

Abstract
Aryl thioethers are tremendously important motifs in various facets of chemical science. Traditional technologies for the precise assembly of aryl thioethers rely on transition-metal-catalyzed cross-coupling of aryl halides; however, despite the continuous advances, the scope of these methods remains limited. Recently a series of reports has advanced an alternative manifold in which thio(esters) are subject to transition-metal-catalyzed decarbonylation, which (1) permits to exploit ubiquitous carboxylic acids as highly desirable and orthogonal precursors to aryl halides; (2) overcomes the issues of high concentration of thiolate anion leading to catalyst poisoning; (3) enables for novel disconnections not easily available from aryl halides; and (4) introduces new concepts in catalysis.
Keywords: sulfur, thioethers, cross-coupling, decarbonylation, aryl exchange
Graphical Abstract

Aryl thioethers represent one of the most important moieties in chemical science, ranging from polymers through functional materials to pharmaceuticals. Recently a series of reports has advanced an alternative cross-coupling manifold in which thio(esters) are subject to transition-metal-catalyzed intramolecular decarbonylation. These emerging technologies address challenges encountered during the traditional synthesis and permit to directly install aryl thioethers on complex APIs, thus advancing horizons for novel previously unattainable synthetic disconnections and introducing new concepts in catalysis.
Aryl thioethers represent one of the most important moieties in chemical science, ranging from polymers through functional materials to pharmaceuticals.[1,2] For example, sulfur-containing polymers, such as aryl polythioethers show enhanced properties over aryl polyethers.[2a,b] Furthermore, aryl thioethers are the key component of sulfur-functionalized metal-organic frameworks, where the soft and polarizable nature of sulfur leads to unique affinity to metal ions.[2c] Perhaps most importantly, the privileged role of sulfur in medicinal chemistry has led to a number of breakthroughs in drug design,[2c–2h] including sulfa drugs,[2i] and now sulfur containing drugs constitute more than 25% of all approved pharmaceuticals (Figure 1). Thus, it comes as no surprise that new methods for the synthesis of aryl thioethers are highly desirable.
Figure 1.

Commercially available APIs featuring aryl thioethers.
Synthetically, the most popular method for the synthesis of aryl thioethers involves transition-metal-catalyzed cross-coupling of aryl halides or pseudohalides with thiols, first reported by Migita in 1978 (Figure 2A).[3] This technology has significantly expanded the horizons available with the transition-metal-free approaches based on SNAr-type reactions.[1a–1d] In the last decades, major advances using Pd–phosphine systems have been established that enable the widespread application of C–S coupling in chemical synthesis.[4–7] In particular, the development of bidentate phosphine systems Josiphos and DiPPF by Hartwig[4a–4c] and Buchwald[4d] groups, the more recent studies on soluble-base approach with designer monodentate biaryl phosphines, such as tBuBrettPhos,[5] as well as those exploiting orthogonal reactivity of the bridged Pd(I) dimer, [(tBu3P)Pd(μ-I)]2,[6] have provided great milestones in the synthesis of aryl thioethers and advanced the efforts to precisely harness the reactivity of thiols in cross-coupling reactions.[7,8]
Figure 2.
