

Abstract
Variola virus (VARV), causing smallpox, is a potential biological weapon. Methods to detect VARV rapidly and to differentiate it from other viruses causing similar clinical syndromes are needed urgently. We have developed a new microarray-based method that detects simultaneously and discriminates four orthopoxvirus (OPV) species pathogenic for humans (variola, monkeypox, cowpox, and vaccinia viruses) and distinguishes them from chickenpox virus (varicella-zoster virus or VZV). The OPV gene C23L/B29R, encoding the CC-chemokine binding protein, was sequenced for 41 strains of seven species of orthopox viruses obtained from different geographical regions. Those C23L/B29R sequences and the ORF 62 sequences from 13 strains of VZV (selected from GenBank) were used to design oligonucleotide probes that were immobilized on an aldehyde-coated glass surface (a total of 57 probes). The microchip contained several unique 13–21 bases long oligonucleotide probes specific to each virus species to ensure redundancy and robustness of the assay. A region approximately 1100 bases long was amplified from samples of viral DNA and fluorescently labeled with Cy5-modified dNTPs, and single-stranded DNA was prepared by strand separation. Hybridization was carried out under plastic coverslips, resulting in a fluorescent pattern that was quantified using a confocal laser scanner. 49 known and blinded samples of OPV DNA, representing different OPV species, and two VZV strains were tested. The oligonucleotide microarray hybridization technique identified reliably and correctly all samples. This new procedure takes only 3 h, and it can be used for parallel testing of multiple samples.
Keywords: Rapid identification, Nucleic acids diagnostics, Variola virus, Monkeypox virus, Chickenpox virus, Vaccinia, Biodefense
1. Introduction
Smallpox is a highly contagious disease with very high mortality. The disease is caused by variola virus (VARV), a member of the genus orthopoxvirus (OPV) of the family Poxviridae, subfamily Chordopoxvirinae (Breman and Henderson, 2002). Variola virus has two subtypes, variola major and variola minor, causing infections that differ in morbidity and mortality. Smallpox is the first and only viral disease in human history to have been eradicated completely; in 1980 the World Health Organization announced that the last known case had occurred in 1977 (Fenner, 1977, Henderson, 1980). After the smallpox eradication campaign ended, routine vaccination was stopped throughout the world. Consequently, a significant portion of the population—probably a large majority—is now completely unprotected from infection with variola virus as well as with related orthopoxviruses. While only two WHO-approved laboratories possess officially stocks of variola virus, there is no certainty that other covert repositories do not exist (Henderson, 2002). Considering the high mortality and transmissibility of smallpox, the absence of immunity in most of the human population, and the availability of an effective vaccine to protect perpetrators who might be tempted to use of smallpox virus as a weapon of bioterrorism, the reintroduction of smallpox has been identified as a real and frightening possibility. Although the overall risk of a deliberate release of variola virus has been considered relatively low (Breman and Henderson, 2002), the consequences of such an event would be enormous.
Apart from the variola virus, a number of other orthopoxviruses and viruses belonging to other families cause diseases with similar symptoms (fever and papulovesicular rash). There is a need to differentiate among them as an important part of any early smallpox detection system (Marennikova and Shchelkunov, 1998). For instance, monkeypox (MPXV), cowpox (CPXV), and buffalopox viruses can infect humans, causing a milder and less contagious illness than smallpox. Vaccinia virus (VACV) itself can also cause severe illness in immunocompromised individuals (Kesson et al., 1997). Camelpox virus (CMLV) and ectromelia virus (ECTV) are important animal pathogens (Fenner, 1995) that also require rapid detection. The high frequency of genomic recombination of OPVs, in conjunction with an exceptional ability to cross animal species barriers, substantially increases their pathogenic potential (Ball, 1987, Marennikova and Shchelkunov, 1998, Turner and Moyer, 1990). Thus, development of reliable and rapid methods for sensitive detection of poxviruses and accurate discrimination between different orthopoxvirus species is an important task.
Several approaches have been used to identify and differentiate orthopoxviruses. The current classification of orthopoxviruses is based both on their biological characteristics and genomic properties, such as DNA restriction maps (Baxby, 1975, Esposito and Knight, 1985), as well as by examination of viral antigens (Marennikova et al., 1988). Other methods to discriminate orthopoxviruses include differentiation of viral protein profiles following separation in sodium dodecyl-sulfate polyacrylamide gel with western blotting (Mukinda et al., 1997) and various other serological assays (Arita and Tagaya, 1977, Obijeski et al., 1973). All these methods require virus isolation and propagation, and clear-cut typing of the viruses is often difficult. More recently, PCR-based methods were developed to detect and type OPVs (Meyer et al., 1994, Mukinda et al., 1997). PCR products are analyzed by size and restriction endonuclease cleavage to identify species-specific nucleotide differences (Ibrahim et al., 1997, Loparev et al., 2001, Ropp et al., 1995, Wienecke et al., 2000). PCR-based methods are sensitive and relatively rapid, allowing detection of orthopoxviruses within 24 h.
Recently, three-dimensional polyacrylamide-gel microchips containing arrays of oligonucleotides specific to the viral CrmB gene were used to discriminate different orthopoxviruses (Lapa et al., 2002). We developed a different kind of oligonucleotide microarray, created on plain glass slides, for rapid detection and discrimination between orthopoxviruses pathogenic for humans based on the viral C23L/B29R gene (VACV Copenhagen strain nomenclature) (Shchelkunov et al., 2002) encoding the CC-chemokine binding protein (Graham et al., 1997, Shchelkunov, 2003, Smith et al., 1997). Our array incorporates diagnostic oligonucleotide probes specific to a number of orthopoxvirus species, and it also includes probes binding to a sequence of varicella-zoster virus (VZV, also known as human herpesvirus type 3 or HHV-3), because severe chickenpox is the infection most often confused with smallpox. Besides its speed and sensitivity, an advantage of the proposed method is its high redundancy; each virus species is identified by several independent probes, significantly increasing the overall robustness of the assay.
2. Materials and methods
2.1. Viruses and DNAs
The orthopoxvirus strains used in this study are summarized in Table 1 . DNAs from these strains were isolated as described previously (Lapa et al., 2002, Shchelkunov et al., 1998). Amplicons of the ORF 62 (124696-123257) gene of VZV were kindly provided by Dr Vladimir Loparev (National VZV Laboratory, CDC, Atlanta, GA).
Table 1.
