

Abstract
The first step in anaerobic ethylbenzene mineralization in denitrifying Azoarcus sp. strain EB1 is the oxidation of ethylbenzene to (S)-(−)-1-phenylethanol. Ethylbenzene dehydrogenase, which catalyzes this reaction, is a unique enzyme in that it mediates the stereoselective hydroxylation of an aromatic hydrocarbon in the absence of molecular oxygen. We purified ethylbenzene dehydrogenase to apparent homogeneity and showed that the enzyme is a heterotrimer (αβγ) with subunit masses of 100 kDa (α), 35 kDa (β), and 25 kDa (γ). Purified ethylbenzene dehydrogenase contains approximately 0.5 mol of molybdenum, 16 mol of iron, and 15 mol of acid-labile sulfur per mol of holoenzyme, as well as a molydopterin cofactor. In addition to ethylbenzene, purified ethylbenzene dehydrogenase was found to oxidize 4-fluoro-ethylbenzene and the nonaromatic hydrocarbons 3-methyl-2-pentene and ethylidenecyclohexane. Sequencing of the encoding genes revealed that ebdA encodes the α subunit, a 974-amino-acid polypeptide containing a molybdopterin-binding domain. The ebdB gene encodes the β subunit, a 352-amino-acid polypeptide with several 4Fe-4S binding domains. The ebdC gene encodes the γ subunit, a 214-amino-acid polypeptide that is a potential membrane anchor subunit. Sequence analysis and biochemical data suggest that ethylbenzene dehydrogenase is a novel member of the dimethyl sulfoxide reductase family of molybdopterin-containing enzymes.
Accidental spills and leaking underground storage tanks, as well as natural petroleum seeps, have released aromatic hydrocarbons into natural environments and led to abundant contamination of water resources (32). Aromatic hydrocarbons comprise one of the least reactive classes of organic molecules. Their relative inertness can be attributed to the absence of a functional group (e.g., hydroxyl, carbonyl, or carboxyl) and the resonance energy stabilization of the aromatic ring. These properties hamper rapid biodegradation of alkylbenzenes in many environments. However, some prokaryotic microorganisms employ intriguing metabolic strategies to activate alkylbenzenes leading to their oxidation in catabolic pathways. Under aerobic conditions, the well-characterized mono- and dioxygenases catalyze the activation of hydrocarbons by introducing a hydroxyl group via oxidative hydroxylation with molecular oxygen as cosubstrate (16). The absence of molecular oxygen in anoxic environments precludes this activation mode, and novel, alternative mechanisms are expected to operate under such conditions.
In recent years, two mechanisms for initiating anaerobic metabolism of alkylbenzenes have emerged: methyl-substituted benzenes, such as toluene or m-xylene, are activated by enzymatic addition to fumarate to form benzylsuccinate or its methyl homolog (5, 6, 9, 25, 28, 34), whereas ethylbenzene (and possibly other alkylbenzenes with carbon chain length of ≥2) is oxidatively activated by an anaerobic dehydrogenation of the benzylic carbon to form 1-phenylethanol (3, 29). Ethylbenzene dehydrogenase is a novel enzyme in that it is the first enzyme shown to catalyze the hydroxylation of an aromatic hydrocarbon in the absence of molecular oxygen (Fig. 1). Stable isotope labeling studies showed that the hydroxyl group of 1-phenylethanol is derived from water (3).
FIG. 1.

Ethylbenzene dehydrogenase reaction as the first step in anaerobic ethylbenzene oxidation.
In vitro studies with Azoarcus sp. strain EB1 demonstrated that ethylbenzene dehydrogenase activity is membrane associated and couples the oxidation of ethylbenzene to the reduction of p-benzoquinone (22). This dehydrogenation reaction is highly stereoselective and solely forms (S)-(−)-1-phenylethanol. The enzyme activity is expressed when Azoarcus sp. strain EB1 is grown anaerobically on ethylbenzene, as well as on 1-phenylethanol or acetophenone, as the sole carbon and electron source, but it is not expressed when cells are grown with benzoate. 1-Phenylethanol, acetophenone, and benzoate are intermediates in anaerobic ethylbenzene oxidation (3, 29).
We report here for the first time on the purification and initial characterization of the novel ethylbenzene dehydrogenase, the cofactor content, and the nucleotide sequence and structure of the genes involved. Based on these findings, as well as on substrate transformation studies, we discuss possible reaction mechanisms for anaerobic ethylbenzene oxidation.
MATERIALS AND METHODS
Materials.
Chemicals and biochemicals of the highest purity available were obtained from Sigma-Aldrich and Bio-Rad. Fast-protein liquid chromatography (FPLC) columns were obtained from Pharmacia.
Growth conditions, preparation of cell extracts, and enzyme assay.
Cells of Azoarcus sp. strain EB1 were grown under denitrifying conditions in minimal medium with ethylbenzene as the sole carbon and electron source as described previously (3). Cell extracts were prepared anaerobically from exponentially growing cells at an optical density at 400 nm of 0.3 as previously described (22) and frozen in 1-ml aliquots of 10 to 20 mg protein per ml at −20°C. Ethylbenzene dehydrogenase activity was determined in a 1-ml 100 mM Tris-HCl (pH 7.5) buffered assay containing 1 μmol of p-benzoquinone as electron acceptor and 1 μmol of ethylbenzene (22). The formation of the product of the reaction, 1-phenylethanol, was measured by gas chromatography (GC)-flame ionization detection following extraction into methylene chloride (22). Standard assays were stopped after 5 to 10 min. For assaying the spectrum of substrates, 1 μmol of the compound to be tested was added to the assay in place of ethylbenzene, and the assay was incubated for 3 h with shaking.
