

Abstract
Background
Modified vaccinia Ankara (MVA) is being developed as a safer smallpox vaccine and is being placed in the US Strategic National Stockpile (SNS) as a liquid formulation for subcutaneous (SC) administration at a dose of 1 × 108 TCID50 in a volume of 0.5 mL. This study compared the safety and immunogenicity of the standard formulation, dose and route with both a more stable, lyophilized formulation and with an antigen-sparing intradermal (ID) route of administration.
Methods
524 subjects were randomized to receive either a full dose of Lyophilized-SC, a full dose of Liquid-SC or 20% (2 × 107 TCID50 in 0.1 mL) of a full dose Liquid-ID MVA on Days 0 and 28. Safety and immunogenicity were followed through 180 days post second vaccination.
Results
Among the 3 groups, the proportion of subjects with moderate/severe functional local reactions was significantly different (P = 0.0013) between the Lyophilized-SC group (30.3%), the Liquid-SC group (13.8%) and Liquid-ID group (22.0%) only after first vaccination; and for moderate/severe measured erythema and/or induration after any vaccination (P = 0.0001) between the Lyophilized-SC group (58.2%), the Liquid-SC group (58.1%) and the Liquid-ID group (94.8%) and the reactions lasted longer in the Liquid-ID group. In the ID Group, 36.1% of subjects had mild injection site skin discoloration lasting ≥6 months.
After second vaccination Day (42–208), geometric mean of peak neutralization titers were 87.8, 49.5 and 59.5 for the Lyophilized-SC, Liquid-SC and Liquid-ID groups, respectively, and the maximum number of responders based on peak titer in each group was 142/145 (97.9%), 142/149 (95.3%) and 138/146 (94.5%), respectively. At 180 days after the second vaccination, geometric mean neutralization titers declined to 11.7, 10.2 and 10.4 with only 54.3%, 39.2% and 35.2% of subjects remaining seropositive for the Lyophilized-SC, Liquid-SC and Liquid-ID groups, respectively. Both the Lyophilized-SC and Liquid-ID groups were considered non-inferior (primary objective) to the Liquid-SC group.
Conclusions
Transitioning to a lyophilized formulation, which has a longer shelf life, will not negatively impact immunogenicity. In a situation where insufficient vaccine is available, ID vaccination could be used, increasing the number of available doses of vaccine in the SNS 5-fold (i.e., from 20 million to 100 million doses).
Keywords: NCT00914732
Keywords: MVA, IMVAMUNE, ELISA, Plaque reduction neutralizing antibody, Smallpox, Variola, Intradermal, Subcutaneous, Lyophilized, Vaccinia-naïve
1. Introduction
Despite the official eradication of smallpox in 1980 [1], the threat of potential emerging zoonotic orthopoxviruses [2] and potential use of variola virus as an agent for biological warfare/terrorism remains a concern. Although cell-culture grown ACAM2000® replaced Dryvax® as the licensed smallpox vaccine in the United States’ Strategic National Stockpile (SNS), its safety profile is similar to Dryvax® [3]. IMVAMUNE® is a modified vaccinia Ankara (MVA)-based smallpox vaccine, derived from MVA-597. IMVAMUNE® is replication-restricted to avian cells [4], [5] and has an improved safety profile [6], [7], [8], [9]; however, its efficacy against variola has not been tested. Both liquid and lyophilized formulations of IMVAMUNE® were tested in clinical trials. The liquid formulation of MVA is currently being added to the SNS and is licensed in Canada and Europe. Due to the need to stockpile IMVAMUNE® for extended periods of time, efforts are underway to transition from the liquid formulation to a lyophilized formulation; stability studies are still ongoing. This study provides a direct comparison of the two formulations to bridge safety and immunogenicity data.
The intradermal (ID) route was historically used for MVA vaccination [10], [11], [12], [13] alone, or as a vector for other antigens [14], [15], [16], [17], [18], [19], [20], [21], as a means to enhance the immunogenicity of vaccines such as BCG, malaria and HIV, and for antigen sparing effects compared to other routes [22], [23], [24], [25], [26], [27], [28], [29]. Though IMVAMUNE® is currently administered subcutaneously (SC), a study with another MVA suggested that ID vaccination can be dose sparing relative to SC and intramuscular routes [28]. Since reducing the vaccination dose would expand the number of doses available in the SNS during an emergency, a formal antigen-sparing assessment of the ID route with IMVAMUNE® was conducted.
2. Methods
2.1. Study design
In this Phase II trial (clinicaltrials.gov NCT00914732), subjects were randomized 1:1:1 to three arms to receive 2 doses of MVA 28 days apart (Fig. 1 ). The Lyophilized-SC (n = 165 subjects) and Liquid-SC (n = 167 subjects) groups received 1 × 108 TCID50 of vaccine virus in a dose of 0.5 mL reconstituted lyophilate and liquid formulation, respectively, administered subcutaneously into the deltoid area. The Liquid-ID (n = 191) group received 2 × 107 TCID50 in 0.1 mL per dose [28] in the volar area of the forearm. Staff was blinded to the formulation used for SC administration, but not to ID administration. Immunogenicity was measured using ELISA and Plaque Reduction Neutralization Titer (PRNT) assays.
Fig. 1.
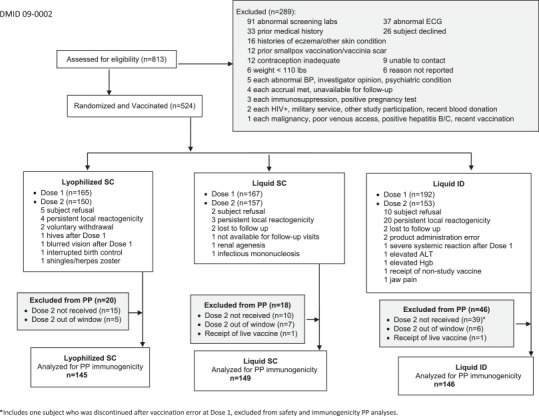
Consort diagram representing the number of subjects assessed for eligibility, randomized to treatment and available for per protocol immunogenicity analysis.
Note: Many subjects, especially in the Liquid-ID arm had ongoing local (injection site) reactogenicity 28 days after the first vaccination. A protocol amendment allowed subjects to receive the second dose in the contralateral arm if only mild erythema and/or induration from the first vaccination were present at Day 28 (previously subjects were excluded if they had any reactogenicity), to exclude subjects with continued moderate/severe erythema and/or induration from receiving dose 2, and to replace subjects who did not receive dose 2 for any reason.
Subjects reported local and systemic reactogenicity, i.e., solicited adverse events (AE), using a memory aid on Days 0–14 after each vaccination. Unsolicited AEs, i.e., non-reactogenicity events, were collected for 56 days following the initial vaccination. Local reactogenicity symptoms that extended beyond the 15-day memory aid period and were stable for 30 days, and unexpected local reactions, i.e., nodules and discoloration at the vaccination site, were reported as unsolicited AEs. Serious AEs were collected during the entire study. Hematologic and chemistry evaluations were obtained at screening and 14 days after each vaccination. Electrocardiograms and troponin 1 levels were evaluated at screening.