OPVs DNA used in orthopox-microarray analysis
| OPV | Account number in genebank | Strains sequenced and used for oligoprobe designs | Strain checked on orthopox-microarray |
|---|---|---|---|
| Discriminated orthopoxvirus species | |||
| VARV | AY102974 | Kuw-5 | Kuw-5 |
| AY102970 | Congo-2 | Congo-2 | |
| AY102972 | Ind-3a | Ind-3a | |
| AY102966 | 12/62 | ||
| AY102969 | Butler | ||
| L22579 | Bangladesh-1975 | ||
| AY102971 | Congo-9 | ||
| X74352 | Garcia-1966 | ||
| AY102978 | Ngami | ||
| U18341 | Somalia-1977 | ||
| Monkeypox viruse | AY102985 | Congo 8 | Congo 8 |
| AY102986 | CDC# 77-666 | CDC# 77-666 | |
| AF380138 | Zaire-96-I-16 | Zaire-96-I-16 | |
| VACV | AY102943 | Wyeth | Wyeth |
| AY102942 | LIVP | LIVP | |
| AY102941 | EM-63 | EM-63 | |
| AY102939 | Elstree 3399 | Elstree 3399 | |
| AY102947 | CVI-78 | CVI-78 | |
| AY102944 | Chambon St-Yves Menard | Chambon St-Yves Menard | |
| AY102938 | Copenhagen | Copenhagen | |
| AY102948 | Buffalopox virus BP-1 | Buffalopox virus BP-1 | |
| U64724 | Rabbitpox virus strain Utrecht | ||
| U94848 | MVA | ||
| M35027 | Copenhagen, clone VC-2 | ||
| AF095689 | Tian tan | ||
| Y17730 | WR | ||
| AY102940 | Elstree 6664 | ||
| Cowpox A | AY102953 | Turk-74 | Turk-74 |
| AY102949 | Brighton | Brighton | |
| Cowpox B | AY102954 | Ep-1 | Ep-1 |
| AY102955 | Ep-2 | Ep-2 | |
| AY102957 | Ep-3 | Ep-3 | |
| AY102958 | Ep-4 | Ep-4 | |
| AY102959 | Ep-5 | Ep-5 | |
| AY102960 | Ep-6 | Ep-6 | |
| AY102961 | Ep-7 | Ep-7 | |
| AY102962 | Ep-8 | Ep-8 | |
| AY102956 | Ep-267 | Ep-267 | |
| AY102951 | Puma M-73 | Puma M-73 | |
| AY102950 | Ham-85 | ||
| AF531439 | RP-9 | ||
| Human herpesvirus 3 (VZV) | |||
| VZV | NC001348 | (1) Human | VZV wild-type |
| AY017047 | (2) VZV-Ellen | VZV-Oka | |
| AY005334 | (3) MSP | ||
| AY01375 | (4) VZV-VSD | ||
| AY016466 | (5) VZV-LAX2 | ||
| AY016460 | (6) VZV-80-2 | ||
| AF314220 | (7) VZV-32 | ||
| AF322638 | (8) VZV-Iceland | ||
| X04370 | (9) Varicella-Zoster virus | ||
| AF325440 | (11) VZV-VIA | ||
| AY016455 | (12) VZV-LAX1 | ||
| AY016449 | (13) VZV-Oka | ||
2.2. PCR amplification
The C23L/B29R gene was amplified from different OPV species using a set of universal primers presented in Table 2 . The standard PCR mixture (50 μl) contained 1.5 mm MgCl2, 50 mM KCl, 10 mM Tris–HCl (pH 8.3), 0.2 mM of each dNTP, 2.5 U Taq polymerase (SibEnzym, Russia), 100 nM of each primer, and 1 μl of viral DNA solution. The PCR was performed in an ABI 6700 thermocycler (Applied Biosystems Inc., Foster City, CA) using the following protocol: 2 min at 94 °C; ten cycles of 45 s at 94 °C, 45 s at 55–45 °C (with a 1 °C decrease after each cycle), and 1.5 min at 72 °C, followed by 25 cycles of 35 s at 94 °C, 45 s at 45 °C, and 1.5 min at 72 °C; and final incubation for 10 min at 72 °C. The resulting PCR products were analyzed by electrophoresis in 1% agarose gel prepared in 1× TAE buffer containing 0.2 μg/ml of ethidium bromide.
Table 2.
Specific primers used for amplification and sequencing of C23L/B29R gene for OPV species and of part of VZV ORF 62
| Primer names | Primer sequences | PCR product size (bp) | Primer sizes | Annealing temperature ( °C) |
|---|---|---|---|---|
| VZV | ||||
| 1099r (Reverse) | GCTGGTGTTGGACGC | 331 | 15 | 47 |
| 769f (Forward) | GTATTCACGGGGCGA | 15 | 45 | |
| Orthopoxviruses | ||||
| bioB29r-U1 (Forward) | GGTAGTTGCGATATACA | 1100 | 17 | 42 |
| ShortB29r-L1 (Reverse) | ACAGAAAAAGAAGAAGT | 17 | 37 | |
| bioB29r-U1 (Forward) | GGTAGTTGCGATATACA | 500 | 17 | 42 |
| B29r-465 (Reverse) | CATTTGGTGAATCCAAA | 17 | 40 | |
| bioB29r-465 (Forward) | TTTGGATTCACCAAATG | 600 | 17 | 40 |
| shortB29r-L1 (Reverse) | ACAGAAAAAGAAGAAGT | 17 | 37 | |
| B29-U1-cpxv (Forward) | CAATCTCTTATCATGTGGGTAATGT | 1177 | 25 | 48 |
| B29-L1-cpxv (Reverse) | ACATATTGAGACAGAAAAAGAAGAAGT | 27 | 48 | |
| B29-U1-mpxv (Forward) | ACGAAACGGATACAATCTCTTATCAT | 1208 | 26 | 51 |
| B29-L1-mpxv (Reverse) | GAGACAAGAGAGAAAGAGAATACGAATAG | 29 | 52 | |
| B29-U2 (Forward) | ATGAACTTTGGATTCACCAAAT | 22 | 46 | |
| B29-L2 (Reverse) | GGGAGAGTCCTTACCTTGTCC | 21 | 46 | |
2.3. Sequencing
DNA amplicons of different orthopoxvirus strains were purified by electrophoresis and eluted from agarose gels using a MinElute Gel Extraction kit (QiaGen, Valencia, CA). Each sequencing reaction was performed with a volume of 5 μl in a programmable GeneAmp PCR-system 6700 (Applied Biosystems) using the following program: 30 cycles of 10 s at 96 °C, 15 s at 46 °C, and 4 min at 60 °C. The reaction mixture contained 2 μl of a Big Dye v.2 kit (Applied Biosystems, USA), 1 pmol of primer, and 0.5 μg DNA. The reaction products were purified by ethanol precipitation. For this purpose, to each reaction mixture was added 4 μl water and 16 μl of 96% ethanol; each mixture was incubated for 15 min at a room temperature and then centrifuged at 20 000 rpm for 20 min. The supernatant was removed and the pellet resuspended in 50 μl of 70% ethanol and centrifuged at 20 000 rpm for 10 min. The supernatant was removed, and the pellet was vacuum-dried for 20 min and dissolved in 20 μl of TSR (Applied Biosystems). After denaturation for 2 min at 95 °C, the sample was placed on ice for 2 min. Nucleotide sequences of the DNA products were determined in an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, USA) using sequencer v. 4.0.5 software (Gene Codes, USA). The sequencing data were deposited in the GenBank Database.
