

Abstract
The control of smallpox was achieved using live vaccinia virus (VV) vaccine, which successfully eradicated the disease worldwide. As the variola virus no longer exists as a natural infection agent, mass vaccination was discontinued after 1980. However, emergence of smallpox outbreaks caused by accidental or deliberate release of variola virus has stimulated new research for second-generation vaccine development based on attenuated VV strains. Considering the closely related animal poxviruses that also arise as zoonoses, and the increasing number of unvaccinated or immunocompromised people, a safer and more effective vaccine is still required. With this aim, new vectors based on avian poxviruses that cannot replicate in mammals should improve the safety of conventional vaccines, and protect from zoonotic orthopoxvirus diseases, such as cowpox and monkeypox. In this study, DNA and fowlpox (FP) recombinants that expressed the VV L1R, A27L, A33R, and B5R genes were generated (4DNAmix, 4FPmix, respectively) and tested in mice using novel administration routes. Mice were primed with 4DNAmix by electroporation, and boosted with 4FPmix applied intranasally. The lethal VVIHD-J strain was then administered by intranasal challenge. All of the mice receiving 4DNAmix followed by 4FPmix, and 20% of the mice immunized only with 4FPmix, were protected. The induction of specific humoral and cellular immune responses directly correlated with this protection. In particular, higher anti-A27 antibodies and IFNγ-producing T lymphocytes were measured in the blood and spleen of the protected mice, as compared to controls. VVIHD-J neutralizing antibodies in sera from the protected mice suggest that the prime/boost vaccination regimen with 4DNAmix plus 4FPmix may be an effective and safe mode to induce protection against smallpox and poxvirus zoonotic infections. The electroporation/intranasal administration routes contributed to effective immune responses and mouse survival.
Keywords: Recombinant vaccines; Fowlpox virus; Prime/boost; OPXV vaccine; L1R, A27L, A33R, B5R VV genes; Intranasal mucosal vaccination
1. Introduction
New infectious diseases are continuously emerging, and the lack of efficient immune prevention requires development of novel vaccines and vaccination strategies. Due to successful eradication of smallpox worldwide, the use of the vaccinia virus (VV) vaccine that was administered by scarification was discontinued or was replaced by VV-derived cultured immunogens (Weltzin et al., 2003). However, serious side effects can result from the traditional vaccine (Ferrier-Rembert et al., 2008), especially in immunocompromised people (Jacobson et al., 2008, Lane and Goldstein, 2003) and in patients with skin diseases (Schulze et al., 2007). Thus, a new generation of attenuated vaccines has been developed to decrease undesired effects, and to face the potential re-emergence in the human population through accidental or deliberate release of orthopoxviruses (OPXVs) (Megid et al., 2012, Vogel et al., 2012, Whitley, 2003). In this regard, although not as lethal as the variola virus, the monkeypox virus (MPXV) also represents a threat to public health, as it causes mortality in underdeveloped countries (Reed et al., 2004) and can become a potential bioweapon if adapted to grow and spread in humans (Lewis-Jones, 2004).
Previous studies have demonstrated that, after the conventional vaccination, neutralizing antibodies were mainly raised against surface proteins of both the VV extracellular virions (e.g., A33, B5) and the intracellular mature virions released after cell lysis (e.g., L1, A27) (Moss, 2011, Roberts and Smith, 2008, Smith et al., 2002). Therefore, subunit vaccines have been designed based on plasmids that express the VV L1R, A27L, A33R, and B5R genes. These have been shown to be protective in mice after intranasal (i.n.) VVIHD-J challenge, and in monkeys after intravenous MPXV inoculation (Buchman et al., 2010, Fogg et al., 2007, Hirao et al., 2011).
Attenuated avipoxviruses, and in particular canarypox and fowlpox (FP) viruses, have also been developed as novel vectors for the construction of recombinant vaccines against several human infectious diseases (Radaelli et al., 1994, Zanotto et al., 2010). These vectors are naturally restricted to avian species for their replication, although they are permissive for entry and transgene expression in most mammalian cells. In these cells, they undergo abortive replication and express early and late viral products, but no mature infectious viruses (Somogyi et al., 1993). Moreover, because of the absence of cross-reactivity with VV, avipoxviruses can also escape neutralization by vector-generated antibodies in smallpox-vaccine-experienced humans (Baxby and Paoletti, 1992).
Previous studies have also shown that systemic delivery of FP-based vaccines in humans is safe and does not cause adverse effects (Skinner et al., 2005). More recently, it was demonstrated that FP is an excellent mucosal delivery vector, compared to recombinant DNA or VV (Ranasinghe et al., 2011, Trivedi et al., 2014), and that mucosal immunization induces better protective efficacy against HIV-1, compared to systemic vaccination (Belyakov et al., 2006, Ranasinghe et al., 2007).
Although most pathogens enter the body through mucosal sites, most vaccines are administered by the parenteral route, and only a few mucosal vaccines have been approved for human use. Vaccination via intramuscular (i.m.) and subcutaneous routes also leads to stimulation of systemic immune responses, but poorly promotes immune protection at mucosal membranes (Riese et al., 2014). Conversely, i.n. mucosal immunization can trigger humoral and cell-mediated immunity both at mucosal sites and systemically (Brandtzaeg, 2010, Holmgren and Czerkinsky, 2005). The presence of high levels of IgAs in nasal lymphoid tissue and in the lungs, which are the respiratory pathways through which OPVXs infect animals and humans, can be fundamental for inhibition of viral attachment to the mucosal epithelium, and provide protection from infection (Pierantoni et al., 2015). Finally, i.n. vaccination is more practical than the i.m. route (Lycke, 2012), and should facilitate mass vaccination campaigns. The development of live-attenuated or inactivated mucosal vaccines should therefore meet the needs for better protection against pathogens that penetrate through mucosal membranes (Neutra and Kozlowski, 2006).
Several studies have demonstrated that combined systemic and mucosal prime/boost immunization can enhance both the humoral and cellular arms of immune responses (Ranasinghe et al., 2006, Srivastava et al., 2008), and different immune outcomes have resulted from combinations of poxvirus vectors using prime/boost vaccination regimens (Ranasinghe et al., 2006, Wijesundara et al., 2014). Moreover, vaccinations in which DNA priming is followed by a recombinant viral vaccine boost can elicit greater immunity when compared to the use of single immunogens (Lu, 2009, Radaelli et al., 2003, Radaelli et al., 2007, Wang et al., 2008). Combined vaccines can also elicit improved antigen-specific antibody responses (Vaine et al., 2010).
In the present study, genetic vaccines were administered using in-vivo electroporation (e.p.) followed by i.n. administration of FP recombinants. After determination of the optimal schedules for these e.p. and i.n. immunizations, the mice were primed with a mix of four different DNA plasmids that carried the VV L1R, A27L, A33R, and B5R genes (4DNAmix) (Pacchioni et al., 2013), and then boosted with FP recombinants that carried the same VV genes (4FPmix). All of the mice primed with 4DNAmix and boosted with 4FPmix were protected after a challenge with the highly pathogenic VVHID-J, which correlated with a neutralizing titer against the VV A27 envelope protein.
2. Materials and methods
2.1. Cells
Primary fibroblasts were prepared from specific-pathogen-free chick embryos (Charles River Laboratories, Wilmington, MA, USA) and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% heat-inactivated calf serum (Gibco Life Technologies, Grand Island, NY, USA), 5% Tryptose Phosphate Broth (Difco Laboratories, Detroit, MI, USA), 100 U/ml penicillin and 100 mg/ml streptomycin. Green monkey kidney (Vero) cells (American Type Culture Collection, Rockville, MD, USA) were grown in DMEM supplemented with 10% heat-inactivated calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin. Splenocytes from BALB/c mice were grown in RPMI with glutamine, 10% heat-inactivated foetal calf serum, and 100 U/ml penicillin and 100 mg/ml streptomycin (complete medium) and frozen in 90% foetal calf serum and 10% dimethylsulfoxide.