The study was approved by the respective institutional review boards. All subjects provided written informed consent.
2.2. Inclusion criteria
Eligible subjects were healthy, ≥18 years of age and born after 1971, not pregnant, and had an acceptable ECG, a ≤10% risk of myocardial infarction or coronary death using the National Cholesterol Education Program's risk assessment tool, and no evidence of a vaccinia scar, history of smallpox vaccination or history of eczema.
2.3. Vaccine
IMVAMUNE® was supplied by Bavarian Nordic (BN) A/S (Kvistgård, Denmark). Liquid-frozen aliquots (2 lots) and lyophilized (freeze-dried) product (1 lot) was supplied as single use vials with an approximate titer of 1 × 108 TCID50 IMVAMUNE® per 0.5 mL. Excipients in the lyophilized product included Tris-buffered saline, Tris, NaCl, sucrose, Dextran, and Glutamic acid monopotassium salt monohydrate. Liquid formulation excipients included Tris (hydroxymethyl)-amino methane and NaCl. Sterile water for injection was used for reconstitution of the lyophilized vaccine.
2.4. Immunogenicity assays
The immunogenicity assays were previously described [8], [30], [31]. BN (primary assays) used vaccinia-Western Reserve (replicating vaccinia) [7] and BN–MVA (non-replicating vaccinia in humans) in the plaque reduction neutralizing antibody (PRNT50) assay [8] and ELISA [8], respectively. Saint Louis University (SLU) (secondary assays) used ATCC MVA (VR-1508) as the assay antigen in the ELISA [30] and PRNT60 [31] assay. Seroconversion was defined as the ELISA or PRNT value above the cut-off value of the assay. Blood samples were obtained on Days 0, 14, 28, 42, 56 and 208 after first vaccination.
2.5. Statistical analysis
The sample size calculations targeted at least 80% power to test non-inferiority for the primary [geometric mean of peak (GMT) of the BN–PRNT50] and secondary (GMT of the BN–ELISA) immunogenicity end points for two investigational arms in reference to a control arm. Assuming a standard deviation of 2.8 for log2 peak PRNT or ELISA titer in each group (default for BN's PRNT50 assay at time of protocol development), a non-inferiority margin of 2.0-fold [32], [33], [34], a type I error rate of 1.25%, and drop-out rate of 10%, 165 subjects in each of the three groups were to be enrolled in this study to achieve at least 148 evaluable subjects. To maintain at least 148 evaluable subjects per arm, an additional 29/151 (15.1%) subjects were enrolled to replace subjects who did not receive their second vaccination. Seventeen subjects [Liquid-ID = 10/191 (5.2%), Liquid-SC = 2/167 (1.2%), Lyophilized-SC = 5/165 (3%)] were discontinued due to subject refusal (P = 0.09).
The number of subjects with safety events and titers above an assay-specific cut-off and ≥4-fold baseline increase were summarized using frequency, proportion and its 95% two sided exact (Clopper–Pearson) confidence interval. Frequency comparisons between treatment groups were carried out using a Fisher's exact test if large sample approximation criteria were not met. Otherwise a Chi-Square test was applied. The peak PRNT and ELISA titer was calculated as the largest titer obtained for post second vaccination measurements. For each investigational arm, the control arm difference in log2 GMT and associated two-sided 97.5% CI was used to evaluate non-inferiority (established if upper CI bound <1) and superiority (established if upper CI bound <0).
Except for non-inferiority testing, alpha was not adjusted for multiple testing. Antibody half-life was calculated by fitting a linear regression model to log2 transformed titers for Days 42, 56, and 208 (negative reciprocal of the slope estimate). The per protocol (PP) population was used for immunogenicity analysis. Subjects excluded from PP (n = 84) are detailed Fig. 1.
3. Results
3.1. Study subjects
A total of 524 subjects were enrolled at 8 sites between February 9, 2010 and September 2, 2010. Excluding one vaccination error, 260 males and 263 females were enrolled. Most subjects were non-Hispanic (93.3%) and white (82.0%). The median age was 26.8 years (range: 18–38 years). There were no significant differences in the distribution of gender (P = 0.54), ethnicity (P = 0.06), race (P = 0.40), and age (one-way ANOVA P = 0.12) between treatment groups.
3.2. Safety outcomes
All abnormal laboratory values associated with vaccination were considered mild to moderate in severity.
3.2.1. Systemic reactogenicity
Five (3.0%), 0 (0.0%), and 6 (3.1%) of the subjects from Lyophilized-SC, Liquid-SC and Liquid-ID groups, respectively, experienced severe systemic AEs after first vaccination (P = 0.04); 3 (2.0%), 3 (1.9%) and 1 (0.7%), respectively, after second vaccination. The rates were significantly higher for both investigational arms after the first vaccination (P ≤ 0.032) (Fig. 2 ). After vaccination 1, the proportions of subjects with any systemic reaction were 108/165 (65.5%), 86/167 (51.5%), and 115/191 (60.2%) for the Lyophilized-SC, Liquid-SC, and Liquid-ID groups, respectively (P = 0.03 among the three groups). In pair-wise comparisons, the proportion of subjects with any AE for the Lyophilized-SC group was significantly higher than the Liquid-SC group (P = 0.01); there was no significant difference between the Liquid-SC and Liquid-ID groups (P = 0.10) after first vaccination. After the second or any vaccination, there were no significant differences among the three groups (Fig. 2) or in pairwise comparisons.
Fig. 2.
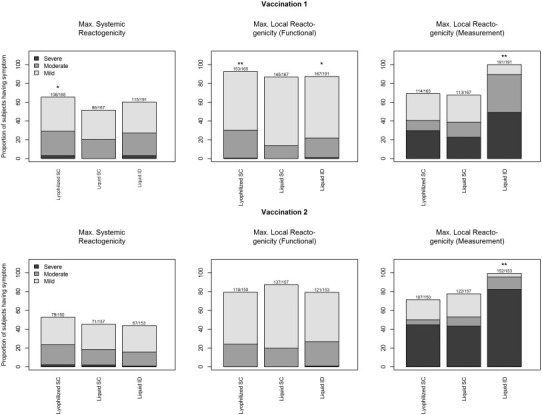
Maximum severity grade for reactogenicity collected by subjects in the Lyophilized-SC, Liquid SC and Liquid-ID groups for 15 days (Days 0–14) after each vaccination. Systemic reactogenicity events were graded using a functional scale of mild (present but easily tolerated), moderate (able to tolerate routine activity with effort), and severe (unable to continue routine activity). Fever grading scale for oral temperature was mild ≥37.8–<38 °C, moderate ≥38–<39 °C, and severe ≥39 °C; fever is included in the systemic reactogenicity. Local injection site reactogenicity events other than erythema and induration were graded using a functional scale of mild (present but easily tolerated), moderate (able to tolerate routine activity with effort), and severe (unable to continue routine activity). Local injection site erythema and induration were measured and graded as mild (<15 mm), moderate (15–30 mm) or severe (>30 mm). * P < 0.05, ** P < 0.01.