2.4. Design of oligoprobes for orthopoxvirus discrimination
The C23L/B29R gene sequences of different orthopoxvirus species were aligned using clustalx software (Thompson et al., 1994) to find variable regions suitable to design species-specific oligoprobes. Another custom computer program was developed in our laboratory (oligoscan, to be published elsewhere) to screen for DNA sequences that identify oligonucleotides common to a group of viruses and not present in genomes of any other known viruses. That program does not require prior alignment of DNA sequences and performs an exhaustive comparison of all possible combinations of oligonucleotides with specified hybridization stringency parameters. Criteria for oligoprobe design were as follows: one or more mismatches with other orthopoxvirus species, length of 13–21 nucleotides, and predicted melting temperatures between 36 and 58 °C. A similar approach was used to design oligoprobes specific to the VZV ORF 62 gene using 13 VZV strain sequences available in GenBank. Sequences of all oligoprobes are listed in Table 3 .
Table 3.
Oligonucleotide probes for detection and discrimination OPV species and their differentiation from human herpesvirus type 3 (VZV)
| OPV species and VZV | Name | Sequence | Tm (°C) |
|---|---|---|---|
| Common to All OPV species | (1) 83POX | TCGTAAAAGTAGAAAATATATTCTAATTTATT | 50 |
| (2) 258POX | ATATGGGAATCGATGTTATTATCAAAGTCAC | 56 | |
| (3) 360pox | ATGATAAGATTTGYCAATC | 40–42 | |
| (4) 713POX | ATGTAGCGAAGAAGAGAAAGACAGC | 56 | |
| (5) 860pox | CGTATMGGAGACATGTG | 45–47 | |
| (6) 900pox | CTTGARGTCAAGGATGG | 45–47 | |
| (7) 1040pox | GGGTTAATCGARCGATT | 42–45 | |
| (8)1100pox | TCCGGTTTRCAAAACAAA | 41–43 | |
| (9) Pox-U1-2 | CCCGGTAGTTGCGATATACA | 52 | |
| (10) Pox-465-2 | ACTTTGGATTCACCAAATG | 45 | |
| Variola | (1) 467VAR | TCCATTATTGGCGGAGG | 47 |
| (2) 570VAR | TGTCCAGTGTGCCCCCAGG | 58 | |
| (3) 840VAR | AAGGAATCGTCTGATCT | 42 | |
| (4) 910var | AAATCGTGTGTCTGAATAAA | 44 | |
| (5) 915VAR-r | TGTGTCTGAATAAATAACTC | 44 | |
| (6) 924var | GTGTACCTGACACTATA | 42 | |
| (7) 974var | GATTTAGWCACACTACGAA | 45 | |
| (8) 982var | TCACACTACGAATTAAA | 37 | |
| (9) 1006var | CCTGACACTATACTCCG | 47 | |
| (10) 1012VAR | ACTATACTCCGGTTTACAAA | 46 | |
| Monkeypox | (1) 31MK-R2 | ACTTATAGTAAGTTTTTTT | 36 |
| (2) 200MK | TGTGCCTAGTGGCAGCTG | 53 | |
| (3) 212MK-R | TATGCCTACTAGTCTTC | 42 | |
| (4) 427mk | GTCGGCGCGGGTC | 48 | |
| (5) 432mk | CGCGGGTCTTAACATG | 46 | |
| (6) 680mk | AGTACTTGGGTCTAACA | 42 | |
| (7) 790mk | ATGTGTTAAGAACCTAGAG | 45 | |
| (8) 830mk | ACATGTGTGAGGAATCA | 42 | |
| (9) 959mk | CAGACACCGCACCAC | 47 | |
| (10)970MK-R | AAAATGGATGAGTTGGGT | 43 | |
| Cowpox A | (1) 7COW | TACTAATTCCAATTCCCA | 41 |
| (2) 14COW | TCCAATTCCCAGTATCC | 45 | |
| (3) 55COW | CCATAAATAAATACAATAATT | 39 | |
| (4) 124COW | TGAGGAAATAGAATCATCT | 42 | |
| (5) 155COW | ATGAAACAAATCGTCCT | 40 | |
| (6) 206COW | GCGGCAGTTGCTATCCC | 52 | |
| (7) 357COW | AGTCTGAAGACGAGTCAG | 47 | |
| (8) 865COW | TCAATTCAGCAAAACTTATA | 42 | |
| (9) 893COW | GAAGATGCAGCCGATGAT | 48 | |
| (10)956COW | CCACAAACTAAAAAAAGTG | 42 | |
| Cowpox B | (1) 970EP | AGATTCGGGTGAGTAGG | 45 |
| (2) 982erc | GCACACCACAAATTAAA | 40 | |
| (3) 1040EP | GCACACCACAAATTAAAA | 41 | |
| Vaccinia | (1) 957Vac-r | CAGGCACACCACGAA | 45 |
| (2) 982vac-r | GCACACCACGAATTAA | 41 | |
| (1) 957Vac | CAGGCACACCACGAAT | 46 | |
| (2) 982vac | GCACACCACGAATTAAA | 42 | |
| VZV | (1) 790VZV | GACGCGGGTCTTGGGGC | 57 |
| (2) 811VZV | GCGGGTACACACGGTGTA | 53 | |
| (3) 847VZV | GCACAAACACAGGGGTTG | 50 | |
| (4) 874VZV | GTACAGGTTGGCAAACGC | 50 | |
| (5) 893VZV | GACGATGCAAACACGGCC | 50 | |
| (6) 932VZV | GTCTCGATACGAGCAAAACT | 53 | |
| (7) 961VZV | TTCTGTGACCGCCGAGTCT | 53 | |
| (8) 984VZV | ATCGGACGACGGCCTGG | 54 | |
| (9) 1028VZV | TACACGTGATACTGAGACA | 57 | |
| (10)1066VZV | CCACCTCTCGAGGGCCA | 54 |
To immobilize oligoprobes on aldehyde-coated slides (Cel Associates Inc., Pearland, TX), the 5′-end of each probe was modified with an amino-link group (TFA amino-link CE reagent, PE Applied Biosystems) during automated oligonucleotide synthesis. The amino-modified oligonucleotides were purified by extraction with an equal volume of chloroform and precipitated by adding 10 volumes of acetone containing 2% lithium chlorate (Daniliuk et al., 1986).
