

Abstract
Protein kinases play critical roles in cell survival, proliferation and motility. Their dysregulation is therefore a common feature in the pathogenesis of a number of solid tumors, including thyroid cancers. Inhibiting activated protein kinases has revolutionized thyroid cancer therapy, offering a promising strategy in treating tumors refractory to radioactive iodine treatment or cytotoxic chemotherapies. However, despite satisfactory early responses, these drugs are not curative and most patients inevitably progress due to drug resistance. This review summarizes up-to-date knowledge on various mechanisms that thyroid cancer cells develop to bypass protein kinase inhibition and outlines strategies that are being explored to overcome drug resistance. Understanding how cancer cells respond to drugs and identifying novel molecular targets for therapy still represents a major challenge for the treatment of these patients.
Keywords: differentiated thyroid cancers, anaplastic thyroid carcinoma, BRAF, RAS, kinase inhibitors, drug resistance, NIS
I. Introduction
Thyroid cancer is the most common endocrine malignancy, with an estimate 44,280 cases in the United States in 2021 (Siegel et al., 2021). They originate from two types of endocrine cells: the thyrocytes, or follicular cells, or the parafollicular C cells. Our understanding of the molecular pathways involved in thyroid tumorigenesis has greatly improved due to the identification of several oncogenic alterations and the availability of agents to target them (Nikiforov and Nikiforova, 2011). These alterations include point mutations as well as gene translocations and their chimeric fusion products, gene amplifications, and deletions. Many of these alterations result in oncogenic drivers that promote cell proliferation, motility, and metastasis (Fagin and Wells, 2016). They often directly or indirectly increase the activity of protein kinases, which are critical for many cellular functions, including regulation of the cell cycle. The identification of these mutations has led to the development of new targeted therapies, mostly protein kinase inhibitors (PKIs), for advanced thyroid cancers (Sipos and Ringel, 2022). So far, four of the FDA–approved PKIs for thyroid cancers are antiangiogenic multi-kinase inhibitors and five are mutation-selective. These novel drugs have revolutionized the treatment of patients with advanced thyroid cancers (Cabanillas et al., 2019). Kinase inhibitors have shown a good efficacy in controlling the disease and are better tolerated than cytotoxic therapies. However, despite an early response, many patients will show transient benefit and progress under treatment, and tumor-specific mortality is nearly universal. The mechanisms leading to acquired drug resistance are far from being completely understood. In this review, we summarize the properties and function of protein kinases and kinase inhibitors, and how these drugs are used to treat thyroid cancers. We then describe some mechanisms of resistance recently identified and which strategies were used to overcome the challenges.
2. Protein Kinases
2.1. Protein kinases and their dysregulation
Protein kinases are enzymes that covalently add phosphate groups to selected proteins. They catalyze the transfer of phosphate from adenosine triphosphate (ATP) to specific amino acid residues of target proteins in a process called phosphorylation. Phosphorylation is considered one of the most abundant post-translational modification of proteins and usually modulates their function by adjusting their activity, modifying their cellular location, or altering their association with other proteins (Seok, 2021). The majority of protein kinases are serine/threonine kinases, which phosphorylate the hydroxyl groups of serines and threonines in their target proteins. Most other kinases are tyrosine kinases, which catalyze the addition of a phosphate group to tyrosine residues. Up to 30% of all human proteins may be modified by kinase activity, and kinases are known to regulate many cellular processes and pathways, especially those involved in signal transduction. Protein kinases may be located at the cell membrane and function as receptors, such as growth factor receptors (EGFR, MET, KIT). They may also reside in the cytoplasm, where their activation is dependent on upstream signaling molecules such as RAS and other kinases functioning in signaling cascades (Figure 1). Phosphorylation through protein kinases regulates key cellular processes that include gene expression, proliferation, differentiation, motility, membrane transport, metabolism, and apoptosis. Therefore, the predominance of dysregulation of protein kinases activity in many cancers is not surprising (Brognard and Hunter, 2011).
Figure 1: Overview of the MAPK and AKT signaling pathways.
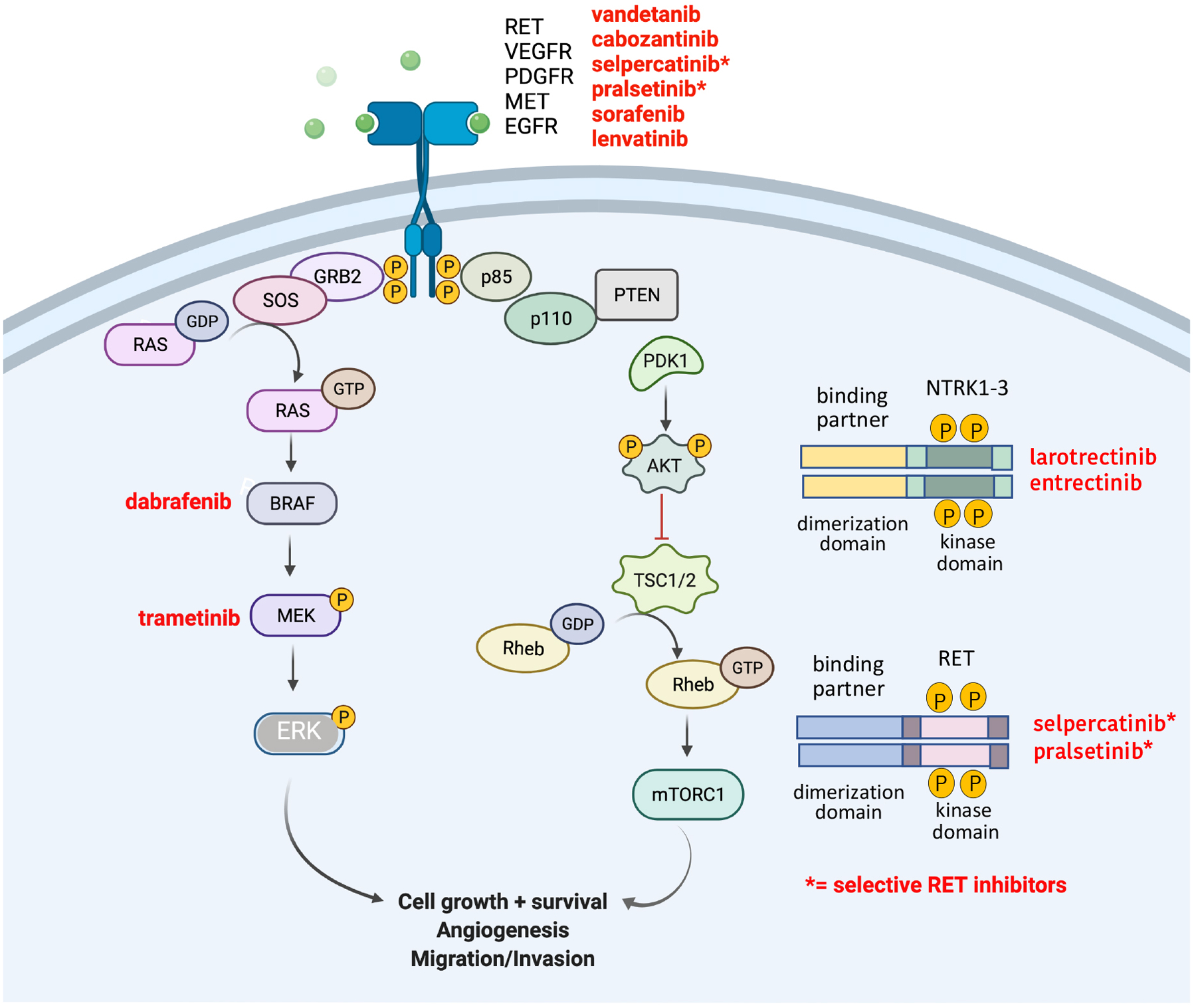
The main protein kinases driving these pathways are depicted. Kinase inhibitors that are presently FDA-approved for thyroid cancers are highlighted.
Alterations of protein kinase activities are indeed a frequent component of the pathogenesis of solid and hematologic malignancies. These alterations are caused by a variety of mechanisms, such as genomic rearrangements (chromosomal translocations and fusions)(Hallberg and Palmer, 2013, Nikiforov, 2002), gene amplification or deletions (Rodriguez-Antona et al., 2010, Lopez-Gines et al., 2010, Cicenas et al., 2018), and missense point mutations (Lahiry et al., 2010). Missense point mutations often arise in the kinase domain of receptor tyrosine kinases or in cytoplasmic serine/threonine protein kinases. For example, a sporadic point mutation in the BRAF gene, which belongs to the MAPK pathway (Figure 1), can constitutively activate the kinase to drive cell proliferation continually, without the presence of growth factors (Wan et al., 2004, Xing, 2005). Most BRAF-activating missense point mutations are found in exons 11 and 15 of the gene, within the kinase domain of the protein. In particular, the c.T1799A mutation is present in over 80% of all BRAF-mutated cancers (Davies et al., 2002). The protein resulting from this mutation is a substitution of valine for glutamic acid at codon 600 (V600E), which leads to constitutive activation of BRAF and its downstream targets MEK and ERK. Activating BRAF mutations are found in 40–60% of melanoma and 50–60% of papillary thyroid carcinoma (Figure 2B) (Xing et al., 2005, Long et al., 2011, Cancer_Genome_Atlas_Research_Network, 2014). BRAF mutations have also been frequently reported in colorectal cancers (Barras, 2015). An example of gene rearrangement/fusion in thyroid cancers is illustrated by somatic juxtapositions of 5’ activating sequences from other genes with 3’ sequences of the RET or NTRK genes encoding the tyrosine kinase domain (Figure 1). Sporadic RET and NTRK rearrangements occur in approximately 16% of thyroid cancers (Ullmann et al., 2022, Nikiforov, 2002, Park et al., 2022)(Figure 2B). In addition to mutations affecting DNA sequences, epigenetic events can also increase or modify the expression of many receptor tyrosine kinases such as EGFR, ERBB2, and MET in many solid tumors (Hoque et al., 2010, Ogunwobi et al., 2013) and untimely activation of the kinases p38, MEK, ERK, AURKA and AKT due to phosphorylation is well known (Cicenas et al., 2018).
Figure 2: Clinical and Molecular Spectrum of Thyroid Cancers.