2.2. Viruses
The highly pathogenic IHD-J strain of VV (VVIHD-J) was supplied by S. Dales (University of Western Ontario, London, Canada) (Wilton et al., 1986), and it was used as the challenging virus (1 × 107 PFU/mouse, i.e., 5-fold the LD50), with i.n. administration. VVIHD-J was grown in Vero cells, then amplified, purified on discontinuous sucrose density gradient, and titrated, as described previously (Pacchioni et al., 2013). The 4FP recombinants, FPL1R, FPA27L, FPA33R, and FPB5R, that expressed the VV L1, A27, A33, and B5 proteins, respectively, were generated in our laboratory by in-vivo homologous recombination (Pozzi et al., 2009). They were then amplified in chick embryo fibroblasts and purified on discontinuous sucrose gradients, as described previously (Soprana et al., 2011). Gene insertion was performed downstream of the VV H6 early/late promoter (Rosel et al., 1986), inside the 3-β-hydroxysteroid dehydrogenase 5-delta 4 isomerase gene, which was interrupted by a multiple cloning site.
2.3. Plasmids
The expression plasmids pcDNA3.1A27L, pcDNA3L1R, pcDNA3A33R, and pcDNA3B5R were constructed by insertion of the same genes used for the generation of the 4FP recombinants, as described previously (Bissa et al., 2013). These were used to excise the genes to be inserted into the pVAX expression plasmid (Invitrogen Corp., San Diego, CA, USA) that contained the kanamicin resistance gene, and generated the pVAXA27L, pVAXL1R, pVAXA33R, and pVAXB5R, respectively, before their amplification. Here, pcDNA3L1R, pcDNA3A33R, and pcDNA3B5R were cut with HindIII/XhoI, whereas pcDNA3.1A27L was cut with HindIII/NotI, with all inserted into the same sites of the previously cut pVAX. Transformation was performed using DH5α competent bacteria. Bacterial selection was performed using the forward L1R V210 (5′ GGG GGG ATC CCA TTT AGT ATC CTA AAA TTG AAT TGT AAT TAT CGA TAA TAA ATG GGT GCC GCA GCA 3′) and reverse V211 (5′ GGG GCT CGA GAG AAA AAC GAG ATT TTC AGT TTT GCA T 3′) primers, the forward A33R V186 (5′ GGG AAG CTT TAT CAT GAT GAC ACC AGA AAA CGA CGA 3′), and reverse V212 (5′ GGG GTC GAC AAT ATT AGT TCA TTG TTT TAA CAC AAA 3′) primers; the forward B5R V206 (5′ GGG GGT CGA CCA TTT AGT ATC CTA AAA TTG AAT TGT AAT TAT CGA TAA TAA ATG AAA ACG ATT TCC 3′) and reverse V207 (5′ GGG GAA GCT TAG AAA AAG GAG ATA TTT ACG GTA GCA A 3′) primers, and the forward A27L V208 (5′ GGG GAG ATC TCA TTT AGT ATC CTA AAA TTG AAT TGT AAT TAT CGA TAA TAA ATG GAC GGA ACT CTT 3′) and reverse V209 (5′ GGG GGT CGA CAG AAA AAG GAG ATA TTT ACT CAT ATG G 3′) primers. Amplifications were performed as described previously (Zanotto et al., 2011), using 2.5 mM MgCl2 and 2 mM MgSO4, with annealing at 57 °C for 30 s (for A33R, B5R) or 61 °C for 30 s (for L1R, A27L), and extension at 72 °C for 45 s (for L1R, A33R, A27L) or 1 min (for B5R). The mixture of equal concentrations of the four recombinants was then prepared (4DNAmix). PcDNA3gagpol was used as an irrelevant negative control, and is called DNAgagpol (Zanotto et al., 2010). Both pcDNA3 and pVAX contain the human CMV promoter, but only pVAX has been approved for use in humans.
2.4. Immunization protocols
Five groups of 8-week-old BALB/c female mice were used (Charles River Laboratories, Wilmington, MA, USA), as seven mice/group. Before each immunization, the mice were anaesthetized by i.m. injection of 30-μl of a mixture of 3.5 μl Rompun (stock, 20 mg/ml; Bayer SpA, Milan, Italy) plus 5.7 μl Zoletil 100 (Virbac Srl, Milan, Italy) and 35.7 μl phosphate-buffered saline without Ca2+ and Mg2+ (PBS−). The vaccination course consisted of priming with two e.p. administrations of the plasmid recombinants (i.m. injections followed by electroporation), and the boost with two i.n. administrations of the FP recombinants. Briefly, for the e.p., two 25-ms transcutaneous low-voltage electric pulses were administered (amplitude, 150 V; interval, 300 ms) at the injection site via a multiple-needle electrode connected to the e.p. apparatus (Cliniporator™; IGEA Srl, Carpi, Italy). Each immunization was performed at two-week intervals. Two weeks after the last immunization, the mice were i.n. challenged with a lethal dose of VVIHD-J.
Five different immunization protocols were followed (Fig. 1 ) using: (i) DNAgagpol plasmid (40 μg/mouse), followed by FPgagpol recombinant (4 × 106 PFU/mouse; G1); (ii) DNAgagpol plasmid (40 μg/mouse), followed by 4FPmix recombinants (1 × 106 PFU of each recombinant/mouse; G2); (iii) 4DNAmix (10 μg of each recombinant/mouse), followed by FPgagpol recombinant (4 × 106 PFU/mouse; G3); (iv) 4DNAmix (10 μg of each recombinant/mouse), followed by 4FPmix recombinants (1 × 106 PFU of each recombinant/mouse; G4); (v) 4FPmix recombinants (1 × 106 PFU of each recombinant/mouse), with no DNA priming; G5). Bleeding was performed from the retro-orbital eye plexus before the first immunization (Fig. 1, T0), before each of the first and second FP boosts (Fig. 1, T1, T2), and just before the challenge (Fig. 1, T3). The plasma fraction was aliquoted and frozen at −80 °C.
Fig. 1.

Immunization protocols. Five different regimens (G1-G5) were followed using 7 mice per group. The four genetic recombinants expressing the VV L1R, A27L, A33R, and B5R genes (4DNAmix) were used for priming, and the viral recombinants expressing the same four genes (4FPmix) were used for the boost. DNAgagpol and FPgagpol recombinants containing HIV-1 gagpol genes were used as irrelevant immunogens. Each plasmid was administered in vivo by e.p. (10 μg/recombinant/mouse), and each virus was administered i.n. (1 × 106 PFU/recombinant/mouse). The VVIHD-J challenge virus was administered i.n. at 1 × 107 PFU/mouse (i.e. 5-fold the LD50 determined for FPgagpol). Mice were bled before the first immunization (T0), before the first and second FP boosts (T1, T2) and just before the VVIHD-J challenge (T3).
All of the mice were maintained according to the Italian National Guidelines and the EU Directive 2010/63/EU for animal experiments. They were observed for signs of disease, weighed daily, and provided with food and water ad libitum. Every effort was made to minimize their suffering, and based on the predetermined criterion of loss of >30% body weight, they were euthanized. Approval for this study was granted by the Ethical Committee of the University of Milan.
2.5. ELISA
The mouse plasma samples were tested for antibodies against the VV-specific L1, A27, A33, and B5 proteins. Mixtures of these L1, A27, A33, and B5 proteins (NIH Biodefense and Emerging Infections Research Resources Repository, NIAID), or alternatively, the individual proteins, were plated as 100 ng of each protein/well in 96-well microtiter plates (MaxiSorp; Nunc, Naperville, IL, USA) in 0.05 M carbonate-bicarbonate buffer, pH 9.6, and incubated overnight at 4 °C. ELISAs were performed in triplicate, essentially as described previously (Radaelli et al., 2010), using the pooled sera of each group of mice from T0, T1, T2, and T3 (see Fig. 1). For the protein mixtures, the sera were diluted 1:500; for the single L1, A33, and B5 proteins, the sera dilutions were 1:100; for the A27 protein, the sera dilutions were 1:500.