When considering severity (Fig. 2) of the systemic reaction (none/mild vs. moderate/severe), there were no significant differences in the proportion of subjects among groups post vaccination 1, 2, or any vaccination. In general, feeling tired was the most common symptom in each group after each vaccination.
One subject each had a moderate fever after first vaccination in the Liquid-SC and Liquid-ID groups, and none after the second vaccination in either group. Two subjects had mild fever after vaccination 1, and 6 subjects had fever (5 mild, 1 moderate) after vaccination 2 in the Lyophilized-SC group.
3.2.2. Local reactogenicity
Four subjects experienced severe local reactions with functional grading (Fig. 2): 1 in the Lyophilized-SC group (pain), and 3 in the Liquid-ID group (itchiness).
The proportions of subjects with moderate/severe functional local reactions post vaccination 1 were 30.3%, 13.8%, 22.0% for the Lyophilized-SC, Liquid-SC, and Liquid-ID groups, respectively (P = 0.0013) (Fig. 2); for the comparison among the three groups, P = 0.0003 for Lyophilized SC compared to Liquid SC, and P = 0.04 for comparison of Liquid ID to Liquid SC). There were no significant differences after vaccination 2 (P = 0.34 among groups). However, in pairwise comparisons, the proportion of subjects with moderate/severe local reactions was significantly higher in the Lyophilized-SC group than the Liquid-SC group (P = 0.02). Pain at the injection site was the most prevalent local reaction in both of the SC groups and itchiness at the injection site was the most prevalent in the ID group.
Following any vaccination, the proportion of subjects with any measured erythema or induration at the injection site differed significantly among groups (P < 0.0001): the proportions were 79.4%, 84.4%, and 100% for the Lyophilized-SC, Liquid-SC, and the Liquid-ID dose groups, respectively (Fig. 2). Of these, 58.2%, 58.1% and 94.8% had severe local reactions (>30 mm), respectively.
Local reactogenicity lasting at least 30 days, unexpected nodules and skin discoloration at the vaccination site, accounted for 389 (80%) of the unsolicited adverse events reported as associated to vaccination and included 50/165 (30.3%), 42/167 (25.1%), and 128/191 (67.0%) for the Lyophilized-SC, Liquid-SC, and Liquid-ID groups, respectively. There was no significant difference between the Liquid-SC and Lyophilized-SC group (P = 0.33). The proportion for the Liquid-ID group was significantly higher than the Liquid-SC group (P < 0.0001).
3.2.3. Protocol amendment
A total of 15, 10 and 39 subjects in the Lyophilized-SC, Liquid-SC, and the Liquid-ID dose groups, respectively (Fig. 1) did not receive second vaccination for any reason. Of these, 4, 3, and 20 subjects in the Lyophilized-SC, Liquid-SC, and the Liquid-ID dose groups, respectively, (Fig. 1) did not receive a second vaccination due to persistent local reactogenicity (mild or greater) at the time of second vaccination. To ensure 148 evaluable subjects per arm, a protocol amendment allowed subjects to receive the second dose in the contralateral arm if only mild erythema and/or induration from the first vaccination was present at Day 28. At the time of the writing of the amendment 152 subjects had been enrolled and no subject had severe local reactogenicity at Visit 3 (Days 13–15) or Visit 4 (Days 26–30).
3.2.4. Unsolicited AEs
Overall, 830 unsolicited AEs within 28 days after each vaccination were reported by 356 (68.1%) subjects. Of these events, 486 (58.6%) were considered associated with vaccine and were experienced by 256 subjects. Sixty-four subjects (38.8%) in the Lyophilized-SC group, 57 (34.1%) in the Liquid-SC group, and 135 (70.1%) in the Liquid-ID group experienced an associated adverse event; none of which were severe. The difference among the three groups was significant (P < 0.0001). The Liquid-ID group had a significantly higher proportion than the Liquid-SC group (P < 0.0001); the difference between the two SC groups was not significant.
One subject in the Lyophilized-SC group reported intermittent moderate chest pain 3 days post vaccination 2. The next day the subject was asymptomatic and had a normal CK, CKMB, Troponin I and ECG.
Four subjects had a serious adverse event (back pain, ischemic colitis, appendicitis and colitis) which were considered not associated with vaccine. All received both vaccinations.
3.3. Immunogenicity
3.3.1. PRNT
BN–PRNT50 (primary assay): There was no significant difference in the number (range 0–2) of subjects with a positive titer (≥15) in each group prior to vaccination 1.
After second vaccination (day 42–208), GMTs (based on peak titer) were 87.8, 49.5 and 59.6 for the Lyophilized-SC, Liquid-SC and Liquid-ID groups, respectively (Table 1a , Fig. 3 ) and the maximum number of responders (based on peak titer) in each group was 142/145/(97.9%), 142/149 (95.3%) and 138/146 (94.5%), respectively. The 97.5% CI for the log2 mean difference between the Liquid-SC and Lyophilized-SC groups and the Liquid-SC and Liquid-ID groups was <1 (Supplementary Table 1). Thus, both the Lyophilized-SC and Liquid-ID groups were considered non-inferior (primary objective) to the Liquid-SC group. For the Lyophilized-SC group, superiority was established (Fig. 4 ).
Table 1a.
BN PRNT per protocol population analysis: summary of number and proportion of responders with titers ≥ 15, peak geometric mean titers (GMT), and number of subjects with ≥4-fold rise by vaccination and visit.