2.5. Microarray fabrication
Oligonucleotide microarrays were printed using a contact micro-spotting robotic system (PixSys 5500, Cartesian Technologies, Inc., Irvine, CA), equipped with a single microspotting pin (SMP7, ArrayIt, Sunnyvale, CA). The spotting solution contained 60 μM of specific oligoprobe and 10 μM of a quality control (QC) oligonucleotide in 0.25 M acetic acid. The average size of spots was 250 μm. The slides were dehydrated for 30 min at 80 °C and treated for 15 min with a freshly-prepared 0.25% solution of NaBH4 to bind the oligoprobes irreversibly to the glass surface. Then the slides were washed once for one min with 0.2% aqueous solution of SDS and twice with distilled water (1 min) to remove unbound oligonucleotides. Non-specific spots marked the array limits (1× Spotting solution, ArrayIt, Sunnyvale, CA in 0.25 M acetic acid).
2.6. Design of orthopoxvirus microarray
Oligoprobes specific to individual species were placed in six separate rows of the microarray. The first row contained ten broadly-specific probes reacting with DNA of all orthopoxvirus species; each of the second, third, and fourth rows included ten probes specific for Variola virus, monkeypox virus, and cowpox virus (group A) viruses, respectively; the first three oligoprobes in the fifth row identified Cowpox virus-B followed by four oligoprobes specific for vaccenia virus only, the last row contained ten oligoprobes to detect VZV (see Fig. 4A).
Fig. 4.

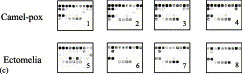
Patterns of detection and discrimination between the OPV species pathogenic for humans and their differentiation from HHV 3 (VZV) (A) composition microarray for OPV species identification. (B) Patterns of discriminated species; B1 VARV strain Congo-2, B2; VARV strain Ind-3A, B3; VARV strain Kuw-5, B4; VACV strain Elstree 3399, B5; VACV strain Copenhagen, B6; VACV strain CVI-78, B7; CPXV-A strain CPV Turk-74, B8; CPXV-A strain CPV Brighton, B9; CPXV-B strain EP-1, B10; CPXV-B strain EP-2, B11; CPXV-B strain EP-7, B12; MPXV strain CDC# 77-666, B13; VZV strain Oka. B14; Pattern of the mixture hybridization of VARV strain Ind-3A and VZV strain Oka. (C) Patterns of unclassified OPVs detected in blinded samples; C1; CmPV CP-5 (Camelpox), C2; CmPV CP-1 (Camelpox), C3; CmPV 1269/95 (Camelpox), C4; CmPV 1231 (Camelpox), C5; ECT. 33221 (Ectomelia), C6; ECT.4908 (Ectomelia), C7; ECT. MP-1 (Ectomelia), C8; ECT. MP-2 (Ectomelia).
2.7. Synthesis of fluorescently labeled DNA samples
The OPV fluorescently-labeled DNA samples for microarray analysis were prepared by PCR in the presence of the Cy5-dCTP (Amersham Bioscience, UK). The standard PCR mixture contained 50 μl of 1×AmpliTaq PCR buffer with 2.5 mM MgCl2 (Qiagen, Chatsworth, CA), 400 nM each of forward and reverse primers, 200 μM each of dATP, dGTP, and dTTP, 20 μM dCTP, 20 μM Cy5-dCTP, and 1 U of AmpliTaq polymerase. Amplification was performed using a Gene Amp 9700 thermocycler (Applied Biosystems Inc.) in the following conditions: initial DNA denaturation at 94 °C for 30 s followed by 35 cycles (94 °C for 30 s, 42 °C for 30 s, 72 °C for 1 min), and a final extension for seven min at 72 °C. The PCR products were analyzed by electrophoresis in 1% agarose gel. The forward primer contained biotin at the 5′-end for separation of DNA strands on streptavidin-coated GenoPrep magnetic beads (GenoVision Inc.,West Chester, PA) according to the manufacturer's protocol. The beads were washed twice with 1× TE, and the single-stranded fluorescent DNA was eluted with 50 μl of 0.1 N NaOH and purified using the QIAquick PCR purification kit (Qiagen, Chatsworth, CA), dried in vacuum, and reconstituted in 5 μl of water.
In some experiments, fluorescently-labeled single-stranded DNA was synthesized by asymmetric PCR. The conditions used for asymmetric PCR were similar to those for the standard reaction except that the concentrations of forward and reverse primers were 60 and 600 nM (1:10 molar ratio), respectively.
The same conditions described above were used to prepare of fluorescent DNA from the VZV ORF 62 gene with 769F (forward) and 1099R (reverse) primers (Table 2), designed to match highly conserved regions of the gene (Sequences were available in GenBank for 13 different VZV strains).
2.8. Hybridization conditions
Hybridization of Cy5-labeled DNA samples with oligonucleotides on the microchip was carried out as described previously (Volokhov et al., 2002). Briefly, the final hybridization mixture contained Cy5-labeled DNA at varying concentrations with 0.5 μM of the Cy3-QC oligonucleotide in 1× hybridization solution (5× Denhardt solution, 6× SSC, 0.1% Tween 20). The mixture was preheated at 95 °C for 30 s to denature dsDNA, followed by chilling on ice. The hybridization mixture was applied to the array and covered with a 5×5-mm plastic cover slip to prevent drying of the probe during incubation in the hybridization cassette (TeleChem International, Inc., Sunnyvale, CA). The microchip was incubated at 45 °C for 30 min in a water bath. After hybridization, the slides were washed once with 6× SSC containing 0.2% Tween 20 for 1 min at room temperature, once with 6× SSC for 1 min, once with 2× SSC, and then dried with an air stream to remove any remaining solution.
