

Abstract
RNA-guided CRISPR-Cas12a endonucleases are promising tools for genome engineering. Here we demonstrate that LbCas12a variants derived from Lachnospiraceae bacterium show a broad PAM preference, recognizing certain non-canonical PAMs with high efficiency. Furthermore, we engineered LbABE8e to carry G532R and/or K595R mutations, altering its original PAM specificities; these variants exhibited superior base editing activity in human cells compared with wild-type LbABE8e at sites with non-canonical PAMs. Based on this finding, we utilized the most effective LbCas12a and LbABE8e variants to demonstrate multiplexed and mutant-allele-specific gene editing in oncogenes, made possible by the variant’s recognition of non-canonical PAMs. Importantly, LbCas12a-G532R/K595R and LbABE8e-G532R/K595R with optimized crRNA arrays targeted to triple oncogenic mutations inhibited colon cancer cell proliferation. Taken together, these results demonstrate the potential of engineered LbCas12a and LbABE8e as tools for targeting sites with alternative PAMs for genome engineering and therapeutic editing in cancer cells.
Keywords: MT: RNA/DNA editing, LbCas12a, LbABE8e, CRISPR, non-canonical PAMs, colorectal cancer, CRC, base editing, gene therapy, oncogenic mutation
Graphical abstract
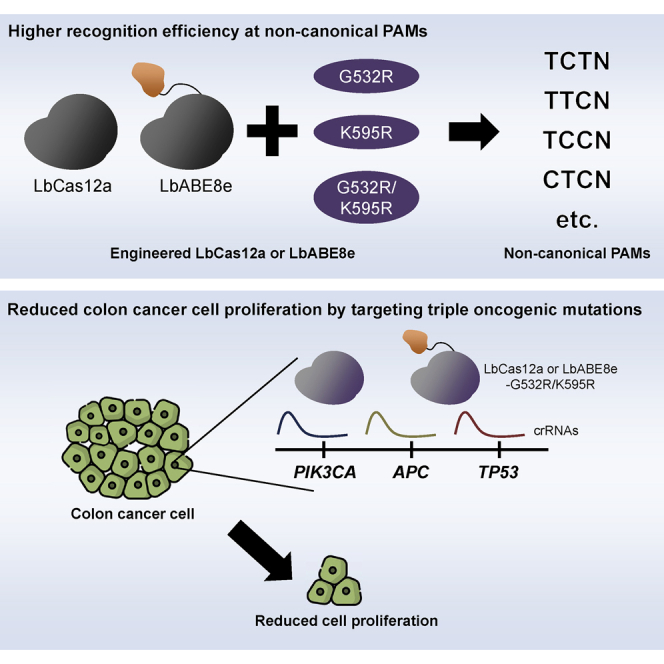
Choi and colleagues expanded the targeting scope of LbCas12a variants by introduction of G532R and/or K595R mutations. Delivery of engineered LbCas12a variants prevents the proliferation of colon cancer cells, indicating that application of LbCas12a variants will be an effective strategy to treat cancers and other polygenic diseases.
Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR)-associated proteins (Cas) system, an adaptive immune system used by bacteria and archaea to defend against viral infections, offers an attractive platform for genome editing and holds promise for therapeutic applications.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 One type of Cas protein, Cas12a endonuclease (also called CRISPR from Prevotella and Francisella 1 [Cpf1]) is derived from a type V CRISPR system and differs from SpCas9 in several respects.11 The Cas12a crRNA array is processed by Cas12a ribonuclease activity to generate mature crRNAs without the need for a tracrRNA.11 Given this characteristic, Cas12a can be used for multiplexed genome engineering, in which multiple sites are simultaneously edited using a CRISPR array that encodes several crRNAs in a single transcript.12 In addition, Cas12a can be used as a gene therapy tool with the advantage of a smaller gene size (3.7 kbp encoding 1,299 amino acids) compared with SpCas9 (4.1 kbp encoding 1,368 amino acids).13 Furthermore, Cas12a recognizes T-rich PAMs, canonically, TTTV at the 5′-end of a protospacer, and generates a staggered double-strand break (DSB) distal to the PAM site.11,14
Recent studies revealed that members of the Cas12a family can also recognize suboptimal PAMs such as TCCV, CTTV, TCTV, and TTCV, in which one or more T nucleotides are replaced with C, although the editing activity is lower at such sites compared with sites with a TTTV PAM.15,16 This behavior occurs because the PAM-binding channels of Cas12a are more loosely aligned than those of SpCas9, causing the channel to open slightly during suboptimal PAM binding.15 Several attempts have been made to engineer Cas12a to improve its ability to recognize suboptimal PAMs; these attempts have been based on studies showing that mutating Cas9 residues that come in close proximity to the PAM-DNA duplex can alter the PAM preference.17, 18, 19, 20 It has been reported that the introduction of S542R/K607R or S542R/K548V/N552R mutations into Acidaminococcus sp. BV3L6 Cas12a [AsCas12a] can extend the PAM preferences of Cas12a, respectively allowing it to recognize TYCV and TATV PAMs in human cells.21,22 In addition, AsCas12a Ultra carrying the M537R/F870L mutations showed enhanced genome editing activity at sites with TTTN PAM.23
Similarly, the introduction of G532R/K595R (RR) or G532R/K538V/Y542R (RVR) mutations into Lachnospiraceae bacterium ND 2006 Cas12a [LbCas12a] altered its PAM specificity, respectively allowing it to recognize TYCV and TATV PAMs.21 Two other reports have demonstrated that LbCas12a-RR, -RVR, and -RVRR variants respectively recognize TYYV, TWTV and TNTN PAMs.24,25 However, the PAM preferences of the LbCas12a variants were mostly studied with PAMs containing V at the fourth position. Further investigation of PAM specificity could expand the targeting scope of LbCas12a.