| Study visit day | Group |
||
|---|---|---|---|
| Lyophilized SC | Liquid SC | Liquid ID | |
| Seroconversion, n/N (%) [95% CI] | Seroconversion, n/N (%) [95% CI] | Seroconversion, n/N (%) [95% CI] | |
| GMT [95% CI] | GMT [95% CI] | GMT [95% CI] | |
| ≥4-fold rise n/N (%) [95% CI] | ≥4-fold rise n/N (%) [95% CI] | ≥4-fold rise n/N (%) [95% CI] | |
| Day 0a | 0/145 (0.0) [0.0, 2.5] | 2/149 (1.3) [0.2, 4.8] | 2/146 (1.4) [0.2, 4.9] |
| 7.5 [,] | 7.7 [7.4, 8.0] | 7.7 [7.4, 7.9] | |
| NA | NA | NA | |
| Day 14 | 60/145 (41.4) [33.3, 49.8]* | 44/149 (29.5) [22.3, 37.5] | 56/146 (38.4) [30.4, 46.8] |
| 10.9 [9.9, 12.0]NIE | 10.0 [9.0, 11.1] | 10.3 [9.3, 11.3]NIE | |
| 6/145 (4.1) [1.5, 8.8] | 3/149 (2.0) [0.4, 5.8] | 2/146 (1.4) [0.2, 4.9] | |
| Day 28b | 61/145 (42.1) [33.9, 50.5]** | 39/149 (26.2) [19.3, 34.0] | 68/146 (46.6) [38.3, 55.0]*** |
| 10.8 [9.9, 11.9]NIE | 9.6 [8.7, 10.6] | 10.8 [9.9, 11.9]NIE | |
| 6/145 (4.1) [1.5, 8.8] | 3/149 (2.0) [0.4, 5.8] | 2/146 (1.4) [0.2, 4.9] | |
| Day 42 | 137/145 (94.5) [89.4, 97.6] | 137/148 (92.6) [87.1, 96.2] | 134/146 (91.8) [86.1, 95.7] |
| 77.6 [62.3, 96.7]NIE | 45.2 [36.4, 56.2] | 54.4 [43.7, 67.8]NIE | |
| 100/145 (69.0) [60.8, 76.4] | 70/148 (47.3) [39.0, 55.7] | 82/146 (56.2) [47.7, 64.4] | |
| Day 56 | 132/144 (91.7) [85.9, 95.6]** | 117/148 (79.1) [71.6, 85.3] | 124/146 (84.9) [78.1, 90.3] |
| 39.4 [31.9, 48.6]NIE | 23.4 [19.4, 28.3] | 33.4 [27.2, 41.0]NIE | |
| 63/144 (43.8) [35.5, 52.3] | 37/148 (25.0) [18.3, 32.8] | 59/146 (40.4) [32.4, 48.8] | |
| Day 208 | 75/138 (54.3) [45.7, 62.8]* | 56/143 (39.2) [31.1, 47.7] | 50/142 (35.2) [27.4, 43.7] |
| 11.7 [10.7, 12.8]NIE | 10.2 [9.4, 11.0] | 10.4 [9.4, 11.5]NIE | |
| 5/138 (3.6) [1.2, 8.3] | 1/143 (0.7) [0.0, 3.8] | 5/142 (3.5) [1.2, 8.0] | |
| Peak post vaccination 2 | 142/145 (97.9) [94.1, 99.6] | 142/149 (95.3) [90.6, 98.1] | 138/146 (94.5) [89.5, 97.6] |
| 87.8 [71.2, 108.3]NIE | 49.5 [40.0, 61.3] | 59.6 [48.1, 74.0]NIE | |
| 105/145 (72.4) [64.4, 79.5] | 75/149 (50.3) [42.0, 58.6] | 86/146 (58.9) [50.5, 67.0] | |
| Half Life [days]c | 69 | 92 | 77 |
NIE: non-inferiority established. PRNT titers ≥15 and <75 were designated a titer of 15 by BN. Titer values of <15 (below limit of detection) were replaced by 7.5 (half the lower limit of detection) for analysis. Seroconversion was defined as PRNT value ≥15.
First vaccination.
Second vaccination.
Based on Day 42, 56, and 208. Accuracy of these 3-point estimates was compromised as many Day 208 observations for BN-PRNT were found below the lower limit of detection.
P < 0.05.
P < 0.01.
P < 0.001.
Fig. 3.
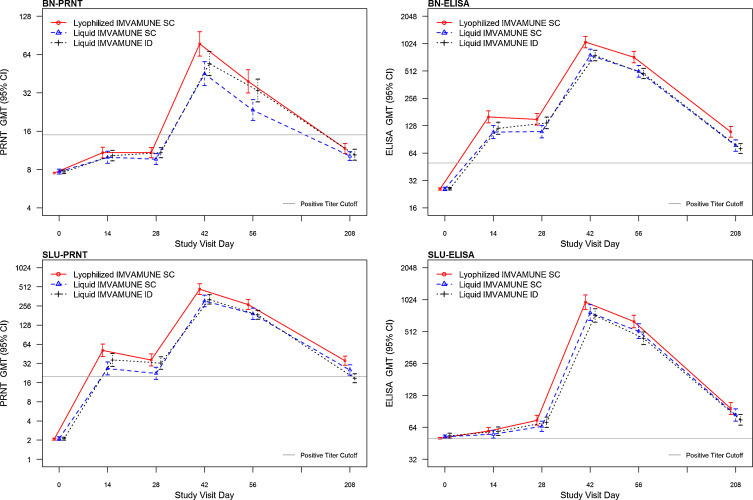
Per protocol Analysis of Geometric Mean Titers (GMT) and 95% Confidence Intervals (CI) by Group and Day Post First Vaccination for (a) BN PRNT, (b) SLU PRNT, (c) BN ELISA and (e) SLU ELISA. PRNT = Plaque reduction neutralizing antibody. ELISA = Enzyme linked immunosorbent assay.
Fig. 4.
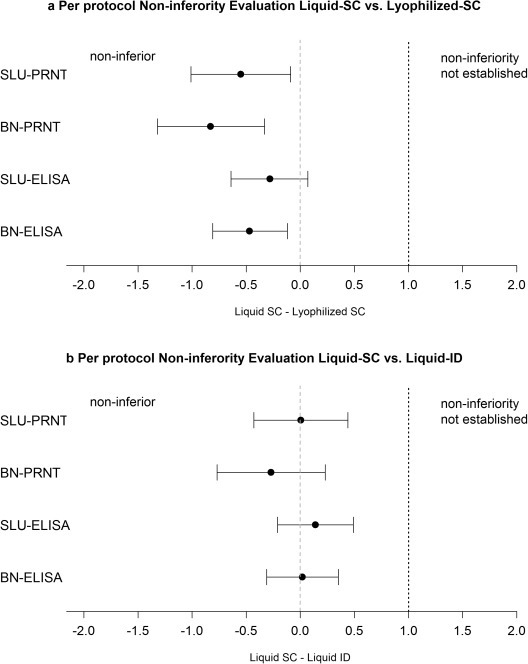
(a) Per protocol Non-inferiority Evaluation of Liquid-SC vs. Lyophilized-SC 97.5% confidence interval (CI) for the log 2 difference in the geometric means of the peak titers (GMT) for the Liquid-SC and Lyophilized-SC group. The black circles mark the point estimate of the difference and the bars mark the upper and lower limits of the 97.5% CI. The dotted black line marks the non-inferiority margin. The grey dashed line marks a zero difference. If the circle is to the right of the grey dashed line, Liquid SC has higher GMTs than the Lyophilized SC group; if the circle is to the left of the dashed line, Liquid SC has lower GMTs than the Lyophilized SC group. For all assays, the Lyophilized SC group obtained higher GMTs. As the upper bounds of the CIs were below 1 (to the left of the black dotted non-inferiority margin), non-inferiority was established. As the upper bound was also below 0 (grey dashed line) for SLU PRNT60, BN PRNT50, and BN ELISA, for these assays, the Lyophilized-SC group was superior to the Liquid-SC group. (b). Per protocol Non-inferiority Evaluation of Liquid-SC vs. Liquid-ID 97.5% confidence interval (CI) for the log2 difference in the geometric means of the peak titers (GMT) for the Liquid-SC and Liquid-ID group. As the upper bounds of the CIs were below 1 (to the left of the black dotted non-inferiority margin), non-inferiority was established. Superiority was not established for any assay as the upper bounds of the CIs were not below 0 (grey dashed line).