2.9. Microarray scanning
Fluorescent microchip images were acquired by scanning the slides with ScanArray 5000 (Perkin–Elmer Life Sciences, Boston, MA) using light of 570 nm (for Cy3) and 694 nm (for Cy5). Images were analyzed using quantarray software (Perkin–Elmer Life Sciences, Boston, MA). Background fluorescence readings obtained from the region surrounding each spot were subtracted, and the absolute value of the Cy5 fluorescence signal from each oligoprobe was divided by the Cy3 signal from the QC probe of the same spot to compensate for possible variations in spot size. Fluorescent signals exceeding the average background by a statistically significant margin (P<0.01) were considered positive.
3. Results
3.1. Design of PCR-primers and oligonucleotide probes
Poxviruses and herpesviruses are two unrelated groups that share no significant homology in their DNA sequences. To develop a microarray-based assay for simultaneous identification and discrimination among orthopoxvirus species and VZV, we chose two different gene products, one specific for orthopoxviruses and the other specific for VZV. The C23L/B29R gene is common to all orthopoxviruses and encodes the viral CC-chemokine binding protein (Graham et al., 1997, Shchelkunov, 2003, Smith et al., 1997). To identify VZV, we selected the ORF 62 gene, a gene present in all herpesviruses (Moriuchi et al., 1994), coding for IE 62 protein—a direct activator of transcription (Cohen et al., 1993, Perera et al., 1993). Within the C23L/B29R gene sequences of different OPV species are several species-specific differences. clustalx analysis of C23L/B29R gene sequences showed that cowpoxviruses are represented on the phylogenic tree by two separate genetically distant subgroups that we designated as cowpox A and cowpox B. Attempts to find regions common to cowpox A and B but different from those of other OPVs failed due to the high genetic variability between these cowpox subgroups. Therefore, we designed two sets of oligoprobe microarrays specific for each cowpox subgroup (Table 3). Combining these two oligoprobe sets allowed us to detect and discriminate different cowpox genetic variants from other orthopoxviruses. The orthopoxvirus group primers, designed BioB29r-U1 and ShortB29r-L1 (Table 2), efficiently amplified the target gene from OPV DNA specimens available in the collection of the State Research Center of Virology and Biotechnology “Vector” (Koltsovo, Russia). These DNA samples were re-amplified using symmetric or asymmetric PCR to generate fluorescent DNA products for microarray hybridization (Fig. 1 ). The sizes of amplicons varied somewhat, depending on the OPV species, but all were about 1 100 base pairs long.
Fig. 1.
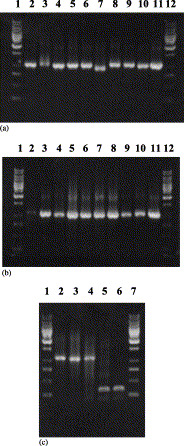
Amplification of C23L/B29R genes of different OPV strains with specific primers bioB29r-U1and shortB29r-L1 and amplification of the region in ORF 62 of chickenpox virus with primers 1099r and 769f. (A) 1 Kb DNA-marker (lane 1), CPXV strains RP-9 (lane 2), MPXV strain CDC# 77-666 (lane 3), VACV strains Wyeth (lane 4); Chambon St-Yves Menard (lane5), EP-267 (lane 6); EM-63 (lane 7), EP-7 (lane 8), EP-5 (lane 9), EP-2 (lane 10), Elstree 3399 (lane 11), 1Kb DNA-marker (lane 12). (B) 1 Kb DNA-marker (lane 1), VARV strain Kuw-5 (lane 2), VACV strains LIVP clone 4 (lane 3), CPXV strains Turk-74 (lane 4), EP-4 (lane 5) CVI-78 (lane 6), Copenhagen (lane 7); EP-3 (lane 8), EP-8 (lane 9), EP-6 (lane 10), EP-1 (lane 11), 1 Kb DNA-marker (lane 12). (C) 1 Kb DNA-marker (lane 1), VARV strains Congo-2 (lane 2), CPXV strain Brighton (lane 3), Ind-3A (lane 4), VZV OKA (lane 5), VZV wild type (lane 6), 1 Kb DNA-marker (lane 7).
3.2. Optimization of microarray assay
Several protocols for microarray fabrication, fluorescent sample preparation, and hybridization conditions were developed and optimized. Fig. 2 A shows the influence of oligoprobe concentration in the spotting mixture on the microarray hybridization. The data showed that essentially complete saturation of the aldehyde-coated glass surface was achieved with a 100-μM concentration of oligoprobe; a further increase in concentration to 200 μM resulted in only a slightly stronger hybridization (17% increase). The oligoprobe spotting concentration used in this study (60 μM) yielded efficient hybridization (about 60% of the maximum value), while reducing the consumption of oligonucleotides.
Fig. 2.
Dependence of fluorescence intensity on concentrations of labeled DNA amplicon and oligoprobe. (A) Dependence of fluorescence intensity on oligoprobe concentrations, (B) dependence of fluorescence intensity on fluorescent DNA concentrations.
Fig. 2B shows the influence of concentration of single-stranded fluorescent DNA in the hybridization mixture on the hybridization efficiency. These data allowed us to estimate the minimal concentration of fluorescent sample that can be confidently detected (detection defined as a DNA generating a corrected fluorescent signal at least twice the background fluorescence), found to be 0.005 μM for a 1000-bp-long ssDNA. The dynamic range of fluorescent sample concentrations was from 0.005 to 0.3 μM. Since the typical hybridization volume was about 5 μM, the minimal amount of ssDNA required for confident detection was 25 pmol.
The predicted melting temperatures of the oligoprobes varied from 37 to 57 °C (Table 3), therefore, we sought an optimal hybridization temperature that would provide efficient hybridization and high specificity for every spot on the array. We tested several hybridization temperatures: 25, 35, 45, and 65 °C. Hybridization at 45 °C discriminated best between OPV species.
3.3. Optimization of fluorescent probe preparation
We compared three different PCR-based approaches for preparing fluorescently-labeled DNA samples: dsDNA products of symmetric PCR, ssDNA products of asymmetric PCR, and ssDNA prepared from dsDNA by strand separation on streptavidin-coated magnetic beads (see above). The fluorescently-labeled DNAs prepared by the three methods were purified by Qiaquick PCR purification kit (Quiagen, CA), dried in a vacuum and diluted in 5 μl of water. One μl of each DNA sample was mixed with 1 μl of hybridization buffer and hybridized with a microarray using the conditions described above. The results of this comparison are presented in Fig. 3 . The hybridization was much lower with fluorescent dsDNA than with ssDNA samples (Fig. 3A–B. DL, BL, AL). This reduction might have resulted from preferential reannealing of labeled ssDNA, preventing its binding to the microchip.
Fig. 3.