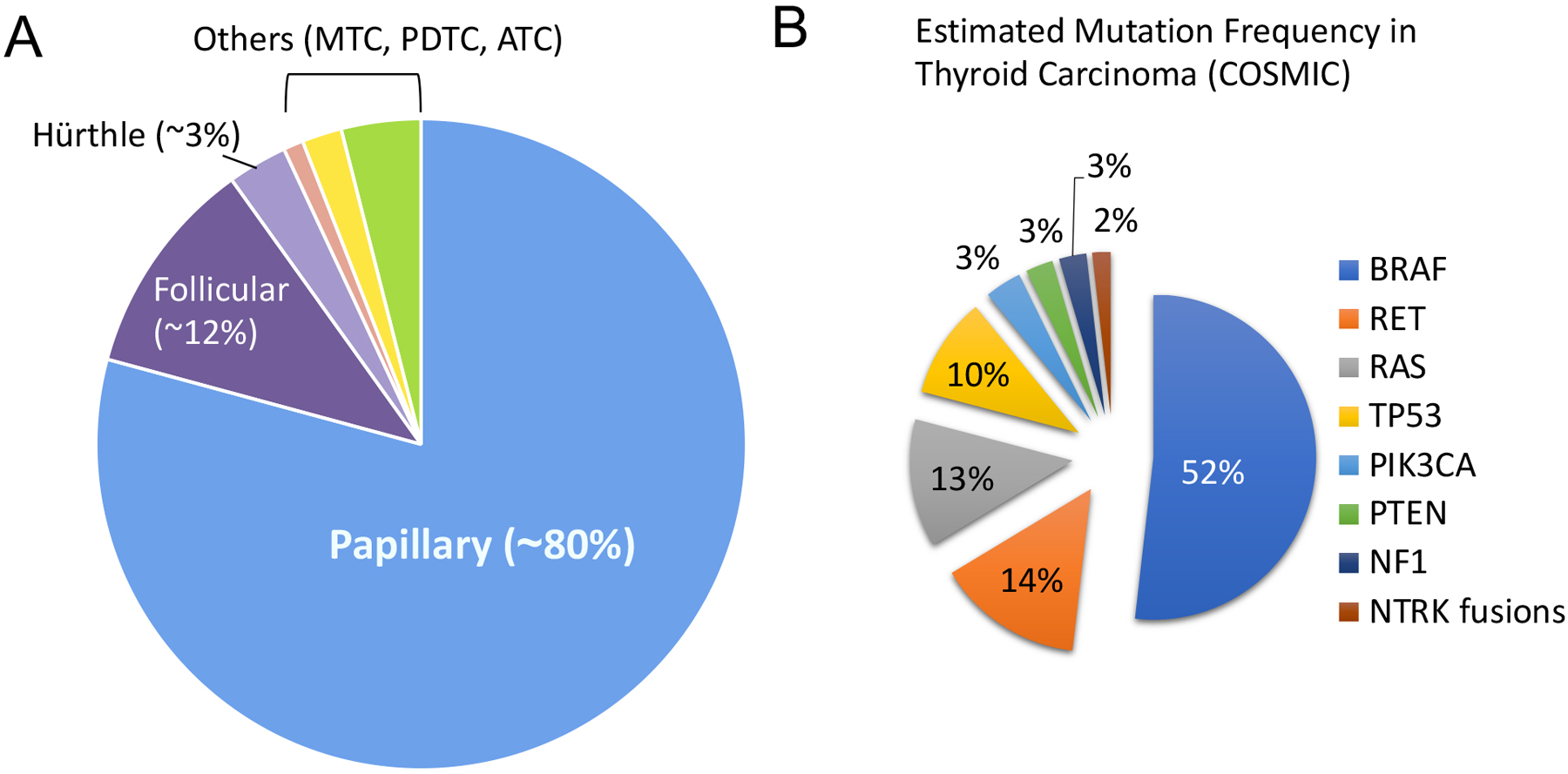
A. Incidence of the main types of thyroid cancers (adapted from Fagin and Wells, 2016). B: Distribution of the top 8 mutated genes in thyroid cancers (from COSMIC, 2022).
2.2. Targeting Protein Kinases
Small molecule kinase inhibitors have been very efficient in targeting specific mutations driving tumorigenesis. These inhibitors are generally categorized according to their mechanism of action. They are classified as types I, II, III (allosteric), IV (substrate directed), V (bivalent), and VI (covalent) inhibitors (Bhullar et al., 2018, Cicenas et al., 2018, Martinez et al., 2020)(Table 1). Type I inhibitors compete for the substrate and bind the DFG (Asp-Phe-Gly) motif in the ATP-binding pocket of the active kinase when the Asp residue is oriented toward bound ATP (“in” conformation)(Peng et al., 2013). Such is the case for crizotinib, dasatinib, erlotinib, lapatinib, pazopanib, sunitinib, vemurafenib, and dabrafenib. Type II inhibitors bind to the ATP pocket of the inactive kinase (DFG-Asp “out” conformation) (imatinib, sorafenib, axitinib, nilotinib). The disadvantages of Type I and II inhibitors reside in their binding to the ATP-binding site, which is conserved in protein kinases. Therefore, developing inhibitors that are specific is particularly challenging (Table 1). This lack of specificity increases the potential for off-target side effects, in particular cardiotoxicity. In contrast, Type III inhibitors such as the MEK1/2 inhibitors trametinib and cobimetinib occupy highly specific allosteric sites close to the ATP binding pocket, and therefore are thought to be more selective than type I and II inhibitors (Zhao et al., 2017). This design might also prevent the occurrence of “gate-keeper” mutations, which can obstruct drug binding by modulating access to the kinase ATP-binding site. More recently, efforts have been made to develop inhibitors that bind to unique structural domains outside the ATP-binding pocket. This led to the synthesis of Type IV inhibitors, which usually induce a conformational change disrupting interactions of the kinases with their downstream targets (JNK, ERK1/2 inhibitors)(Martinez et al., 2020). This approach again offers more selectivity by blocking only the kinase function associated with a particular disease, while preserving other kinase functions that have potential benefits. To further increase selectivity, Type V, or bivalent inhibitors, were developed that can target both the ATP-binding site and a unique structural feature found on the kinase (lenvatinib and specific c-SRC, ERK5 and PLK1 inhibitors)(Lee et al., 2021, Okamoto et al., 2015)(Table 1). Finally, type VI inhibitors bind covalently and irreversibly to their target for a longer duration of the effect (Martinez et al., 2020, Weisner et al., 2019). This is the case for afatinib (an EGFR inhibitor), ibrutinib (a BTK inhibitor), and neratinib (a HER1-HER4 inhibitor).
Table 1:
Classification of some small molecule kinase inhibitors (adapted from Bulhar et al., Kannaiyan et al., Martinez et al, and Lee et al)
| Class | Mechanism of Action | Names | Target Protein Kinases |
|---|---|---|---|
| Type I inhibitors | Compete for the substrate and binds in the ATP-binding pocket of the active kinase conformation (DFG-Asp “in”) | Bosutinib | BCR-ABL, Src |
| Cabozantinib | VEGFR, PDGFR, KIT, MET, FLT3, RET | ||
| Ceritinib | ALK-EML4 | ||
| Crizotinib | ALK-EML4, MET, ROS-1 | ||
| Dabrafenib | BRAFV600E mutant kinase | ||
| Gefitinib | EGFR | ||
| Papozanib | VEGFR, PDGFR, KIT, FGFR3 | ||
| Vandetanib | VEGFR, RET, MET | ||
| Vemurafenib | BRAFV600E mutant kinase | ||
| Encorafenib | BRAFV600E mutant kinase | ||
| Larotrectinib | NTRK fusions | ||
| Entrectinib | NTRK fusions | ||
| Type II inhibitors | Compete for the substrate and binds in the ATP-binding pocket of the inactive kinase conformation (DFG-Asp “out”) | Imatinib | BCR-ABL, KIT, PDGFR |
| Sorafenib, | VEGFR, PDGFR, BRAF, FTL3, RET, KIT | ||
| Axitinib, | VEGFR | ||
| Nilotinib | BCR-ABL, KIT, PDGFR | ||
| Type III (allosteric inhibitors) | Occupy a site next to the ATP-binding pocket so that both ATP and the allosteric inhibitor simultaneously bind to the protein kinase. Offer more selectivity against the targeted kinases and avoid the development of ATP-binding site gatekeeper mutations | Trametinib | MEK1/2 |
| Cobimetinib | MEK1/2 | ||
| Selumetinib | MEK1/2 | ||
| Binimetinib | MEK1/2 | ||
| GnF2 | BCR-ABL | ||
| Type IV (substrate directed inhibitors) | Bind to a site distal from the ATP pocket and induce a conformational change. Offer more selectivity against the targeted kinases | ONO12380 | BCR-ABL |
| Type V (bivalent inhibitors) | Consist of a small molecule that targets the ATP-binding site coupled to a peptide representing the substrate targeted by the specific kinase | Lenvatinib | VEGFR1–3, FGFR1–4, PDGFRα, KIT, RET |
| ARC-1411 | PKA | ||
| ARC-3140 | CK2 | ||
| ERK5.1 | ERK5 | ||
| Rapalink-1 | mTORC1 | ||
| BI 2536 | PLK1 | ||
| BI 53 | c-SRC | ||
| Type VI (covalent inhibitors) | Irreversibly bind the targeted kinase | Afatinib | EGFR |
| Ibrutinib | BTK | ||
| Neratinib | HER1, HER2, HER3, HER4 |
3. Thyroid Cancers
The thyroid lies within the neck and consists of two connected lobes. The functional unit of the gland is the spherical thyroid follicle lined with thyrocytes, or follicular cells, that secrete triiodothyronine (T3) and thyroxine (T4). T3 and T4 have a wide range of effects including control of metabolism, regulation of the cardiac rate, and control of growth during development. Parafollicular cells, or C cells, are located adjacent to the thyroid follicles and secrete calcitonin, which contributes to the control of blood calcium levels. The most common primary thyroid cancers originate from thyrocytes and are classified as differentiated thyroid cancers (DTCs). DTCs include papillary thyroid carcinoma (PTC), follicular thyroid carcinoma (FTC), and Hürthle cell thyroid carcinoma (HTC). DTCs usually harbor single driver mutations such as RAS or BRAF, which are mutually exclusive. Poorly differentiated thyroid carcinoma (PDTC) and anaplastic thyroid carcinoma (ATC) are rare and aggressive tumors that also originate from thyrocytes (Figure 2A). They both show a de-differentiated phenotype, however ATCs harbor a higher number of mutations than PDTCs and PTCs (Landa et al., 2016). Medullary thyroid carcinoma (MTC) are tumors derived from neuroendocrine parafollicular C-cells and are often driven by a RET receptor mutation. In thyroid cancers, most somatic driver mutations are found in receptor tyrosine kinases or in serine/threonine kinases belonging to the mitogen activated protein kinase (MAPK) signaling pathway, regardless of the histologic subtype (Figure 1).
3.1. Molecular pathology of thyroid cancers
Papillary thyroid carcinomas (PTC) represent about 80% of thyroid cancers and often harbor somatic mutations in the MAPK pathway (Figure 2A+B). Activation of the pathway is mainly due to BRAF or RAS mutations, or re-arrangements of the RET or NTRK genes. Primary PTCs often display single mutations, such as the BRAFV600E mutation mentioned above (60% of the cases), and these tumors harbor one of the lowest mutation densities of cancers that have been studied according to whole-exome sequencing (WES) data (Fagin and Wells, 2016). About 10–20% of thyroid cancers are due to RET mutations or rearrangements, and another 10–20% are due to RAS point mutations (Figure 2B). In classical PTCs, RET fusions account for 6–8% of the cases (RET/PTC rearrangements) and the presence of RAS mutations is low (1–6%) (Cancer_Genome_Atlas_Research_Network, 2014, Song and Park, 2019). If the PTC tumor is small and localized within the thyroid, the prognosis is excellent, especially in younger patients. However, larger tumors that may extend outside the thyroid, and have invaded lymph nodes and/or distant organs carry a poorer prognosis.
Follicular thyroid carcinomas (FTC) account for ~12% of thyroid cancers (Figure 2A+B). FTCs usually harbor NRAS, HRAS or KRAS gene mutations (40–50% of the cases). Another common mutation is the PAX8/PPARG rearrangement (30–35% of the cases), while point mutations in the PTEN and PIK3CA genes are observed in about 5–10% of the cases. The prognosis of FTC depends on the age of the patients, the size and stage of the tumors, the completeness of surgery, the presence of distance metastases, and the responsiveness to radioactive iodine therapy (Sobrinho-Simoes et al., 2011).
Hürthle cell thyroid carcinomas (HTC) amount to 2–3% of thyroid cancers (Figure 2A+B). HTCs exhibit a more aggressive behavior compared with PTC and FTC and the frequency of distant metastases is high (Mcfadden and Sadow, 2021). A fraction of HTCs harbor mutations in the RAS, TP53, PTEN, E1F1AX, and PAX8-PPARγ genes, which are often found in other thyroid cancers. Mutations in the TERT promoter, as well as the DAXX and ATRX genes that cause alternate telomere elongation, are found in more aggressive forms of HTCs (Ganly et al., 2018). In addition, an interesting feature of HTCs is their accumulation of abnormally shaped mitochondria, probably due to mutations in the mitochondrial DNA (mtDNA) leading to impairments in proteins of the electron transport chain, in particular subunits of the complex I and III (Bonora et al., 2006). Further, chromosomes gains and losses are often observed, with near haploidization in certain cases, or selective gains of chromosomes 7, 12 and 17 (Ganly et al., 2018).
