975;48:47–61. doi: 10.1007/BF01320565. [DOI] [PubMed] [Google Scholar]
- Shishkov S., Pancheva S. The synergistic antiviral effect of acyclovir and ribavirin against the herpes simplex type-1 virus and the pseudorabies virus in vitro. Acta Microbiol. Bulg. 1990;25:69–75. [PubMed] [Google Scholar]
- Smee D.F., Bailey K.W., Sidwell R.W. Treatment of cowpox virus respiratory infections in mice with ribavirin as a single agent or followed sequentially by cidofovir. Antiviral Chem. Chemother. 2000;11:303–309. doi: 10.1177/095632020001100406. [DOI] [PubMed] [Google Scholar]
- Smee D.F., Bailey K.W., Wong M., Sidwell R.W. Intranasal treatment of cowpox virus respiratory infections in mice with cidofovir. Antiviral Res. 2000;47:171–177. doi: 10.1016/s0166-3542(00)00105-4. [DOI] [PubMed] [Google Scholar]
- Smee D.F., Bailey K.W., Wong M., Sidwell R.W. Effects of cidofovir on the pathogenesis of a lethal vaccinia virus respiratory infection in mice. Antiviral Res. 2001;52:55–62. doi: 10.1016/s0166-3542(01)00159-0. [DOI] [PubMed] [Google Scholar]
- Smee D.F., Sidwell R.W., Kefauver D., Bray M., Huggins J.W. Characterization of wild-type and cidofovir-resistant strains of camelpox cowpox monkeypox and vaccinia viruses. Antimicrob. Agents Chemother. 2002;46:1329–1335. doi: 10.1128/AAC.46.5.1329-1335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tignor G.H., Kende M., Hanham C.A. Chemotherapeutic prevention of complications caused by vaccinia virus-vectored immunogen. Ann. N.Y. Acad. Sci. U.S.A. 1992;653:334–343. doi: 10.1111/j.1749-6632.1992.tb19660.x. [DOI] [PubMed] [Google Scholar]