Recently, several DNA base editors have been developed that enable the direct conversion of a targeted single base in cells and organisms with great potential for targeted base mutagenesis. Among them, LbABE8e, which consists of an evolved version of Escherichia coli tRNA deaminase (a monomeric TadA-8e variant) fused with catalytically dead LbCas12a, showed efficient adenine base editing at sites with TTTV PAMs.26 However, its narrow targeting window and limited PAM specificity restricts the use of this base editor for therapeutic editing. Expanding the targeting range of the LbABE8e base editor by altering its PAM specificities would broaden its therapeutic usefulness.
Colorectal cancer (CRC) is the third most common malignancy, accounting for about 10% of all cancers.27 Several genomic alterations that cause CRC have been identified; in one study of 145 patients, 97.2% showed somatic mutations with high frequency in genes including TP53 (70%), APC (60%), KRAS (49%), and PIK3CA (23%).28,29 It has been reported that the development of most cancers require carcinogenic mutations in more than two genes.4 In CRC patients, different combinations of mutation types were observed, affecting RNA expression levels, the tumor environment, and immunocyte infiltration.5 These multiple CRC-associated mutations could be targetable using either LbCas12a or LbABE8e, given their ability for multiplexed editing, if their PAM specific could be broadened so that non-canonical PAMs were recognized.
Here, we demonstrated an extended PAM specificity for LbCas12a and its variants with no preference for T or V nucleotides in the fourth position of non-canonical PAMs, as well as PAM-dependent enhanced activity of these variants. In addition, we engineered LbABE8e to carry G532R and/or K595R mutation(s) for altering its PAM preference. We then utilized LbCas12a and LbABE8e variants with multiplexed crRNAs to simultaneously edit three oncogenic mutations, in the TP53, APC, and PIK3CA genes, associated with non-canonical PAMs in cells derived from CRC patients. These findings suggest the potential of engineered LbCas12a and LbABE8e as alternative genome editing and therapeutic tools in the treatment of cancers and other polygenic disorders.
Results
Non-canonical PAM-dependent activity of LbCas12a
In our previous study, we demonstrated nuclease-digested whole-genome sequencing (Digenome-seq) for profiling genome-wide off-target sites of LbCas12a30 and found that ∼10% of in vitro cleavage sites (4/41 sites) involved non-canonical PAMs, whereas 90% of such sites (37/41 sites) involved TTTN PAMs (Table S1). Based on this finding, we further examined the genome editing activity of LbCas12a at sites containing various non-canonical PAMs. We generated a total of 60 crRNAs targeting the TP53, APC, and PIK3CA genomic loci at sites containing non-canonical PAMs (TCTN, TTCN, TCCN, and CTCN) and canonical TTTN PAMs (Table S2). Then, plasmids encoding LbCas12a and the corresponding crRNAs were transfected into HEK293T cells, and the resulting genome editing efficiencies were measured. Targeted deep sequencing determined that LbCas12a exhibits activity at sites with TCTN, TTCN, TCCN, CTCN, and TTTN PAMs at frequencies of 2.7% ± 1.0%, 10.1% ± 3.6%, 2.1% ± 0.7%, 0.4% ± 0.1%, and 29.9% ± 3.5%, respectively (Figure 1A). Interestingly, among the tested non-canonical PAMs, LbCas12a recognized TTCN the best.
Figure 1.

Activity of wild-type LbCas12a at sites with non-canonical and canonical PAMs
(A) LbCas12a-induced indel frequencies at 60 endogenous target sites containing non-canonical and canonical PAMs in HEK293T cells. Indel frequencies were measured by targeted deep sequencing. Black arrows indicate target sites containing TTTT PAM. Data points (n = 12) are plotted as dots representing the crRNAs indicated in Table S2. Each dot represents the mean of biologically independent triplicates. (B) Genome-wide Circos plot representing DNA cleavage scores. Intact genomic DNA (gray) and genomic DNA digested with LbCas12a (blue) were subjected to whole-genome sequencing and Digenome-seq analysis. (C) On-target and off-target sites identified by Digenome-seq analysis. The chromosome location and DNA sequences at in vitro cleavage sites are shown together with the DNA cleavage score. PAM sequences are shown in blue. Target sequences that hybridize with the crRNA are underlined and mismatched nucleotides are shown in red. (D) Comparison of the activity of LbCas12a at sites containing TTTA, TCCA, and GTTT sequences in HEK293T cells. Indel frequencies were measured by targeted deep sequencing. Each dot represents an individual data point. Error bars indicate SEM (n = 3).