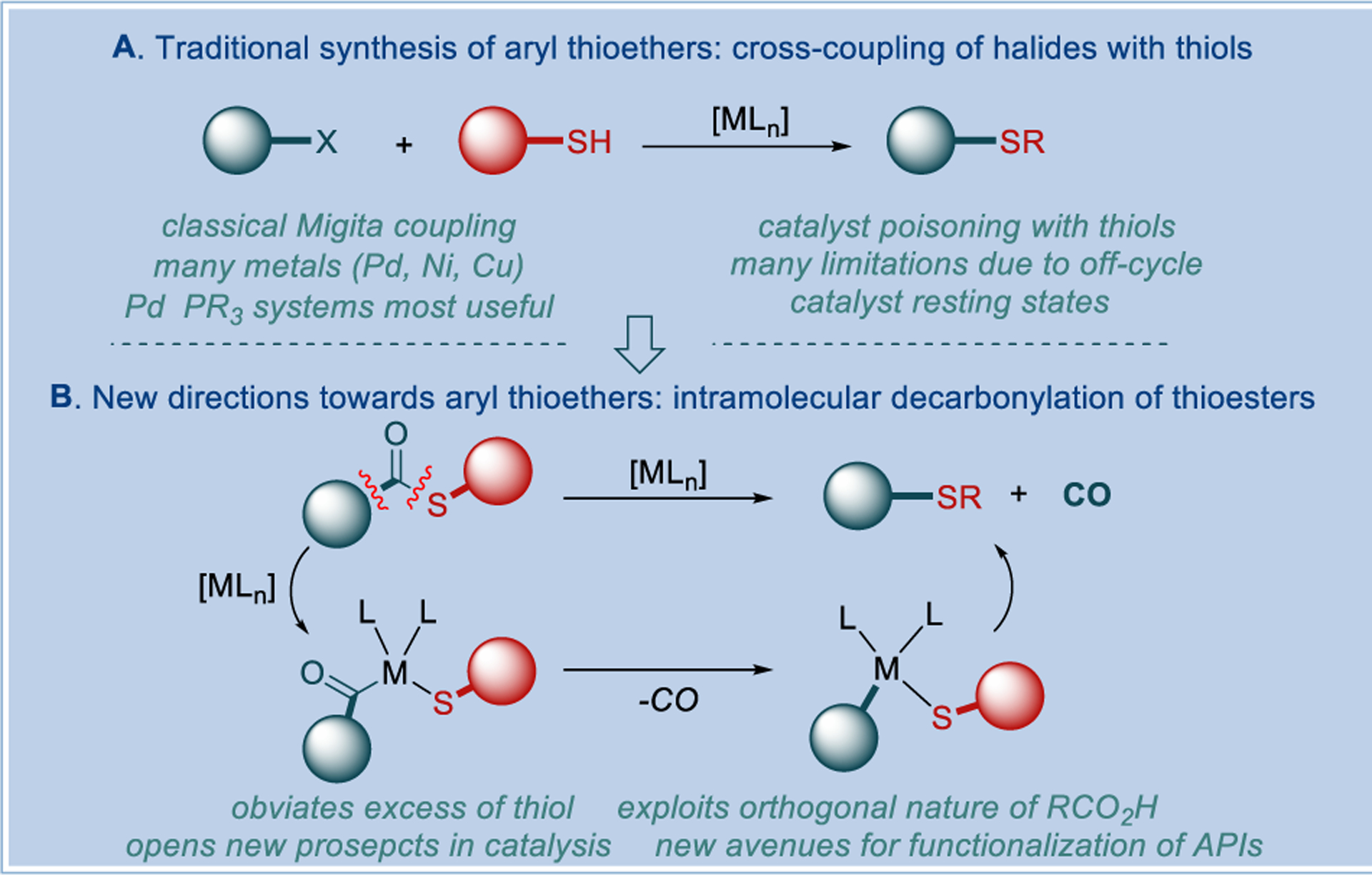
Synthesis of aryl thioethers via transition-metal-catalysis.
However, the development of cross-coupling methods for the synthesis of aryl thioethers has been slower than expected.[3–7] One major impediment is that metal catalysts are typically poisoned by thiols, which leads to off-cycle resting states induced by the presence of reactive thiolates.[4b] This complexation of palladium can result in either ancillary ligand displacement or formation of bridging thiolate complexes that undergo slow reductive elimination, which necessitates design of complex and sterically hindered ligands to protect the metal center. This in turn is less compatible with facile oxidative addition, which curtails many reactions to specific activated substrates, while methods using the most desirable aryl chlorides as coupling partners are still limited in scope.
Recently a series of reports has advanced an alternative manifold in which thio(esters) are subject to transition-metal-catalyzed intramolecular decarbonylation[9–14] (Figure 2B). This alternative approach offers new perspectives for the synthesis of aryl sulfides and most broadly (1) permits to engage ubiquitous and innately present in many APIs carboxylic acids as highly desirable and orthogonal precursors to aryl halides;[15] (2) circumvents the high concentration of thiolate anion[4–6] by diverting the coupling to an intramolecular decarbonylative process; (3) enables for novel disconnections not easily amenable using aryl halides; and (4) introduces new potentially broadly applicable concepts in catalysis.