Poorly differentiated thyroid carcinomas (PDTCs) are more aggressive than PTC and FTC tumors and represent approximately 2–3% of thyroid cancers. Radioiodine therapy is of limited benefit and these tumors can readily metastasize to the neck lymph nodes, lungs, and bones. They are associated with a mean survival of 3.2 years (Landa et al., 2016). Molecular pathology data indicate that these tumors retain the driver mutations seen in differentiated PTCs or FTCs (BRAFV600E or RAS), but that they have acquired additional alterations such as PTEN loss of function (Volante et al., 2021). In addition, while TERT promoter mutations are found in about 5–15% of PTCs and FTCs, this mutation is significantly more frequent in PDTCs (20–50%)(Volante et al., 2021). Thus, acquisition of a TERT promoter mutation appears to be a key transitional step in the microevolution of these tumors (Landa et al., 2016).
Anaplastic thyroid carcinomas (ATCs) are rare but are among the most aggressive tumors known, with historic median survival of 3–5 months, although more recent studies have demonstrated improved survival (Maniakas et al., 2020). The current accepted model considers that ATCs derive from PTCs, FTCs or HTCs through the acquisition of additional genomic alterations (Figure 3). BRAF, RAS, and RET mutations are prevalent, however de novo acquisition of TERT promoter, PIK3CA and TP53 mutations (70% of the cases) are a hallmark of these malignancies (Qin et al., 2021, Landa et al., 2016, Pozdeyev et al., 2018). Mutations in the PTEN and NF1 genes are also occasionally seen. In general, ATC tumors harbor a higher mutation burden than PTC. ATC tumors easily invade neck structures, metastasize to distant organs, and are resistant to conventional chemotherapy and radiation therapy. Capdevilla and colleagues sought to understand more in detail how, and if, ATCs truly derive from PTCs by performing whole exome sequencing (WES) of 14 ATC samples, including from three patients with concomitant ATC and PTC (Capdevila et al., 2019). Interestingly, in the cases of concomitant ATC and PTC, most mutations identified in the ATC component differed from the ones in PTC. Phylogenic tree analyses indicated that ATC and PTC might diverge early in tumor development and evolve independently. Mutational analysis of 44 ATCs and 401 PTC/FTC from public databases confirmed that most of the high incidence lesions were not the same between ATCs and PTCs or FTCs. For example, despite the fact that BRAF and NRAS are frequently mutated in these tumor types, BRAF mutations have a higher incidence in PTC (60%) than ATC (37%), and NRAS mutations are more frequent in FTC (30–45%) than ATC (21%), supporting early divergence.
Figure 3. Genetic Evolution of Thyroid Cancers.
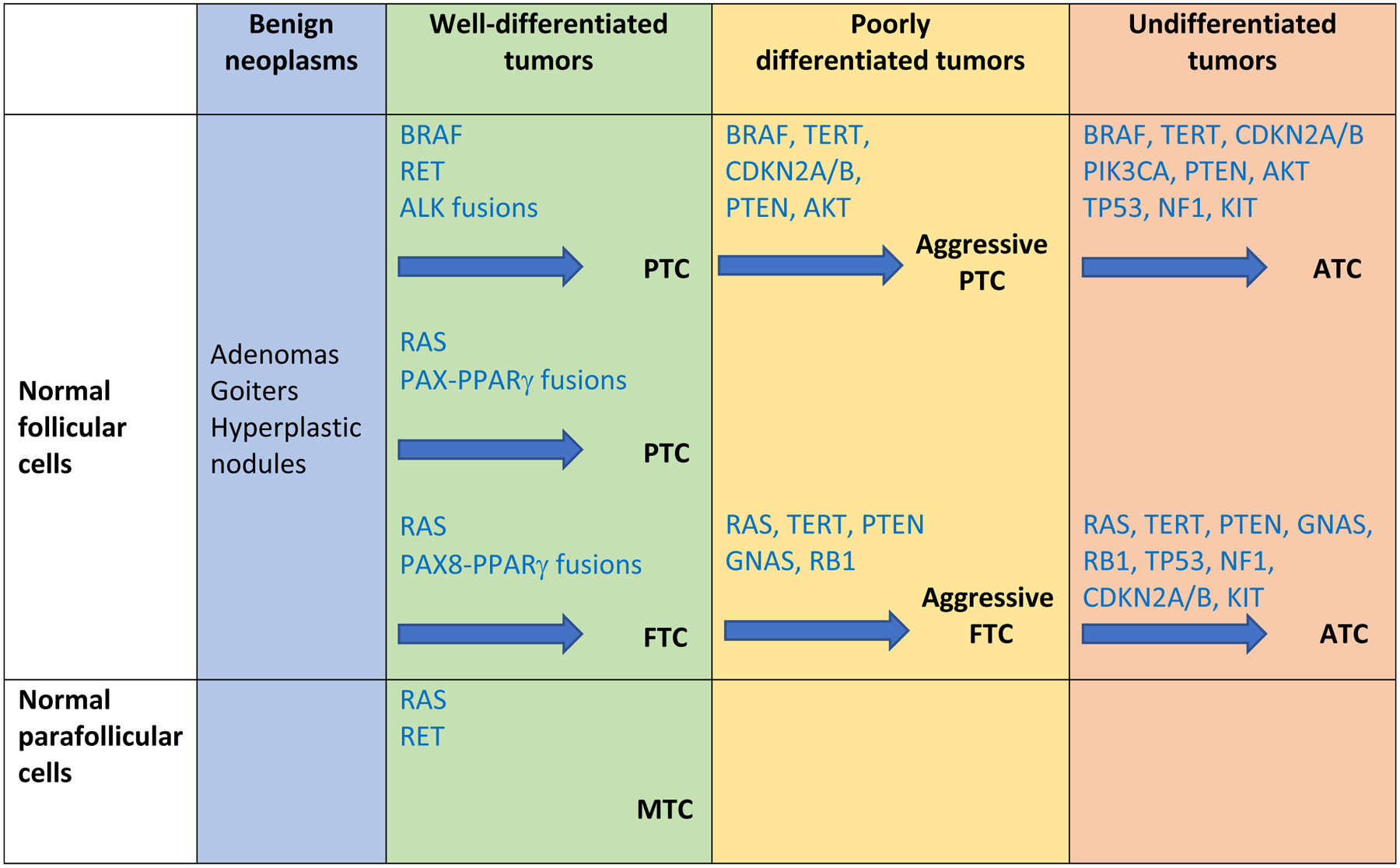
The diagram postulates that transformation from normal thyroid cells is associated with accumulation of an increasing number of mutated genes, starting with early driver mutations in the BRAF, RAS or RET genes. Tumors can then evolve to a less differentiated morphology and more aggressive behavior in poorly differentiated thyroid cancers (PDTC) with acquisition of TERT promoter and/or PTEN mutations. Ultimately the tumors can develop into ATC through acquisition of additional mutations in TP53, NF1 and PIK3CA. Adapted from Pozdeyev et al., 2018.
Medullary thyroid carcinomas (MTCs) originate from the interstitial C cells of the thyroid and account for 2–3% of thyroid cancers. Most cases are sporadic but familial cases exist that are inherited in an autosomal dominant manner and account for approximately 20% of the cases. Familial MTCs are most of the time associated with gain of function germline RET receptor mutations. Somatic RET mutations occur often in sporadic MTCs (30–60%), but RAS mutations can also be found. Evidence suggests that RET and RAS mutations very rarely overlap (Ciampi et al., 2019). Further, RET mutations are generally associated with an elevated risk for metastasis, tumor recurrence and patient mortality while the association of RAS mutations with tumor aggressiveness in sporadic MTC is unclear (Vuong et al., 2018).
3.2. Molecular Targeted Therapies in Thyroid Cancers
While more than 90% of localized, well-differentiated carcinomas of the thyroid such as PTC and FTC can be cured by surgery combined with TSH suppressive thyroid hormone therapy and occasional use of radioactive iodine, cancers that persist or recur following these regimens have a poorer prognosis. Cytotoxic chemotherapy or external beam radiotherapy show poor efficacy in these patients. In addition, since MTC tumor cells have a neuroendocrine origin, they do not respond to radioiodine or TSH suppression. Therefore, the advent of therapies targeting growth factor receptors, or constitutively activated protein kinases represented a welcomed additional option. This led to significant improvements in the treatment of advanced cases of radioiodine refractory PTC and FTC, as well as PDTC and ATC. In the United States, there are currently eight drugs and one drug combination that are FDA-approved for advanced thyroid cancers. Three antiangiogenic drugs, sorafenib lenvatinib, and cabozantinib, are FDA-approved for differentiated thyroid cancers (Schlumberger et al., 2015, Brose et al., 2014, Brose et al., 2021). The combination of dabrafenib and trametinib is FDA-approved for BRAFV600E-mutated ATC (Subbiah et al., 2018b, Wang et al., 2019a, Subbiah et al., 2022). The NTRK inhibitors larotrectinib and entrectinib are approved for NTRK fusion solid tumors, regardless of tumor histology. In MTCs, two antiangiogenic drugs, vandetanib and cabozantinib, are FDA-approved. Finally, the selective RET inhibitors pralsetinib and selpercatinib are approved for RET-altered thyroid cancers (Wells et al., 2012, Elisei et al., 2013, Subbiah et al., 2018a, Subbiah et al., 2018c) (Figure 1). All these drugs have resulted in remarkable improvement in progression-free survival, in particular in ATC. The anti-angiogenic drugs inhibit VEGFR activity, but they are not specific; whereas some secondary targets may also be anti-angiogenic, such as FGFR, others can lead to “off target” toxicities (Table 1). This lack of target specificity might cause side effects such as fatigue, diarrhea and weight loss, as well as poor wound healing, hypertension and congestive heart failure. However, the selective RET inhibitors pralsetinib and selpercatinib are new generation drugs that show a safer toxicity profile probably due to lower inhibition of other tyrosine kinases, including VEGFR (Wirth et al., 2020, Subbiah et al., 2020).
The majority of mutations found in thyroid cancers affect components of the MAPK pathway, with most alterations found in the RAS and BRAF genes (Figure 2B). Targeting RAS mutations in cancers has proven challenging but recent advances in targeting KRASG12C with inhibitors such as sotorasib and adagrasib have been promising (Huang et al., 2021). These drugs form an irreversible, covalent bond with the cysteine residue of KRASG12C, holding the protein in its inactive form and blocking downstream signaling. Unfortunately, KRASG12C mutations are very rare in thyroid cancers. As mentioned above, 60% of PTCs and 37% of ATCs harbor a BRAF mutation, most often the BRAFV600E mutation. Dabrafenib is a BRAFV600E-specific drug, which is FDA-approved alone or in combination with a MEK inhibitor such as trametinib for melanoma (Long et al., 2014). This combination was recently approved for most solid tumors in patients who have progressed following prior treatment and have no satisfactory alternative treatment options. In thyroid cancers, dabrafenib is only approved for BRAFV600E-mutated ATCs in combination with trametinib (Subbiah et al., 2018b). The combination dabrafenib-trametinib is also used as neoadjuvant therapy followed by surgery in cases of advanced unresectable disease (Wang et al., 2019a). This regimen represents a significant progress in the treatment of ATC as long-term survival of these patients is now significantly increased (Subbiah et al., 2022, Maniakas et al., 2020).