Next, we investigated whether a single crRNA together with LbCas12a could target both canonical and non-canonical PAMs, simultaneously. First, we designed a 23-nucleotide crRNA that includes an extra guanine (G) relative to the target site because crRNAs are transcribed under the control of the U6 promoter, which requires a guanine at the 5′ end; the remaining nucleotides in the crRNA hybridize with a 23-nucleotide region downstream of a TTTA PAM, and a 20-nucleotide region downstream of a TCCA PAM. Cell-free human genomic DNA was digested in vitro using an LbCas12a-crRNA ribonucleoprotein complex targeting the sites with TTTA and TCCA PAMs and then subjected to Digenome-seq. LbCas12a-mediated cleavage occurred at the sites containing the TTTA and TCCA PAMs and at an additional site downstream of a GTTT sequence (Figures 1B and 1C). To examine the cleavage activity at these three sites in cells, we transfected plasmids encoding LbCas12a and the crRNA into HEK293T cells and sequenced the target loci. LbCas12a induced indels with frequencies of 96.6% ± 0.8% and 12.7% ± 1.1% at the sites containing the TTTA and TCCA PAM sequences, respectively, but not at the site containing the GTTT sequence (Figure 1D). Note that a high DNA cleavage score does not mean a high mutation frequency. For example, the DNA cleavage score was high in OT-II site containing the GTTT PAM, but no mutation frequency was observed, indicating a discrepancy between in vitro cleavage and cellular mutation. These results demonstrate that the canonical and non-canonical PAMs were both recognized by a single crRNA together with LbCas12a, despite the lower activity at the site with the non-canonical versus the canonical PAM.
Non-canonical PAM-dependent enhanced genome editing by engineered LbCas12a variants
We then investigated whether the increased genome editing activities caused by LbCas12a-engineering are PAM dependent. To generate LbCas12a variants, we introduced mutation(s) into human codon-optimized LbCas12a (LbCas12a harboring the G532R mutation [LbCas12a-G532R], the K595R mutation [LbCas12a-K595R], or G532R/K595R double mutations [LbCas12a-G532R/K595R]).21 Then, we transfected plasmids encoding the LbCas12a variants and the corresponding crRNAs into HEK293T cells and measured mutation frequencies at 60 endogenous target sites containing non-canonical PAMs (TCTN, TTCN, TCCN, and CTCN) and canonical TTTN PAMs (Table S2). LbCas12a-G532R showed enhanced genome editing activities at sites with TCTN, TTCN, TCCN, and CTCN PAMs (indel frequencies of 5.5% ± 1.4%, 23.5% ± 5.1%, 13.9% ± 3.1%, and 6.9% ± 2.0%, respectively) (Figures 2A–2D). In particular, LbCas12a-G532R-induced indel frequencies at sites containing TCCN PAMs were increased by 6.8-fold compared with indel frequencies induced by wild-type LbCas12a (Figure 2C). LbCas12a-K595R also exhibited enhanced genome editing at sites with TCTN PAMs, compared with wild-type LbCas12a (Figure 2A). In addition, the effects of the double mutations in LbCas12a-G532R/K595R were additive at sites with TCTN PAMs (indel frequencies of 12.4% ± 2.6% for LbCas12a-G532R/K595R, 5.5% ± 1.4% for LbCas12a-G532R, and 9.6% ± 2.5% for LbCas12a-K595R) (Figure 2A). Nevertheless, the indel frequencies induced by LbCas12a-G532R/K595R at target sites containing TTCN, TCCN, and CTCN PAMs were similar to those induced by LbCas12a-G532R (Figures 2B–2D). Interestingly, introduction of mutation(s) into wild-type LbCas12a did not enhance TTTN PAM recognition (Figure 2E). Taken together, these results indicate that the G532R and/or K595R mutations in the engineered LbCas12a variants seem to alter nucleotide preferences in non-canonical PAMs, particularly at the second position (Table S3), a result similar to previously published findings.24,25
Figure 2.

Activity of engineered LbCas12a variants at sites with non-canonical and canonical PAMs
(A–E) Comparison of the genome editing activities of wild-type LbCas12a and LbCas12a variants harboring G532R, K595R, or G532R/K595R mutations measured by targeted deep sequencing at 60 endogenous target sites in HEK293T cells. Data points (n = 12) are plotted as dots representing the crRNAs indicated in Table S2. Each dot represents the mean of biologically independent triplicates. (A) TCTN, (B) TTCN, (C) TCCN, (D) CTCN, and (E) TTTN PAMs. The heatmap shows the mean indel frequencies at 60 target sites with TCTN, TTCN, TCCN, CTCN, and TTTN PAMs.
LbCas12a variants exhibit no preference for the fourth nucleotide in non-canonical PAMs
We next examined whether the fourth nucleotide of the PAM affects the activities of LbCas12a and its variants. We included two additional target sites with TTTT PAMs to identify more statistically meaningful differences in PAM preferences. In line with previous reports,11,14 we found that wild-type LbCas12a exhibited a preference for TTTV versus TTTT PAMs (Figure 3A). However, neither LbCas12a-G532R nor -G532R/K595R exhibited a preference for the fourth nucleotide in canonical PAMs. Moreover, LbCas12a variants showed higher activity than wild-type LbCas12a at sites containing all tested non-canonical PAMs with a T at the fourth position (Figure 3A). More interestingly, LbCas12a and its three variants showed no preference for the fourth nucleotide in non-canonical PAMs (Figure 3B).
Figure 3.

