

Abstract
Mathematical models are able to reflect biological processes and to capture epidemiologic data. Thus, they may help elucidate roles of risk factors in disease progression. We propose to account for smoking, hypertension, and dyslipidemia in a previously published process-oriented model that describes the development of atherosclerotic lesions resulting in myocardial infarction (MI). The model is sex-specific and incorporates individual heterogeneity. It was applied to population-based individual risk factors and MI rates (Cooperative Health Research in the Region of Augsburg (KORA) study) together with subclinical atherosclerotic lesion data (Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study). Different model variants were evaluated, testing the association of risk factors with different disease processes. Best fits were obtained for smoking affecting a late-stage disease process, suggesting a thrombogenic role. Hypertension was mainly related to complicated, vulnerable lesions. Dyslipidemia was consistent with increasing the number of initial lesions. By accounting for heterogeneity, individual hazard ratios differ from the population average. The mean individual hazard ratio for smoking was twice the population-based hazard ratio for men and even more for women. Atherosclerotic lesion progression and MI incidence data can be related in a mathematical model to illuminate how risk factors affect different phases of this pathological process.
Keywords: atherosclerosis, biological models, cardiovascular risk factors, computer simulation, myocardial infarction, survival analysis
Abbreviations
- AIC
Akaike information criterion
- CVD
cardiovascular disease
- HDL
high-density lipoprotein
- HR
hazard ratio
- KORA
Cooperative Health Research in the Region of Augsburg
- MI
myocardial infarction
- PDAY
Pathobiological Determinants of Atherosclerosis in Youth
Editor's note: An invited commentary on this article appears on page 1776, and the authors' response appears on page 1781.
Risk factors for a disease are established essentially by two different kinds of observations. On the one hand, statistical associations between risk factors and disease are derived in epidemiologic studies. On the other hand, the pathophysiological mechanism of the risk factor on the disease can only be understood with biological experiments. Both independent approaches are crucial for inference of relevant, causal effects. Here, we want to demonstrate that both the epidemiologic approach and the biological approach can be combined in a process-oriented mathematical risk model.
On the one hand, integrated, process-oriented modeling can be informative for risk prediction. To give a simple example, low-density lipoprotein is strongly involved in the development of atherosclerotic lesions. The process of atherosclerosis takes decades from first lesion development to the first clinical manifestation, such as myocardial infarction (MI). Therefore, present cholesterol levels have impact on the risk later in life, and a process-oriented model can provide a reasonable age-risk pattern beyond what can be derived from epidemiologic data alone. On the other hand, risk factors may affect chronic disease development via several biological mechanisms, and process-oriented modeling can help to identify the most relevant ones with regard to the observed risk. Thus, process-oriented models have the potential to inform schemes of risk stratification across the life span and thus identify individuals who can benefit most from therapeutical intervention.
Process-oriented models have long tradition in particular for cancer (1–5). They are popular in radiation research but have also been applied to other risk factors (6, 7). The present study investigates roles of risk factors for MI, applying a previously published model of atherosclerosis and subsequent MI (8). The general approach is inspired by earlier cancer modeling but there is a main difference: In the present study, we strongly take advantage of the fact that there is data on atherosclerotic lesions in an unselected population. The model is thus calibrated not only on epidemiologic cohort data but also on data on subclinical coronary plaques. Our approach allows us to investigate in the same model the role of risk factors in risk and in atherosclerotic lesion formation.
METHODS
The basic process-oriented model
Our model is based on the process-oriented model of atherosclerosis and subsequent risk of MI proposed in Simonetto et al. (8). Briefly, the model characterizes the state of atherosclerosis progression by the area of the coronary artery involved with lesions of increasing severity: fatty streaks, fibrous plaques, and complicated lesions. At birth, the coronary artery is assumed to be free from any lesion. New lesions are supposed to form with rate 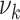 , where
, where 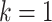 refers to fatty streaks,
refers to fatty streaks, 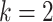 to fibrous plaques, and
to fibrous plaques, and 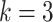 to complicated lesions. Fibrous plaques can only form within an area of fatty streaks and complicated lesions can only form from fibrous plaques. Lesion formation is modeled as a stochastic process and with a probability proportional to the susceptible area. The size of new lesions is denoted by
to complicated lesions. Fibrous plaques can only form within an area of fatty streaks and complicated lesions can only form from fibrous plaques. Lesion formation is modeled as a stochastic process and with a probability proportional to the susceptible area. The size of new lesions is denoted by 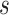 .