The decarbonylative thioetherification is not a new process. In 1987, in the cross-coupling timeline almost as early as Migita’s report, Yamamoto first established the capacity of the decarbonylation of thioesters by catalytic Pd or stoichiometric Rh but the key unsolved problem was catalyst turnover.[9a] Shortly afterwards, in 1991, Wenkert verified a decarbonylative thioether synthesis using stoichiometric Ni and Zn as a co-reductant.[9b] Moreover, in 2006, Kambe reported a related method using Pd; however, the scope was limited to ortho-substituted S–Ar thioesters.[10]
In 2018, three concurrent reports by Sanford, our group and Yamaguchi provided the first insight that the catalytic decarbonylative synthesis of thioesters to furnish aryl thioethers with broad and synthetically useful scope is feasible (Figure 3).[11] The reported catalytic systems were complementary using either Pd or Ni catalysts: Pd[(P(o-tol)3]2 (10 mol%)/PAd2Bn (20 mol%), toluene, 150 °C or Ni(cod)2 (10 mol%)/PCy3 (20 mol%), p-xylene, 130 °C by Sanford; Ni(dppp)Cl2 (10 mol%)/Na2CO3 (1.5 equiv), dioxane, 160 °C by our group; and Ni(OAc)2 (5 mol%)/PnBu3 (20 mol%) or dppb (10 mol%)/Na2CO3 (2 equiv), DMF, 150 °C by Yamaguchi. These methods were noteworthy as the first general methods for the synthesis of thioethers via intramolecular decarbonylation. The application to the direct decarbonylative thioetherification of pharmaceuticals, such as Probenecid or Flufenamic acid,[11a,b] highlighted the functional group tolerance previously unattainable using other systems for decarbonylation, and underscored the utility of orthogonal presence of aryl carboxylic acid moiety that can be exploited for a facile generation of novel aryl thioethers from APIs.
Figure 3.

Timeline of intramolecular decarbonylation of thioesters.
Recently, Sanford group expanded this methodology to decarbonylative synthesis of fluoroalkyl aryl thioethers (Figure 4).[12] This process readily accomplishes the synthesis of Ar–SRF motifs in a formal fluoroalkylthiolation by exploiting fluoroalkyl carboxylic acids as the intramolecular source of fluoroalkyl groups after thioesterification. The catalytic system (Ni(cod)2 (10 mol%)/dppf (12 mol%), THF, 130 °C) is highly general and permits for –SRF installation of a variety of fluoroalkyl fragments on aromatic rings. Most notable is functional group tolerance to drugs (Captopril) and natural products (Thioglucose) and selectivity of metal insertion into the S–C(O) bond vs the electronically-activated C(O)–RF bond.
Figure 4.

Intramolecular decarbonylation of fluoroalkyl thioesters by Sanford.
Furthermore, the decarbonylative platform for the synthesis of aryl thioethers has been streamlined to Pd and Rh catalysis,[13a,b] both as increasingly more efficient systems in this field (Pd(OAc)2 (3 mol)%/dppb (6 mol%), toluene, 160 °C and [Rh(cod)Cl]2 (2 mol%), toluene, 160 °C) (not shown). Likewise, Wei group investigated an intriguing catalyst-controlled process for mono- or double-decarbonylation of α-ketothioesters (not shown).[13c] Thus, the use of Ni(cod)2/PPh3 promoted single CO de-insertion, while Ni(cod)2 /IPrMe system furnished double CO de-insertion, providing a further alternative to the Migita coupling from α-ketoacids as the ultimate precursors.