The upper limits of the individual 97.5% CIs were <1 for the mean difference between Lyophilized-SC and Liquid-SC groups and for the Liquid-ID group and the Liquid-SC groups for all days (Day 14, 28, 42, 56, 208) individually (exploratory analysis). However, the proportions of responders for the Lyophilized-SC group were significantly larger than the Liquid-SC group on Days 14, 28, 56 and 208 (P: 0.04, 0.0046, 0.028 and 0.01) and the proportions of responders for the Liquid-ID group were significantly larger than the Liquid-SC group on Day 28 only (P = 0.006). Per timepoint results are summarized in Table 1a.
SLU–PRNT60 (exploratory assay): Although GMTs (based on peak titer) of the SLU–PRNT60 were higher compared to the BN–PRNT50 (Supplementary Table 2a, Fig. 3), non-inferiority results were similar (Supplementary Table 1, Fig. 4). The maximum number of subjects seroconverting (titer ≥20) in each group after second vaccination was 144/145 (99.3%), 146/149 (98.0%) and 146/146 (100%) for the Lyophilized-SC, Liquid-SC and Liquid-ID groups, respectively. The Lyophilized-SC group also was considered superior to the Liquid-SC group (exploratory analysis).
3.3.2. ELISA
BN–ELISA (secondary assay): There was no significant difference in the number (range 3–6) of subjects with a positive titer (≥50) in each group prior to vaccination (Table 1b ). After second vaccination (day 42–208), GMTs (based on peak titer) were 1062.4, 769.3, 757.9 for the Lyophilized-SC, Liquid-SC and Liquid-ID groups, respectively (Table 1b, Fig. 3) and the maximum number of subjects seroconverting (>50) in each group (based on peak titer) after vaccination 2 was 145/145 (100%), 148/149 (99.3%) and 146/146 (100%), respectively. The upper limit of the CI was 1 for the mean difference between the Liquid-SC and Lyophilized-SC group and between the Liquid-SC group and Liquid-ID group (Supplementary Table 1). Thus both the Lyophilized-SC and Liquid-ID groups were considered non-inferior (secondary objective) to the Liquid-SC (Fig. 4). The Lyophilized-SC group was also considered superior to the Liquid-SC group.
Table 1b.
BN ELISA per protocol population analysis: summary of number and proportion of responders with titers ≥50, peak geometric mean titers (GMT), and number of subjects with ≥4-fold rise by vaccination and visit.
| Study visit day | Group |
||
|---|---|---|---|
| Lyophilized SC | Liquid SC | Liquid ID | |
| Seroconversion, n/N (%) 95% CI | Seroconversion, n/N (%) 95% CI | Seroconversion, n/N (%) 95% CI | |
| GMT (95% CI) | GMT (95% CI) | GMT (95% CI) | |
| Day 0a | 5/145 (3.4) [1.1, 7.9] | 3/149 (2.0) [0.4, 5.8] | 6/146 (4.1) [1.5, 8.7] |
| 25.7 [25.1, 26.4] | 25.8 [24.8, 26.9] | 26.0 [25.1, 26.8] | |
| NA | NA | NA | |
| Day 14 | 138/145 (95.2) [90.3, 98.0]* | 130/149 (87.2) [80.8, 92.1] | 135/146 (92.5) [86.9, 6.2] |
| 159.9 [137.3, 186.1]NIE | 108.8 [92.4, 128.0] | 119.9 [102.7, 139.9]NIE | |
| 115/145 (79.3) [71.8, 85.6] | 92/149 (61.7) [53.4, 69.6] | 95/146 (65.1) [56.7, 72.8] | |
| Day 28b | 137/145 (94.5) [89.4, 97.6] | 133/149 (89.3) [83.1, 93.7] | 140/146 (95.9) [91.3, 8.5]* |
| 150.4 [129.8, 174.4]NIE | 110.0 [93.9, 128.8] | 137.4 [118.6, 159.2]NIE | |
| 113/145 (77.9) [70.3, 84.4] | 93/149 (62.4) [54.1, 70.2] | 107/146 (73.3) [65.3, 80.3] | |
| Day 42 | 145/145 (100.0) [97.5, 100.0] | 147/148 (99.3) [96.3, 100.0] | 146/146 (100.0) [97.5, 100.0] |
| 1061.9 [920.4, 1225.2]NIE | 764.6 [657.4, 889.2] | 756.9 [663.4, 863.5]NIE | |
| 145/145 (100.0) [97.5, 100.0] | 147/148 (99.3) [96.3, 100.0] | 146/146 (100.0) [97.5, 100.0] | |
| Day 56 | 143/143 (100.0) [97.5, 100.0] | 147/148 (99.3) [96.3, 100.0] | 146/146 (100.0) [97.5, 100.0] |
| 725.5 [627.8, 838.4]NIE | 506.8 [437.6, 586.9] | 478.5 [420.3, 544.7]NIE | |
| 142/143 (99.3) [96.2, 100.0] | 145/148 (98.0) [94.2, 99.6] | 144/146 (98.6) [95.1, 99.8] | |
| Day 208 | 129/138 (93.5) [88.0, 97.0]** | 118/142 (83.1) [75.9, 88.9] | 123/142 (86.6) [79.9, 91.7] |
| 109.5 [95.8, 125.3]NIE | 77.4 [66.9, 89.5] | 71.7 [63.3, 81.1]NIE | |
| 91/138 (65.9) [57.4, 73.8] | 67/142 (47.2) [38.8, 55.7] | 57/142 (40.1) [32.0, 48.7] | |
| Peak post vaccination 2 | 145/145 (100.0) [97.5, 100.0] | 148/149 (99.3) [96.3, 100.0] | 146/146 (100.0) [97.5, 100.0] |
| 1062.4 [920.8, 1225.8]NIE | 769.3 [661.8, 894.2] | 757.9 [664.4, 864.6]NIE | |
| 145/145 (100.0) [97.5, 100.0] | 148/149 (99.3) [96.3, 100.0] | 146/146 [100.0) (97.5, 100.0] | |
| Half Life [days]c | 53 | 52 | 51 |
NIE: non-inferiority established.
First vaccination.
Second vaccination; titer results below limit of detection (50) are assigned a value of 25. Seroconversion was defined as ELISA value ≥50.
Based on Day 42, 56, and 208.
P < 0.05.
P < 0.01.
***P < 0.001.
Per-visit, exploratory analysis showed that there were no significant differences in GMTs at baseline (Table 1b). The Liquid-ID group was considered non-inferior to the Liquid-SC group while the Lyophilized SC group was not only considered non-inferior but also superior to the Liquid-SC group at each timepoint, respectively (Fig. 4).
However, the proportions of responders in the Lyophilized-SC group (95.2%, 93.5%) were significantly larger than the Liquid-SC group (87.2%, 83.1%) on Days 14 and 208 (P = 0.02 and 0.0089) (Table 1b). In the Liquid-ID group, the proportions were significantly larger than the Liquid-SC group on Day 28 (P = 0.0003).