.
Logistic growth is assumed with growth rates 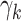 to ensure that lesions of grade
to ensure that lesions of grade 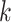 can only progress within the area involved with lesions of grade
can only progress within the area involved with lesions of grade  . Lesion growth rates are assumed to be normally distributed in the population. Finally, risk of MI was modeled to be proportional to the area involved with complicated lesions, with a proportionality factor
. Lesion growth rates are assumed to be normally distributed in the population. Finally, risk of MI was modeled to be proportional to the area involved with complicated lesions, with a proportionality factor 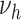 . A schematic model representation is presented in Figure 1 and an exemplary sequence sketched in Figure 2.
. A schematic model representation is presented in Figure 1 and an exemplary sequence sketched in Figure 2.
Figure 1.
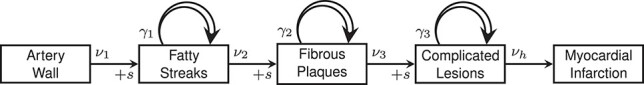
Schematic representation of the model of atherosclerosis development and subsequent myocardial infarction. New lesions emerge with rates 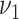 ,
, 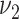 , and
, and 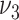 and size
and size 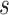 from lower-grade lesions or the normal artery wall. Existing lesions grow with rates
from lower-grade lesions or the normal artery wall. Existing lesions grow with rates 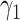 ,
, 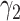 , and
, and 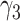 . With rate
. With rate 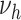 , complicated lesions give rise to myocardial infarction. Adapted from Simonetto et al. (8).
, complicated lesions give rise to myocardial infarction. Adapted from Simonetto et al. (8).
Figure 2.
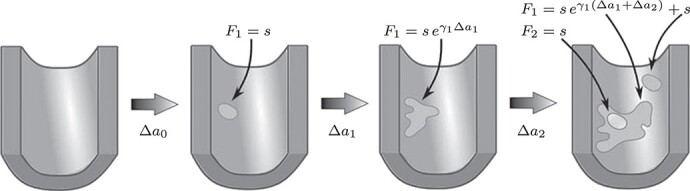
Sketch of possible early plaque development in the model. A first fatty streak appears after some time 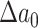 with area
with area 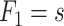 . Subsequently it expands with growth rate
. Subsequently it expands with growth rate 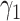 . At the last depicted time point, part of the fatty streak has become a fibrous plaque, with area
. At the last depicted time point, part of the fatty streak has become a fibrous plaque, with area 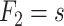 . The intimal surface area involved with fatty streaks and more advanced lesions results from further growth of the first fatty streak and from the origin of a second fatty streak. Adapted from Simonetto et al. (8).
. The intimal surface area involved with fatty streaks and more advanced lesions results from further growth of the first fatty streak and from the origin of a second fatty streak. Adapted from Simonetto et al. (8).
Epidemiologic data
Cross-sectional epidemiologic data was retrieved from the Cooperative Health Research in the Region of Augsburg (KORA) S3 survey (n = 4,856), conducted in 1994/1995, and S4 survey (n = 4,261), conducted in 1999/2001. Further details on the surveys are given in Löwel et al. (9). Briefly, participants aged 25 to 74 years were randomly sampled from the region of Augsburg, Southern Germany. All participants underwent a standardized interview and medical examination. Based on self-reported smoking behavior, participants were categorized as current, former, or never smokers. In the S3 study, data on the age of smoking initiation and cessation were also recorded. Furthermore, participants were asked about medication intake, including antihypertensive and lipid lowering medication. Blood pressure was measured under strictly standardized conditions with 3 recordings at least 3 minutes apart. The arithmetic mean of the second and third recording was used as the final value. Total serum cholesterol was measured by an enzymatic method. High-density lipoprotein (HDL) was measured after precipitation of the apoprotein B–containing lipoproteins with phosphotungstate/magnesium2+. Self-reported MI cases were validated by the treating physician, review of medical records, and the official MI registry of the Klinikum Augsburg. We excluded participants with prevalent MI at baseline and participants with missing information on MI, smoking, blood pressure, or serum cholesterol. Epidemiologic data were available at the individual level.