The reactions were revealed using a 1:2000 dilution of goat anti-mouse horseradish-peroxidase-conjugated serum (DakoCytomation, Glostrup, Denmark) and tetramethylbenzidine substrate (Sigma). The pre-immunization mouse sera (Fig. 1, T0) were used as negative controls. The absorbance of each well was read at 450 nm using a Microplate Reader 550 (Bio-Rad, Hercules, CA, USA).
2.6. Splenocyte preparation
Two out of the seven mice per group were sacrificed by neck dislocation two weeks after the last vaccination and their spleen was removed; an exception was for G3, where only one mouse was used, as two in this group died before the challenge for nonexperimental reasons. Briefly, the spleen was laid on a 40-μm nylon cell strainer (Corning Incorporated, NY, USA) and mechanically disrupted for 2 min with a flat plastic piston. The cells were passed through the filter using 6 ml RPMI complete medium. After centrifugation at 400 × g for 10 min at 4 °C, the supernatant was removed, and the pelleted cells were aliquoted at 2 × 106/vial for the interferon-γ (IFNγ) ELISPOT assay.
2.7. ELISPOT assay
Splenocytes from the immunized mice (1 × 106) were plated in triplicate into nitrocellulose 96-well plates (HTS IP; Millipore, Bedford, MA, USA) that had been pre-coated with 5 μg/ml rat anti-mouse IFNγ antibody (clone R4–6A2; BD Biosciences Pharmingen, San Diego, CA, USA). The cells were stimulated for 48 h at 37 °C in RPMI complete medium containing 10 μg/ml of each of the A27, A33, B5, and L1 proteins individually. Unstimulated cells were used as the negative control, and 2 μg/ml concanavalin A (Sigma-Aldrich) as the positive control. The plates were developed according to the manufacturer instructions (BD™ ELISPOT; BD Biosciences). The specific spots were enumerated using a reader (Transtec 1300 ELISPOT Reader; AMI Bioline, Buttigliera Alta, Turin, Italy), and analyzed using the ImmunoSpot image analysis software (A.EL.VIS GmbH, Hannover, Germany). IFNγ-secreting spot-forming cells (SFCs) were counted, as the mean numbers per million assessed, including subtraction of the number of SFCs in the absence of stimulation.
2.8. Determination of VVIHD-J LD50 for mice i.n. challenge
Preliminary tests were performed with female BALB/c mice to evaluate the VVIHD-J LD50 after administration of different concentrations of the DNAgagpol and FPgagpol recombinants, carrying the irrelevant gagpol gene of HIV-1 (Table 1 , LD50 tests 1–4). The immunization protocols were followed by challenges with different amounts of VVIHD-J, to evaluate the lowest dose that killed 50% of the mice (i.e., LD50). The VVIHD-J challenge virus was administered i.n. in 30 μl PBS− through a plastic pipette tip after anesthetizing the mice, as described previously (Bissa et al., 2013). All of the mice were followed daily, with measurements of their weight, and monitoring for disease symptoms.
Table 1.
Determination of LD50 by inoculation of different doses of VVIHD-J in immunized mice.
| LD50 test | Immunogen | Route of administration | Concentration | VVIHD-J i.n. challenge (PFU/mouse) | Survival % |
|---|---|---|---|---|---|
| 1 | Prime 4DNAmixa | e.p. | 40 μg | 1 × 106 | 100 |
| Boost 4FPmixa | i.n. | 4 × 107 PFU/mouse | |||
| 2 | DNAgagpol | e.p. | 40 μg | 2 × 105 | 50 |
| 3 | FPgagpol | i.n. | 1 × 107 PFU/mouse | 5 × 106 | 50 |
| 4 | FPgagpol | i.n. | 4 × 106 PFU/mouse | 2 × 106 | 50 |
Mix comprising equal amounts of each of the four components (VV L1R, A27L, A33R, B5R).
2.9. Virus neutralization assays
The neutralizing activities of the mice sera obtained before the challenge were determined by measuring the extent of in-vitro inhibition of VVIHD-J infectivity. The assays were performed by pre-incubation of an equal volume of VVIHD-J with heat-inactivated mouse serum, used at different dilutions in 48-well plates, for 1 h at 37 °C. The viral titer was adjusted to provide approximately 4 × 102 PFU VVIHD-J/ml in the assays. The infection was performed in duplicate on Vero cells, and was allowed to proceed for 1 h at 37 °C. The same amount of virus incubated with DMEM was used as the control. Three days later, 1.5% neutral red was added, and the plaques were counted the next day, as described previously (Pacchioni et al., 2013). In the preliminary assays, sera from the T3 bleeding were pooled to perform the neutralization tests for each group. For the sera from G2 and G4, in which some or all of the mice were protected, these were also tested individually. Neutralization was expressed as the percentage of inhibition of infectivity compared to the control, where the virus was incubated with DMEM only.
2.10. Statistical analysis
Statistical analysis was performed using one-way ANOVA parametric tests and Bonferroni analysis of variance, with the GraphPad Prism software, version 2.0. The statistical significance was set as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
3. Results
3.1. Specific humoral and cellular immunity is elicited in mice primed with 4DNAmix and boosted with 4FPmix
With the aim to develop a protective vaccination strategy against OPXV infections, five different immunization protocols were compared for their ability to induce antibodies against the VV L1, A27, A33, and B5 proteins expressed by DNA or FP recombinants administered alone or in combination. The specific humoral responses were measured by ELISA, using pooled sera from immunized mice and either a mix of all of these proteins (Fig. 2 A) or the individual proteins (Fig. 2B) as the plate-bound antigens. As expected, the control mice of G1 did not show any specific antibody response against any of the proteins tested (Fig. 2A). In contrast, the mice vaccinated with 4DNAmix plus 4FPmix (G4) showed significantly higher antibody titer against the pooled VV proteins, as compared to the other experimental groups at all of the bleeding times (p < 0.001). Interestingly, a significant increase in antibody titers was observed after the FP boost (i.e., T2 and T3 vs. T1; p < 0.001). The single antigens were then plated to test the specificity of the antibodies for each protein that were induced by these vaccinations (Fig. 2B). None of the groups showed humoral responses against L1. In contrast, both G3 and G4 showed humoral responses against A27, which was significantly greater for G4 (G4 vs. G3; p < 0.001), where a further significant increase was also observed after the FP boosts (i.e., T2 and T3 vs. T1; p < 0.001). For A33, with G2 and G4, humoral responses were measurable only at T3, which were significantly greater for G2 than the other groups (p < 0.001), although it never reached the level attained with A27. Against B5, there was a significantly greater response only at T3 after the second FP boost for G2, G4, and G5 (p < 0.001).
Fig. 2.