The mean log2 transformed peak titers (positive response >50) were similar for the SLU–ELISA (exploratory assay) results (Supplementary Table 2b). The Lyophilized-SC and Liquid-ID groups were considered non-inferior to the Liquid-SC group (Fig. 4). See Supplementary Table 2b for per-visit results.
3.3.3. Day 42–Day 208 antibody decline
Antibody concentrations declined rapidly between Days 42 and Day 208 (Fig. 3) with half-life estimates ranging between 42 and 54 days for BN–ELISA, SLU–PRNT60, SLU–ELISA, and between 69 and 92 days for BN–PRNT50 (Supplementary Fig. 1). Accuracy of these 3-point estimates was compromised for BN–PRNT50 and SLU–ELISA assays as many Day 208 observations (>46% and >52%, respectively) were below the lower limit of detection.
4. Discussions
This trial provides the first direct clinical comparison of the liquid and lyophilized IMVAMUNE® formulations. The immunogenicity and safety profiles of both formulations delivered SC were shown to be similar to each other and consistent with prior clinical trial data [8], [35]. It is interesting that the proportion of subjects with moderate/severe functional local reactions after the first vaccination was significantly greater in the Lyophilized-SC group compared to the Liquid-SC group. The Lyophilized-SC group also had a superior immune response in three out of the four assays and had higher peak titers and proportion of responders at individual timepoints post vaccination. It is possible that the additional excipients present in the lyophilized formulation increased local reactogenicity and enhanced the immune response. The Liquid-ID arm was immunologically non-inferior to the Liquid-SC arm. The diminished antibody responses 6 months after second dose in all groups are similar to other studies using SC and ID routes [9], [28].
As shown with another MVA vaccine [28], the local site reactogenicity was significantly higher after each dose in the ID arm compared to the SC arm but not reactogenic enough to limit its use in the event of a smallpox release; all subjects in the Liquid-ID group had at least one solicited local reaction after any vaccination. In addition, local reactogenicity extending beyond the 15 day memory aid period was significantly greater for the ID group than the SC groups. At Day 180, greater than a third of subjects in the ID group continued to have minimal induration or erythema present on exam. The response seen in the ID group was likely due the large number of cells in the dermis that respond to foreign antigen. The finding of nodules and discoloration that was present for extended periods of time was an unexpected outcome not identified in the recent MVA route comparison study, though it has been identified with other vaccines [25], [29]. Possible reasons for this difference between the studies include vaccine formulation, vaccine administration technique, or assessment by study staff.
A protocol amendment allowed subjects to receive the second dose of vaccine in the contralateral arm if only none or mild erythema and/or induration from the first vaccination were present and to replace subjects who did not receive dose 2 for any reason. Most of the subjects excluded from receiving second vaccination due to erythema/induration had measurements graded as mild at the time of discontinuation.
Although a vaccinia neutralization titer of 20 to 32 [36], [37], [38], depending on the study, is thought by some to be protective against smallpox, a MVA neutralization titer that correlates with protection has not been defined. However, a two-dose regimen of IMVAMUNE® has been shown to provide a similar antibody response as Dryvax® [9], reduced the size of replicating vaccinia takes [9], [28] and was as effective if not better than Dryvax® in producing peak 90% variola virus neutralization GMTs [39], [40]. The two-dose SC MVA regimen remains the standard against which other regimens/doses are compared until more data are available. Of interest, the half-life, although limited, for the BN neutralizing titers was longer than the half-life of the SLU neutralizing titers. However, the accuracy of the BN half-life was compromised as many of the results at the third time point were below the lower limit of detection. Additional studies are required to more accurately determine half-life.
5. Conclusion
The demonstration of immunogenic non-inferiority (and superiority in 3 of the 4 immunogenicity assays) and safety equivalence of the lyophilized formulation compared to the liquid formulation provides bridging data for the transition from a liquid to a lyophilized formulation of IMVAMUNE®. Additionally, the ID route provided an equivalent immune response to the SC route using 80% less antigen. This antigen sparing effect could significantly increase the number of vaccine doses available in the event of an emergency by 5-fold. Although the ID route had increased local reactogenicity events, the lack of clinical significance of these events makes the route suitable in an emergency situation.
Funding
This project was funded fully or in part by: National Institute of Allergy and Infectious Diseases (NIAID) Contract HHSN272200800003C (Saint Louis University); NIH National Center for Advancing Translational Sciences through the Clinical and Translational Science Awards Program (CTSA) grants UL1TR000423, KL2TR000421 and TL1TR000422 (University of Washington Institute of Translational Health Science); NIAID Contract HHSN272200800005C (Emory University); NIAID Contract HHSN272200800004C (Group Health Cooperative); NIH Grant UL1RR024979 and NIAID Contract HHSN272200800008C (University of Iowa); NIAID Contract HHSN272200800002C (Baylor College of Medicine); NIAID Contract HHSN272200800001C (University of Maryland); NIAID Contract HHSN272200800007C (Vanderbilt University); and NIAID Contract HHSN272200800013C (EMMES); NIAID Contract HHSN266200400072C (Bavarian Nordic).
Acknowledgements
The authors would like to thank Sharon Irby-Moore, Mahendra Mandava and Tammy Blevins, Irene Graham, Edwin Anderson and the staff of Saint Louis University's Vaccine and Treatment Evaluation Unit (VTEU); Christine Johnston of University of Washington; the staff of the Group Health VTEU; Paul Spearman, Andi Shane, Ildefonso Tellez (deceased) and the staff at the Hope Clinic of the Emory Vaccine Center including William Emery, Eileen Osinski, Rebecca Gerkin, Beverly Weaver, Susan Rogers, and Nayoka Rimann; the staff of the Emory Children's Center Vaccine Clinic including Kathy Stephens, and Brooke Hartwell, Karen Pierce and Theda Gajadhar; Nancy Wagner and the staff of the University of Iowa Vaccine and Treatment Evaluation Unit; Celsa Tajonera, Tracey Lanford, Semahat Eiswirth, Connie Rangel, Janet Brown and Jose Diaz of the Baylor College of Medicine; Melissa Billington, Lisa Chrisley, Brenda Dorsey, Alyson Kwon, Robin Barnes, Sandra Getlein, Maria Johnson, Panagiota Komninou, Mary-Lou Mullen, Mardi Reymann, Kim Rincavage, Inna Ruslanova, Jennifer Russell, Giovionna Yanez, JoAnna Becker and Jeffrey Floyd at the University of Maryland; Sara Anderson, Shanda Phillips, Gayle Johnson and the staff of Vanderbilt University VTEU; and Aruna Acharyya and Ying Zhang of The EMMES Corporation; Robert Johnson, Marianne R. Baker, Stephen Heyse and Suzanne Murray of the Division of Microbiology and Infectious Diseases, NIAID, NIH; and the volunteers who participated in the study.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.landusepol.2015.07.020.
Appendix A. Supplementary data
The following are the supplementary data to this article:
Supplementary Figure 1.