4. Resistance to Kinase Inhibitors
Despite a large amount of promising data and early therapeutic successes, blocking signaling pathways with kinase inhibitors appears to be ultimately ineffective and most patients progress. To overcome this issue, second generation BRAF and MEK inhibitors are presently studied in thyroid cancers, such as encorafenib (type I BRAF inhibitor) and binimetinib (type III MEK inhibitor), which showed significant benefit in metastatic colorectal cancer and advanced melanoma (Table I)(Tabernero et al., 2021, Dummer et al., 2018). However, many tumors display multiple coexisting cellular subclones that confer a high degree of heterogeneity, therefore single targeting approaches are limited because new clones may be selected that become dominant. Inherent genomic instability such as found in ATC predispose the tumor cells to rapidly acquire additional alterations that increase their fitness in a changing microenvironment. Efforts have been made to combine targeted therapies with immune checkpoint inhibitors. In differentiated thyroid cancers, treatments with immunotherapy are associated with low response rates (Mehnert et al., 2019). Side effects of immunotherapy have led to treatment discontinuations in RET-mutated tumors (Hegde et al., 2020). In ATC, where both PD-L1 positive cells and tumor infiltrating lymphocytes (TILs) are present in high frequency, kinase inhibitors combined with immunotherapy are useful only in a subset of patients, but the reasons are unclear (Iyer et al., 2018, Wang et al., 2019a, Capdevila et al., 2020).
4.1. Resistance to VEGFR inhibitors
Molecular targeted therapies inhibiting the VEGFR tyrosine kinase have dominated the field of differentiated thyroid cancer therapeutics. Because this approach also targets other receptor tyrosine kinases such as RET, FGFR, PDGFR, KIT, and MET, it is likely to remain important in the treatment of this disease. Although some patient subsets show prolonged overall survival (Elisei et al., 2013, Brose et al., 2017), rates of cure are low, responses are only partial, and patients remain on drug for prolonged periods of time causing long term toxicity. Acquired resistance to anti-VEGF therapy often involves escape mechanisms that activates parallel signaling pathways. For example, upregulation of fibroblast growth factor (FGF2) signaling is often observed in anti-VEGF-resistant tumors, especially in tumors that are exposed to a hypoxic environment (Casanovas et al., 2005). While preclinical studies showed that dual blockade of VEGF and FGF signaling pathways was beneficial, this did not translate in clinical studies (Semrad et al., 2017). Acquired resistance to sorafenib can also be due to activation of the PI3K/AKT pathway or reactivation of the JAK-STAT pathway and STAT3 phosphorylation (Zhu et al., 2017). Further, epigenetic mechanisms might also explain sorafenib resistance. For example, the group of Wang and colleagues demonstrated that miR-124 and miR-506 were significantly reduced in the serum of sorafenib-resistant thyroid cancer patients and that their common target, EZH2, was upregulated in these tumors and thyroid cancer cell lines in comparison with controls (Wang et al., 2019b). EZH2 is a well-known histone modification factor that repress transcription of specific genes. Its upregulation is associated with poor clinical outcomes in many cancer types and EZH2 inhibitors are presently tested in clinical trials (Duan et al., 2020, Li et al., 2021a). Acquired resistance to lenvatinib in thyroid cancers has been also studied, mainly in preclinical models (Bertol et al., 2022, Khan et al., 2019). Khan and colleagues performed long term cultures of ATC cells exposed to lenvatinib for 72 days. They demonstrated significant changes of lenvatinib-resistant cells toward a mesenchymal morphology. Transcriptomic and proteomic analyses uncovered upregulation of SNAIL and ZEB1 genes/proteins, activation of pro-survival signaling pathways, activation of the nuclear exporter protein exportin 1 (XPO1), and activation of the RAC/RHO GTPase effector p21 activated kinases (PAK). When combined with lenvatinib, XPO1 and PAK4 inhibitors showed anti-tumor activity that was superior to lenvatinib alone, both in vitro and in sub-cutaneous xenografts (Khan et al., 2019). This and similar studies may pave the way for future novel therapies.
4.2. Resistance to BRAFV600E inhibitors
Acquired (or secondary) resistance to BRAFV600E inhibitors presents a significant therapeutic challenge in thyroid cancer patients. Vemurafenib and dabrafenib monotherapies increase progression-free survival, but responses are incomplete (Brose et al., 2016, Dadu et al., 2015, Falchook et al., 2015, Kim et al., 2013). Resistance mechanisms have been extensively studied in melanoma and colon cancers. Sequence analysis of the mutated BRAF gene in vemurafenib-resistant melanoma samples demonstrated that secondary mutations within this gene are rare (Nazarian et al., 2010). Only one case of an additional mutation in the kinase domain was found (Wagenaar et al., 2014). Therefore, other mechanisms, including the activation of parallel signaling pathways, may be responsible for acquired resistance.
4.2.1. Over-Expression of Membrane Receptors
Several studies have now demonstrated that cancer cells adapt to BRAF inhibitor therapies by overexpressing growth factor receptors at their surface. This is often the case for KIT, MET, EGFR, and PDGFRβ. This increase of expression may be epigenetically directed since no secondary activating mutation nor DNA amplification of these genes were detected (Nazarian et al., 2010, Prahallad et al., 2012, Corcoran et al., 2012). Corcoran and colleagues showed that in colorectal cancers, resistance to vemurafenib was mediated by the epigenetic upregulation of EGFR, which reactivated RAS and CRAF, and the downstream phosphorylation of ERK1/2 (Corcoran et al., 2012). Similarly, Montero-Conde and colleagues demonstrated that BRAF-mutated PTC cell lines acquire resistance to vemurafenib by over-expressing the ERBB or FGF receptor families, therefore reactivating the MAPK pathway downstream of activated RAS (Montero-Conde et al., 2013). Resistance to vemurafenib in melanoma can also be caused by over-expression of EPHA2, a member of the Ephrin receptor family of tyrosine kinases (Miao et al., 2015). These mechanisms of resistance driven by membrane receptor over-expression imply that the corresponding ligands must be present in sufficient quantity in the cellular microenvironment for their activation. Often, ligand production is indeed upregulated in an autocrine manner in cancer cells, and experimental data have shown that most cells can bypass receptor tyrosine kinase inhibitors by simply exposing them to one or more receptor tyrosine kinase ligands (Wilson et al., 2012). For example, exposure to hepatocyte growth factor (HGF) from the microenvironment will promote intrinsic resistance to BRAF inhibitors in melanoma, colorectal cancer, and glioblastoma cells overexpressing the MET receptor (Straussman et al., 2012). These results underscore the importance of the cellular microenvironment for drug resistance.
4.2.2. Gene Amplification
Gain of wild-type gene copy numbers has been associated with resistance to BRAF inhibitors. For example, several copies of the gene MCL1, a BCL2 family member and apoptosis modulator, have been associated with resistance to vemurafenib treatment in PTC (Duquette et al., 2015). Our group recently demonstrated that resistance to BRAF inhibitors can also be caused by acquired chromosomal polyploidy. In one case of PTC that dedifferentiated into ATC during dabrafenib treatment, we observed triploidy of chromosome 7, which harbors the EGFR, RAC1, MET and BRAF genes. Copy numbers and expression of these protooncogenes were consequently increased in the dedifferentiated sample, probably causing tumor progression (Bagheri-Yarmand et al., 2021). It is now evident that over-expression of any non-mutated receptor tyrosine kinase has the potential to reactivate the MAPK pathway (Yadav et al., 2012) or the PI3K pathway (Chapman et al., 2011, Atefi et al., 2011, Greger et al., 2012, Delmas et al., 2015, Byeon et al., 2017) provided that the ligand is present in sufficient quantity.
4.2.3. Point Mutations
Acquired resistance to BRAF inhibitors in melanoma and colorectal cancers have been often attributed to secondary mutations in the NRAS or KRAS genes (Nazarian et al., 2010, Monsma et al., 2015, Kopetz et al., 2015). Our group sought to identify acquisition of secondary resistance mechanisms in thyroid cancers by exposing BRAFV600E-mutated PTC cells to long-term treatment to vemurafenib for a period of 5 months (Danysh et al., 2016). This treatment induced up-regulation of the MET, EGF AND ERBB receptors and activation of the PI3K/AKT and MAPK pathways, as previously described by others (Montero-Conde et al., 2013, Byeon et al., 2017). Using whole exome sequencing (WES), we also identified a subpopulation of cells harboring a secondary KRAS mutation (KRASG12D), conferring them significant proliferative and invasive advantages. This data, obtained with a cell line, was later validated by the observation that about 50% of our BRAFV600E-mutated patients progressing under vemurafenib or dabrafenib treatment (with/without MEK inhibitors) harbored a secondary RAS mutation (NRAS or KRAS) after WES analysis (Table 2) (Cabanillas et al., 2020, Owen et al., 2019). These studies were the first to establish that acquired resistance to kinase inhibitors in thyroid cancers can be RAS-driven. In addition to RAS point mutations, secondary mutations that possibly conferred drug resistance were often found in the PTEN, NF1, NF2, TP53, and CDKN2A genes (Table 2). We recently analyzed a secondary mutation in the RAC1 gene, RAC1P34R, found in a lymph node metastasis of a PTC patient who progressed under dabrafenib treatment. A cell line was established that showed dabrafenib resistance in vitro, and sensitivity to the drug was restored with a RAC1 activity inhibitor (Bagheri-Yarmand et al., 2021). RAC1 is a small GTPase involved in cell motility and cytoskeletal reorganization, as well as cell proliferation through its canonical targets, PAK1–3 (Mack et al., 2011). The RAC1P29S mutation, which affects the same protein domain as RAC1P34R, is a known driver mutation in primary and drug-resistant melanoma (Cannon et al., 2020, Watson et al., 2014). Similar to RAS, efforts to target RAC1 itself has been challenging, therefore approaches to target downstream effectors such as PAK1–3 are underway. Understanding tumor heterogeneity and mutational patterns emerging under drug pressure is therefore fundamental to improving therapies.
Table 2:
Mutations in PTC, PDTC and ATC patients at baseline and at progression.
| Patient | Histology | Mutation(s) at baseline | Kinase inhibitor(s) | Immune checkpoint inhibitor | Additional mutations at progression |
|---|---|---|---|---|---|
| 1 | PTC | BRAFV600E | D+T | ND | BRCAD237A PTENE40* |
| 2 | ATC | BRAFV600E PIK3CAE525K TP53Q331* |
D+T | ND | NF1R1276Q |
| 3 | PDTC | BRAFV600E | D | ND | CDKN2AD84N TP53R273H |
| 4 | ATC | BRAFV600E | V | ND | KITM541L |
| 5 | ATC | BRAFV600E | D+T | ND | NF2K40fs*3 |
| 6 | ATC | BRAFV600E | D+T | ND | NF2Y144* |
| 7 | ATC | BRAFV600E PIK3CAH1047R TP53R273S |
D+T | ND | METR1005Q NOTCH1P2138R NOTCH1E360K |
| 8 | ATC | BRAFV600E TP53R175H EGFRG322S |
D+T | ND | NRASQ61K |
| 9 | PDTC | BRAFV600E ATMI1986V |
D+T | ND | KRASG12V |
| 10 | PDTC | BRAFV600E PIK3CAE545K |
V+C | A | KRASG12D |
| 11 | ATC | BRAFV600E PIK3CAN345K TP53R213* |
V+C | A | NRASQ61K |
| 12 | PTC | BRAFV600E | V | ND | KRASG12V |
| 13 | PTC | BRAFV600E | D+T | ND | KRASG12V |
| 14 | PTC | BRAFV600E | D+T | P | NRASG13D |
D=dabrafenib, T=trametinib, V=vemurafenib, C=cobimetinib, A=atezolizumab, P= pembrolizumab, ND = no immunotherapy
4.3. Super-enhancers
Enhancers are short sequences of DNA that are located near or far from promoter regions and can be bound by specific transcription factors to increase transcription. In a broad range of human cell types, super-enhancers (SEs) are large clusters of enhancers with high levels of transcription factor binding (Tang et al., 2020). SEs are critical for driving the expression of genes that control cell identity. They also regulate and increase the expression of key oncogenes such as c-MYC in many tumor cells, and their development drives drug resistance (Li et al., 2021b). Therefore, disrupting SE structure or inhibiting SE protein co-factors can be utilized as therapeutic strategies. For example, BET bromodomain inhibitors can repress c-MYC expression by decreasing the binding of bromodomain-containing-protein 4 (BRD4) to c-MYC SE regions (Delmore et al., 2011). SE-dependent transcription also utilizes cyclin-dependent kinase 7 (CDK7) at the initiation complex (TFIIH), which can be inhibited by the covalent inhibitor THZ1 (Chipumuro et al., 2014). Inhibitors of BET and CDK7 were recently tested in preclinical studies and significantly reduced tumor growth in cancer cell lines and tumor-bearing mice (Nakamura et al., 2017, Mertz et al., 2011). The effects of selective CDK7 and BET inhibitors in advanced solid tumors are now studied in clinical trials. In thyroid cancers cells, BET inhibitors repressed MYC expression in ATC, and the CDK7 inhibitor THZ1 repressed RET expression in MTC cells (Zhu et al., 2019, Cao et al., 2019, Valenciaga et al., 2018).