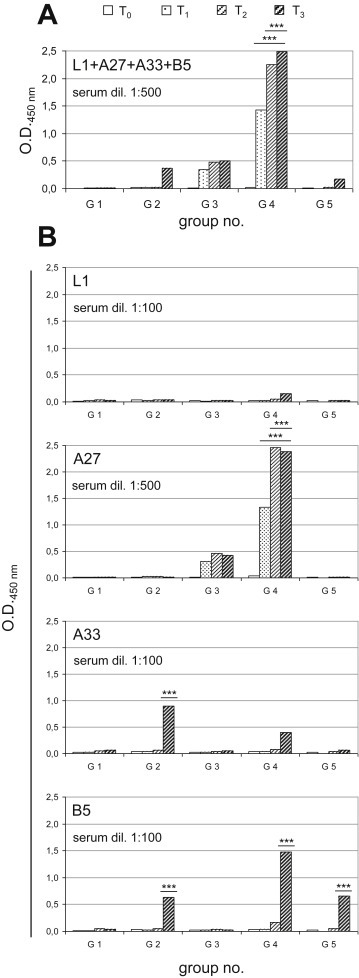
Analysis of specific humoral responses by ELISA. The sera of the mice of the different groups were examined at different times post immunization as the pooled sera diluted 1:500 for the protein mixture (A) and the sera diluted 1:100 for the individual L1, A33, and B5 proteins and 1:500 for the individual A27 protein (B). (A) Anti-L1, -A27, -A33 and -B5 antibody levels were determined using a mixture of all of the proteins (L1 + A27 + A33 + B5) as plate-bound antigens. Data are means of the animal sera for each group. G4 mice showed significant increases in specific antibody titers from T1. (B) The individual proteins were used as plate-bound antigens. None of the groups showed humoral responses against L1. For A27, the humoral response was significantly greater for G4 (G4 vs. G3; p < 0.001) and significantly increased over time (G4: T2 and T3vs. T1; p < 0.001). For A33, the humoral response was significantly greater for G2 than the other groups (p < 0.001). For B5, the humoral response was significantly greater only at T3 for G2, G4, and G5 (G2, G4, G5 vs. G3; p < 0.001). Statistical differences are shown (one-way ANOVA parametric tests, Bonferroni analysis of variance): ***, p < 0.001.
To determine the vaccine-induced cell-mediated immunity, the secretion of IFNγ by splenocytes from mice that were immunized following the different immunization regimens was assessed using the IFNγ-ELISPOT assay. Following the stimulation with A27, the immunized mice from G2, G3, G4, and G5 showed significantly greater numbers of SFCs (Fig. 3 ), as compared to those observed using splenocytes from the control mice (G1). Interestingly, the number of SFCs was significantly greater in the mice immunized with 4DNAmix + 4FPmix (G4 vs. G2; p < 0.05). Conversely, stimulation with A33, B5, and L1 did not result in any significant increase in the numbers of SFCs in any of these experimental groups (data not shown).
Fig. 3.
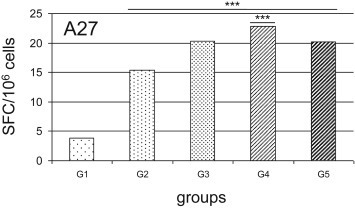
Analysis of the functional virus-specific T-cell responses to A27 antigen using IFNγ−ELISPOT assays. IFNγ secretion by splenocytes was measured in mice immunized following the different immunization regimens shown in Fig. 1. Stimulation with A33, B5, and L1 did not result in any significant increase in spot-forming cells SFCs (data not shown). Only when stimulation was performed with A27, there were significant increases in IFNγ SFCs in G2 to G5, compared to G1 as negative control. Comparison among the different groups also showed significant increase of G4 vs. G2 (p < 0.001). Statistical differences are shown (one-way ANOVA parametric tests, Bonferroni analysis of variance): ***, p < 0.001. Data are means of the SFC/106 splenocytes for mice of each group.
3.2. The VVIHD-J challenge shows higher LD50 when the FP vaccination is performed i.n
As previous data from our laboratory were obtained from BALB/c mice immunized i.m. and subcutaneously, preliminary challenge tests were performed to determine the VVIHD-J LD50 when administered i.n. To determine whether the DNA e.p. and the FP i.n. administration routes affect the LD50 of VVIHD-J, two groups of BALB/c mice were vaccinated twice with the irrelevant DNAgagpol or FPgagpol (e.p., i.n., respectively) and challenged with increasing VVIHD-J doses. Here, the LD50 remained unvaried at 2 × 105 VVIHD-J PFU/mouse when using either the previously determined i.m. immunization with 100 μg of each recombinant DNA (Bissa et al., 2013) or the e.p. immunization with 40 μg of the DNAgagpol recombinant (Table 1, LD50 test 2). Conversely, in mice immunized i.n. with FPgagpol (1 × 107 PFU), the LD50 increased to 5 × 106 PFU/mouse (Table 1, LD50 test 3), 25-fold greater compared to the LD50 observed by immunization with the same FP recombinants given i.m. (Bissa et al., 2013). To reduce the amount of the VVIHD-J challenge, other tests were performed by reducing the titer of the FPgagpol immunogen from 1 × 107 PFU to 4 × 106 PFU (Table 1, LD50 test 4). Using this FPgagpol dose, the LD50 was reduced from 5 × 106 PFU/mouse to 2 × 106 PFU/mouse. The challenge for the different immunization protocols was then performed using 5-fold the LD50 determined for FP (i.e., 1 × 107 VVIHD-J PFU/mouse).
3.3. Priming with 4DNAmix and boosting with 4FPmix protects all of the mice from the challenge
To determine the protective efficacy of the vaccine-induced immune responses, the mice of G1 to G5 that were not sacrificed for spleen removal were taken beyond T3 to the challenge with VVIHD-J, after which they were monitored for weight loss and survival. Soon after the experimental challenge, all of the mice progressively lost 25%–30% of their weight, up to days 4–5 post challenge (p.c.) (Fig. 4 A). This weight loss progressed with no relevant differences among G1, G3, and G5, and all of the mice of these three groups died between day 4 p.c. and day 14 p.c. Conversely, 100% of the mice in G4, and 20% of those in G2, regained weight after day 3 p.c. and day 5 p.c., respectively, and thus survived the i.n. challenge with a 5-fold increase in the LD50 (Fig. 4B). These data demonstrate that DNA priming and FP boosting is the most effective way to induce effective for these mice against VVIHD-J.
Fig. 4.
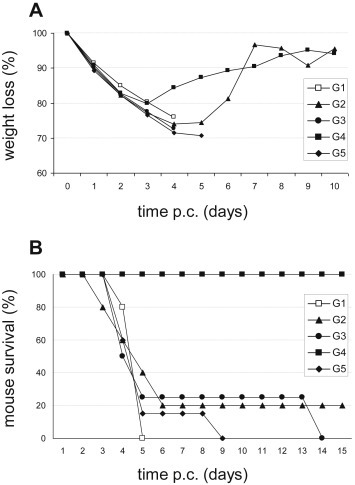
Effects on weight loss and protective efficacy against VVIHD-Jchallenge induced by the different immunization regimens. All of the mice challenged with VVIHD-J were monitored daily for the post-challenge (p.c.) percentage weight loss (A) and survival (B). (A) Data are means of percentage weight loss of each group. Mice progressively lost weight until day 4 p.c., as 25%–30%. There were no relevant differences among most of the groups, but G4 mice and one mouse of G2 started to regain weight after days 3 and 5 p.c., respectively. (B) All of the G4 mice and one mouse of G2 were protected and survived, whereas the remaining mice of G2, G3, and G5 died from days 5–14 p.c. All of the control G1 mice died on days 4 or 5 p.c.
3.4. Pre-challenge neutralizing activity against VVIHD-J correlates with post-challenge mouse survival
To determine the putative pre-challenge immune correlates of this protection against VVIHD-J, viral neutralization assays were performed using the sera from T0 and T3 (Fig. 5 ). This included both pooled sera from the mice of each experimental group and the sera from each mouse of the G2 and G4 protocols, where protection was obtained for 20% and 100% of the mice, respectively.
Fig. 5.