Purification of ethylbenzene dehydrogenase.
Ethylbenzene dehydrogenase activity was purified from cell extracts of Azoarcus sp. strain EB1 cells grown as described above. Cell extracts were amended with EDTA to a final concentration of 2.8 mM to solubilize the enzyme activity. Ethylbenzene dehydrogenase activity was then precipitated from the EDTA containing cell extract by dropwise addition of cold acetone to a final concentration of 45 to 55% (vol/vol) with stirring at −15°C in an ice-salt water bath. The precipitate was collected by centrifugation at 15,000 × g for 15 min (−15°C) and resuspended in ice-cold 20 mM Tris (pH 7.5). The solution was recentrifuged (15,000 × g) at 4°C for 15 min. The supernatant was then passed through a 0.45-μm (pore-size) filter, diluted twofold with 50 mM morpholineethanesulfonic acid (MES; pH 6.5), and loaded onto a 1-ml MonoS HR 5/5 (Pharmacia) cation-exchange column equilibrated with 50 mM MES (pH 6.5). After the column was washed with 5 volumes of the MES buffer, the protein was eluted with a linear gradient of 0 to 1 M NaCl (20 column volumes) in 50 mM MES buffer (pH 6.5). The flow rate was 1 ml/min, and 1-ml fractions were collected. The elution of protein was monitored by measuring the absorbance at 280 nm. Ethylbenzene dehydrogenase activity was detected in the fraction eluting at 90 mM NaCl. FPLC was performed at 15°C in an anaerobic glove box (atmosphere of 90% nitrogen and 10% hydrogen). All other purification steps were conducted under aerobic conditions. Enzyme activity was assayed immediately, or else the fractions were stored at −20°C for further characterization.
Gel electrophoresis.
Denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out by standard procedures using 12% acrylamide separating and 4% acrylamide stacking gels (15). Native PAGE was performed with 5 to 12% acrylamide separating and 4% acrylamide stacking gels in the absence of SDS and β-mercaptoethanol (1). Denaturing and native molecular mass standards were obtained from Bio-Rad and Sigma, respectively. Proteins were visualized by Coomassie blue R-250 staining or silver staining.
Gel filtration.
A Superose 6 HR 10/30 chromatography column (Pharmacia) was used to determine the native molecular mass of ethylbenzene dehydrogenase by gel filtration. The column was equilibrated with 50 mM phosphate buffer (pH 7) containing 0.15 M NaCl at a flow rate of 0.5 ml/min. The following molecular mass standards (Bio-Rad) were used: thyroglobulin (Mr = 670,000), bovine gamma globulin (Mr = 158,000), chicken ovalbumin (Mr = 44,000), equine myoglobin (Mr = 17,000), and vitamin B12 (Mr = 1,350). Blue dextran was used as a void volume marker. Acetone was used as a total liquid volume marker. A single peak was detected at 17 ml, assayed for ethylbenzene dehydrogenase, and analyzed by SDS-PAGE.
Analytical ultracentrifugation.
Velocity sedimentation analysis was performed at the University of Texas Health Science Center, San Antonio's Center for Analytical Ultracentrifugation of Macromolecular Assemblies.
N-terminal sequence analysis.
The three polypeptide bands of ethylbenzene dehydrogenase were blotted from an SDS-polyacrylamide gel onto a polyvinylidene fluoride membrane (Bio-Rad) using a Mini Trans Blot transfer cell (Bio-Rad) according to the manufacturer's specifications. The three bands were excised from the membrane, and the N-terminal amino acid sequence was determined (PAN Facility; Stanford University, Stanford, Calif.).
Metal analysis.
Iron was determined by atomic absorption spectroscopy (30) with modified ashing and atomization temperatures of 600°C and 2,500°C, respectively. Acid-labile sulfur was determined by the method of Beinert (4). Molybdenum was determined with an Hewlett-Packard 4500 Series inductively coupled plasma mass spectrometer. Background levels for molybdenum, cobalt, iron, nickel, copper, zinc, and tungsten were approximately <10 nM, 50 nM, 70 nM, 20 nM, 1.5 μM, 2.1 μM, and 2 nM, respectively. Protein concentrations ranged from 0.04 to 0.3 mg/ml. Molybdopterin cofactor identification was performed as described in Johnson and Rajagopalan (21).
Chemical analysis.
1-Phenylethanol was quantified by GC-flame ionization detection after extraction from the ethylbenzene dehydrogenase assay mixture with methylene chloride as described in Johnson and Spormann (22). Methylene chloride extractions of the assay mixtures containing substrates other than ethylbenzene were analyzed by GC-mass spectrometry in electron impact ionization mode (GC-MS) as previously described (3). To screen for potential products from assays containing 3-methyl-1-pentene and 3-methyl-2-pentene, the assay mixture was heated to 80°C, and 0.5 ml of the headspace was removed for GC-MS analysis. Retention times and mass spectra were compared to those of standards if the compounds were commercially available. Protein concentration was determined by the method of Bradford (13) with a commercially available dye-binding assay (Bio-Rad). Bovine serum albumin was used as the standard.
Cloning and DNA manipulations.