Activity of engineered LbCas12a variants at sites with non-canonical and canonical PAMs with a T or V nucleotide at the fourth position
Representative graphs show the activities of LbCas12a and its variants at 62 target sites according to the type of nucleotide (V or T) at the fourth position in (A) canonical and (B) non-canonical PAMs. (C) Activities of LbCas12a and its variants at 32 target sites containing a T at the fourth position in non-canonical PAMs. (D) The heatmap shows mean indel frequencies measured by targeted deep sequencing. Mean indel frequencies ± SEM are shown in the graphs. Data points (n = 4–36) are plotted as dots representing the crRNAs indicated in Tables S2 and S4. Each dot represents the mean of biologically independent triplicates. p values were derived from a Student’s two-tailed t test. ∗p < 0.05; NS, not significant.
We further investigated the editing activities at 32 sites in TP53, APC, CTNNB1, and SRSF3 genes with non-canonical PAMs harboring a T at the fourth position (Table S4). We did not analyze editing in the PIK3CA gene further, because the editing efficiency was low at all tested target sites. With the exception of the CCCT PAM, the remaining seven non-canonical PAMs (TCCT, TTCT, TCTT, CTCT, CCTT, CTTT, and TTTT) were recognized by wild-type LbCas12a, which induced mean indel frequencies ranging from 1.8% to 21% (Figures 3C and 3D). Strikingly, LbCas12a-G532R/K595R induced mean indel frequencies ranging from 8.7% to 35.8%; at sites with CCCT PAMs, the activity of this variant was increased 51-fold compared with that of wild-type LbCas12a (Figures 3C and 3D).
LbABE8e variants induce efficient adenine base editing at sites with non-canonical PAMs
Next, we investigated whether LbABE8e can induce adenine base editing at sites with non-canonical PAMs. Plasmids encoding wild-type LbABE8e and the corresponding crRNAs were transfected into HEK293T cells to determine the resulting base editing efficiencies at the 22 endogenous target sites at which indels were generated by LbCas12a-G532R/K595R at frequencies above 20% (Table S5). The base editing efficiency of wild-type LbABE8e ranged from 0% to 5.3% at these sites with non-canonical PAMs (Figure 4A).
Figure 4.

Adenine base editing with engineered LbABE8e variants at sites with non-canonical PAMs
Adenine base editing efficiencies induced by (A) wild-type LbABE8e and engineered LbABE8e containing (B) G532R, (C) K595R, or (D) G532R/K595R mutations at 22 endogenous target sites, at which indels were generated by LbCas12a-G532R/K595R at frequencies above 20%, measured by targeted deep sequencing in HEK293T cells. Mean base editing frequencies ± SEM are shown in the graphs. (E) A representative heatmap shows mean base editing frequencies measured by targeted deep sequencing. (F) The base editing window within the protospacer at six genomic sites in HEK293T cells for wild-type LbABE8e and engineered LbABE8e containing G532R, K595R, or G532R/K595R mutations. (G) The enhanced editing window of LbABE8e variants at site 13. Each dot represents an individual data point. Error bars indicate SEM (n = 3).
To overcome the low efficiency of wild-type LbABE8e in recognizing non-canonical PAMs, the PAM preferences were altered by suitable amino acid changes that loosen PAM constraints. The G532R and/or K595R mutations were incorporated into human codon-optimized LbABE8e to generate LbABE8e-G532R, LbABE8e-K595R, and LbABE8e-G532R/K595R. Introduction of a G532R mutation into wild-type LbABE8e increased base editing efficiencies at sites with several different PAMs (Figures 4B). However, the effect of the K595R mutation in LbABE8e was minimal (Figure 4C). Incorporation of double mutations to generate LbABE8e-G532R/K595R further increased base editing efficiencies, up to 14-fold compared with that of wild-type LbABE8e, at sites with all tested PAM sequences (Figures 4A, 4D, and 4E). The effect of the double G532R/K595R mutations was striking for the site with a TTCC PAM; the wild-type LbABE8e-induced editing efficiency at this site was low (3.8% ± 0.9%), but LbABE8e-G532R/K595R showed an editing efficiency of 15.3% ± 1.3% (Figures 4D and 4G). Next, we examined the base editing window of LbABE8e variants at the six genomic sites at which A-to-G conversions were generated by LbABE8e-G532R/K595R at frequencies above 5%. The editing window for LbABE8e-G532R/K595R is similar to that of wild-type LbABE8e: the protospacer spans positions 8–14 (counting downstream of the PAM) (Figure 4F and Table S6). Of special interest, LbABE8e-G532R/K595R, which induced the highest frequency of base editing among all LbABE8e variants tested, was used for correcting oncogenic mutations in the studies described below.
Editing of triple oncogenic mutations using engineered LbCas12a and LbABE8e
To investigate oncogenic mutations present in HCT-15 CRC cells, we isolated genomic DNA from the cells and analyzed the sequences of cancer-associated genes using targeted deep sequencing. We found missense mutations in APC (6496C-to-T), PIK3CA (1633G-to-A), and TP53 (722C-to-T) that were present in 48% ± 0.2%, 41% ± 1.2%, and 50% ± 0.8% of the alleles, respectively (Figures 5A and S1). To evaluate LbCas12a-mediated editing of these triple oncogenic mutations, in each case we designed several crRNAs that would hybridize with these mutant target sequences located downstream of various non-canonical and canonical PAMs; editing occurred at different frequencies depending on the PAM (Figure S2 and Table S7). Each crRNA that resulted in the highest indel frequencies in the APC, PIK3CA, and TP53 genes was transfected into HCT-15 cells along with LbCas12a-G532R, LbCas12a-K595R, or LbCas12a-G532R/K595R to compare genome editing efficiencies of the LbCas12a variants (Figure S2). Of note, LbCas12a-G532R/K595R complexed with crRNAs induced mutant-allele-specific gene editing at frequencies up to 47% ± 0.9% (Figure S2).