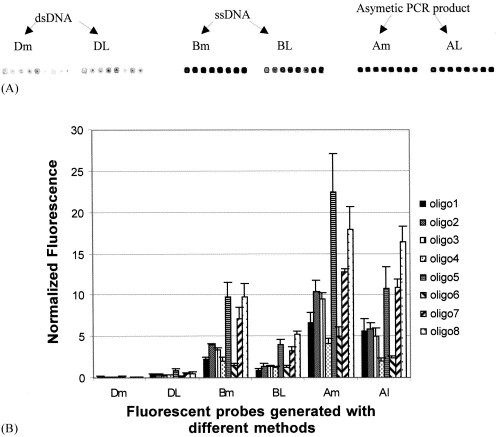
Comparison of the intensity of hybridization with fluorescent DNA amplicon generated by different methods from OPV C23L/B29R gene (A) Image of microarray profile (B) Quantification of fluorescence by quantarray Software. Fluorescent DNA amplicons used in this study were; Dm, dsDNA generated with multiplex PCR; DL, dsDNA PCR Product; Bm, ssDNA separated from dsDNA multiplex PCR product on streptavidin beads; BL, ssDNA separated from dsDNA PCR product on streptavidin beads; Am, product of multiplex asymmetric PCR; Al, product of asymmetric PCR.
The size of fluorescent DNA was also found to be important for efficient hybridization. After early experiments amplifying the complete 1100 sequence in a single reaction, we tried amplifying the C23L/ B29R gene as two DNA segments of 500 nucleotides and 600 nucleotides, using a combination of two primer pairs, bioB29r-U1 (For) / B29r-465 (Rev) and bioB29r-465 (For) / shortB29r-L1 (Rev) instead of the single pair of primers bioB29r-U1 (For) and shortB29r-L1 used for full-length amplification (Table 2). The two-amplicon approach yielded hybridization signals approximately twice those of the full-length PCR product (Fig. 3A–B. Dm, Bm, Lm). This two-amplicon approach was then used for subsequent orthopoxvirus discrimination experiments.
3.4. Evaluation of orthopoxvirus microarrays using reference viruses
Fluorescent DNAs were generated by asymmetric PCR (see above) using 26 reference OPV strains (Table 1) and hybridized with the chip as described above. The hybridization pattern for each orthopoxvirus species is shown in Fig. 4 B (images 1–12). These results demonstrate that the oligonucleotide microarray simultaneously and unambiguously detected and discriminated four orthopoxvirus species pathogenic for humans (VARV, MPXV, CPXV, and VACV). Analysis of hybridization patterns showed low but consistent cross hybridization of vaccinia-specific oligoprobes with DNAs of some other orthopoxvirus species (Fig. 4B). That might be explained by the fact that those sequences differed by only a single nucleotide in the region of oligoprobe binding. Although this cross-hybridization slightly complicated the interpretation of microarray data, the discrimination of VACV was nevertheless unambiguous.
To hybridize VZV strains, the fluorescent DNAs were generated by asymmetric PCR (as described above) from VZV-Oka vaccine and VZV wild-type strains (Table 1) using primers 1099r (Rev) and 769f (For) (Table 2). The hybridization of VZV DNAs showed that the microarray detected chickenpox virus and differentiated it from orthopoxviruses (Fig. 4B, image 13).
To evaluate the ability of the microarray to detect and discriminate orthopoxviruss and VZV simultaneously, fluorescently-labeled DNA was prepared by amplification of an artificial mixture containing amplicons of orthopoxviruses and VZV using a mixture of primers. The products of this reaction were purified and hybridized with the microchip. As shown in Fig. 4B (image 14), the microarray detected both orthopox and chickenpox viruses present in a mixture.
3.5. Identification of coded orthopoxvirus samples
To validate the ability of the microarray assay to discriminate OPV species, 23 amplicons of the C23L/B29R gene generated from orthopoxvirus templates from different OPVs were prepared and submitted in a coded format for analysis. The identity of the samples was revealed to the operator only after the final results of microarray identification were recorded. The results of this study are shown in Table 4 . Almost all coded DNA samples were unambiguously identified by hybridization profile. However, a few samples gave patterns of hybridization that did not match those of the samples tested previously (Fig. 4C, Images 1–8). The ambiguous samples were identified as OPV by their hybridization with oligoprobes common for all viruses of this genus. Patterns of some samples resembled those of VACV, but they showed hybridized with oligoprobes specific for other orthopoxvirus species. After the codes were revealed, all those samples were confirmed to have been derived from orthopoxvirus species not tested during the development of the microarray (Table 4).
Table 4.
Identification of coded OPV samples using oligonucleotide-microarray
| Sample codes | Species identification | Genbank accession number | Strains names |
|---|---|---|---|
| 1 | Variola | AY102969 | Butler |
| 2 | Variola | AY102973 | India 378 |
| 3 | Monkeypox | AF380138 | Zaire-96-I-16 |
| 4 | Monkeypox | AY102985 | Congo 8 |
| 5 | Vaccinia | AY102943 | Weyth |
| 6 | Vaccinia | AY102942 | LIVP |
| 7 | Vaccinia | AY102938 | Copenhagen |
| 8 | Cowpox B | N\A | OPV-90/1 |
| 9 | Cowpox B | AY102958 | EP-4 |
| 10 | Cowpox B | N\A | OPV-91/1 |
| 11 | Cowpox B | N\A | OPV-98/5 |
| 12 | Cowpox B | N\A | OPV 89/4 |
| 13 | Cowpox A | AF531438 | OPV-88 |
| 14 | Cowpox A | N\A | OPV 89/3 |
| 15 | Cowpox A | N\A | OPV-90/4 |
| 16 | Unclassified Orthopoxviruses | AY102987 | ECTV MP-1 |
| 17 | AY102988 | ECTV MP-2 | |
| 18 | AY102991 | ECTV #33221 | |
| 19 | AY102989 | ECTV #4908 | |
| 20 | AY102983 | CMLV CP-5 | |
| 21 | AY102980 | CMLV CP-1 | |
| 22 | AY102982 | CMLV 1260 | |
| 23 | AY102981 | CMLV 1231 |
4. Discussion
Poxviruses are considered to be among the most dangerous microorganisms that might be used for a bioterrorist attack (Breman and Henderson, 1998, Orent, 1998). Thus, development of rapid and accurate methods to detect, identify and genotype pox viruses and other viruses that cause similar illnesses is important. Such methods would help to initiate a rapid defensive response, prevent the possible spread of smallpox, and avoid false alarms. Several PCR-based methods to detect and discriminate orthopoxviruses have been reported (Meyer et al., 1994, Mukinda et al., 1997), most of them based on an analysis of the molecular sizes of multiple PCR products. The main disadvantage of such an approach is that the sizes and restriction endonuclease patterns of DNAs amplified from OPVs are potentially variable. The situation becomes even more complicated when multiple primers are used to amplify several genes simultaneously (multiplex PCR). As a general rule, increased complexity of a PCR primer mixture leads to generation and amplification of non-specific DNA products (resulting from occasional mispriming Elnifro et al., 2000) more often than does amplification with a single primer pair. That may ultimately result in erroneous interpretation of PCR data. In addition, the discriminating power of most PCR-based methods may be insufficient to distinguish between viruses with a low level of genetic divergence. Thus, a better sequence-specific approach may be helpful. In the present study we developed a combined PCR-microarray method that substantially improved specificity of the assay.