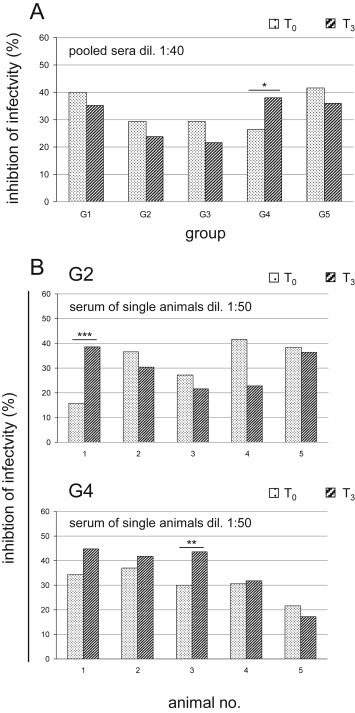
Inhibition of viral infectivity by the different immunization protocols. Viral neutralization assays were performed on Vero cells using sera before the first immunization (T0) and just before the challenge (T3). Plaque reduction was quantified, and expressed as percentages of inhibition of infectivity. (A) When using pooled sera from all of the groups of mice, only those from G4 showed significant inhibition of infectivity (G4 vs. G2, G3, G5; p < 0.05). (B) Only the sera from G2 (no. 1 survived) and G4 (all protected) mice were separately analyzed for correlation between neutralizing activity and survival. For the G2 mice, only animal no. 1 showed significantly higher viral inhibition (T3vs. T0; p < 0.001). Most of the G4 mice showed inhibition of infectivity, which was significantly greater for one mouse (no. 3: T3vs. T0, p < 0.01). Statistical differences are shown (one-way ANOVA parametric tests, Bonferroni analysis of variance): *, p < 0.05; **, p < 0.01; ***, p < 0.001.
For the pooled sera, inhibition of viral infectivity was generally higher in the pre-immune serum (T0) than after the third immunization (T3), except for G4, where the mice were all seen to be protected. The sera from these G4 mice showed low, although significant, inhibition of infectivity at T3 at 1:40 dilution (T3 vs. T0; p < 0.05) (Fig. 5A). As one of the five challenged mice from G2 was also protected, the sera from both G2 and G4 were separately analyzed at T0 and T3 for each of the five challenged mice of these groups, to define any correlation between the pre-challenge viral neutralizing activity and the p.c. mice survival (Fig. 5B). The pre-immune sera of all of the mice of G2 showed higher neutralizing activity at T0 than T3, except for mouse no. 1, which was the only one that survived the challenge, and showed significantly higher viral inhibition at 1:50 dilution (T3 vs. T0, p < 0.001). Conversely, most of the challenged mice of G4 showed higher inhibition of infectivity at T3 than T0 with the exception of mouse no. 5 (Fig. 5B).
4. Discussion
The lack of preventive vaccines against some infectious diseases and the emergence of new pathogens underlines the need for new and more effective immunogens. In particular, safer vaccines against OPXV infection of animals and humans are still an important issue, as a result of the reduction in the ‘herd immunity’ following discontinuation of the smallpox vaccination campaign. At present, the development of safer vaccines against OPXV (Artenstein, 2008, Poland, 2005, Wiser et al., 2007) has also been encouraged by increased human MPXV zoonotic infections (Hutin et al., 2012) or by problems that might arise if there is a deliberate release of variola virus for terrorist purposes.
It has already been shown that different viral vectors and their combinations can significantly influence vaccine efficacy (Ranasinghe et al., 2011) and enhance immune responses, depending on inoculation site and recruitment of antigen-presenting cells (Hervouet et al., 2014, Trivedi et al., 2014). Local administration of vaccines to mucosal tissues can indeed elicit IgAs and cytotoxic T lymphocytes, which can have pivotal roles in neutralizing viruses at their natural port of entry (Brandtzaeg, 2007), and in elimination of infected cells.
In the present study, the four DNA and four FP recombinants all contained the VV L1R, A27L, A33R, and B5R genes and were administered following e.p. (for DNA) and i.n. (for FP recombinants) routes and heterologous prime/boost immunization regimens. Our aim was to compare different vaccination protocols and to evaluate the humoral and cell-mediated responses, as well as protection for mice challenged with the highly pathogenic VVIHD-J. Here, we have demonstrated that: (i) the specific humoral response correlates with protection; (ii) only protected mice show specific VVIHD-J neutralizing antibodies; (iii) after i.n. FP vaccination, mice are protected against higher VVIHD-J challenges; (iv) the putative protective role of the cellular immune response appears to be ascribed to only the A27 protein; and (v) all of the mice were protected when primed with 4DNAmix and boosted with 4FPmix.
Although still controversial, the critical role of the antigen-specific humoral response against OPXV has already been described (Edghill-Smith et al., 2005, Panchanathan et al., 2006), and passive transfer of VV-specific sera was shown to confer protection in both mice and monkeys (Golden et al., 2011). In the present study, although the magnitude of the humoral response was highly variable, a correlation with mice survival was shown soon after the first immunization. In particular, the A27 antigen was the most immunogenic, as also demonstrated previously for the MPXV A29 ortholog of VV A27 (Heraud et al., 2006), whereas no response was elicited by L1 for all of the protocols used here.
The protective efficacy of genetic immunization was demonstrated previously both in mice and nonhuman primates (Hooper et al., 2003, Hooper et al., 2007). However, in the present study, 4DNAmix of G3 and 4FPmix of G5 elicited antibodies against A33 and B5, although they did not protect the mice. Surprisingly, irrelevant DNA in G2 increased the 4FPmix antibody response against A33, which was not shown in G5, when only 4FPmix was used.
Neutralization of infectivity generally correlates with the level of antibodies against the viral surface antigens, and is usually a direct indication of vaccine efficacy and protection. In the present study, natural in-vitro virus-neutralizing antibodies were present before immunization, and these might be a characteristic of this animal species. However, although we cannot provide a reason for this neutralizing activity by pre-immune sera, only the protected mice showed increased neutralization titers before the challenge.
When 10 μg DNA of each antigen was administered e.p., this appeared to be as efficient as 100 μg DNA administered i.m. (Bissa et al., 2013), as the VVIHD-J challenge dose remained unvaried. In contrast, an increase in the dose of the VVIHD-J challenge was necessary after i.n. immunization with FP recombinants, which also provided an advantageous reduction in the amount of immunogen. Indeed, although the 2 × 106 PFU VVIHD-J challenge was previously found to be lethal after i.m. immunization with all of the FP recombinants, i.n. administration of the same immunogens raised the LD50 here by 25-fold, which indicates that the efficacy of FP vaccines can increase remarkably when administered by this route. This might be due to the same i.n. administration used for both the vaccine and the challenge virus, which would indicate that this i.n. vaccine can induce prominent mucosal immunity that is effective against the incoming VVIHD-J. It has already been shown that, compared to modified vaccinia Ankara and VV, FP recombinants can better promote recruitment of dendritic cells and induce CD8+ T-cell–mediated immunity. Their i.n. delivery can recruit unique antigen-presenting cells to the lung mucosa, when compared to other recombinant poxvirus vectors (Trivedi et al., 2014), by eventually conferring a different T-cell functionality (Furuhashi et al., 2012). Similarly, canarypox recombinants can elicit qualitatively distinct cytokine and chemokine profiles compared to attenuated VV vectors in rhesus macaques (Teigler et al., 2014). Moreover, viral interference and competition for penetration cannot be excluded, as FP-based recombinants might bind to the same poxvirus receptors and hamper VVIHD-J penetration through the airway mucosa (Laliberte and Moss, 2014).
The protective role of the cytotoxic T-lymphocyte response after OPXV vaccination is still debated (Buchman et al., 2010), although vaccines that target T-cell epitopes also appear to be protective (Goulding et al., 2013, Moise et al., 2011, Snyder et al., 2004). Moreover, vaccination with VV was also effective when there was dysfunction in the humoral response, although not in patients with T-cell–related immunodeficiencies (Golden and Hooper, 2013). Our data also confirm the efficacy of both DNA and FP recombinants for stimulation of CD8/IFNγ cell-mediated immunity, with high specific response induced by the A27 antigen. In particular, cellular immunity induced by recombinant genetic and viral vaccines administered alone was lower than that observed when these vaccines were administered in combination, and the cellular immune responses against A27 shown here for G2, G3, and G5 were not significantly different from that of G1. In contrast, significantly greater numbers of IFNγ-producing SFCs were measured for the G4 mice, which were all protected. This survival was also found to be inversely correlated with the weight decrease, which was initially similar in all of the groups after the VVIHD-J challenge, but all of the G4 mice recovered their weight, as also for the only protected mouse of G2.