The most important recent breakthrough was reported by Yamaguchi and co-workers in establishing an inventive Ni-catalyzed aryl sulfide synthesis by aryl exchange (Figure 5).[14a] This process contrasts with the previous methods for decarbonylative synthesis of aryl thioethers in that 2-pyridyl sulfides are used as a sulfur donor in the reactions with aromatic esters as the preferred class of acyl electrophiles. The process involves a simultaneous oxidative addition of 2-py–SR and ArC(O)–OR’ bonds to a nickel catalyst, followed by ligand exchange and decarbonylation to furnish Ar–Ni–SR, which undergoes reductive elimination to afford the aryl thioether product. After an extensive screening of the reaction conditions, the authors identified a catalyst system based on Ni(cod)2 (10 mol%)/dcypt (15 mol%)/Zn (1 equiv), toluene, 150 °C. The role of zinc is to regenerate Ni(0) catalyst by decomposition of Ar–Ni–OAr complex. A range of aryl alkyl thioethers was synthesized using this methodology, including impressive applications to the synthesis of bioactive thioethers from APIs (Probenecid, Estrone, Indomethacin, Ticlopidine, Haloperidol, to name a few). Mechanistically, the authors established viability of the catalytic cycle by isolating reactive intermediates and showing reversibility of aryl exchange. In a broader context, the method by Yamaguchi not only opens new vistas to the synthesis of aryl thioethers by exploiting neutral sulfide donors, but also advances recent breakthroughs in decarbonylative aryl exchange[16] reactions to sulfur containing electrophiles.
Figure 5.

Decarbonylative synthesis of aryl thioethers by aryl exchange by Yamaguchi.
A related method for decarbonylative synthesis of aryl thioethers from carboxylic acid and thioesters by cross-over C–S activation has also been reported (not shown),[14b] bolstering the access to thioethers from carboxylic acids.[15]
To put the recent methods in a critical perspective, although there are still several challenges that must be addressed, these recent technologies address the major issue of metal poisoning by thiols inherent to the traditional cross-couplings by circumventing the use of an excess of thiol by an intramolecular decarbonylative process. With respect to the reaction conditions, although CO de-insertion inherently requires high temperature for the efficient decarbonylation to occur, the reported conditions are broadly functional group tolerant as demonstrated by late-stage derivatization of complex pharmaceuticals on multiple occasions (Figure 6). Finally, it is interesting to compare the orthogonal nature of carboxylic acids and aryl halides, which have already been exploited as powerful synthetical handles to functionalize a range of synthetic molecules.
Figure 6.

Late-stage applications to APIs.
In summary, a range of straightforward methods has been developed for the intramolecular decarbonylation of thioesters. These endeavors have been fueled by the major importance of aryl thioethers, which span a diverse range of applications in chemical science.[1,2] The interplay of facile oxidative addition of the ArC(O)–SR’ bond and resulting intramolecular Ar–[M]–SR’ coupling after CO de-insertion represents an attractive strategy for the construction of aryl thioethers. Broad substrate scope and excellent functional group tolerance have been demonstrated with different catalyst systems, sulfur nucleophiles and cross-coupling manifolds. Alternative methods using readily available carboxylic acids as substrates bode well for orthogonal manipulation of complex APIs and facile synthesis of previously unattainable aryl thioethers. Some of these decarbonylation strategies are base-free in nature and could allow valuable applications to base sensitive functionalities, exemplified by Captopril that contains acidic hydrogen attached to a stereogenic center and showed minimal stereochemical erosion.[12] Undoubtedly, future advances will focus on the development of more efficient catalysts and milder reaction conditions to boost this already efficient approach to become widely applicable in organic synthesis. [17]
Acknowledgements
We thank the NIH (1R35GM133326), the NSF (CAREER CHE-1650766), Rutgers University and Nanjing University of Information Science and Technology for generous financial support. Additional support was provided by the Rutgers Graduate School in the form of a Dean’s Dissertation Fellowship.
Biographies

Chengwei Liu was born and raised in Yantai, China. He received his B.Eng. degree from Zaozhuang University in 2011 and his M.Sc. degree from Soochow University in 2014. In 2020, he received his Ph.D. from Rutgers University with Prof. Michal Szostak. After postdoctoral stints at University of Oxford with Prof. Stephen P. Fletcher, in 2021, he joined the faculty at Nanjing University of Information Science and Technology. His research group is focused on decarbonylative cross-coupling of amides, thioesters and carboxylic acids.