5. Overcoming resistance
5.1. Additional Kinase Inhibitors
Overcoming acquired resistance in BRAF-mutated PTC and ATC has been difficult because targeting MEK downstream of BRAFV600E with MEK inhibitors such as selumetinib, cobimetinib and trametinib, alone or in combination with BRAF inhibitors, rarely prevents progression (Table 2)(Wagle et al., 2014, Fofaria et al., 2015, Cabanillas et al., 2020). In addition, BRAF and/or MEK inhibitors might reactivate ERK1/2 activation/phosphorylation through parallel signaling pathways (Samatar and Poulikakos, 2014). Therefore, recent efforts have focused on targeting ERK1/2/ itself (Germann et al., 2017, Roskoski, 2019). Drugs showing promising preclinical activities such as BVD-523 (ulixertinib) are still in clinical trials, but the FDA recently granted expanded access to ulixertinib for treatment of stage IIb through Stage IV BRAF-mutant melanoma. Unfortunately, resistance to ERK1/2 inhibitors have already been identified, with activation of MEK5-ERK5 as a compensatory mechanism (Tubita et al., 2021).
In a recent study, Iyer and colleagues showed that ATC patients treated with lenvatinib, or dabrafenib combined with trametinib, showed additional response when given the PD-L1 inhibitor pembrolizumab after initial progression. Therefore, in certain cases, immunotherapy can be used as an effective therapy to improve the benefits of kinase inhibitors (Iyer et al., 2018).
5.2. Redifferentiation Therapies
The uptake of iodine by thyroid follicular cells is mediated by a sodium iodide symporter (NIS) located in the basolateral membrane of follicular cells. NIS spatiotemporal expression and function depends on a number of modulators. Thyroid stimulating hormone (TSH, thyrotropin) upregulates NIS expression and stimulates its transport to the plasma membrane (Kogai et al., 1997, Riedel et al., 2001). PIGU, a subunit of GPI transamidase that attaches GPI-anchors to proteins, ensures proper anchoring of NIS at the endoplasmic reticulum, and therefore at the plasma membrane after its vesicular transport, itself stimulated by ARF4 (Amit et al., 2017, Eisenhaber et al., 1998, Fletcher et al., 2020). These NIS effectors are counterbalanced by inhibitors of NIS function, expression, and localization. For example, at the plasma membrane, the NIS inhibitor PTTG-binding factor (PBF) inhibits the uptake of iodide by binding NIS (Smith et al., 2009). Further, β-catenin seems to control the availability of NIS since its overexpression leads to the decrease of NIS expression and its transfer from the plasma membrane to an intracellular location close to the nucleus (Lan et al., 2017a, Lan et al., 2017b). In thyroid cancer cells, the balance between positive and negative NIS modulating factors is dysregulated. Cellular dedifferentiation, which increases with the accumulation of BRAF, RAS, TERT or other gene mutations, correlates with downregulation of NIS expression at the plasma membrane and its relocation in the cytoplasm, which is not its proper functional location (Martin et al., 2019, Tavares et al., 2018). Thus, redifferentiation therapies often aim at redistributing NIS properly at the plasma membrane to improve NIS-mediated RAI treatment.
Increasing the level of serum thyrotropin (TSH) before RAI therapy, either by inducing hypothyroidism or administering recombinant human TSH, will significantly increase the uptake of RAI in differentiated tumors with intact iodine uptake mechanisms (Klubo-Gwiezdzinska et al., 2012, Haugen et al., 2016). However, additional modalities are required to re-differentiate tumors that have lost the capability to uptake iodine. Approaches have been attempted to increase NIS expression at the plasma membrane. For example, retinoic acid (RA) has been used to redifferentiate cancer cells for decades. RA is the bioactive metabolite of vitamin A and plays a critical role in cellular differentiation during mammalian development. Retinoic acid indeed may increase the expression of NIS mRNA in human FTC cells (Schmutzler et al., 2002, Lan et al., 2017a), however a recent meta-analysis reported that only a minority of patients with RAI-refractory DTC responded to RA treatment (Pak et al., 2018). This poor response might be explained by the fact that RA treatment induces growth inhibition through retinoic acid receptor beta (RARB) in cells where it is expressed, rather than through an increase of NIS at the plasma membrane (Elisei et al., 2005). Similarly, PPARγ agonists and histone deacetylase (HDAC) inhibitors, which showed good effects in preclinical studies, offered disappointing results in clinical trials (Park et al., 2005, Rosenbaum-Krumme et al., 2012, Cheng et al., 2016, Nilubol et al., 2017, Sherman et al., 2013, Woyach et al., 2009). Further, PBF phosphorylation levels could be reduced using a c-SRC kinase inhibitor, at least in vitro (Smith et al., 2013). Other preclinical studies indicated that treatment with MEK inhibitors rescued PIGU expression in PTC cells (Amit et al., 2017). Recently, Fletcher et al. demonstrated that clotrimazole, an antifungal medication and ebastine, an antihistaminic, also enhanced RAI uptake in normal mouse thyrocytes and human thyroid cancer cells, due to their allosteric inhibition of Valosin-containing protein (VCP/p97). They also demonstrated the importance of ARF4 in the trafficking of NIS to the plasma membrane (Fletcher et al., 2020). Further, clinically relevant redifferentiation was previously obtained in small cohorts of patients treated with MAPK pathway/BRAF inhibitors. In particular, dabrafenib, trametinib, and selumetinib, seemed to improve the iodine uptake rate of thyroid cancer cells by improving NIS expression and function (Ho et al., 2013, Rothenberg et al., 2015, Jaber et al., 2018, Leboulleux et al., 2021, Dunn et al., 2019). However, a recently completed phase III clinical trial showed that addition of selumetinib to adjuvant RAI did not significantly improve complete remission rate in DTC patients with metastatic disease (Ho et al., 2022). Finally, redifferentiation of NTRK-rearranged, RAI-resistant thyroid tumors was possible with larotrectinib (Groussin et al., 2022), and selpercatinib restored iodine uptake in almost all metastases in a patient with a RET rearrangement/fusion as a sole mutation (Groussin et al., 2021). While these results are encouraging, they were obtained from case studies. Therefore, these drugs should not yet be used as routine short-term pretreatments before RAI in differentiated thyroid cancers.
6. Conclusions
Protein kinases play critical roles in signaling pathways that transfer information from receptors located at the plasma membrane down to the nucleus, eventually modifying and adapting cell behavior to the environment. Mutational changes can dysregulate the function of many of these kinases, which become constitutively active and lead to cancer cell proliferation and metastases. Targeted therapies use kinase inhibitors that often target the ATP binding pocket. These novel drugs are better tolerated than chemotherapies. However, resistance to these inhibitors is a challenge that will need to be overcome. Multiple molecular strategies will be required to thoroughly understand mechanisms of resistance and devise effective targeted treatments, including disruption of the transcriptional machinery of specific genes and cellular redifferentiation. Finally, there is also a need to identify biomarkers that will help predict response and resistance to improve the selection of an optimal targeted therapy for each patient.
Funding
This work was supported by the National Cancer Institute (NCI) Grants P30CA016672, R21CA259839, R21CA259839, R01CA227847 as well as the MD Anderson Cancer Center Petrick Thyroid Cancer Fund.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported. Mark Zafereo has research (clinical trials) from Merck and Eli Lilly. Maria E. Cabanillas has research grant funding from Merck, Eisai, Exelixis and Genentech, and has participated in advisory boards for Exelixis, Blueprint, Ignyta, Bayer and LOXO/Eli Lilly. Stephen Y. Lai is a medical affairs consultant for Cardinal Health.