Overall, this protection appears to have been mainly determined by the humoral response, which was endowed with a specific virus-neutralizing activity. This was the case for all of the mice immunized with 4DNAmix followed by 4FPmix, thus showing the efficiency of this prime/boost vaccination regimen, and the fundamental contribution of 4FPmix. As our antibody determination was performed on peripheral blood, it could not have discriminated among the different IgG, IgM, and IgA isotypes to estimate the contribution of the mucosal IgAs. This isotype is mainly present at the i.n. inoculation site, and it might have been the main effector of this protection, considering that both the 4FPmix boost and the VVIHD-J challenge were performed using the same administration route.
This combined use of the L1 and A27 envelope proteins of the intracellular mature virions and the A33 and B5 proteins of the extracellular virions has already been shown to protect mice better than the same proteins administered alone (Hooper et al., 2003). However, in the present study, the humoral, neutralizing, and cellular responses were mainly raised against the A27 surface protein, and thus it would be interesting to determine whether protection can also be obtained by administration of only DNAA27 followed by FPA27, using this prime/boost immunization protocol.
Conflict of interest statement
The authors declare that they have no competing interests, and that the manuscript has been approved by all authors for publication in its present form.
Authors' contributions
MB performed the construction and purification of poxvirus recombinants, neutralization assays, mice immunization and challenge; EQ performed mice immunizations by EP, ELISPOT assays; splenocyte preparation; CZ performed molecular cloning, prepared the primary cell cultures, performed statistical analyses and revised the Figures; EI helped in virus preparation, ELISA, and immunofluorescence; VR helped in mice immunization and ELISPOT assays; SP performed Western blotting, immunofluorescence, ELISA; CDGM, FC, AR conceptualized, designed, and supervised the whole study, and prepared the article. All of the authors have read and approved the manuscript.
Acknowledgements
This work was partially founded by the University of Milano Transition Grant, code no. 18498, CUP G42I14001030001 and by grants to FC from Fondazione Ricerca Molinette Onlus and Fondazione CRT, Torino, Italy, grant no. 2015-2518. The following reagents were obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, (NIAID, NIH): vaccinia virus (WR) L1R protein with C-terminal histidine tag, recombinant from baculovirus, NR-2625; vaccinia virus (WR) A27L protein with C-terminal histidine tag, recombinant from baculovirus, NR-2622; vaccinia virus (WR) A33R protein with C-terminal histidine tag, recombinant from baculovirus, NR-545; vaccinia virus (WR) B5R protein with N-terminal histidine tag, recombinant from baculovirus, NR-546. The authors also thank Dr. Christopher P. Berrie for editorial assistance with the manuscript.
References
- Artenstein A.W. New generation smallpox vaccines: a review of preclinical and clinical data. Rev. Med. Virol. 2008;18:217–231. doi: 10.1002/rmv.571. [DOI] [PubMed] [Google Scholar]
- Baxby D., Paoletti E. Potential use of nonreplicating vectors as recombinant vaccines. Vaccine. 1992;10:8–9. doi: 10.1016/0264-410x(92)90411-c. [DOI] [PubMed] [Google Scholar]
- Belyakov I.M., Kuznetsov V.A., Kelsall B., Klinman D., Moniuszko M., Lemon M., Markham P.D., Pal R., Clements J.D., Lewis M.G., Strober W., Franchini G., Berzofsky J.A. Impact of vaccine-induced mucosal high-avidity CD8-CTLs in delay of AIDS viral dissemination from mucosa. Blood. 2006;107:3258–3264. doi: 10.1182/blood-2005-11-4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissa M., Pacchioni S., Zanotto C., De Giuli Morghen C., Illiano E., Granucci F., Zanoni I., Broggi A., Radaelli A. Systemically administered DNA and fowlpox recombinants expressing four vaccinia virus genes although immunogenic do notprotect mice against the highly pathogenic IHD-J vaccinia strain. Virus Res. 2013;178:374–382. doi: 10.1016/j.virusres.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandtzaeg P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine. 2007;25:5467–5484. doi: 10.1016/j.vaccine.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Brandtzaeg P. Function of mucosa-associated lymphoid tissue in antibody formation. Immunol. Invest. 2010;39:303–355. doi: 10.3109/08820131003680369. [DOI] [PubMed] [Google Scholar]
- Buchman G.W., Cohen M.E., Xiao Y., Richardson-Harman N., Silvera P., DeTolla L.J., Davis H.L., Eisenberg R.J., Cohen G.H., Isaacs S.N. A protein-based smallpox vaccine protects non-human primates from a lethal monkeypox virus challenge. Vaccine. 2010;28:6627–6636. doi: 10.1016/j.vaccine.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edghill-Smith Y., Golding H., Manischewitz J., King L.R., Scott D., Bray M., Nalca A., Hooper J.W., Whitehouse C.A., Schmitz J.E., Franchini G. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat. Med. 2005;11:740–747. doi: 10.1038/nm1261. [DOI] [PubMed] [Google Scholar]
- Ferrier-Rembert A., Drillien R., Tournier J.N., Garin D., Crance J.M. Short- and long-term immunogenicity and protection induced by non-replicating smallpox vaccine candidates in mice and comparison with the traditional 1st generation vaccine. Vaccine. 2008;26:1794–1804. doi: 10.1016/j.vaccine.2007.12.059. [DOI] [PubMed] [Google Scholar]
- Fogg C., Americo J.L., Lustig S., Huggins J.W., Smith S.K., Damon I.K., Resch W., Earl P.L., Klinman D.M., Moss B. Adjuvant enhanced antibody responses to recombinant proteins correlates with protection of mice and monkeys to orthopoxvirus challenges. Vaccine. 2007;25:2787–2799. doi: 10.1016/j.vaccine.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi K., Suda T., Hasegawa H., Suzuki Y., Hashimoto D., Enomoto N., Fujisawa T., Nakamura Y., Inui N., Shibata K., Nakamura H., Chida K. Mouse lung CD103 þ and CD11 b high dendritic cells preferentially induce distinct CD4þ T-cell responses. Am. J. Respir. Cell Mol. Biol. 2012;46:165–172. doi: 10.1165/rcmb.2011-0070OC. [DOI] [PubMed] [Google Scholar]
- Golden J.W., Hooper J.W. The strategic use of novel smallpox vaccines in the post-eradication world. Expert Rev. Vaccines. 2013;10:1021–1035. doi: 10.1586/erv.11.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden J.W., Zaitseva M., Kapnick S., Fisher R.W., Mikolajczyk M.G., Ballantyne J., Golding H., Hooper J.W. Polyclonal antibody cocktails generated using DNA vaccine technology protect in murine models of orthopoxvirus disease. Virol. J. 2011;8:441. doi: 10.1186/1743-422X-8-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding J., Bogue R., Tahiliani V., Croft M., Salek-Ardakani S. CD8 T cells are essential for recovery from a respiratory vaccinia virus infection. J. Immunol. 2013;189:2432–2440. doi: 10.4049/jimmunol.1200799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heraud J.M., Edghill-Smith Y., Ayala V., Kalisz I., Parrino J., Kalyanaraman V.S., Manischewitz J., King L.R., Hryniewicz A., Trindade C.J., Hassett M., Tsai W.P., Venzon D., Nalca A., Vaccari M., Silvera P., Bray M., Graham B.S., Golding H., Hooper J.W., Franchini G. Subunit recombinant vaccine protects against monkeypox. J. Immunol. 2006;177:2552–2564. doi: 10.4049/jimmunol.177.4.2552. [DOI] [PubMed] [Google Scholar]