Standard protocols were used for DNA cloning and transformations. Clones were generated in pUC19 or pBluescript II KS (Stratagene) and maintained in Escherichia coli DH5α. Plasmid DNA purification was performed using Qiaprep spin columns (Qiagen). Southern blot analysis was performed using dioxigenin labeled probes following Genius kit protocol (Boehringer Mannheim). Degenerate oligonucleotides were designed for PCR amplification based on the N-terminal amino acid sequence of the 35- and 25-kDa subunits of the purified ethylbenzene dehydrogenase. The sequence of the oligonucleotides is as follows: 35N-forward, 5′-GGGGAAGCTTGTNCARGAYGGNAAYAAG-3′; 35N-reverse, 5′-GGGGAAGCTTYTTRTTNCCRTCYTGNAC-3′; 25N-forward, 5′-GGGGAAGCTTGTNCCNGGNGGNAARGAG-3′; and 25N-reverse, 5′-GGGGAAGCTTYTCYTTNCCNCCNGGNAC-3′; Primers sets 35N-forward+25N-reverse and 35N-reverse+25N-forward were used for PCR amplification with Azoarcus sp. strain EB1 chromosomal DNA as a template. Amplification products were digested with HindIII and cloned into pUC19. These cloned fragments were used to generate probes for Southern blot analysis (probes for ebdA and ebdB).
Southern blot analysis identified a 5.0-kb ClaI fragment and a 2.7-kb SphI fragment that hybridized to the ebdB probe. Subsequently, inverse PCR (26) was used to clone DNA flanking ebdB. Briefly, Azoarcus sp. strain EB1 chromosomal DNA (200 ng) was digested with ClaI or SphI overnight in a 50-μl reaction. The reaction was extracted with phenol-chloroform-isoamyl alcohol and then ethanol precipitated. The digested DNA was resuspended in 30 μl water and used in a 50-μl ligation reaction with T4 DNA ligase (Epicentre Technologies) overnight at room temperature. The ligation reaction was precipitated with ethanol and resuspended in 50 μl of water, and 10 μl was used for PCR amplification. Primers for PCR amplification were designed based on previously determined DNA sequence. The oligonucleotides 100R6 (5′-GTACAACTCGTCGTACCG-3′) and 35F3 (5′-GGTTCTGCACACTGAGC-3′) were used with the ClaI-digested Azoarcus sp. strain EB1 DNA to amplify a 3.2-kb fragment which was digested with ClaI and XmaI to generate a 3.1-kb fragment that was ligated into pBluescript II vector digested with the same two enzymes to generate the clone pDPS100-13. The oligonucleotides 100R7 (5′-CCGTTCATGAAGTAGTAGCG-3′) and 35F3 were used to amplify a 1.8-kb fragment from the SphI-digested Azoarcus sp. strain EB1 DNA. This product was digested overnight with ClaI and SphI and ligated into pUC19 digested with AccI and SphI to generate the clone pDPS25-1.
Cloned fragments were sequenced with custom oligonucleotides synthesized by Operon Inc. DNA sequence was determined using cycle sequencing with Big-Dye terminator and analyzed using an ABI Prism 310 capillary DNA sequencer and genetic analyzer. DNA sequence reads were assembled, and open reading frames (ORFs) were identified using the DNA star software package. Similar sequences were identified using the BLAST network services at the National Center for Biotechnology Information (2). Motif searches were conducted using the Pfam database 5.5 at the Washington University, St. Louis, Mo.
DNA sequences were submitted to GenBank with accession number AF337952.
RESULTS
Purification of ethylbenzene dehydrogenase.
Ethylbenzene dehydrogenase was purified from cell extract of Azoarcus sp. strain EB1 grown on ethylbenzene as the sole carbon and electron source under denitrifying conditions (Table 1). Ethylbenzene dehydrogenase activity was previously found to be membrane associated and to partition equally between the membrane and cytoplasmic fractions (22). When cell extract was separated by ultracentrifugation, 30% of the total activity was lost. Thus, isolation of the membrane fraction was not pursued as the first purification step. The membrane-associated ethylbenzene dehydrogenase activity was solubilized in total cell extract by the addition of EDTA, which resulted in solubilization of 95% of the total enzyme activity (data not shown). Ethylbenzene dehydrogenase activity was purified to apparent homogeneity by standard protein purification methods as summarized in Table 1. The purity of ethylbenzene dehydrogenase was analyzed by SDS-PAGE (Fig. 2). The overall purification scheme yielded an approximately sevenfold purification (Table 1), with a resulting specific activity of 47 nmol min−1 mg of protein−1. For the following sections, the enzyme obtained following cation-exchange chromatography is referred to as the purified enzyme.
TABLE 1.
Purification of ethylbenzene dehydrogenase from Azoarcus sp. strain EB1
| Purification step | Protein amt (mg) | Total activity (nmol min−1) | Sp act (nmol min−1 mg of protein−1) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Cell extract | 16.5 | 109 | 6.6 | 100 | 1 |
| Solubilization and acetone precipitation | 7.4 | 36 | 4.8 | 33 | 0.7 |
| Cation-exchange chromatography | .08 | 3.8 | 47 | 3.5 | 6.8 |
FIG. 2.