A similar approach was recently described to discriminate OPVs based on differences of a few nucleotides in the CrmB gene, detected using three-dimensional polyacrylamide gel pads (Lapa et al., 2002). However, pad-based microchips have a major disadvantage: the longer time needed for the oligonucleotides to hybridize with fluorescently-labeled DNA samples due to the slow diffusion of DNA molecules into the gel. In contrast, the immobilization of oligoprobes onto a plain aldehyde-coated glass surface formed high-density monolayers, easily accessible for hybridization with DNA samples. Such microarrays allow reactions between fluorescently-labeled DNA samples and oligoprobes to proceed quickly, reducing the overall time needed to complete an assay to about 3 h. That speed favorably distinguishes the assay from earlier procedures used to detect poxviruses (Baxby, 1975, Lapa et al., 2002, Marennikova et al., 1988, Meyer et al., 1994, Mukinda et al., 1997, Obijeski et al., 1973, Ropp et al., 1995). Amplification of the C23L/B29R gene of OPVs allowed us to increase substantially the number oligoprobes used to identify each orthopoxvirus species (see Table 3). Selecting several oligoprobes of high specificity for portions of a single gene enabled simultaneous detection of more than one species of orthopoxvirus in the same sample—something that proved impossible to do with a chip based on the CrmB gene and combinations of oligoprobes to detect each species. Incorporation of VZV-specific oligoprobes into the same microchip design also enabled us to discriminate chickenpox virus from orthopoxvirus quickly, even when both viruses were present in the same sample. The potential importance of rapidly detecting both VZV and an orthopoxvirus was demonstrated dramatically by a recent situation in which simultaneous outbreaks of human monkeypox and chickenpox occurred in the same population (Hutin et al., 2001).
The simplicity of the proposed microarray protocol, together with its use of a large number of species-specific oligoprobes and its ability to analyze multiple samples in a short time, offers clear advantages. The use of asymmetric PCR to generate fluorescent samples simplified the process further and reduced the time needed for sample preparation.
The high sensitivity provided by use of PCR to prepare labeled DNA and the ability of the oligonucleotide microarray to detect and unambiguously discriminate quickly among four orthopoxvirus species most dangerous to human health demonstrated that this method may provide a valuable tool for defending the population against infections with smallpox and other poxviruses.
Acknowledgements
This work was supported by grants from the International Scientific and Technology Center to S.S. (#1987p, #2504p), and from the Defense Advanced Research Project Agency (DARPA) to K.C. We thank Dr Svetlana Marennikova for providing OPV strains, Dr Hermann Meyer and Dr Joseph Esposito for some OPV genomic DNAs, and Dr Vladimir Loparev for VZV DNA amplicons.
References
- Arita M., Tagaya I. Structural polypeptides of several strains of orthopoxvirus. Microbiol. Immunol. 1977;21(6):343–346. doi: 10.1111/j.1348-0421.1977.tb00298.x. [DOI] [PubMed] [Google Scholar]
- Ball L.A. High-frequency homologous recombination in vaccinia virus DNA. J. Virol. 1987;61(6):1788–1795. doi: 10.1128/jvi.61.6.1788-1795.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxby D. Identification and interrelationships of the variola/vaccinia subgroup of poxviruses. Prog. Med. Virol. 1975;19:215–246. [PubMed] [Google Scholar]
- Breman J.G., Henderson D.A. Poxvirus dilemmas—monkeypox, smallpox, and biologic terrorism. New Engl. J. Med. 1998;339(8):556–559. doi: 10.1056/NEJM199808203390811. [DOI] [PubMed] [Google Scholar]
- Breman J.G., Henderson D.A. Diagnosis and management of smallpox. New Engl. J. Med. 2002;346(17):1300–1308. doi: 10.1056/NEJMra020025. [DOI] [PubMed] [Google Scholar]
- Cohen J.I., Heffel D., Seidel K. The transcriptional activation domain of varicella-zoster virus open reading frame 62 protein is not conserved with its herpes simplex virus homolog. J. Virol. 1993;67(7):4246–4251. doi: 10.1128/jvi.67.7.4246-4251.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniliuk N.K., Iastrebov S.I., Artamonova T.P., Popov S.G. A simplified variant of the Maxam-Gilbert method for determining the primary structure of oligonucleotides and DNA fragments. Bioorg. Khim. 1986;12(9):1185–1188. [PubMed] [Google Scholar]
- Elnifro E.M., Ashshi A.M., Cooper R.J., Klapper P.E. Multiplex PCR: optimization and application in diagnostic virology. Clin. Microbiol. Rev. 2000;13(4):559–570. doi: 10.1128/cmr.13.4.559-570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito J.J., Knight J.C. Orthopoxvirus DNA: a comparison of restriction profiles and maps. Virology. 1985;143(1):230–251. doi: 10.1016/0042-6822(85)90111-4. [DOI] [PubMed] [Google Scholar]
- Fenner F. The eradication of smallpox. Prog. Med. Virol. 1977;23:1–21. [PubMed] [Google Scholar]
- Fenner F. In: Fields B.N., Knipe D.M., Howley P.M., editors. Vol. 2. Lippincott-Raven; Philadelphia: 1995. Poxviruses; pp. 2673–2702. (Fields Virology). [Google Scholar]
- Graham K.A., Lalani A.S., Macen J.L., Ness T.L., Barry M., Liu L.Y., Lucas A., Clark-Lewis I., Moyer R.W., McFadden G. The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology. 1997;229(1):12–24. doi: 10.1006/viro.1996.8423. [DOI] [PubMed] [Google Scholar]
- Henderson D.A. The history of smallpox eradication. Henry E. Sigerist Suppl. Bull. Hist. Med. 1980;(4):99–114. [PubMed] [Google Scholar]