- Hervouet C., Luci C., Bekri S., Juhel T., Bihl F., Braud V.M., Czerkinsky C., Anjuere F. Antigen-bearing dendritic cells from the sublingual mucosa recirculate to distant systemic lymphoid organs to prime mucosal CD8 Tcells. Mucosal Immunol. 2014;7:280–291. doi: 10.1038/mi.2013.45. [DOI] [PubMed] [Google Scholar]
- Hirao L.A., Draghia-Akli R., Prigge J.T., Yang M., Satishchandran A., Wu L., Hammarlund E., Khan A.S., Babas T., Rhodes L., Silvera P., Slifka M., Sardesai N.Y., Weiner D.B. Multivalent smallpox DNA vaccine delivered by intradermal electroporation drives protective immunity in nonhuman primates against lethal monkeypox challenge. J. Infect. Dis. 2011;203:95–102. doi: 10.1093/infdis/jiq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren J., Czerkinsky C. Mucosal immunity and vaccines. Nat. Med. 2005;11:S45–S53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- Hooper J.W., Custer D.M., Thompson E. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicitis appropriate antibody responses in nonhuman primates. Virology. 2003;306:181–195. doi: 10.1016/S0042-6822(02)00038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper J.W., Golden J.W., Ferro A.M., King A.D. Smallpox DNA vaccine delivered by novel skin electroporation device protects mice against intranasal poxvirus challenge. Vaccine. 2007;25:1814–1823. doi: 10.1016/j.vaccine.2006.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutin Y.J., Williams R.J., Malfait P., Pebody R., Loparev V.N., Ropp S.L., Rodriguez M., Knight J.C., Tshioko F.K., Khan A.S., Szczeniowski M.V., Esposito J.J. Outbreak of human monkeypox, democratic republic of Congo, 1996 to 1997. Emerg. Infect. Dis. 2012;7:434–438. doi: 10.3201/eid0703.010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson I.G., Smith T.C., Smith B., Wells T.S., Ryan M.A. US military service members vaccinated against smallpox in 2003 and 2004 experience a slightly higher risk of hospitalization postvaccination. Vaccine. 2008;26:4048–4056. doi: 10.1016/j.vaccine.2008.05.044. [DOI] [PubMed] [Google Scholar]
- Laliberte J.P., Moss B. A novel mode of poxvirus superinfection exclusion that prevents fusion of the lipid bilayers of viral and cellular membranes. J. Virol. 2014;88:9751–9768. doi: 10.1128/JVI.00816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J.M., Goldstein J. Adverse events occurring after smallpox vaccination. Semin. Pediatr. Infect. Dis. 2003;14:189–195. doi: 10.1016/s1045-1870(03)00032-3. [DOI] [PubMed] [Google Scholar]
- Lewis-Jones S. Zoonotic poxvirus infections in humans. Curr. Opin. Infect. Dis. 2004;17:81–89. doi: 10.1097/00001432-200404000-00003. [DOI] [PubMed] [Google Scholar]
- Lu S. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 2009;21:346–351. doi: 10.1016/j.coi.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycke L. Recent progress in mucosal vaccine development: potential and limitations. Nat. Immunol. 2012;12:592–605. doi: 10.1038/nri3251. [DOI] [PubMed] [Google Scholar]
- Megid J., Borges I.A., Trindade G.S., Appolinário C.M., Ribeiro M.G., Allendorf S.D., Antunes J.M., Silva-Fernandes A.T., Kroon E.G. Vaccinia virus zoonotic infection, São Paulo State, Brazil. Emerg. Infect. Dis. 2012;18:189–191. doi: 10.3201/eid1801.110692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise L., Buller R.M.L., Schriewer J., Lee J., Frey S.E., Martin W., De Groot A.S. VennVax, a DNA-prime, peptide-boost multi-T-cell epitope poxvirus vaccine, induces protective immunity against vaccinia infection by T cell response alone. Vaccine. 2011;29:501–511. doi: 10.1016/j.vaccine.2010.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Smallpox vaccines: targets of protective immunity. Immunol. Rev. 2011;239:8–26. doi: 10.1111/j.1600-065X.2010.00975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutra M.R., Kozlowski P.A. Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- Pacchioni S., Bissa M., Zanotto C., De Giuli Morghen C., Illiano E., Radaelli A. L1R, A27L, A33R and B5R vaccinia virus genes expressed by fowlpox recombinants as putative novel orthopoxvirus vaccines. J. Transl. Med. 2013;11:95. doi: 10.1186/1479-5876-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchanathan V., Chaudhri G., Karupiah G. Protective immunity against secondary poxvirus infection is dependent on antibody but not on CD4 or CD8 T-cell function. J. Virol. 2006;80:6333–6338. doi: 10.1128/JVI.00115-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierantoni A., Esposito M.L., Ammendola V., Napolitano F., Grazioli F., Abbate A., Del Sorbo M., Siani L., D'Alise A.M., Taglioni A., Perretta G., Siccardi A., Soprana E., Panigada M., Thom M., Scarselli E., Folgori A., Colloca S., Taylor G., Cortese R., Nicosia A., Capone S., Vitelli A. Mucosal derivery of a vectored RSV vaccine is safe and elicits protective immunity in rodens and nonhuman primates. Mol. Ther. Methods Clin. Dev. 2015;2:15018. doi: 10.1038/mtm.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland G.A. Smallpox vaccines: from first to second to third generation. Vaccine. 2005;365:362–363. doi: 10.1016/S0140-6736(05)17840-4. [DOI] [PubMed] [Google Scholar]
- Pozzi E., Basavecchia V., Zanotto C., Pacchioni S., De Giuli Morghen C., Radaelli A. Construction and characterization of recombinant fowlpox viruses expressing human papilloma virus E6 and E7 oncoproteins. J. Virol. Methods. 2009;158:184–189. doi: 10.1016/j.jviromet.2009.01.021. [DOI] [PubMed] [Google Scholar]
- Radaelli A., Bonduelle O., Beggio P., Mahe B., Pozzi E., Elli V., Paganini M., Zanotto C., De Giuli Morghen C., Combadière B. Prime-boost immunization with DNA, recombinant fowlpox virus and VLP(SHIV) elicit both neutralizing antibodies and IFNgamma-producing T cells against the HIV-envelope protein in mice that control env-bearing tumour cells. Vaccine. 2007;25:2128–2138. doi: 10.1016/j.vaccine.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Radaelli A., Gimelli M., Cremonesi C., Scarpini C., De Giuli Morghen C. Humoral and cell mediated immunity in rabbits immunized with live non replicating avipox recombinants expressing the HIV-1sf2 env gene. Vaccine. 1994;12:1110–1117. doi: 10.1016/0264-410x(94)90181-3. [DOI] [PubMed] [Google Scholar]
- Radaelli A., Nacsa J., Tsai W.P., Edghill-Smith Y., Zanotto C., Elli V., Venzon D., Tryniszewska E., Markham P., Mazzara G.P., Panicali D.L., De Giuli Morghen C., Franchini G. Prior DNA immunization enhances immune response to dominant and subdominant viral epitopes induced by a fowlpox-based SIVmac vaccine in long-term slow-progressor macaques infected with SIVmac251. Virology. 2003;312:181–195. doi: 10.1016/s0042-6822(03)00184-3. [DOI] [PubMed] [Google Scholar]