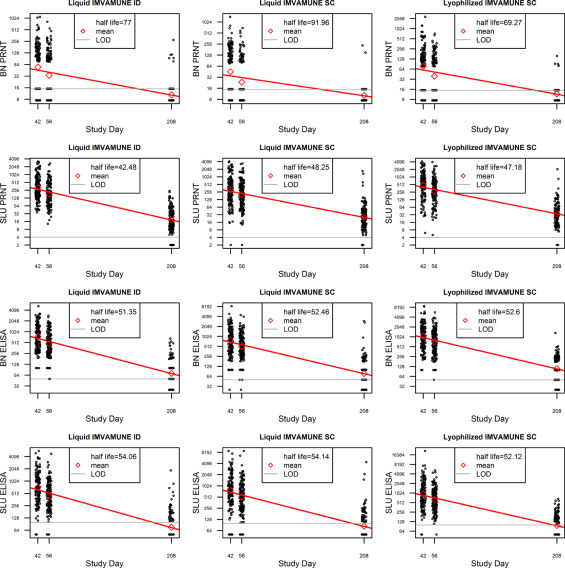
The antibody half-life is shown for each assay (PRNT and ELISA) by site (SLU and BN) for each of the three vaccines. The mean antibody is represented by the red triangle. LOD = level of detection. Individual antibody titers are provided for each of the three timepoints. The half-life in days is provided in the legend box.
References
- 1.Fenner F., Henderson D.A., Arita I., Jezek Z., Ladnyi I.D. World Health Organization; Geneva: 1988. Smallpox and its eradication; pp. 1–1460. [Google Scholar]
- 2.Shchelkunov S.N. Emergence and reemergence of smallpox: the need in development of a new generation smallpox vaccine. Vaccine. 2011;29(Suppl. 4):D49–D53. doi: 10.1016/j.vaccine.2011.05.037. [DOI] [PubMed] [Google Scholar]
- 3.Acambis Inc. 2007. ACAM2000TM smallpox vaccine, vaccines and related biological products advisory committee (VRBPAC) briefing document April 18. http://www.fda.gov/ohrms/dockets/ac/07/briefing/2007-4292b2-02.pdf. [Google Scholar]
- 4.Mayr A., Hochstein-Mintzel V., Stickl H. Passage history, properties, and use of attenuated vaccinia virus strain MVA. Infection. 1975;3:6–14. [Google Scholar]
- 5.Suter M., Meisinger-Henschel C., Tzatzaris M., Hülsemann V., Lukassen S., Wulff N.H., et al. Modified vaccinia Ankara strains with identical coding sequences actually represent complex mixtures of viruses that determine the biological properties of each strain. Vaccine. 2009;27:7442–7450. doi: 10.1016/j.vaccine.2009.05.095. [DOI] [PubMed] [Google Scholar]
- 6.Walsh S.R., Wilck M.B., Dominguez D.J., Zablowsky E., Bajimaya S., Gagne L.S., et al. Safety and immunogenicity of modified vaccinia ankara in hematopoietic stem cell transplant recipients: a randomized, controlled trial. J Infect Dis. 2013;207:1888–1897. doi: 10.1093/infdis/jit105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg R.N., Overton E.T., Haas D.W., Frank I., Goldman M., von Krempelhuber A., et al. Safety, immunogenicity, and surrogate markers of clinical efficacy for modified vaccinia Ankara as a smallpox vaccine in HIV-infected subjects. J Infect Dis. 2013;207:749–758. doi: 10.1093/infdis/jis753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Von Krempelhuber A., Vollman J., Pokorny R., Rapp P., Wulff N., Petzold B., et al. A randomized, double-blind, dose-finding phase II study to evaluate immunogenicity and safety of the third generation smallpox vaccine candidate IMVAMUNE®. Vaccine. 2010;28:1209–1216. doi: 10.1016/j.vaccine.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frey S.E., Newman F.K., Kennedy J.S., et al. Clinical and immunologic responses to multiple doses of IMVAMUNE® modified vaccinia Ankara followed by Dryvax® challenge. Vaccine. 2007;25:8562–8573. doi: 10.1016/j.vaccine.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayr A., Stickl H., Müller H.K., Danner K., Singer H. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defense mechanism. Zentralbl Bakteriol. 1978;167:375–390. [PubMed] [Google Scholar]
- 11.Stickl H., Hochstein M. Intracutaneous smallpox vaccination with a weak pathogenic vaccinia virus (MVA virus) Münch Med Wochenschr. 1971;113:1149–1153. [PubMed] [Google Scholar]
- 12.Schroeter W., Stickl H., Idel H., Schlipkoter H.A. MVA-vaccine in primovaccination against smallpox after the age of three. Zentralbl Bakteriol Mikrobiol Hyg B. 1980;171:309–319. [PubMed] [Google Scholar]
- 13.Hoffmann F., Poduschnik H. Richter KH Local reaction and neutralization test in smallpox. MMW Munshener Med Wochenschr. 1978;120:989–992. [PubMed] [Google Scholar]
- 14.Moorthy V.S., Imoukhuede E.B., Keating S., Pinder M., Webster D., Skinner M.A., et al. Phase I evaluation of 3 highly immunogenic prime-boost regimens, including a 12-month reboosting vaccination, for malaria vaccination in Gambian men. J Infect Dis. 2004;189:2213–2219. doi: 10.1086/421118. [DOI] [PubMed] [Google Scholar]
- 15.Cebere I., Dorrell L., McShane H., Simmons A., McCormack S., Schmidt C., et al. Phase I clinical safety of DNA- and modified virus Ankara-vectored human immunodeficiency virus type 1 (HIV-1) vaccines administered alone and in a prime-boost regime to healthy HIV-1-uninfected volunteers. Vaccine. 2006;24:417–425. doi: 10.1016/j.vaccine.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 16.McShane H., Pathan A.A., Sander C.R., Keating S.M., Gilbert S.C., Huygen K., et al. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycrobial immunity in humans. Nat Med. 2004;10:1240–1244. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- 17.Moorthy V.S., Imoukhuede E.B., Milligan P., Bojang K., Keating S., Kaye P., et al. A randomized, double-blind, controlled vaccine efficacy trial of DNA/MVA ME-TRAP against malaria infection in Gambian adults. PLoS Med. 2004;1:128–136. doi: 10.1371/journal.pmed.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moorthy V.S., Pinder M., Reece W.H., Watkins K., Atabani S., Hannan C., et al. Safety and immunogenicity of DNA/Modified vaccinia Ankara malaria vaccination in African adults. J Infect Dis. 2008;188:1239–1244. doi: 10.1086/378515. [DOI] [PubMed] [Google Scholar]
- 19.Mwau M., Cebere I., Sutton J., Chikoti P., Winstone N., Wee E.G., et al. A human immunodeficiency virus 1 (HIV-1) clade A vaccine in clinical trials: stimulation of HIV-specific T-cell responses by DNA and recombinant modified vaccinia virus Ankara (MVA) vaccines in humans. J Gen Virol. 2004;85:911–919. doi: 10.1099/vir.0.19701-0. [DOI] [PubMed] [Google Scholar]