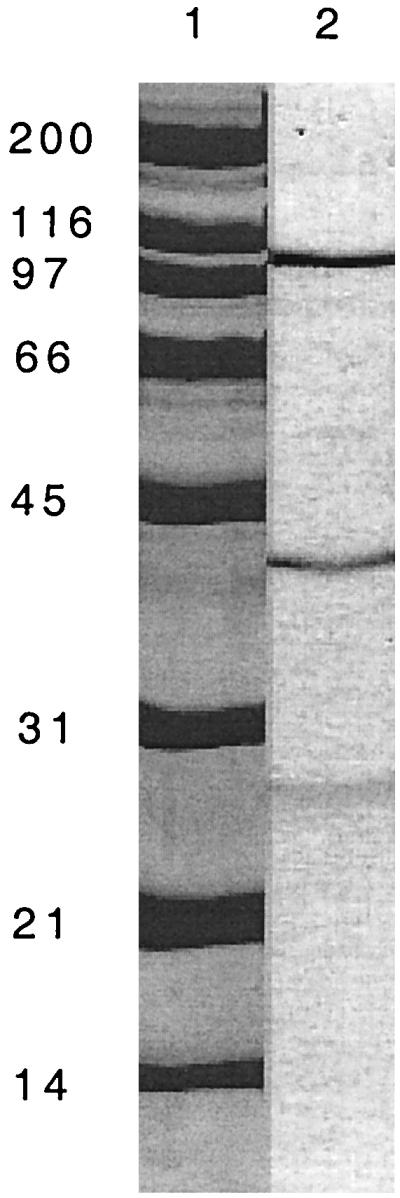
SDS-PAGE of the purified ethylbenzene dehydrogenase from Azoarcus sp. strain EB1. Lane 1, molecular mass standards in kilodaltons; lane 2, 0.5 μg of ethylbenzene dehydrogenase protein recovered from a cation-exchange column.
Subunit composition and native size.
SDS-PAGE analysis of purified ethylbenzene dehydrogenase revealed three major bands with relative molecular masses of 100, 35, and 25 kDa (Fig. 2). When the purified enzyme was subjected to gel filtration, only one protein peak, at a relative molecular mass of approximately 70 kDa, was observed to elute from the column (data not shown). The fraction containing the protein peak contained 70% of the ethylbenzene dehydrogenase activity that was applied to the column. When this fraction was analyzed by SDS-PAGE, the presence of three bands with relative molecular masses of 100, 35, and 25 kDa was revealed, suggesting that these bands were the same as the three bands observed with the purified enzyme. Native PAGE of the purified enzyme resulted in a single band with a relative mass of greater than 545 kDa. Use of EDTA (2.5 mM), urea (1.25 M), CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} (1 to 2%), or deoxycholate (1 to 2%) did not alter the mobility of the single band under native conditions. Analytical ultracentrifugation was also employed to determine the native molecular mass and revealed that 80% of the sample derived from the cation-exchange fraction had a sedimentation coefficient of 8 to 9 S corresponding to a molecular mass of approximately 120 kDa. A total of 20% of the sample sedimented at lower S values, suggesting partial proteolysis of the sample. Taken together, the SDS-PAGE, analytical ultracentrifugation, and gene sequence data (see below) suggest that native ethylbenzene dehydrogenase is a heterotrimer of approximately 160 kDa, composed of a 100-kDa α, a 35-kDa β, and a 25-kDa γ subunit.
Cofactor composition of ethylbenzene dehydrogenase.
Analysis of purified ethylbenzene dehydrogenase for total iron and acid-labile sulfur revealed 16.3 (standard deviation [SD] = 0.3) mol of iron and 15 (SD = 4) mol of acid-labile sulfur per mol of enzyme (based on an Mr of 160,000), suggesting the presence of iron sulfur clusters. Analysis of the purified enzyme by inductively coupled plasma MS revealed 0.49 (SD = 0.07) mol of molybdenum per mol of enzyme (based on an Mr of 160,000). Cobalt, nickel, copper, zinc, and tungsten were not significantly elevated above background levels and, if present, were at levels of less than 0.015, 0.08, 0.06, 0.2, and 0.0003 mol per mol of enzyme, respectively. The presence of a molybdopterin cofactor was investigated by extracting the purified enzyme in the presence (form A) or absence (form B) of iodine (Fig. 3). The shift in the local fluorescence maxima between the two forms was characteristic of molybdopterin cofactor-containing proteins (21).
FIG. 3.

Fluorescence spectra of purified ethylbenzene dehydrogenase (1 mg ml−1) prepared in the presence (Aex and Aem, solid lines) or absence (Bex and Bem, dashed lines) of iodine. The emission spectra (Aem and Bem) were recorded at 375 nm. The excitation spectra (Aex and Bex) were recorded at 470 nm.
Substrate spectrum of ethylbenzene dehydrogenase.
A variety of compounds, including aromatic hydrocarbons and alkenes that carry structural similarity to some region of ethylbenzene, were examined for transformation by purified ethylbenzene dehydrogenase (Table 2). No transformation products from toluene, 4-ethylphenol, 2-phenylethanol, ethylcyclohexane, and 3-methyl-1-pentene were detected by GC-MS analysis. Interestingly, no transformation products were detected when propylbenzene was tested as substrate, although propylbenzene conversion to 1-phenyl-1-propanol and propiophenone was previously detected in cell extract at near-detection levels (22). Consistent with findings in the cell extract, 4-fluoro-ethylbenzene was transformed (22). When 3-methyl-2-pentene (FW = 84) was added as a substrate, a product with a molecular ion of m/z = 100 was detected by GC-MS. No product was detected when the electron acceptor, p-benzoquinone, was absent from the assay mixture. These findings suggest that 3-methyl-2-pentene was oxidatively hydroxylated such that a hydroxyl group replaced a hydrogen atom, resulting in an increase in the product mass by 16 amu. When ethylidenecyclohexane (FW = 110) was tested as a substrate, three small peaks were detected by GC. Each peak contained a compound with a molecular ion of m/z = 126 as determined by MS. As with 3-methyl-2-pentene, the findings suggest that the double bond of ethylidenecyclohexane was retained and that a hydroxyl group replaced a hydrogen atom. The hydroxylated products hypothesized to be formed are not commercially available and, thus, could not be identified and quantified.