Figure 5.

Editing of triple oncogenic mutations by engineered LbCas12a inhibits abnormal cancer cell proliferation
(A) Schematic diagram of oncogenic mutations in HCT-15 colon cancer cells. Oncogenic mutations are shown in red, and PAM sequences are shown in blue. (B) Schematic representation of vectors encoding LbCas12a-G532R/K595R, LbABE8e-G532R/K595R, and a CRISPR array with direct repeats; multiplexed crRNA-V1, -V2, and -V3 are shown. Multiplexed crRNA-V2 and -V3 contain separators between spacer sequences as shown. In all three versions, expression of a single transcript is controlled by the U6 promoter. (C) Activities of LbCas12a-G532R/K595R with multiplexed crRNA-V3 targeting APC, PIK3CA, and TP53 and of (D) LbABE8e-G532R/K595R with multiplexed crRNA-V3 targeting APC, PIK3CA, and TP53 in HCT-15 cells. Mean base editing frequencies in the protospacer are shown. (E) Cell viabilities were determined using an MTT assay after transfection of plasmids encoding LbCas12a-G532R/K595R or LbABE8e-G532R/K595R and a corresponding crRNA. “Mock” refers to cells treated with plasmids encoding LbCas12a-G532R/K595R or LbABE8e-G532R/K595R and a non-targeting multiplexed crRNA-V3. Error bars indicate SEM (n = 3). p values were derived from a Student’s two-tailed t test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; NS, not significant.
Next, we combined the respective crRNA-encoding sequences that resulted in the highest indel frequencies in each oncogene (Table S7) to generate a CRISPR array targeting APC, PIK3CA, and TP53 simultaneously. The three crRNA-encoding sequences were arranged according to the GC content in the spacers, from lowest to highest: that targeting the APC (35% GC content), PIK3CA (39%), and TP53 (52%) genes. The first CRISPR array we generated, multiplexed crRNA-V1, contains three spacer sequences; multiplexed crRNA-V2 contains separators31 between the spacers. We further designed multiplexed crRNA-V3 that includes an extra separator downstream of the promoter (Figure 5B and Table S8). After CRISPR array- and LbCas12a-G532R/K595R-encoding plasmids were transfected into HCT-15 cells, we found that multiplexed crRNA-V3 was associated with the highest activity, resulting in indel frequencies of 25.9% ± 2.4%, 32.6% ± 2.9%, and 43.3% ± 0.5% in mutant APC, PIK3CA, and TP53 alleles, respectively, representing a 1.7-fold and 1.3-fold increase compared with the activity associated with multiplexed crRNA-V1 and -V2, respectively (Figure 5C). However, indels were also generated in the corresponding wild-type alleles with frequencies ranging between 0.9% and 3.3%.
In addition, treatment of HCT-15 cells with multiplexed crRNA-V3 and LbABE8e-G532R/K595R-encoding plasmids induced adenine base editing with frequencies of 2.3% ± 0.5% in APC, 12.8% ± 2.9% in PIK3CA, and 3.3% ± 0.4% in TP53, resulting in correction of oncogenic missense mutations (Figure 5D and Table S6). These results suggest that LbCas12a- or LbABE8e-G532R/K595R together with a multiplexed CRISPR array with three separators can successfully induce multiplexed gene editing.
Inhibition of cancer cell proliferation mediated by LbCas12a- and LbABE8e-G532R/K595R
To investigate whether LbCas12a-induced indels or LbABE8e-induced base editing of these triple oncogenic mutant alleles could reduce cancer cell proliferation, we conducted an MTT cell proliferation assay. Treatment of HCT-15 cells with multiplexed crRNA-V3- and LbCas12a-G532R/K595R-encoding plasmids led to killing of HCT-15 cells, with an 11.1% reduction in cell viability compared with that in a mock transfection experiment (Figure 5E). Moreover, cells treated with LbABE8e-G532R/K595R showed a greater decrease in viability (a 39.1% decrease compared with mock) (Figure 5E). Taken together, these data show that LbABE8e-mediated correction of multiple oncogenic mutations with a multiplexed CRISPR array efficiently inhibits abnormal cancer cell proliferation.
Discussion
In this report, we have shown that LbCas12a and LbABE8e variants can recognize a diversity of suboptimal PAMs and induce efficient gene editing in human cells. The ability of LbCas12a and LbABE8e variants to recognize alternative PAMs with high efficiency offers the advantage of being able to correct a broader range of disease-causing mutations. Based on this finding, we suggest a novel therapeutic strategy that uses these nucleases and alternative PAMs to target multiple oncogenic mutations for the treatment of CRC.
We previously showed that LbCas12a induces precise editing of targeted oncogenes in non-small cell lung cancer cells and tumor xenografts, leading to efficient tumor regression.8 We speculated that the development of a mutant form of LbABE8e consisting of dLbCas12a fused to a deaminase would potently augment the therapeutic effect. Moreover, the unique ability of LbABE8e to induce multiplexed base editing offers the possibility of targeting various genetic alterations with a single CRISPR array, an ability that would be particularly useful for treating polygenic disorders such as cancers, which are primarily caused by multiple genetic mutations.32 However, the ability of LbCas12a or LbABE8e to edit combinations of mutations in therapeutic application is limited by their requirement for a TTTV PAM at the desired position. Broadening its PAM specificity would extend usefulness for such an approach.