Michal Szostak received his Ph.D. from the University of Kansas in 2009. After postdoctoral stints at Princeton University and the University of Manchester, in 2014, he joined the faculty at Rutgers University, where he is currently Professor of Chemistry. His research group is focused on the development of new synthetic methodology based on transition metal catalysis, amide bonds, NHCs, C–H, C–O, and C–N activation, decarbonylative coupling, and application to the synthesis of biologically active molecules. He is the author of over 200 peer-referred publications.
References
- [1].a) Smith MB, March J, Advanced Organic Chemistry, Wiley, 2006; [Google Scholar]; b) Ackermann L, Modern Arylation Methods, Wiley, Hoboken, 2009; [Google Scholar]; c) Terrier F, Modern Nucleophilic Aromatic Substitution, Wiley, 2013; [Google Scholar]; d) Colacot TJ, New Trends in Cross-Coupling, RSC, 2015; [Google Scholar]; e) Yet L, Privileged Structures in Drug Discovery, Wiley, 2018. [Google Scholar]
- [2].For studies on sulfur-containing polymers and materials, see:; a) Park NH, dos Passos Gomes G, Fevre M, Jones GO, Alabugin IV, Hedrick JL, Nat. Comm 2017, 8, 166; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vo CD, Kilcher G, Tirelli N, Macromol. Rapid Commun 2009, 30, 299–315; [DOI] [PubMed] [Google Scholar]; c) Li X, Ma W, Li H, Zhang Q, Liu H, Coord. Chem. Rev 2020, 408, 213191; [Google Scholar]; For studies on sulfur-containing drugs, see:; d) Ilardi EA, Vitaku E, Njardarson JT, J. Med. Chem 2014, 57, 2832–2842; [DOI] [PubMed] [Google Scholar]; e) Beno BR, Yeung KS, Bartberger MD, Pennington LD, Meanwell NA, J. Med. Chem 2015, 58, 4383–4438; [DOI] [PubMed] [Google Scholar]; f) Feng M, Tang B, Liang SH, Jiang X, Curr. Top. Med. Chem 2016, 16, 1200–1216; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Scott KA, Njardarson JT, Top. Curr. Chem 2018, 376, 5; [DOI] [PubMed] [Google Scholar]; h) Ertl P, Altmann E, McKenna JM, J. Med. Chem 2020, 63, 8408–8418; [DOI] [PubMed] [Google Scholar]; For a perspective on sulfa drugs, see:; i) Drews J, Science 2000, 287, 1960–1964. [DOI] [PubMed] [Google Scholar]
- [3].Kosugi M, Shimizu T, Migita T, Chem. Lett 1978, 7, 13–14. [Google Scholar]
- [4].a) Fernandez-Rodriguez MA, Shen Q, Hartwig JF, J. Am. Chem. Soc 2006, 128, 2180–2181; [DOI] [PubMed] [Google Scholar]; b) Alvaro E, Hartwig JF, J. Am. Chem. Soc 2009, 131, 7858–7868; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hartwig JF, Nature 2008, 455, 314–322; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Murata M, Buchwald SL, Tetrahedron 2004, 60, 7397–7403. [Google Scholar]
- [5].Xu J, Liu RY, Yeung CS, Buchwald SL, ACS Catal 2019, 9, 6461–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Scattolin T, Senol E, Yin G, Guo Q, Schoenebeck F, Angew. Chem. Int. Ed 2018, 57, 12425–12429; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 130, 12605–12609. [Google Scholar]
- [7].For select applications of Migita coupling, see:; a) Dahl T, Tornøe CW, Bang-Andersen B, Nielsen P, Jørgensen M, Angew. Chem. Int. Ed 2008, 47, 1726–1728; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 1750–1752; [Google Scholar]; b) Bryan CS, Braunger JA, Lautens M, Angew. Chem. Int. Ed 2009, 48, 7064–7068; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 7198–7202. [Google Scholar]