- Henderson D.A. Countering the posteradication threat of smallpox and polio. Clin. Infect. Dis. 2002;34(1):79–83. doi: 10.1086/323897. [DOI] [PubMed] [Google Scholar]
- Hutin Y.J., Williams R.J., Malfait P., Pebody R., Loparev V.N., Ropp S.L., Rodriguez M., Knight J.C., Tshioko F.K., Khan A.S., Szczeniowski M.V., Esposito J.J. Outbreak of human monkeypox, Democratic Republic of Congo, 1996 to 1997. Emerg. Infect. Dis. 2001;7(3):434–438. doi: 10.3201/eid0703.010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M.S., Esposito J.J., Jahrling P.B., Lofts R.S. The potential of 5′ nuclease PCR for detecting a single-base polymorphism in orthopoxvirus. Mol. Cell Probes. 1997;11(2):143–147. doi: 10.1006/mcpr.1996.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesson A.M., Ferguson J.K., Rawlinson W.D., Cunningham A.L. Progressive vaccinia treated with ribavirin and vaccinia immune globulin. Clin. Infect. Dis. 1997;25(4):911–914. doi: 10.1086/515534. [DOI] [PubMed] [Google Scholar]
- Lapa S., Mikheev M., Shchelkunov S., Mikhailovich V., Sobolev A., Blinov V., Babkin I., Guskov A., Sokunova E., Zasedatelev A., Sandakhchiev L., Mirzabekov A. Species-level identification of orthopoxviruses with an oligonucleotide microchip. J. Clin. Microbiol. 2002;40(3):753–757. doi: 10.1128/JCM.40.3.753-757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loparev V.N., Massung R.F., Esposito J.J., Meyer H. Detection and differentiation of old world orthopoxviruses: restriction fragment length polymorphism of the crmB gene region. J. Clin. Microbiol. 2001;39(1):94–100. doi: 10.1128/JCM.39.1.94-100.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marennikova S.S., Nagieva F.G., Matsevich G.R., Shelukhina E.M., Khabakhpasheva A.A., Platonova G.M. Monoclonal antibodies to monkey pox virus: preparation and application. Acta Virol. 1988;32(1):19–26. [PubMed] [Google Scholar]
- Marennikova S.S., Shchelkunov S.N. Orthopoxviruses Pathogenic for Humans. KMK Scientific Press Ltd; Moscow, Russia: 1998. [Google Scholar]
- Meyer H., Pfeffer M., Rziha H.J. Sequence alterations within and downstream of the A-type inclusion protein genes allow differentiation of Orthopoxvirus species by polymerase chain reaction. J. Gen. Virol. 1994;75(Pt 8):1975–1981. doi: 10.1099/0022-1317-75-8-1975. [DOI] [PubMed] [Google Scholar]
- Moriuchi M., Moriuchi H., Straus S.E., Cohen J.I. Varicella-zoster virus (VZV) virion-associated transactivator open reading frame 62 protein enhances the infectivity of VZV DNA. Virology. 1994;200(1):297–300. doi: 10.1006/viro.1994.1190. [DOI] [PubMed] [Google Scholar]
- Mukinda V.B., Mwema G., Kilundu M., Heymann D.L., Khan A.S., Esposito J.J. Re-emergence of human monkeypox in Zaire in 1996. Monkeypox epidemiologic working group. Lancet. 1997;349(9063):1449–1450. doi: 10.1016/S0140-6736(05)63725-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obijeski J.F., Palmer E.L., Gafford L.G., Randall C.C. Polyacrylamide gel electrophoresis of fowlpox and vaccinia virus proteins. Virology. 1973;51(2):512–516. doi: 10.1016/0042-6822(73)90454-6. [DOI] [PubMed] [Google Scholar]
- Orent W. Escape from Moscow. Sciences (New York) 1998;38:26–31. [Google Scholar]
- Perera L.P., Mosca J.D., Ruyechan W.T., Hayward G.S., Straus S.E., Hay J. A major transactivator of varicella-zoster virus, the immediate-early protein IE62, contains a potent N-terminal activation domain. J. Virol. 1993;67(8):4474–4483. doi: 10.1128/jvi.67.8.4474-4483.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropp S.L., Jin Q., Knight J.C., Massung R.F., Esposito J.J. PCR strategy for identification and differentiation of small pox and other orthopoxviruses. J. Clin. Microbiol. 1995;33(8):2069–2076. doi: 10.1128/jcm.33.8.2069-2076.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchelkunov S.N. Immunomodulatory proteins of orthopoxviruses. Mol. Biol. (Moscow) 2003;37:37–48. [PubMed] [Google Scholar]
- Shchelkunov S.N., Safronov P.F., Totmenin A.V., Petrov N.A., Ryazankina O.I., Gutorov V.V., Kotwal G.J. The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology. 1998;243(2):432–460. doi: 10.1006/viro.1998.9039. [DOI] [PubMed] [Google Scholar]
- Shchelkunov S.N., Totmenin A.V., Safronov P.F., Mikheev M.V., Gutorov V.V., Ryazankina O.I., Petrov N.A., Babkin I.V., Uvarova E.A., Sandakhchiev L.S., Sisler J.R., Esposito J.J., Damon I.K., Jahrling P.B., Moss B. Analysis of the monkeypox virus genome. Virology. 2002;297(2):172–194. doi: 10.1006/viro.2002.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.A., Smith T.D., Smolak P.J., Friend D., Hagen H., Gerhart M., Park L., Pickup D.J., Torrance D., Mohler K., Schooley K., Goodwin R.G. Poxvirus genomes encode a secreted, soluble protein that preferentially inhibits beta chemokine activity yet lacks sequence homology to known chemokine receptors. Virology. 1997;236(2):316–327. doi: 10.1006/viro.1997.8730. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner P.C., Moyer R.W. The molecular pathogenesis of poxviruses. Curr. Top. Microbiol. Immunol. 1990;163:125–151. doi: 10.1007/978-3-642-75605-4_5. [DOI] [PubMed] [Google Scholar]
- Volokhov D., Rasooly A., Chumakov K., Chizhikov V. Identification of listeria species by microarray-based assay. J. Clin. Microbiol. 2002;40(12):4720–4728. doi: 10.1128/JCM.40.12.4720-4728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienecke R., Wolff H., Schaller M., Meyer H., Plewig G. Cowpox virus infection in an 11-year-old girl. J. Am. Acad. Dermatol. 2000;42(5 Pt.):892–894. doi: 10.1016/s0190-9622(00)90265-2. [DOI] [PubMed] [Google Scholar]