Here, we provide an in-depth demonstration that variants of LbCas12a and LbABE8e exhibit an enhanced ability to recognize suboptimal PAMs compared with the wild-type version. Furthermore, we use these variants to edit previously non-targetable mutations in CRC.
We observed, in contrast to previous studies,21,24,25 that LbCas12a and its variants exhibit no preference for the fourth nucleotide in non-canonical PAMs. In addition, the enhancement of activity exhibited by engineered LbCas12a was shown at target sites containing TCTN, TTCN, TCCN, and CTCN PAMs. Of note, LbCas12a variants recognize CTCN PAMs with highly improved efficiency, broadening target availability. Interestingly, this enhanced effect caused by LbCas12a engineering was not observed at sites with canonical PAMs. Based on this finding, we engineered LbABE8e by introducing the identified PAM-interacting mutation (G532R and/or K595R) to alter its non-canonical PAM specificity and showed an improvement in base editing activity compared with that of wild-type LbABE8e. Most notably, we observed superior base editing efficiencies when G532R/K595R double mutations were incorporated into wild-type LbABE8e. The activity of this variant was increased up to 14-fold compared with that of LbABE8e at sites containing these non-canonical PAMs; the base editing window remained the same. We also improved the effectiveness of crRNA array sequences by introducing separators31 between the spacers, resulting in an improvement in genome editing efficacy compared with that obtained with crRNAs with fewer than three separators.
We then utilized this system to induce multiplexed and mutant-allele-specific gene editing by targeting three oncogenes using LbCas12a or LbABE8e variants in cells derived from CRC patients. Interestingly, mutant-allele-specific knockout by LbCas12a-G532R/K595R reduced abnormal cancer cell proliferation. However, it should be also considered that the tolerance of LbCas12a for mismatched spacer sequences could lead to the induction of DSBs in the wild-type alleles, although at low frequencies. Interestingly, correction of triple oncogenic mutations by LbABE8e-G532R/K595R led to a more substantial reduction of cancer cell proliferation compared with that seen with LbCas12a-G532R/K595R, despite the lower frequencies of gene editing induced by the LbABE8e variant. These results are in line with those from previous studies, showing that a low level of base editing can lead to substantial reduction of protein levels and altered disease phenotypes in cells.33, 34, 35 We speculated that the correction of triple oncogenic mutations, albeit at a low frequency, would be sufficient to reduce abnormal cancer cell proliferation. Further studies are needed to evaluate the synergetic effects of multiplexed compared with single base editing to correct oncogenic mutations in vitro and in vivo. The small sizes of LbCas12a and LbABE8e confer the advantage of efficient delivery using adeno-associated viral or adenoviral vectors in vivo. As a next step, the therapeutic effects of multiplexed gene editing using LbCas12a or LbABE8e variants delivered by viral vectors in an animal model should be thoroughly investigated. Our results support that application of LbCas12 or LbABE8e variants as a gene editing tool will be an effective strategy for treating cancers and other diseases associated with mutations.
Materials and methods
Cell lines and cell culture
HEK293T cells (a human embryonic kidney cell line) were purchased from American Type Culture Collection (CRL-3216, Manassas, VA, USA) and HCT-15 cells (a human colon cancer cell line) were purchased from Korean Cell Line Bank (KCLB, No. 10225). HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Welgene, Korea) supplemented with 10% fetal bovine serum (FBS, Welgene, Korea) and 1% antibiotics (100 units/mL of penicillin and 100 μg/mL streptomycin) (Welgene, Korea). HCT-15 cells were cultured in RPMI-1640 medium with 10% FBS and 1% antibiotics. All cells were maintained at 37°C in a humidified 5% CO2 atmosphere.
Construction of plasmids encoding LbCas12a, LbABE8e, and crRNA
A human codon-optimized LbCas12a coding sequence, derived from Lachnospiraceae bacterium ND 2006, was cloned into a pcDNA3.1 vector plasmid. A plasmid encoding LbABE8e was purchased from Addgene (plasmid #138504). LbCas12a and LbABE8e sequences were modified to encode G532R, K595R, and G532R/K595R mutations via site-directed mutagenesis (Q5 Site-Directed Mutagenesis Kit, New England Biolabs). Plasmid DNA was extracted using an Exprep Plasmid SV miniprep kit (Geneall) and a NucleoBond Xtra Midi EF kit (MACHEREY-NAGEL) according to the manufacturer’s instructions. Sequences of the crRNAs targeting theTP53, APC, PIK3CA, CTNNB1, and SRSF3 genes are listed in Tables S1, S2, S4, S5, and S7. The crRNA-encoding sequences were subcloned into the pU6-Lb-crRNA vector plasmid (Addgene, #78957) digested with BsmB1 (New England Biolabs). The crRNAs were transcribed under the control of the U6 promoter, and LbCas12a and LbABE8e expression was controlled by the cytomegalovirus promoter.
Transfection and genomic DNA extraction
HEK293T and HCT-15 cells were seeded into 24-well plates 1 day prior to transfection and then transfected with the crRNA plasmid (1,500 ng) and the LbCas12a or LbABE8e variant plasmid (500 ng) using 4 μL of jetPRIME (Polyplus). Cells were maintained in DMEM or RPMI-1640 supplemented with 10% FBS. After 48 h of transfection, genomic DNA was extracted using an AccuPrep Genomic DNA Extraction Kit (Bioneer, Korea). Extracted genomic DNA was then used for targeted deep sequencing.