TABLE 2.
Substrates tested for transformation by purified ethylbenzene dehydrogenase
| Compound tested | Structure | Oxidation product detecteda |
|---|---|---|
| Ethylbenzene |  |
+ |
| Toluene | − | |
| Propylbenzene | − | |
| 4-Ethylphenol | − | |
| 2-Phenylethanol | − | |
| 4-Fluoro-ethylbenzene | + | |
| Ethylcyclohexane | − | |
| Ethylidenecyclohexane | + | |
| 3-Methyl-2-pentene | + | |
| 3-Methyl-1-pentene | − |
Detection limit approximately 1 nmol. The assay was incubated for 3 h with shaking and analyzed as described in Materials and Methods.
Cloning of the genes encoding ethylbenzene dehydrogenase.
Degenerate oligonucleotide primers were derived from the N-terminal amino acid sequence of the 35-kDa β subunit (x-x-Val-Gln-Asp-Gly-Asn-Lys-Ser-Glu-Leu-x-Lys-Ala-Lys-x-Gln-Leu-Val; primers 35N-forward and 35N-reverse) and the 25-kDa γ subunit (x-Pro-Ala-Lys-x-Val-Pro-Gly-Gly-Lys-Glu-Leu-Leu-Leu-Asp-Leu; primers 25N-forward and 25N-reverse) of purified ethylbenzene dehydrogenase. Assuming that the three genes encoding ethylbenzene dehydrogenase are organized in an operon, one primer pair (e.g., 35N-forward plus 25N-reverse) should yield a product of at least the size predicted to encode one subunit (e.g., the β subunit), while the other primer pair should yield no amplification product. PCR amplification using primer pair 35N-forward plus 25N-reverse and Azoarcus sp. strain EB1 chromosomal DNA yielded a single product of 1.1 kb. Amplification with primer pair 35N-reverse plus 25N-forward also yielded a single product of 0.9 kb. The two PCR products were cloned into the HindIII site of pUC19 to generate plasmids pDPS35-4 and pDPS100-1, respectively. The nucleotide sequence of the cloned PCR amplification products was determined. Analysis of the DNA sequence identified potential ORFs in both cloned fragments. The predicted N-terminal amino acid sequence of the ORF identified in the 1.1-kb fragment matches the amino acid sequence obtained for the N terminus of the β subunit of purified ethylbenzene dehydrogenase. The predicted amino acid sequence of the ORF identified in the 0.9-kb fragment is identical to the predicted carboxy terminus of the EbdA protein. Therefore, this 0.9-kb PCR product must have resulted from mispriming of the degenerate primer 25N-forward during PCR amplification. Southern blot analysis of the Azoarcus sp. strain EB1 chromosomal DNA and subsequent inverse-PCR technique was used to clone and sequence DNA fragments flanking the identified region of the 1.1-kb fragment (see Materials and Methods). Using the sequence data obtained from the inverse-PCR-amplified products, PCR primers were designed that enabled us to successfully amplify and sequence an approximately 6-kb region of Azoarcus sp. strain EB1 chromosomal DNA. Sequence analysis revealed three closely spaced ORFs which we designated ebdA, ebdB, and ebdC (Fig. 4).
FIG. 4.

Organization of the genes encoding anaerobic ethylbenzene dehydrogenase from Azoarcus sp. strain EB1. Arrows indicate the direction of transcription.
The ebdA gene is predicted to encode a polypeptide of 974 amino acids with a molecular mass of 111,030.4 Da. The N-terminal amino acid sequence of the 100-kDa subunit of purified ethylbenzene dehydrogenase (X-X-K-A-P-G-Y/V-A-S) matches amino acids 55 to 63 of the predicted EbdA (G-T-K-A-P-G-Y-A-S), indicating that the predicted polypeptide is posttranslationally cleaved. The predicted ebdA gene product contains a proposed twin-arginine leader peptide sequence based on its similarity to known twin-arginine leader sequences (8). Twin-arginine leader sequences are common for proteins containing complex redox factors that may be translocated to the periplasm by the Sec-independent twin-arginine translocation (TAT) pathway (7). The mature EbdA is predicted to have a molecular mass of 104,226.8 Da. EbdA has amino acid identities of 34, 31, 28, and 23%, respectively, to SerA, the α subunit of selenate reductase of Thauera selenatis; NarG, the α subunit of nitrate reductase 1 of E. coli; DmsA, the α subunit of dimethyl sulfoxide (DMSO) reductase of E. coli; and FdoG, the α subunit of formate dehydrogenase-O of E. coli (10, 11, 24, 27). Multiple sequence alignments indicate amino acid sequence similarity to those proteins throughout the entire length of EbdA (data not shown). All of these proteins identified are members of the DMSO reductase family of molybdopterin-containing enzymes (23). A search of the Pfam database with EbdA identified two sequence motifs, a prokaryotic molybdopterin oxidoreductase domain at amino acids 631 to 691 (PF00384) and a molybdopterin-binding domain at amino acids 843 to 955 (PF01568).