- 20.Peters B.S., Jaoko W., Vardas E., Panayotakopoulos G., Fast P., Schmidt C., et al. Studies of a prophylactic HIV-1 vaccine candidate based on modified vaccinia virus Ankara (MVA) with and without DNA priming: effects of dosage and route on safety and immunogenicity. Vaccine. 2007;25:2120–2127. doi: 10.1016/j.vaccine.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Webster D.P., Dunachie S., McConkey S., Poulton I., Moore A.C., Walther M., et al. Safety of recombinant fowlpox strain FP9 and modified vaccinia virus Ankara vaccines against liver-stage P. falciparum malaria in nonimmune volunteers. Vaccine. 2006;24:3026–3034. doi: 10.1016/j.vaccine.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 22.Belshe R.B., Newman F.K., Cannon J., Duane C., Treanor J., Van Hoecke C., et al. Serum antibody responses after intradermal vaccination against influenza. N Engl J Med. 2004;351:2286–2294. doi: 10.1056/NEJMoa043555. [DOI] [PubMed] [Google Scholar]
- 23.Coleman P.J., Shaw F.E., Jr., Serovich J., Hadler S.C., Margolis H.S. Intradermal hepatitis B vaccination in a large hospital employee population. Vaccine. 1991;9:723–727. doi: 10.1016/0264-410x(91)90287-g. [DOI] [PubMed] [Google Scholar]
- 24.Kenney R.T., Frech S.A., Muenz L.R., Villar C.P., Glenn G.M. Dose sparing with intradermal injection of influenza vaccine. N Engl J Med. 2004;351:2295–2301. doi: 10.1056/NEJMoa043540. [DOI] [PubMed] [Google Scholar]
- 25.Henderson E.A., Louie T.J., Ramotar K., Ledgerwood D., Hope K.M., Kennedy A. Comparison of higher-dose intradermal hepatitis B vaccination to standard intramuscular vaccination of healthcare workers. Infect Control Hosp Epidemiol. 2000;21:264–269. doi: 10.1086/501756. [DOI] [PubMed] [Google Scholar]
- 26.Sabchareon A., Chantavanich P., Pasuralertsakul S., Pojjaroen-Anat C., Prarinyanupharb V., Attanath P., et al. Persistence of antibodies in children after intradermal or intramuscular administration of preexposure primary and booster immunizations with purified Vero cell rabies vaccine. Pediatr Infect Dis J. 1998;17:1001–1007. doi: 10.1097/00006454-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Seaman M.S., Wilck M.B., Baden L.R., Walsh S.R., Grandpre L.E., Devoy C., et al. Effect of vaccination with modified vaccinia Ankara (ACAM3000) on subsequent challenge with Dryvax. J Infect Dis. 2010;201:1353–1360. doi: 10.1086/651560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilck M.B., Seaman M.S., Baden L.R., Walsh S.R., Grandpre L.E., Devoy C., et al. Safety and immunogenicity of modified vaccinia Ankara (ACAM3000): effect of dose and route of administration. J Infect Dis. 2010;201:1361–1370. doi: 10.1086/651561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson E.A.S., Lam H.S., Choi K.C., HO W.C.S., Fung L.W.E., Cheng F.W.T. A pilot randomized study to assess immunogenicity, reactogenticity, safety and tolerability of two human papillomavirus vaccines administered intramuscularly and intradermally to females aged 18–26 years. Vaccine. 2013;31:3452–3460. doi: 10.1016/j.vaccine.2013.06.034. [DOI] [PubMed] [Google Scholar]
- 30.Iacono-Connors L.C., Novak J., Rossi C., Mangiafico J., Ksiazek T. Enzyme-linked immunosorbent assay using a recombinant baculovirus-expressed Bacillus anthracis protective antigen (PA): measurement of human anti-PA antibodies. Clin Diagn Lab Immunol. 1994;1:78–82. doi: 10.1128/cdli.1.1.78-82.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newman F.K., Frey S.E., Blevins T.P., Mandava M., Bonifacio A., Jr., Yan L., et al. Improved assay to detect neutralizing antibody following vaccination with diluted or undiluted vaccinia (Dryvax) vaccine. J Clin Microbiol. 2003;41:3154–3157. doi: 10.1128/JCM.41.7.3154-3157.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W.W.B., Mehrotra D.V., Chan I.S.F., Heyse J.F. Statistical considerations for noninferiority/equivalence trials in vaccine development. J Biopharm Stat. 2006;16:429–441. doi: 10.1080/10543400600719251. [DOI] [PubMed] [Google Scholar]
- 33.Diez-Domingo J., Gurtman A., Bernaola E., Gimenez-Sanchez F., Martinon-Torres F., Pineda-Solas V., et al. Evaluation of 13-valent pneumococcal conjugate vaccine and concomitant meningococcal group C conjugate vaccine in healthy infants and toddlers in Spain. Vaccine. 2013;31:5486–5494. doi: 10.1016/j.vaccine.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 34.Frenck R.W., Jr., Gurtman A., Rubinoc J., Smith W., van Cleeffe M., Jayawardene D., et al. Randomized, controlled trial of a 13-valent pneumococcal conjugate vaccine administered concomitantly with an influenza vaccine in healthy adults. Clin Vaccine Immunol. 2012;19:1296–1303. doi: 10.1128/CVI.00176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frey S.E., Winokur P.L., Salata R.A., El-Kamary S.S., Turley C.B., Walter E.B., Jr., et al. Safety and immunogenicity of IMVAMUNE® smallpox vaccine using different strategies for a post event scenario. Vaccine. 2013;31:3025–3033. doi: 10.1016/j.vaccine.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarkar J.K., Mitra A.C., Mukherjee M.K. The minimum protective level of antibodies in smallpox. Bull World Health Organ. 1975;52:307–311. [PMC free article] [PubMed] [Google Scholar]
- 37.Mack T.M., Noble J., Thomas D.B. A prospective study of serum antibody and protection against smallpox. Am J Trop Med Hyg. 1972;21:214–218. doi: 10.4269/ajtmh.1972.21.214. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy R.B., Lane J.M., Henderson D.A., Poland G.A. In: Vaccines. Plotkin S., Orenstein W., Offit P., editors. Saunders, Elsevier Inc.; Philadelphia, PA: 2013. Smallpox and vaccinia; pp. 718–745. [Google Scholar]
- 39.Damon I.K., Davidson W.B., Hughes C.M., Olson V.A., Smith S.K., Holman R.C., et al. Evaluation of smallpox vaccines using variola neutralization. J Gen Virol. 2009;90:1962–1966. doi: 10.1099/vir.0.010553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hughes C., Newman F., Davidson W., Olson V., Smith S., Holman R., et al. Analysis of variola and vaccinia neutralization assays for smallpox vaccines. Clin Vaccine Immunol. 2012;19:1116–1118. doi: 10.1128/CVI.00056-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.