Digenome sequencing
Digenome-seq was performed as described previously.36 Genomic DNA was isolated using a DNeasy Tissue kit (Qiagen) according to the manufacturer’s instructions. Genomic DNA (8 μg) was mixed with LbCas12a protein (300 nM) and crRNA (900 nM) in a 400-μL reaction volume (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, and 100 μg/mL BSA), and the mixture was incubated for 8 h at 37°C. Digested genomic DNA was then incubated with RNase A (50 μg/mL) for 30 min at 37°C and purified again with a DNeasy Tissue kit (Qiagen). Digested DNA was fragmented using the Covaris system and ligated with adaptors for library formation. DNA libraries were subjected to whole-genome sequencing using an Illumina HiSeq X Ten Sequencer at Macrogen. We used the Isaac aligner to generate a Bam file using the following parameters: ver. 01.14.03.12; Mouse genome reference, mm10 from UCSC; Base quality cutoff, 15; Keep duplicate reads, yes; Variable read length support, yes; Realign gaps, no; and Adaptor clipping, yes (adaptor: AGATCGGAAGAGC∗, ∗GCTCTTCCGATCT).
Mutation analysis
On-target sites were amplified from genomic DNA using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific Korea) for targeted deep sequencing. The region of interest was amplified using the primer pairs listed in Table S9. Illumina TruSeq HT dual index primers were used to label each PCR amplicon. The PCR products were purified using Expin PCR SV (Geneall Biotechnology, Korea). Pooled libraries were sequenced using MiniSeq (Illumina, San Diego, CA). Substitutions and indel frequencies were calculated by MAUND, which is available at https://github.com/ibs-cge/maund. Wild-type and mutant sequences were discriminated based on the presence of the single nucleotide missense mutation in the allele. Sequences carrying indels were counted as genome edited sequences. Indel-harboring sequences that could not be discriminated as wild-type or mutant sequences were counted as mutant sequences, because the percentage of wild-type-specific indels among all alleles was low. A-to-G conversion ratios in the mutant sequences were determined to calculate base editing frequency.
MTT assay
To evaluate the effect of editing mediated by LbCas12a or LbABE8e variants on cell proliferation, HCT-15 cells were plated onto a 24-well plate and then transfected with plasmids encoding an LbCas12a variant and the triplex crRNA or a non-targeting crRNA in the mock control. At 72 h after treatment, the MTT assay was carried out. 200 μL of a solution of MTT (Sigma, St. Louis, MO, USA) in phosphate-buffered saline (2 mg/mL) was added to each well. After a 4 h incubation at 37°C, the supernatant was discarded, and the precipitate was dissolved in 1 mL of dimethyl sulfoxide. The absorbance of the samples at 540 nm was then measured on a microplate reader.
Statistical analysis
All results are expressed as the mean ± SEM unless indicated otherwise. Statistical significance as compared with untreated controls is denoted with ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 in the figures and figure legends. Statistical analysis was performed in GraphPad Prism 9.1.1. p values were derived from a Student’s two-tailed t test.
DATA AVAILABILITY
The deep sequencing data have been deposited in the NCBI Sequence Read Archive database under BioProject accession number PRJNA784339.
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea, South Korea [2022R1A2C1013352 and 2020R1A4A1016142; T Koo and 2020R1A2C2101714; D Kim] and a grant from Ministry of Food and Drug Safety, South Korea in 2022 [21153MFDS601].
Author contributions
T.K. and D.K. supervised the research. T.K., E.C., and D.K. wrote the manuscript. E.C., H.H., and E.K. performed the experiments.
Declaration of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.09.005.
Contributor Information
Daesik Kim, Email: dskim89@skku.edu.
Taeyoung Koo, Email: taeyoungkoo@khu.ac.kr.