Following the translational stop codon of ebdA is a 36-bp intergenic region. The second ORF, designated ebdB, is predicted to encode a 352-amino-acid polypeptide with a molecular mass of 39,643.3 Da. The N-terminal amino acid sequence of the 35-kDa subunit of purified ethylbenzene dehydrogenase (X-X-V-Q-D-G-N-K-S-E-L-X-K-A-K-X-Q-L-V) matched amino acids 2 to 20 of predicted EbdB (M-T-Y-V-Q-D-G-N-K-S-E-L-R-K-A-K-R-Q-L-V). EbdB has amino acid identities of 57, 45, 35, and 23%, respectively, to SerB, the β subunit of selenate reductase of T. selenatis; NarH, the β subunit of nitrate reductase 1 of E. coli; DmsB, the β subunit of DMSO reductase of E. coli; and FdoH, the β subunit of formate dehydrogenase-O of E. coli (10, 11, 24, 27). A motif search identified three potential 4Fe-4S binding domains (PF00037) in EbdB (amino acids 21 to 43, 144 to 169, and 177 to 200), and a fourth iron-sulfur cluster is possible based on amino acid similarities to NarH and DmsB. The β subunit of E. coli DMSO reductase contains four 4Fe-4S clusters (14), and the β subunit of nitrate reductase contains three 4Fe-4S and one 3Fe-4S cluster (17). The iron sulfur clusters in ethylbenzene dehydrogenase may be involved in the transfer of electrons from the molybdopterin cofactor to a quinone or they may be involved in protein stability.
An 18-bp intergenic region follows the translational stop codon of ebdB. The final ORF, designated ebdC, is predicted to encode a 214-amino-acid polypeptide with a molecular mass of 23,061.6 Da. The N-terminal amino acid sequence of the 25 kDa subunit of purified ethylbenzene dehydrogenase (X-P-A-K-X-V-P-G-G-K-E-L-L-L-D-L) matched amino acids 1 to 16 of predicted EbdC (M-K-A-K-R-V-P-G-G-K-E-L-L-L-D-L). Homology searches of the protein databases revealed that EbdC has amino acid similarity only to SerC (27% identity). SerC is the γ subunit of selenate reductase of T. selenatis and has been proposed to carry a cytochrome b (24). The function of the γ subunit of ethylbenzene dehydrogenase may be to couple electron flow from the Fe-S clusters to a quinone.
DISCUSSION
Ethylbenzene dehydrogenase catalyzes the anaerobic dehydrogenation of ethylbenzene to 1-phenylethanol as the first step in the anaerobic ethylbenzene mineralization pathway. Ethylbenzene dehydrogenase is the first enzyme known to oxidize an aromatic hydrocarbon in the absence of molecular oxygen. We purified ethylbenzene dehydrogenase to apparent homogeneity and showed that the heterotrimeric enzyme contains approximately 16 mol of iron, 15 mol of acid-labile sulfur, 0.5 mol of molybdenum per mol of holoenzyme, and a molybdopterin cofactor. The predicted amino acid sequence of the putative ebd genes indicates strong homology to other molybdenum containing enzymes. Thus, ethylbenzene dehydrogenase is a molybdenum-iron-sulfur protein.
Mononuclear molybdenum enzymes catalyze redox reactions that typically involve the transfer of an oxygen atom (19). These molybdenum-containing enzymes are categorized into three or four families, based on the structure of the molybdenum center or on amino acid sequence similarity, respectively (19, 20, 23). The heterotrimeric subunit composition of ethylbenzene dehydrogenase, the genetic arrangement of the three encoding genes, and the predicted amino acid sequence are typical of Mo enzymes of the DMSO reductase family, such as selenate reductase of T. selenatis (31) and DMSO reductase and respiratory nitrate reductase of E. coli (11, 33). It has been shown that for enzymes of the DMSO reductase family that have three subunits, the α-subunit contains the molybdopterin, the β-subunit contains the iron sulfur clusters, and the γ subunit is membrane associated (19).
Ethylbenzene is a relatively unreactive aromatic hydrocarbon. A chemical substitution reaction of this hydrocarbon is most likely to occur at the benzylic carbon because of the stabilization of a reaction intermediate (e.g., radical or ion) by delocalization via the aromatic ring. Ethylbenzene dehydrogenase catalyzes the enzymatic oxidation of the benzylic carbon of ethylbenzene and the stereoselective transfer of a water-derived hydroxyl group (3, 22). Substrate transformation studies with ethylbenzene dehydrogenase provided some insight into what structural or functional aspects of the substrate molecule are important for reactivity. In addition to ethylbenzene and 4-fluoro-ethylbenzene, purified ethylbenzene dehydrogenase transformed two nonaromatic compounds: 3-methyl-2-pentene and ethylidenecyclohexane. 3-Methyl-2-pentene was converted to a product, which contains 16 amu more than the substrate. The additional mass is equivalent to that of an oxygen atom, and the transformation was dependent on the presence of p-benzoquinone. This finding suggests that 3-methyl-2-pentene was oxidized and hydroxylated, presumably at the C-4 carbon. Introduction of the oxygen atom by the addition of water to the double bond is unlikely since it would have yielded a product with an additional mass of 17 amu and would not have been dependent on the presence of an electron acceptor. Also, from the previously observed lack of styrene transformation, it was concluded that ethylbenzene oxidation may not proceed via a two-step mechanism involving the addition of water to a free, unsaturated intermediate (22). The results of these transformation studies can be interpreted in the following way: ethylbenzene and 4-fluoro-ethylbenzene oxidation may proceed via a reaction intermediate, possibly a carbocation, which is stabilized by delocalization by a vicinal aryl group. In the case of 3-methyl-2-pentene as substrate, the reaction intermediate may be stabilized by the vicinal allyl group. This notion is consistent with 3-methyl-1-pentene not being oxidized by ethylbenzene dehydrogenase. Preliminary characterization of products formed from ethylidenecyclohexane by ethylbenzene dehydrogenase identified three compounds, each with a molecular ion of m/z = 126 as determined by GC-MS. It is tempting to speculate that ethylidenecyclohexane transformation may have also proceeded via oxidative hydroxylation at an allylic carbon (e.g., C-2 or C-5 of the cyclohexane ring) or at some other position if the substrate may have rearranged during transformation. Further studies are required to determine the chemical identity of these interesting reaction products.