References
- Amit M, Na’ara S, Francis D, Matanis W, Zolotov S, Eisenhaber B, Eisenhaber F, Weiler Sagie M, Malkin L, Billan S, Charas T & Gil Z 2017. Post-translational Regulation of Radioactive Iodine Therapy Response in Papillary Thyroid Carcinoma. J Natl Cancer Inst, 109. [DOI] [PubMed] [Google Scholar]
- Atefi M, Von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, Mischel PS, Lo RS & Ribas A 2011. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One, 6, e28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Busaidy NL, Mcbeath E, Danysh BP, Evans KW, Moss TJ, Akcakanat A, Ng PKS, Knippler CM, Golden JA, Williams MD, Multani AS, Cabanillas ME, Shaw KR, Meric-Bernstam F, Shah MH, Ringel MD & Hofmann MC 2021. RAC1 Alterations Induce Acquired Dabrafenib Resistance in Association with Anaplastic Transformation in a Papillary Thyroid Cancer Patient. Cancers (Basel), 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barras D 2015. BRAF Mutation in Colorectal Cancer: An Update. Biomark Cancer, 7, 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertol BC, Bales ES, Calhoun JD, Mayberry A, Ledezma ML, Sams SB, Orlicky DJ, Donadi EA, Haugen BR & French JD 2022. Lenvatinib Plus Anti-PD-1 Combination Therapy for Advanced Cancers: Defining Mechanisms of Resistance in an Inducible Transgenic Model of Thyroid Cancer. Thyroid, 32, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhullar KS, Lagaron NO, Mcgowan EM, Parmar I, Jha A, Hubbard BP & Rupasinghe HPV 2018. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer, 17, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora E, Porcelli AM, Gasparre G, Biondi A, Ghelli A, Carelli V, Baracca A, Tallini G, Martinuzzi A, Lenaz G, Rugolo M & Romeo G 2006. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res, 66, 6087–6096. [DOI] [PubMed] [Google Scholar]
- Brognard J & Hunter T 2011. Protein kinase signaling networks in cancer. Curr Opin Genet Dev, 21, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, Sherman SI & Sherman EJ 2016. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol, 17, 1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, De La Fouchardiere C, Pacini F, Paschke R, Shong YK, Sherman SI, Smit JW, Chung J, Kappeler C, Pena C, Molnar I, Schlumberger MJ & Investigators D 2014. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet, 384, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose MS, Robinson B, Sherman SI, Krajewska J, Lin CC, Vaisman F, Hoff AO, Hitre E, Bowles DW, Hernando J, Faoro L, Banerjee K, Oliver JW, Keam B & Capdevila J 2021. Cabozantinib for radioiodine-refractory differentiated thyroid cancer (COSMIC-311): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol, 22, 1126–1138. [DOI] [PubMed] [Google Scholar]
- Brose MS, Worden FP, Newbold KL, Guo M & Hurria A 2017. Effect of Age on the Efficacy and Safety of Lenvatinib in Radioiodine-Refractory Differentiated Thyroid Cancer in the Phase III SELECT Trial. J Clin Oncol, 35, 2692–2699. [DOI] [PubMed] [Google Scholar]
- Byeon HK, Na HJ, Yang YJ, Ko S, Yoon SO, Ku M, Yang J, Kim JW, Ban MJ, Kim JH, Kim DH, Kim JM, Choi EC, Kim CH, Yoon JH & Koh YW 2017. Acquired resistance to BRAF inhibition induces epithelial-to-mesenchymal transition in BRAF (V600E) mutant thyroid cancer by c-Met-mediated AKT activation. Oncotarget, 8, 596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas ME, Dadu R, Iyer P, Wanland KB, Busaidy NL, Ying A, Gule-Monroe M, Wang JR, Zafereo M & Hofmann MC 2020. Acquired Secondary RAS Mutation in BRAF(V600E)-Mutated Thyroid Cancer Patients Treated with BRAF Inhibitors. Thyroid, 30, 1288–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas ME, Ryder M & Jimenez C 2019. Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and Beyond. Endocr Rev, 40, 1573–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer_Genome_Atlas_Research_Network 2014. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon AC, Uribe-Alvarez C & Chernoff J 2020. RAC1 as a Therapeutic Target in Malignant Melanoma. Trends Cancer, 6, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Dang L, Zheng X, Lu Y, Lu Y, Ji R, Zhang T, Ruan X, Zhi J, Hou X, Yi X, Li MJ, Gu T, Gao M, Zhang L & Chen Y 2019. Targeting Super-Enhancer-Driven Oncogenic Transcription by CDK7 Inhibition in Anaplastic Thyroid Carcinoma. Thyroid, 29, 809–823. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Mayor R, Mancuso FM, Iglesias C, Caratu G, Matos I, Zafon C, Hernando J, Petit A, Nuciforo P, Cameselle-Teijeiro JM, Alvarez CV, Recio JA, Tabernero J, Matias-Guiu X, Vivancos A & Seoane J 2019. Early evolutionary divergence between papillary and anaplastic thyroid cancers. Ann Oncol, 30, 1843. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Wirth LJ, Ernst T, Ponce Aix S, Lin CC, Ramlau R, Butler MO, Delord JP, Gelderblom H, Ascierto PA, Fasolo A, Fuhrer D, Hutter-Kronke ML, Forde PM, Wrona A, Santoro A, Sadow PM, Szpakowski S, Wu H, Bostel G, Faris J, Cameron S, Varga A & Taylor M 2020. PD-1 Blockade in Anaplastic Thyroid Carcinoma. J Clin Oncol, 38, 2620–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanovas O, Hicklin DJ, Bergers G & Hanahan D 2005. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell, 8, 299–309. [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, Mcarthur GA & Group B-S 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med, 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Liu R, Zhu G, Wang H & Xing M 2016. Robust Thyroid Gene Expression and Radioiodine Uptake Induced by Simultaneous Suppression of BRAF V600E and Histone Deacetylase in Thyroid Cancer Cells. J Clin Endocrinol Metab, 101, 962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, Perez-Atayde A, Wong KK, Yuan GC, Gray NS, Young RA & George RE 2014. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell, 159, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampi R, Romei C, Ramone T, Prete A, Tacito A, Cappagli V, Bottici V, Viola D, Torregrossa L, Ugolini C, Basolo F & Elisei R 2019. Genetic Landscape of Somatic Mutations in a Large Cohort of Sporadic Medullary Thyroid Carcinomas Studied by Next-Generation Targeted Sequencing. iScience, 20, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicenas J, Zalyte E, Bairoch A & Gaudet P 2018. Kinases and Cancer. Cancers (Basel), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, Flaherty KT, Piris A, Wargo JA, Settleman J, Mino-Kenudson M & Engelman JA 2012. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov, 2, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadu R, Shah K, Busaidy NL, Waguespack SG, Habra MA, Ying AK, Hu MI, Bassett R, Jimenez C, Sherman SI & Cabanillas ME 2015. Efficacy and tolerability of vemurafenib in patients with BRAF(V600E) -positive papillary thyroid cancer: M.D. Anderson Cancer Center off label experience. J Clin Endocrinol Metab, 100, E77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysh BP, Rieger EY, Sinha DK, Evers CV, Cote GJ, Cabanillas ME & Hofmann MC 2016. Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget, 7, 30907–30923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR & Futreal PA 2002. Mutations of the BRAF gene in human cancer. Nature, 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Delmas A, Cherier J, Pohorecka M, Medale-Giamarchi C, Meyer N, Casanova A, Sordet O, Lamant L, Savina A, Pradines A & Favre G 2015. The c-Jun/RHOB/AKT pathway confers resistance of BRAF-mutant melanoma cells to MAPK inhibitors. Oncotarget, 6, 15250–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, Mckeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE & Mitsiades CS 2011. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell, 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan R, Du W & Guo W 2020. EZH2: a novel target for cancer treatment. J Hematol Oncol, 13, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, Chiarion-Sileni V, Dutriaux C, De Groot JWB, Yamazaki N, Loquai C, Moutouh-De Parseval LA, Pickard MD, Sandor V, Robert C & Flaherty KT 2018. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol, 19, 603–615. [DOI] [PubMed] [Google Scholar]
- Dunn LA, Sherman EJ, Baxi SS, Tchekmedyian V, Grewal RK, Larson SM, Pentlow KS, Haque S, Tuttle RM, Sabra MM, Fish S, Boucai L, Walters J, Ghossein RA, Seshan VE, Ni A, Li D, Knauf JA, Pfister DG, Fagin JA & Ho AL 2019. Vemurafenib Redifferentiation of BRAF Mutant, RAI-Refractory Thyroid Cancers. J Clin Endocrinol Metab, 104, 1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette M, Sadow PM, Husain A, Sims JN, Antonello ZA, Fischer AH, Song C, Castellanos-Rizaldos E, Makrigiorgos GM, Kurebayashi J, Nose V, Van Hummelen P, Bronson RT, Vinco M, Giordano TJ, Dias-Santagata D, Pandolfi PP & Nucera C 2015. Metastasis-associated MCL1 and P16 copy number alterations dictate resistance to vemurafenib in a BRAFV600E patient-derived papillary thyroid carcinoma preclinical model. Oncotarget, 6, 42445–42467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P & Eisenhaber F 1998. Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng, 11, 1155–1161. [DOI] [PubMed] [Google Scholar]
- Elisei R, Schlumberger MJ, Muller SP, Schoffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V, Kreissl MC, Niederle B, Cohen EE, Wirth LJ, Ali H, Hessel C, Yaron Y, Ball D, Nelkin B & Sherman SI 2013. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol, 31, 3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elisei R, Vivaldi A, Agate L, Ciampi R, Molinaro E, Piampiani P, Romei C, Faviana P, Basolo F, Miccoli P, Capodanno A, Collecchi P, Pacini F & Pinchera A 2005. All-trans-retinoic acid treatment inhibits the growth of retinoic acid receptor beta messenger ribonucleic acid expressing thyroid cancer cell lines but does not reinduce the expression of thyroid-specific genes. J Clin Endocrinol Metab, 90, 2403–2411. [DOI] [PubMed] [Google Scholar]
- Fagin JA & Wells SA Jr. 2016. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med, 375, 1054–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook GS, Millward M, Hong D, Naing A, Piha-Paul S, Waguespack SG, Cabanillas ME, Sherman SI, Ma B, Curtis M, Goodman V & Kurzrock R 2015. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid, 25, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher A, Read ML, Thornton CEM, Larner DP, Poole VL, Brookes K, Nieto HR, Alshahrani M, Thompson RJ, Lavery GG, Landa I, Fagin JA, Campbell MJ, Boelaert K, Turnell AS, Smith VE & Mccabe CJ 2020. Targeting Novel Sodium Iodide Symporter Interactors ADP-Ribosylation Factor 4 and Valosin-Containing Protein Enhances Radioiodine Uptake. Cancer Res, 80, 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fofaria NM, Frederick DT, Sullivan RJ, Flaherty KT & Srivastava SK 2015. Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget, 6, 40535–40556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganly I, Makarov V, Deraje S, Dong Y, Reznik E, Seshan V, Nanjangud G, Eng S, Bose P, Kuo F, Morris LGT, Landa I, Carrillo Albornoz PB, Riaz N, Nikiforov YE, Patel K, Umbricht C, Zeiger M, Kebebew E, Sherman E, Ghossein R, Fagin JA & Chan TA 2018. Integrated Genomic Analysis of Hurthle Cell Cancer Reveals Oncogenic Drivers, Recurrent Mitochondrial Mutations, and Unique Chromosomal Landscapes. Cancer Cell, 34, 256–270 e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann UA, Furey BF, Markland W, Hoover RR, Aronov AM, Roix JJ, Hale M, Boucher DM, Sorrell DA, Martinez-Botella G, Fitzgibbon M, Shapiro P, Wick MJ, Samadani R, Meshaw K, Groover A, Decrescenzo G, Namchuk M, Emery CM, Saha S & Welsch DJ 2017. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol Cancer Ther, 16, 2351–2363. [DOI] [PubMed] [Google Scholar]
- Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L & Gilmer TM 2012. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther, 11, 909–920. [DOI] [PubMed] [Google Scholar]
- Groussin L, Bessiene L, Arrondeau J, Garinet S, Cochand-Priollet B, Lupo A, Zerbit J, Clerc J & Huillard O 2021. Letter to the Editor: Selpercatinib-Enhanced Radioiodine Uptake in RET-Rearranged Thyroid Cancer. Thyroid, 31, 1603–1604. [DOI] [PubMed] [Google Scholar]