Supplemental information
References
- 1.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 3.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esvelt K.M., Mali P., Braff J.L., Moosburner M., Yaung S.J., Church G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R.J., Joung J.K. Efficient in vivo genome editing using RNA-guided nucleases woong. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501.Efficient. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon A.R., Jung B.K., Choi E., Chung E., Hong J., Kim J.S., Koo T., Yun C.O. CRISPR-Cas12a with an oAd induces precise and cancer-specific genomic reprogramming of EGFR and efficient tumor regression. Mol. Ther. 2020;28:2286–2296. doi: 10.1016/j.ymthe.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jo D.H., Koo T., Cho C.S., Kim J.H., Kim J.S., Kim J.H. Long-term effects of in vivo genome editing in the mouse retina using Campylobacter jejuni Cas9 expressed via adeno-associated virus. Mol. Ther. 2019;27:130–136. doi: 10.1016/j.ymthe.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 11.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Van Der Oost J., Regev A., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zetsche B., Heidenreich M., Mohanraju P., Fedorova I., Kneppers J., Degennaro E.M., Winblad N., Choudhury S.R., Abudayyeh O.O., Gootenberg J.S., et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017;35:31–34. doi: 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koo T., Park S.W., Jo D.H., Kim D., Kim J.H., Cho H.Y., Kim J., Kim J.H., Kim J.S. CRISPR-LbCpf1 prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Nat. Commun. 2018;9:1855. doi: 10.1038/s41467-018-04175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H.K., Song M., Lee J., Menon A.V., Jung S., Kang Y.M., Choi J.W., Woo E., Koh H.C., Nam J.W., Kim H. In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat. Methods. 2017;14:153–159. doi: 10.1038/nmeth.4104. [DOI] [PubMed] [Google Scholar]
- 15.Yamano T., Zetsche B., Ishitani R., Zhang F., Nishimasu H., Nureki O. Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Mol. Cell. 2017;67:633–645.e3. doi: 10.1016/j.molcel.2017.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu R., Qin R., Li H., Li J., Yang J., Wei P. Enhanced genome editing in rice using single transcript unit CRISPR-LbCpf1 systems. Plant Biotechnol. J. 2019;17:553–555. doi: 10.1111/pbi.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleinstiver B.P., Prew M.S., Tsai S.Q., Topkar V.V., Nguyen N.T., Zheng Z., Gonzales A.P.W., Li Z., Peterson R.T., Yeh J.R.J., et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinstiver B.P., Prew M.S., Tsai S.Q., Nguyen N.T., Topkar V.V., Zheng Z., Joung J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015;33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirano S., Nishimasu H., Ishitani R., Nureki O. Structural basis for the altered PAM specificities of engineered CRISPR-cas9. Mol. Cell. 2016;61:886–894. doi: 10.1016/j.molcel.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 20.Anders C., Bargsten K., Jinek M. Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol. Cell. 2016;61:895–902. doi: 10.1016/j.molcel.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao L., Cox D.B.T., Yan W.X., Manteiga J.C., Schneider M.W., Yamano T., Nishimasu H., Nureki O., Crosetto N., Zhang F. Engineered Cpf1 variants with altered PAM specificities. Nat. Biotechnol. 2017;35:789–792. doi: 10.1038/nbt.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimasu H., Yamano T., Gao L., Zhang F., Nureki O., Sciences C. Structural Basis for the Altered PAM Recognition by Engineered CRISPR-Cpf1. Mol. Cell. 2018;67:139–147. doi: 10.1016/j.molcel.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L., Zuris J.A., Viswanathan R., Edelstein J.N., Turk R., Thommandru B., Rube H.T., Glenn S.E., Collingwood M.A., Bode N.M., et al. AsCas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat. Commun. 2021;12:3908. doi: 10.1038/s41467-021-24017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tóth E., Varga É., Kulcsár P.I., Kocsis-Jutka V., Krausz S.L., Nyeste A., Welker Z., Huszár K., Ligeti Z., Tálas A., Welker E. Improved LbCas12a variants with altered PAM specificities further broaden the genome targeting range of Cas12a nucleases. Nucleic Acids Res. 2020;48:3722–3733. doi: 10.1093/NAR/GKAA110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tóth E., Czene B.C., Kulcsár P.I., Krausz S.L., Tálas A., Nyeste A., Varga É., Huszár K., Weinhardt N., Ligeti Z., et al. Mb- and FnCpf1 nucleases are active in mammalian cells: activities and PAM preferences of four wild-type Cpf1 nucleases and of their altered PAM specificity variants. Nucleic Acids Res. 2018;46:10272–10285. doi: 10.1093/nar/gky815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E., et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 28.Lee C.S., Song I.H., Lee A., Kang J., Lee Y.S., Lee I.K., Song Y.S., Lee S.H. Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci. Rep. 2021;11:8154–8226. doi: 10.1038/s41598-021-87486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhuang Y., Wang H., Jiang D., Li Y., Feng L., Tian C., Pu M., Wang X., Zhang J., Hu Y., Liu P. Multi gene mutation signatures in colorectal cancer patients: predict for the diagnosis, pathological classification, staging and prognosis. BMC Cancer. 2021;21:380. doi: 10.1186/s12885-021-08108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D., Kim J., Hur J.K., Been K.W., Yoon S.H., Kim J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016;34:863–868. doi: 10.1038/nbt.3609. [DOI] [PubMed] [Google Scholar]
- 31.Magnusson J.P., Rios A.R., Wu L., Qi L.S. Enhanced cas12a multi-gene regulation using a crispr array separator. Elife. 2021;10:e66406–e66420. doi: 10.7554/eLife.66406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim C.K.W., Gapinske M., Brooks A.K., Woods W.S., Powell J.E., Zeballos C M.A., Winter J., Perez-Pinera P., Gaj T., Gaj T. Treatment of a mouse model of ALS by in vivo base editing. Mol. Ther. 2020;28:1177–1189. doi: 10.1016/j.ymthe.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chadwick A.C., Wang X., Musunuru K. In vivo base editing of PCSK9 (proprotein convertase subtilisin/kexin type 9) as a therapeutic alternative to genome editing. Arterioscler. Thromb. Vasc. Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L., Xue W., Zhang H., Gao R., Qiu H., Wei J., Zhou L., Lei Y.N., Wu X., Li X., et al. Eliminating base-editor-induced genome-wide and transcriptome-wide off-target mutations. Nat. Cell Biol. 2021;23:552–563. doi: 10.1038/s41556-021-00671-4. [DOI] [PubMed] [Google Scholar]
- 36.Kim D., Bae S., Park J., Kim E., Kim S., Yu H.R., Hwang J., Kim J.I., Kim J.S. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods. 2015;12:237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The deep sequencing data have been deposited in the NCBI Sequence Read Archive database under BioProject accession number PRJNA784339.