- Radaelli A., Pozzi E., Pacchioni S., Zanotto C., De Giuli Morghen C. Fowlpox virus recombinants expressing HPV-16 E6 and E7 oncogenes for the therapy of cervical carcinoma elicit humoral and cell-mediated responses in rabbits. J. Transl. Med. 2010;8:40. doi: 10.1186/1479-5876-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranasinghe C., Eyers F., Stambas J., Boyle D.B., Ramshaw I.A., Ramsay A.J. A comparative analysis of HIV-specific mucosal/systemic T cell immunity and avidity following rDNA/rFPV and poxvirus-poxvirus prime boost immunisations. Vaccine. 2011;29:3008–3020. doi: 10.1016/j.vaccine.2011.01.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranasinghe C., Medveczky J.C., Woltring D., Gao K., Thomson S., Coupar B.E., Boyle D.B., Ramshaw I.A. Evaluation of fowlpox-vaccinia virus prime-boost vaccine strategies for high-level mucosal and systemic immunity against HIV-1. Vaccine. 2006;24:5881–5895. doi: 10.1016/j.vaccine.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Ranasinghe C., Turner S.J., McArthur C., Sutherland D.B., Kim J.H., Doherty P.C., Ramshaw I.A. Mucosal HIV-1 poxvirus prime-boost immunization induces high-avidity CD8 T cells with regime-dependent cytokine/granzyme B profiles. J. Immunol. 2007;178:2370–2379. doi: 10.4049/jimmunol.178.4.2370. [DOI] [PubMed] [Google Scholar]
- Reed K.D., Melski J.W., Graham M.B., Regnery R.L., Sotir M.J., Wegner M.V., Kazmierczak J.J., Stratman E.J., Li Y., Fairley J.A., Swain G.R., Olson V.A., Sargent E.K., Kehl S.C., Frace M.A., Kline R., Foldy S.L., Davis J.P., Damon I.K. The detection of monkeypox in humans in the Western Hemisphere. New Engl. J. Med. 2004;350:342–350. doi: 10.1056/NEJMoa032299. [DOI] [PubMed] [Google Scholar]
- Riese P., Sakthivel P., Trittel S., Guzmán C.A. Intranasal formulations: promising strategy to deliver vaccines. Expert Opin. Drug Deliv. 2014;11:1618–1634. doi: 10.1517/17425247.2014.931936. [DOI] [PubMed] [Google Scholar]
- Roberts K.L., Smith G.L. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 2008;16:472–479. doi: 10.1016/j.tim.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Rosel J.L., Earl P.L., Weir J., Moss B. Conserved TAAATG sequence at the transcriptional and translational initiation sites of vaccinia virus late genes deduced by structural and functional analysis of the hindlll H genome fragment. J. Virol. 1986;60:436–449. doi: 10.1128/jvi.60.2.436-449.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze C., Alex M., Schirrmeier H., Hlinak A., Engelhardt A., Koschinski B., Beyreiss B., Hoffmann M., Czerny C.P. Generalized fatal Cowpox virus infection in a cat with transmission to a human contact case. Zoonoses Public Health. 2007;54:31–37. doi: 10.1111/j.1863-2378.2007.00995.x. [DOI] [PubMed] [Google Scholar]
- Skinner M.A., Laidlaw S.M., Eldaghayes I., Kaiser P., Cottingham M.G. Fowlpox virus as a recombinant vaccine vector for use in mammals and poultry. Expert Rev. Vaccines. 2005;4:63–76. doi: 10.1586/14760584.4.1.63. [DOI] [PubMed] [Google Scholar]
- Smith G.L., Vanderplasschen A., Law M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 2002;83:2915–2931. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- Snyder J.T., Belyakov I.M., Dzutsev A., Lemonnier F., Berzofsky J.A. Protection against lethal vaccinia virus challenge in HLA-A2 transgenic mice by immunization with a single CD8+ T-cell peptide epitope of vaccinia and variola viruses. J. Virol. 2004;78:7052–7060. doi: 10.1128/JVI.78.13.7052-7060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P., Frazier J., Skinner M.A. Fowlpox virus host range restriction: gene expression, DNA replication, and morphogenesis in nonpermissive mammalian cells. Virology. 1993;197:439–444. doi: 10.1006/viro.1993.1608. [DOI] [PubMed] [Google Scholar]
- Soprana E., Panigada M., Knauf M., Radaelli A., Vigevani L., Palini A., Villa C., Malnati M., Cassina G., Kurth R., Norley S., Siccardi A.G. Joint production of prime/boost pairs of Fowlpox Virus and Modified Vaccinia Ankara recombinants carrying the same transgene. J. Virol. Methods. 2011;174:22–28. doi: 10.1016/j.jviromet.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Srivastava I., Goodsell A., Zhou F., Sun Y., Burke B., Barnett S., Vajdy M. Dynamics of acute and memory of mucosal and systemic immune responses against HIV-1 envelope following immunizations through single or combinations of mucosal and systemic routes. Vaccine. 2008;26:2796–2806. doi: 10.1016/j.vaccine.2007.11.083. [DOI] [PubMed] [Google Scholar]
- Teigler J.E., Phogat S., Franchini G., Hirsch V.M., Michael N.L., Barouch D.H. The canarypox virus vector ALVAC induces distinct cytokine response scompared to the vaccinia virus-based vectors MVA and NYVAC in rhesus monkeys. J. Virol. 2014;88:1809–1814. doi: 10.1128/JVI.02386-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi S., Jackson R.J., Ranasinghe C. Different HIV pox viral vector-based vaccines and adjuvants can induce unique antigen presenting cells that modulate CD8 T cell avidity. Virology. 2014;468–470:479–489. doi: 10.1016/j.virol.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Vaine M., Wang S., Hackett A., Arthos J., Lu S. Antibody responses elicited through homologous or heterologous prime-boost DNA and protein vaccinations differ in functional activity and avidity. Vaccine. 2010;28:2999–3007. doi: 10.1016/j.vaccine.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel S., Sardy M., Glos K.K.H.C., Ruzicka T., Wollenberg A. The Munich outbreak of cutaneous cowpox infection: transmission by infected pet rats. Acta Derm. Venereol. 2012;92:126–131. doi: 10.2340/00015555-1227. [DOI] [PubMed] [Google Scholar]
- Wang S., Parker C., Taaffe J., Solorzano A., Garcia-Sastre A., Lu S. Heterologous HA DNA vaccine prime–inactivated influenza vaccine boost is more effective than using DNA or inactivated vaccine alone in eliciting antibody responses against H1 or H3 serotype influenza viruses. Vaccine. 2008;26:3626–3633. doi: 10.1016/j.vaccine.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weltzin R., Liu J., Pugachev K.V., Myers G.A., Coughlin B., Blum P.S., Nichols R., Johnson C., Cruz J., Kennedy J.S., Ennis F.A., Monath T.P. Clonal vaccinia virus grown in cell culture as a new smallpox vaccine. Nat. Med. 2003;9:1125–1130. doi: 10.1038/nm916. [DOI] [PubMed] [Google Scholar]
- Whitley R.J. Smallpox: a potential agent of bioterrorism. Antivir. Res. 2003;57:7–12. doi: 10.1016/s0166-3542(02)00195-x. [DOI] [PubMed] [Google Scholar]
- Wijesundara D.K., Ranasinghe C., Jackson R.J., Lidbury B.A., Parish C.R., Quah J.C. Use of an in vivo FTA assay to assess the magnitude, functional avidity and epitope variant cross-reactivity of T cell responses following HIV1-recombinant poxvirus vaccination. PLoS One. 2014:9. doi: 10.1371/journal.pone.0105366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton S., Gordon J., Dale S. Identification of antigenic determinants by polyclonal and hybridoma antibodies induced during the course of infection by vaccinia virus. Virology. 1986;148:84–96. doi: 10.1016/0042-6822(86)90405-8. [DOI] [PubMed] [Google Scholar]
- Wiser I., Balicer R.D., Cohen D. An update on smallpox vaccine candidates and their role in bioterrorism related vaccination strategies. Vaccine. 2007;25:976–984. doi: 10.1016/j.vaccine.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Zanotto C., Pozzi E., Pacchioni S., Bissa M., De Giuli Morghen C., Radaelli A. Construction and characterisation of a recombinant fowlpox virus that expresses the human papilloma virus L1 protein. J. Transl. Med. 2011;9:190–200. doi: 10.1186/1479-5876-9-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto C., Pozzi E., Pacchioni S., Volonté L., De Giuli Morghen C., Radaelli A. Canarypox and fowlpox viruses as recombinant vaccine vectors: a biological and immunological comparison. Antivir. Res. 2010;88:53–63. doi: 10.1016/j.antiviral.2010.07.005. [DOI] [PubMed] [Google Scholar]