- Groussin L, Theodon H, Bessiene L, Bricaire L, Bonnet-Serrano F, Cochand-Priollet B, Leroy K, Garinet S, Pasmant E, Zerbit J, Seban R, Goldwasser F, Clerc J, Cottereau AS & Huillard O 2022. Redifferentiating Effect of Larotrectinib in NTRK-Rearranged Advanced Radioactive-Iodine Refractory Thyroid Cancer. Thyroid. [DOI] [PubMed] [Google Scholar]
- Hallberg B & Palmer RH 2013. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer, 13, 685–700. [DOI] [PubMed] [Google Scholar]
- Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, Pacini F, Randolph GW, Sawka AM, Schlumberger M, Schuff KG, Sherman SI, Sosa JA, Steward DL, Tuttle RM & Wartofsky L 2016. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid, 26, 1–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde A, Andreev-Drakhlin AY, Roszik J, Huang L, Liu S, Hess K, Cabanillas M, Hu MI, Busaidy NL, Sherman SI, Dadu R, Grubbs EG, Ali SM, Lee J, Elamin YY, Simon GR, Blumenschein GR Jr., Papadimitrakopoulou VA, Hong D, Meric-Bernstam F, Heymach J & Subbiah V 2020. Responsiveness to immune checkpoint inhibitors versus other systemic therapies in RET-aberrant malignancies. ESMO Open, 5, e000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AL, Dedecjus M, Wirth LJ, Tuttle RM, Inabnet WB 3rd, Tennvall J, Vaisman F, Bastholt L, Gianoukakis AG, Rodien P, Paschke R, Elisei R, Viola D, So K, Carroll D, Hovey T, Thakre B, Fagin JA & Group AI 2022. Selumetinib Plus Adjuvant Radioactive Iodine in Patients With High-Risk Differentiated Thyroid Cancer: A Phase III, Randomized, Placebo-Controlled Trial (ASTRA). J Clin Oncol, 40, 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, Pentlow KS, Zanzonico PB, Haque S, Gavane S, Ghossein RA, Ricarte-Filho JC, Dominguez JM, Shen R, Tuttle RM, Larson SM & Fagin JA 2013. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med, 368, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque MO, Brait M, Rosenbaum E, Poeta ML, Pal P, Begum S, Dasgupta S, Carvalho AL, Ahrendt SA, Westra WH & Sidransky D 2010. Genetic and epigenetic analysis of erbB signaling pathway genes in lung cancer. J Thorac Oncol, 5, 1887–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Guo Z, Wang F & Fu L 2021. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther, 6, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer PC, Dadu R, Gule-Monroe M, Busaidy NL, Ferrarotto R, Habra MA, Zafereo M, Williams MD, Gunn GB, Grosu H, Skinner HD, Sturgis EM, Gross N & Cabanillas ME 2018. Salvage pembrolizumab added to kinase inhibitor therapy for the treatment of anaplastic thyroid carcinoma. J Immunother Cancer, 6, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber T, Waguespack SG, Cabanillas ME, Elbanan M, Vu T, Dadu R, Sherman SI, Amit M, Santos EB, Zafereo M & Busaidy NL 2018. Targeted Therapy in Advanced Thyroid Cancer to Resensitize Tumors to Radioactive Iodine. J Clin Endocrinol Metab, 103, 3698–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan HY, Ge J, Nagasaka M, Aboukameel A, Mpilla G, Muqbil I, Szlaczky M, Chaker M, Baloglu E, Landesman Y, Mohammad RM, Azmi AS & Sukari A 2019. Targeting XPO1 and PAK4 in 8505C Anaplastic Thyroid Cancer Cells: Putative Implications for Overcoming Lenvatinib Therapy Resistance. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, Nolop K, Lee RJ & Sherman SI 2013. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid, 23, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klubo-Gwiezdzinska J, Burman KD, Van Nostrand D, Mete M, Jonklaas J & Wartofsky L 2012. Radioiodine treatment of metastatic thyroid cancer: relative efficacy and side effect profile of preparation by thyroid hormone withdrawal versus recombinant human thyrotropin. Thyroid, 22, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogai T, Endo T, Saito T, Miyazaki A, Kawaguchi A & Onaya T 1997. Regulation by thyroid-stimulating hormone of sodium/iodide symporter gene expression and protein levels in FRTL-5 cells. Endocrinology, 138, 2227–2232. [DOI] [PubMed] [Google Scholar]
- Kopetz S, Desai J, Chan E, Hecht JR, O’dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, Issa JP, Gibbs P, James B, Powis G, Nolop KB, Bhattacharya S & Saltz L 2015. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol, 33, 4032–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiry P, Torkamani A, Schork NJ & Hegele RA 2010. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat Rev Genet, 11, 60–74. [DOI] [PubMed] [Google Scholar]
- Lan L, Basourakos S, Cui D, Zuo X, Deng W, Huo L, Chen H, Zhang G, Deng L, Shi B & Luo Y 2017a. ATRA increases iodine uptake and inhibits the proliferation and invasiveness of human anaplastic thyroid carcinoma SW1736 cells: Involvement of beta-catenin phosphorylation inhibition. Oncol Lett, 14, 7733–7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L, Basourakos S, Cui D, Zuo X, Deng W, Huo L, Chen L, Zhang G, Deng L, Shi B & Luo Y 2017b. Inhibiting beta-catenin expression promotes efficiency of radioiodine treatment in aggressive follicular thyroid cancer cells probably through mediating NIS localization. Oncol Rep, 37, 426–434. [DOI] [PubMed] [Google Scholar]
- Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP, Xu B, Schultz N, Berger MF, Sander C, Taylor BS, Ghossein R, Ganly I & Fagin JA 2016. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest, 126, 1052–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboulleux S, Do Cao C, Zerdoud S, Attard M, Bournaud C, Benisvy D, Taieb D, Bardet S, Terroir-Cassou-Mounat M, Betrian S, Lion G, Schiazza A, Sajous C, Garcia ME, Schlumberger MJ, Godbert Y & Borget I 2021. MERAIODE: A Redifferentiation Phase II Trial With Trametinib and Dabrafenib Followed by Radioactive Iodine Administration for Metastatic Radioactive Iodine Refractory Differentiated Thyroid Cancer Patients With a BRAFV600E Mutation. J Endocrine Soc, 5, A876. [Google Scholar]
- Lee S, Kim J, Jo J, Chang JW, Sim J & Yun H 2021. Recent advances in development of hetero-bivalent kinase inhibitors. Eur J Med Chem, 216, 113318. [DOI] [PubMed] [Google Scholar]
- Li C, Wang Y, Gong Y, Zhang T, Huang J, Tan Z & Xue L 2021a. Finding an easy way to harmonize: a review of advances in clinical research and combination strategies of EZH2 inhibitors. Clin Epigenetics, 13, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GH, Qu Q, Qi TT, Teng XQ, Zhu HH, Wang JJ, Lu Q & Qu J 2021b. Super-enhancers: a new frontier for epigenetic modifiers in cancer chemoresistance. J Exp Clin Cancer Res, 40, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA & Kefford RF 2011. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol, 29, 1239–1246. [DOI] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, Demarini DJ, Irani JG, Casey M, Ouellet D, Martin AM, Le N, Patel K & Flaherty K 2014. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med, 371, 1877–1888. [DOI] [PubMed] [Google Scholar]
- Lopez-Gines C, Gil-Benso R, Ferrer-Luna R, Benito R, Serna E, Gonzalez-Darder J, Quilis V, Monleon D, Celda B & Cerda-Nicolas M 2010. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod Pathol, 23, 856–865. [DOI] [PubMed] [Google Scholar]
- Mack NA, Whalley HJ, Castillo-Lluva S & Malliri A 2011. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle, 10, 1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniakas A, Dadu R, Busaidy NL, Wang JR, Ferrarotto R, Lu C, Williams MD, Gunn GB, Hofmann MC, Cote G, Sperling J, Gross ND, Sturgis EM, Goepfert RP, Lai SY, Cabanillas ME & Zafereo M 2020. Evaluation of Overall Survival in Patients With Anaplastic Thyroid Carcinoma, 2000–2019. JAMA Oncol, 6, 1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Geysels RC, Peyret V, Bernal Barquero CE, Masini-Repiso AM & Nicola JP 2019. Implications of Na(+)/I(−) Symporter Transport to the Plasma Membrane for Thyroid Hormonogenesis and Radioiodide Therapy. J Endocr Soc, 3, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R, Defnet A & Shapiro P 2020. Avoiding or Co-Opting ATP Inhibition: Overview of Type III, IV, V, and VI Kinase Inhibitors. In: SHAPIRO P (ed.) Next Generation Kinase Inhibitors Cham: Springer. [Google Scholar]
- McFadden DG & Sadow PM 2021. Genetics, Diagnosis, and Management of Hurthle Cell Thyroid Neoplasms. Front Endocrinol (Lausanne), 12, 696386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert JM, Varga A, Brose MS, Aggarwal RR, Lin CC, Prawira A, De Braud F, Tamura K, Doi T, Piha-Paul SA, Gilbert J, Saraf S, Thanigaimani P, Cheng JD & Keam B 2019. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer, 19, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L & Sims RJ 3rd 2011. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A, 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao B, Ji Z, Tan L, Taylor M, Zhang J, Choi HG, Frederick DT, Kumar R, Wargo JA, Flaherty KT, Gray NS & Tsao H 2015. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov, 5, 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsma DJ, Cherba DM, Eugster EE, Dylewski DL, Davidson PT, Peterson CA, Borgman AS, Winn ME, Dykema KJ, Webb CP, Mackeigan JP, Duesbery NS, Nickoloff BJ & Monks NR 2015. Melanoma patient derived xenografts acquire distinct Vemurafenib resistance mechanisms. Am J Cancer Res, 5, 1507–1518. [PMC free article] [PubMed] [Google Scholar]
- Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA, Rosen N & Fagin JA 2013. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov, 3, 520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Hattori N, Iida N, Yamashita S, Mori A, Kimura K, Yoshino T & Ushijima T 2017. Targeting of super-enhancers and mutant BRAF can suppress growth of BRAF-mutant colon cancer cells via repression of MAPK signaling pathway. Cancer Lett, 402, 100–109. [DOI] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, Mcarthur G, Sosman JA, Ribas A & Lo RS 2010. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature, 468, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov YE 2002. RET/PTC rearrangement in thyroid tumors. Endocr Pathol, 13, 3–16. [DOI] [PubMed] [Google Scholar]
- Nikiforov YE & Nikiforova MN 2011. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol, 7, 569–580. [DOI] [PubMed] [Google Scholar]
- Nilubol N, Merkel R, Yang L, Patel D, Reynolds JC, Sadowski SM, Neychev V & Kebebew E 2017. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin Endocrinol (Oxf), 86, 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunwobi OO, Puszyk W, Dong HJ & Liu C 2013. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS One, 8, e63765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Ikemori-Kawada M, Jestel A, Von Konig K, Funahashi Y, Matsushima T, Tsuruoka A, Inoue A & Matsui J 2015. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med Chem Lett, 6, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DH, Konda B, Sipos J, Liu T, Webb A, Ringel MD, Timmers CD & Shah MH 2019. KRAS G12V Mutation in Acquired Resistance to Combined BRAF and MEK Inhibition in Papillary Thyroid Cancer. J Natl Compr Canc Netw, 17, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak K, Shin S, Kim SJ, Kim IJ, Chang S, Koo P, Kwak J & Kim JH 2018. Response of Retinoic Acid in Patients with Radioactive Iodine-Refractory Thyroid Cancer: A Meta-Analysis. Oncol Res Treat, 41, 100–104. [DOI] [PubMed] [Google Scholar]
- Park JC, Ashok A, Liu C & Kang H 2022. Real-World Experience of NTRK Fusion-Positive Thyroid Cancer. JCO Precis Oncol, 6, e2100442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Zarnegar R, Kanauchi H, Wong MG, Hyun WC, Ginzinger DG, Lobo M, Cotter P, Duh QY & Clark OH 2005. Troglitazone, the peroxisome proliferator-activated receptor-gamma agonist, induces antiproliferation and redifferentiation in human thyroid cancer cell lines. Thyroid, 15, 222–231. [DOI] [PubMed] [Google Scholar]
- Peng YH, Shiao HY, Tu CH, Liu PM, Hsu JT, Amancha PK, Wu JS, Coumar MS, Chen CH, Wang SY, Lin WH, Sun HY, Chao YS, Lyu PC, Hsieh HP & Wu SY 2013. Protein kinase inhibitor design by targeting the Asp-Phe-Gly (DFG) motif: the role of the DFG motif in the design of epidermal growth factor receptor inhibitors. J Med Chem, 56, 3889–3903. [DOI] [PubMed] [Google Scholar]
- Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, French JD, Borre PV, Labarbera DV, Tan AC, Schweppe RE, Fishbein L, Ross JS, Haugen BR & Bowles DW 2018. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin Cancer Res, 24, 3059–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A & Bernards R 2012. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature, 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Qin Y, Wang JR, Wang Y, Iyer P, Cote GJ, Busaidy NL, Dadu R, Zafereo M, Williams MD, Ferrarotto R, Gunn GB, Wei P, Patel K, Hofmann MC & Cabanillas ME 2021. Clinical Utility of Circulating Cell-Free DNA Mutations in Anaplastic Thyroid Carcinoma. Thyroid, 31, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel C, Levy O & Carrasco N 2001. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem, 276, 21458–21463. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Antona C, Pallares J, Montero-Conde C, Inglada-Perez L, Castelblanco E, Landa I, Leskela S, Leandro-Garcia LJ, Lopez-Jimenez E, Leton R, Cascon A, Lerma E, Martin MC, Carralero MC, Mauricio D, Cigudosa JC, Matias-Guiu X & Robledo M 2010. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis. Endocr Relat Cancer, 17, 7–16. [DOI] [PubMed] [Google Scholar]
- Rosenbaum-Krumme SJ, Freudenberg LS, Jentzen W, Bockisch A & Nagarajah J 2012. Effects of rosiglitazone on radioiodine negative and progressive differentiated thyroid carcinoma as assessed by (1)(2)(4)I PET/CT imaging. Clin Nucl Med, 37, e47–52. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. 2019. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol Res, 142, 151–168. [DOI] [PubMed] [Google Scholar]
- Rothenberg SM, Mcfadden DG, Palmer EL, Daniels GH & Wirth LJ 2015. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin Cancer Res, 21, 1028–1035. [DOI] [PubMed] [Google Scholar]
- Samatar AA & Poulikakos PI 2014. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov, 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, Habra MA, Newbold K, Shah MH, Hoff AO, Gianoukakis AG, Kiyota N, Taylor MH, Kim SB, Krzyzanowska MK, Dutcus CE, De Las Heras B, Zhu J & Sherman SI 2015. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med, 372, 621–630. [DOI] [PubMed] [Google Scholar]
- Schmutzler C, Schmitt TL, Glaser F, Loos U & Kohrle J 2002. The promoter of the human sodium/iodide-symporter gene responds to retinoic acid. Mol Cell Endocrinol, 189, 145–155. [DOI] [PubMed] [Google Scholar]
- Semrad TJ, Kim EJ, Tanaka MS, Sands J, Roberts C, Burich RA, Li Y, Gandara DR, Lara P Jr. & Mack PC 2017. Phase II Study of Dovitinib in Patients Progressing on Anti-Vascular Endothelial Growth Factor Therapy. Cancer Treat Res Commun, 10, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok SH 2021. Structural Insights into Protein Regulation by Phosphorylation and Substrate Recognition of Protein Kinases/Phosphatases. Life (Basel), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman EJ, Su YB, Lyall A, Schoder H, Fury MG, Ghossein RA, Haque S, Lisa D, Shaha AR, Tuttle RM & Pfister DG 2013. Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid, 23, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Fuchs HE & Jemal A 2021. Cancer Statistics, 2021. CA Cancer J Clin, 71, 7–33. [DOI] [PubMed] [Google Scholar]
- Sipos JA & Ringel MD 2022. Molecular testing in thyroid cancer diagnosis and management. Best Pract Res Clin Endocrinol Metab, 101680. [DOI] [PubMed] [Google Scholar]
- Smith VE, Read ML, Turnell AS, Watkins RJ, Watkinson JC, Lewy GD, Fong JC, James SR, Eggo MC, Boelaert K, Franklyn JA & Mccabe CJ 2009. A novel mechanism of sodium iodide symporter repression in differentiated thyroid cancer. J Cell Sci, 122, 3393–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith VE, Sharma N, Watkins RJ, Read ML, Ryan GA, Kwan PP, Martin A, Watkinson JC, Boelaert K, Franklyn JA & Mccabe CJ 2013. Manipulation of PBF/PTTG1IP phosphorylation status; a potential new therapeutic strategy for improving radioiodine uptake in thyroid and other tumors. J Clin Endocrinol Metab, 98, 2876–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrinho-Simoes M, Eloy C, Magalhaes J, Lobo C & Amaro T 2011. Follicular thyroid carcinoma. Mod Pathol, 24 Suppl 2, S10–18. [DOI] [PubMed] [Google Scholar]
- Song YS & Park YJ 2019. Genomic Characterization of Differentiated Thyroid Carcinoma. Endocrinol Metab (Seoul), 34, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA & Golub TR 2012. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature, 487, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, Hu W, Cao Q, Sheets MP, Wilson D, Wilson KJ, Dipietro L, Fleming P, Palmer M, Hu MI, Wirth L, Brose MS, Ou SI, Taylor M, Garralda E, Miller S, Wolf B, Lengauer C, Guzi T & Evans EK 2018a. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov, 8, 836–849. [DOI] [PubMed] [Google Scholar]
- Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski C, Cabanillas ME, Urbanowitz G, Mookerjee B, Wang D, Rangwala F & Keam B 2018b. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J Clin Oncol, 36, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski CC, Cabanillas ME, Boran A, Ilankumaran P, Burgess P, Romero Salas T & Keam B 2022. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: updated analysis from the phase II ROAR basket study. Ann Oncol, 33, 406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, Wirth LJ, Stock S, Smith S, Lauriault V, Corsi-Travali S, Henry D, Burkard M, Hamor R, Bouhana K, Winski S, Wallace RD, Hartley D, Rhodes S, Reddy M, Brandhuber BJ, Andrews S, Rothenberg SM & Drilon A 2018c. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol, 29, 1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Yang D, Velcheti V, Drilon A & Meric-Bernstam F 2020. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J Clin Oncol, 38, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, Desai J, Ciardiello F, Loupakis F, Hong YS, Steeghs N, Guren TK, Arkenau HT, Garcia-Alfonso P, Elez E, Gollerkeri A, Maharry K, Christy-Bittel J & Kopetz S 2021. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J Clin Oncol, 39, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Yang Z, Tan Y & Li Y 2020. Super-enhancer function and its application in cancer targeted therapy. NPJ Precis Oncol, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares C, Coelho MJ, Eloy C, Melo M, Da Rocha AG, Pestana A, Batista R, Ferreira LB, Rios E, Selmi-Ruby S, Cavadas B, Pereira L, Sobrinho Simoes M & Soares P 2018. NIS expression in thyroid tumors, relation with prognosis clinicopathological and molecular features. Endocr Connect, 7, 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubita A, Tusa I & Rovida E 2021. Playing the Whack-A-Mole Game: ERK5 Activation Emerges Among the Resistance Mechanisms to RAF-MEK1/2-ERK1/2- Targeted Therapy. Front Cell Dev Biol, 9, 647311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann TM, Thiesmeyer JW, Lee YJ, Beg S, Mosquera JM, Elemento O, Fahey TJ 3rd, Scognamiglio T & Houvras Y 2022. RET Fusion-Positive Papillary Thyroid Cancers are Associated with a More Aggressive Phenotype. Ann Surg Oncol. [DOI] [PubMed] [Google Scholar]
- Valenciaga A, Saji M, Yu L, Zhang X, Bumrah C, Yilmaz AS, Knippler CM, Miles W, Giordano TJ, Cote GJ & Ringel MD 2018. Transcriptional targeting of oncogene addiction in medullary thyroid cancer. JCI Insight, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volante M, Lam AK, Papotti M & Tallini G 2021. Molecular Pathology of Poorly Differentiated and Anaplastic Thyroid Cancer: What Do Pathologists Need to Know? Endocr Pathol, 32, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong HG, Odate T, Ngo HTT, Pham TQ, Tran TTK, Mochizuki K, Nakazawa T, Katoh R & Kondo T 2018. Clinical significance of RET and RAS mutations in sporadic medullary thyroid carcinoma: a meta-analysis. Endocr Relat Cancer, 25, 633–641. [DOI] [PubMed] [Google Scholar]
- Wagenaar TR, Ma L, Roscoe B, Park SM, Bolon DN & Green MR 2014. Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment Cell Melanoma Res, 27, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, Johannessen CM, Mckenna A, Cibulskis K, Kryukov G, Hodis E, Lawrence DP, Fisher S, Getz G, Gabriel SB, Carter SL, Flaherty KT, Wargo JA & Garraway LA 2014. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov, 4, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R & Cancer Genome P 2004. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell, 116, 855–867. [DOI] [PubMed] [Google Scholar]
- Wang JR, Zafereo ME, Dadu R, Ferrarotto R, Busaidy NL, Lu C, Ahmed S, Gule-Monroe MK, Williams MD, Sturgis EM, Goepfert RP, Gross ND, Lai SY, Gunn GB, Phan J, Rosenthal DI, Fuller CD, Morrison WH, Iyer P & Cabanillas ME 2019a. Complete Surgical Resection Following Neoadjuvant Dabrafenib Plus Trametinib in BRAF(V600E)-Mutated Anaplastic Thyroid Carcinoma. Thyroid, 29, 1036–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Dai J, Yan J, Zhang Y & Yin Z 2019b. Targeting EZH2 as a novel therapeutic strategy for sorafenib-resistant thyroid carcinoma. J Cell Mol Med, 23, 4770–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, Li L, Cabeceiras PK, Mahdavi M, Gutschner T, Genovese G, Wang G, Fang Z, Tepper JM, Stemke-Hale K, Tsai KY, Davies MA, Mills GB & Chin L 2014. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res, 74, 4845–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisner J, Landel I, Reintjes C, Uhlenbrock N, Trajkovic-Arsic M, Dienstbier N, Hardick J, Ladigan S, Lindemann M, Smith S, Quambusch L, Scheinpflug R, Depta L, Gontla R, Unger A, Muller H, Baumann M, Schultz-Fademrecht C, Gunther G, Maghnouj A, Muller MP, Pohl M, Teschendorf C, Wolters H, Viebahn R, Tannapfel A, Uhl W, Hengstler JG, Hahn SA, Siveke JT & Rauh D 2019. Preclinical Efficacy of Covalent-Allosteric AKT Inhibitor Borussertib in Combination with Trametinib in KRAS-Mutant Pancreatic and Colorectal Cancer. Cancer Res, 79, 2367–2378. [DOI] [PubMed] [Google Scholar]
- Wells SA Jr., Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR, Read J, Langmuir P, Ryan AJ & Schlumberger MJ 2012. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol, 30, 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R & Settleman J 2012. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature, 487, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, Worden F, Brose M, Patel J, Leboulleux S, Godbert Y, Barlesi F, Morris JC, Owonikoko TK, Tan DSW, Gautschi O, Weiss J, De La Fouchardiere C, Burkard ME, Laskin J, Taylor MH, Kroiss M, Medioni J, Goldman JW, Bauer TM, Levy B, Zhu VW, Lakhani N, Moreno V, Ebata K, Nguyen M, Heirich D, Zhu EY, Huang X, Yang L, Kherani J, Rothenberg SM, Drilon A, Subbiah V, Shah MH & Cabanillas ME 2020. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N Engl J Med, 383, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach JA, Kloos RT, Ringel MD, Arbogast D, Collamore M, Zwiebel JA, Grever M, Villalona-Calero M & Shah MH 2009. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J Clin Endocrinol Metab, 94, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M 2005. BRAF mutation in thyroid cancer. Endocr Relat Cancer, 12, 245–262. [DOI] [PubMed] [Google Scholar]
- Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ, Carson KA, Vasko V, Larin A, Tallini G, Tolaney S, Holt EH, Hui P, Umbricht CB, Basaria S, Ewertz M, Tufaro AP, Califano JA, Ringel MD, Zeiger MA, Sidransky D & Ladenson PW 2005. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab, 90, 6373–6379. [DOI] [PubMed] [Google Scholar]
- Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, Buchanan S, Henry JR, Starling JJ & Peng SB 2012. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem, 287, 28087–28098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Xie L & Bourne PE 2017. Insights into the binding mode of MEK type-III inhibitors. A step towards discovering and designing allosteric kinase inhibitors across the human kinome. PLoS One, 12, e0179936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Park S, Lee WK & Cheng SY 2019. Potentiated anti-tumor effects of BETi by MEKi in anaplastic thyroid cancer. Endocr Relat Cancer, 26, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YJ, Zheng B, Wang HY & Chen L 2017. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin, 38, 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]