Several reaction mechanisms for anaerobic ethylbenzene oxidation by ethylbenzene dehydrogenase involving molybdenum and the Fe-S clusters are conceivable. In its unbound state, ethylbenzene dehydrogenase could contain Mo(VI) with a water-derived oxo or hydroxy group. In one model, ethylbenzene oxidation is initiated by transfer of electrons from the benzylic C-H to Mo(VI), forming Mo(IV). A direct hydride transfer has also been proposed for the molybdenum enzyme formate dehydrogenase H of E. coli (12). The nucleophilic, Mo-activated oxo group could then hydroxylate the carbocation at the benzylic carbon. In an alternative mechanism, Mo(VI) could be reduced to Mo(IV) by the bound oxo oxygen. This reverse of polarity of a nucleophilic oxo group could then induce a direct attack of the electrophilic oxygen at the benzylic C-H, resulting in a hydroxylation of this carbon and in formation of Mo(IV). A similar reaction mechanism has been proposed for the Mo-enzyme sulfite oxidase (18). Regardless of the mechanism for anaerobic ethylbenzene oxidation, it is conceivable that, if Mo(IV) is formed as intermediate, the reoxidation of Mo(IV) could involve an electron transfer via the Fe-S clusters to a quinone. Future structure-function analyses in conjunction with electron paramagnetic resonance spectroscopy are expected to distinguish between these and other possible mechanisms and to provide new insights into the mode of action of this novel enzyme.
ACKNOWLEDGMENTS
We thank Virgil Sclurf and Jeff Hansen from the University of Texas Health Science Center for analytical ultracentrifugation analysis and Rizlene Bencheikh-Latmani for technical expertise with the metal analysis.
Funding for this project was provided by grants from the U.S. Environmental Protection Agency through the Western Region Hazardous Substance Research Center and the National Science Foundation, MCB 9733535. H.A.J. was the recipient of an NIH Biotechnology Training Fellowship. D.A.P. was supported by a National Science Foundation postdoctoral research fellowship in microbial biology.
ADDENDUM
While this report was in review, a study by Kniemeyer and Heider (23a) on the isolation of ethylbenzene dehydrogenase from a microorganism closely related to Azoarcus sp. strain EB1 was in press.
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1998. [Google Scholar]
- 2.Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ball H A, Johnson H A, Reinhard M, Spormann A M. Initial reactions in anaerobic ethylbenzene oxidation by denitrifying bacterium, strain EB1. J Bacteriol. 1996;178:5755–5761. doi: 10.1128/jb.178.19.5755-5761.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beinert H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron sulfur proteins. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 5.Beller H R, Spormann A M. Anaerobic activation of toluene and o-xylene by addition to fumarate in denitrifying strain T. J Bacteriol. 1997;179:670–676. doi: 10.1128/jb.179.3.670-676.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beller H R, Spormann A M. Benzylsuccinate formation as a means of anaerobic toluene activation by sulfate-reducing strain PRTOL1. Appl Environ Microbiol. 1997;63:3729–3731. doi: 10.1128/aem.63.9.3729-3731.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berks B C, Sargent F, Palmer T. The TAT protein export pathway. Mol Microbiol. 2000;35:260–274. doi: 10.1046/j.1365-2958.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- 8.Berks B C. A common export pathway for proteins binding complex redox factors. Mol Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- 9.Biegert T, Fuchs G, Heider J. Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur J Biochem. 1996;238:661–668. doi: 10.1111/j.1432-1033.1996.0661w.x. [DOI] [PubMed] [Google Scholar]
- 10.Bilous P T, Cole S T, Anderson W F, Weiner J H. Nucleotide sequence of the dmsABC operon encoding the anaerobic dimethylsulphoxide reductase of Escherichia coli. Mol Microbiol. 1988;2:785–795. doi: 10.1111/j.1365-2958.1988.tb00090.x. [DOI] [PubMed] [Google Scholar]
- 11.Blasco F, Iobbi C, Giordano G, Chippaux M, Bonnefoy V. Nitrate reductase of Escherichia coli: completion of the nucleotide sequence of the nar operon and reassessment of the role of the alpha and beta subunits in iron binding and electron transfer. Mol Gen Genet. 1989;218:249–256. doi: 10.1007/BF00331275. [DOI] [PubMed] [Google Scholar]
- 12.Boyington J C, Gladyshev V N, Khangulov S V, Stadtman T C, Sun P D. Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an FeS cluster. Science. 1997;275:1305–1308. doi: 10.1126/science.275.5304.1305. [DOI] [PubMed] [Google Scholar]