- [8].For recent studies using photoredox systems, see:; a) Jiang M, Li H, Yang H, Fu H, Angew. Chem. Int. Ed 2017, 56, 874–879; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 892–897; [Google Scholar]; b) Oderinde MS, Frenette M, Robbins DW, Aquila B, Johannes JW, J. Am. Chem. Soc 2016, 138, 1760–1763; [DOI] [PubMed] [Google Scholar]; c) Liu B, Lim CH, Miyake GM, J. Am. Chem. Soc 2017, 139, 13616–13619; [DOI] [PMC free article] [PubMed] [Google Scholar]; For methods using other metals, see:; d) Beletskaya IP, Ananikov VP, Chem. Rev 2011, 111, 1596–1636 [DOI] [PubMed] [Google Scholar]
- [9].a) Osakada K, Yamamoto T, Yamamoto A, Tetrahedron Lett 1987, 28, 6321–6324; [Google Scholar]; b) Wenkert E, Chianelli D, J. Chem. Soc., Chem. Commun 1991, 627–628. [Google Scholar]
- [10].a) Kato T, Kuniyasu H, Kajiura T, Minami Y, Ohtaka A, Kinomoto M, Terao J, Kurosawa H, Kambe N, Chem. Commun 2006, 8, 868–870; [DOI] [PubMed] [Google Scholar]; b) Minami Y, Kuniyasu H, Miyafuji K, Kambe N, Chem. Commun 2009, 21, 3080–3082. [DOI] [PubMed] [Google Scholar]
- [11].a) Ichiishi N, Malapit CA, Wozniak L, Sanford MS, Org. Lett 2018, 20, 44–47; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu C, Szostak M, Chem. Commun 2018, 54, 2130–2133; [DOI] [PubMed] [Google Scholar]; c) Ishitobi K, Isshiki R, Asahara KK, Lim C, Muto K, Yamaguchi J, Chem. Lett 2018, 47, 756–759. [Google Scholar]
- [12].Brigham CE, Malapit CA, Lalloo N, Sanford MS, ACS Catal 2020, 10, 8315–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Cao H, Liu X, Bie F, Shi Y, Han Y, Yan P, Szostak M, Liu C, Org. Chem. Front 2021, 8, 1587–1592; [DOI] [PubMed] [Google Scholar]; b) Cao H, Liu X, Bie F, Shi Y, Han Y, Yan P, Szostak M, Liu C, J. Org. Chem 2021, 86, 10829–10837; [DOI] [PubMed] [Google Scholar]; c) Zheng ZJ, Jiang C, Shao PC, Liu WF, Zhao TT, Xu PF, Wei H, Chem. Commun 2019, 55, 1907–1910. [DOI] [PubMed] [Google Scholar]
- [14].a) Isshiki R, Kurosawa MB, Muto K, Yamaguchi J, J. Am. Chem. Soc 2021, 143, 10333–10340; [DOI] [PubMed] [Google Scholar]; b) Liu C, Szostak M, Org. Chem. Front 2021, 8, DOI: 10.1039/d1qo00824b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Gooßen LJ, Rodriguez N, Gooßen K, Angew. Chem. Int. Ed 2008, 47, 3100; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 3144 [Google Scholar]; b) Liu C, Ji CL, Hong X, Szostak M, Angew. Chem. Int. Ed 2018, 57, 16721–16726; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 16963–16968. [Google Scholar]
- [16].a) Delcaillau T, Boehm P, Morandi B, J. Am. Chem. Soc 2021, 143, 3723–3728; [DOI] [PubMed] [Google Scholar]; b) Lian Z, Bhwal BN, Yu P, Morandi B, Science 2017, 356, 1059–1063. [DOI] [PubMed] [Google Scholar]
- [17]. Recently, an aryl exchange method between amides and thioesters to afford aryl sulfides through double decarbonylation was reported: Org. Lett. 2021, DOI: . This methodology bears similarly to ref 14a using amides as C-N aryl exchange partners (cf. aryl esters). [DOI]