- 13.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Cammack R, Weiner J H. Electron paramagnetic resonance spectroscopic characterization of dimethyl sulfoxide reductase of Escherichia coli. Biochemistry. 1990;29:8410–8416. doi: 10.1021/bi00488a030. [DOI] [PubMed] [Google Scholar]
- 15.Garfin D E. One-dimensional gel electrophoresis. In: Deutscher M P, editor. Methods in enzymology: guide to protein purification. San Diego, Calif: Academic Press; 1990. pp. 425–441. [DOI] [PubMed] [Google Scholar]
- 16.Gibson D T, Subramanian V. Microbial degradation of aromatic hydrocarbons. In: Gibson D T, editor. Microbial degradation of organic compounds. New York, N.Y: Marcel Dekker, Inc; 1984. pp. 181–252. [Google Scholar]
- 17.Guigliarelli B, Asso M, More C, Augier V, Blasco F, Pommier J, Giordano G, Bertrand P. EPR and redox characterization of iron-sulfur centers in nitrate reductases A and Z from Escherichia coli. evidence for a high-potential class and their relevance in the electron-transfer mechanism. Eur J Biochem. 1992;207:61–68. doi: 10.1111/j.1432-1033.1992.tb17020.x. [DOI] [PubMed] [Google Scholar]
- 18.Hille R. The reaction mechanism of oxomolybdenum enzymes. Biochim Biophys Acta. 1994;1184:143–169. doi: 10.1016/0005-2728(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 19.Hille R. The mononuclear molybdenum enzymes. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 20.Hille R, Rétey J, Bartlewski-Hof U, Reichenbecher W, Schink B. Mechanistic aspects of molybdenum-containing enzymes. FEMS Microbiol Rev. 1999;22:489–501. doi: 10.1111/j.1574-6976.1998.tb00383.x. [DOI] [PubMed] [Google Scholar]
- 21.Johnson J, Rajagopalan K. Structural and metabolic relationship between the molybdenum cofactor and urothione. Proc Natl Acad Sci USA. 1982;79:6856–6860. doi: 10.1073/pnas.79.22.6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson H A, Spormann A M. In vitro studies on the initial reactions of anaerobic ethylbenzene mineralization. J Bacteriol. 1999;181:5662–5668. doi: 10.1128/jb.181.18.5662-5668.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kisker C, Schindelin H, Baas D, Retey J, Mechenstock R U, Kroneck P M H. A structural comparison of molybdenum cofactor-containing enzymes. FEMS Microbiol Rev. 1999;22:503–521. doi: 10.1111/j.1574-6976.1998.tb00384.x. [DOI] [PubMed] [Google Scholar]
- 23a.Kniemeyer, O., and J. Heider. Ethylbenzene dehydrogenase, a novel hydrocarbon-oxidizing molybdenum/iron-sulfur/heme enzyme. J. Biol. Chem., in press. [DOI] [PubMed]
- 24.Krafft T, Bowen A, Theis F, Macy J M. Cloning and sequencing of the genes encoding the periplasmic-cytochrome b-containing selenate reductase of Thauera selenatis. DNA Sequence. 2000;10:365–377. doi: 10.3109/10425170009015604. [DOI] [PubMed] [Google Scholar]
- 25.Krieger C J, Beller H R, Reinhard M, Spormann A M. Initial reactions in anaerobic oxidation of m-xylene by the denitrifying bacterium Azoarcus sp. strain T. J Bacteriol. 1999;181:6403–6410. doi: 10.1128/jb.181.20.6403-6410.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochman H, Gerber A S, Hartl D L. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plunkett G, III, Burland V, Daniels D L, Blattner F R. Analysis of the Escherichia coli genome. III. DNA sequence of the region from 87.2 to 89.2 minutes. Nucleic Acids Res. 1993;21:3391–3398. doi: 10.1093/nar/21.15.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rabus R, Heider J. Initial reactions of anaerobic metabolism of alkylbenzenes in denitrifying and sulfate-reducing bacteria. Arch Microbiol. 1998;170:377–384. [Google Scholar]
- 29.Rabus R, Widdel F. Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch Microbiol. 1995;163:96–103. doi: 10.1007/BF00381782. [DOI] [PubMed] [Google Scholar]
- 30.Rothery E. Analytical methods for graphite tube atomizers. Mulgrave, Victoria, Australia: Varian; 1988. [Google Scholar]
- 31.Schröder I, Rech S, Krafft T, Macy J M. Purification and characterization of the selenate reductase from Thauera selenatis. J Biol Chem. 1997;272:23765–23768. doi: 10.1074/jbc.272.38.23765. [DOI] [PubMed] [Google Scholar]
- 32.U.S. Environmental Protection Agency. Underground motor fuel storage tanks: a national survey EPA560/5-86-013. U.S. Washington, D.C.: Environmental Protection Agency; 1986. [Google Scholar]
- 33.Weiner J H, MacIsaac D P, Bishop R E, Bilous P T. Purification and properties of Escherichia coli dimethyl sulfoxide reductase, and iron-sulfur molybdoenzyme with broad substrate specificity. J Bacteriol. 1988;170:1505–1510. doi: 10.1128/jb.170.4.1505-1510.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zengler K, Heider J, Rosselló-Mora R, Widdel F. Phototrophic utilization of toluene under anoxic conditions by a new strain of Blastochloris sulfoviridis. Arch Microbiol. 1999;172:204–212. doi: 10.1007/s002030050761. [DOI] [PubMed] [Google Scholar]