Subclinical data
Subclinical data was derived from the large US pathology study, Pathobiological Determinants of Atherosclerosis in Youth (PDAY) (10), for which arteries from 1,532 persons were collected during the years 1987 to 1990. Arteries were retrieved from persons aged 15–34 years who had died from external causes. The types of lesions in the right coronary artery were quantified as well as their extent, measured as the total percentage of intimal surface area involved. Data were evaluated in 5-year age categories according to sex and ethnicity. Prevalence of lesions, prevalence of significant lesions, and mean area and their standard deviations were published for all lesions and for raised lesions. For complicated lesions, only the prevalence was published. For model calibration, we used only data on those with White ethnicity, for comparability with the KORA studies. The association of lesion extent with several risk factors was published in subsequent studies (11–14). However, these data were published only in an adjusted and aggregate form. Therefore, they were not used for calibration of the process-oriented model. Instead, they were used to juxtapose the model results with actual data.
Fitting procedure and inclusion of risk factors
For each member of the epidemiologic cohort, the process-oriented model was simulated 1,000 times. Individual levels of smoking, blood pressure, HDL-, and non-HDL cholesterol were taken into account as these factors had the most significant impact on risk in a preliminary analysis described in Web Appendix 1. The risk factors might affect atherosclerosis development at several stages. In the model, this means that any of the parameters 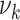 ,
, 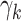 ,
, 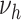 (with
(with 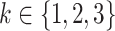 ) can be modified. For simplicity, we tested for each risk factor only one parameter at a time. Transition rates
) can be modified. For simplicity, we tested for each risk factor only one parameter at a time. Transition rates 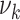 ,
, 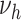 are positive and risk factor dependence is thus modeled with an exponential factor. As growth rates
are positive and risk factor dependence is thus modeled with an exponential factor. As growth rates 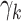 can be negative in principle, risk factors were assumed to affect growth rates linearly. Smoking was included as a binary variable. Risk factor levels were assessed only at the time point of examination but the simulations cover the entire life span. Therefore, the age-dependence of risk factors was imputed as described in Web Appendix 2.
can be negative in principle, risk factors were assumed to affect growth rates linearly. Smoking was included as a binary variable. Risk factor levels were assessed only at the time point of examination but the simulations cover the entire life span. Therefore, the age-dependence of risk factors was imputed as described in Web Appendix 2.
As a measure of goodness-of-fit to both the epidemiologic and subclinical data, a total deviance was calculated from the simulations. The goodness-of-fit related to the epidemiologic data was based on an individual likelihood. Goodness-of-fit to the subclinical data was based on all simulations, averaging over different risk factor levels. Technical details are explained in Web Appendix 3. Of note, fits with risk factor-dependent growth rates 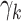 always revealed a similar or worse goodness-of-fit compared with a model variant with risk factor-dependence in an adjacent transition parameter. Thus, results on model variants with risk factor-dependent growth rates are not presented.
always revealed a similar or worse goodness-of-fit compared with a model variant with risk factor-dependence in an adjacent transition parameter. Thus, results on model variants with risk factor-dependent growth rates are not presented.
When comparing different model variants, the number of model parameters remains unchanged and differences in the deviance correspond to differences in the Akaike information criterion (AIC). A model variant is disfavored at the 95% confidence level if its deviance is more than 5.9 points above the model variant with minimal deviance (15). Due to the finite number of samples, there is some uncertainty in the deviance calculation (see Web Appendix 4). To benchmark the process-oriented model, a traditional descriptive model was used (see Web Appendix 5). The process-oriented model provides a link between the inter-individual heterogeneity in the extent of lesions, and the inter-individual heterogeneity in risk. This enables calculation of so-called conditional hazard ratios (HRs) (see Web Appendix 6).
Penalized cubic splines were applied to indicate age dependencies inherent in the data. Analysis with the process-oriented model relies on a purpose-built C++ (https://isocpp.org/) software (8). The preliminary and comparative descriptive analyses were performed with MATLAB R2020a (MathWorks, Natick, Massachusetts).
RESULTS
Studies
The final sample size of the epidemiologic data consists of 8,499 participants with 132,268 person-years and 454 incident cases of MI up to age 80 years. The mean follow-up for MI was 17.7 years and 14.4 years within the S3 and S4 study, respectively. Sex-stratified, age-dependent cohort means and standard deviations of systolic blood pressure, HDL cholesterol, and non-HDL cholesterol are provided in Web Table 1 (available at https://doi.org/10.1093/aje/kwac038). Prevalence of smoking, hypertension, and unfavorable levels of HDL and non-HDL cholesterol levels are shown in Table 1 together with the corresponding values from the PDAY study. In the subsample younger than 35 years, prevalence of most risk factors in the KORA study is similar to PDAY. However, there is a notable difference in the prevalence of unfavorable HDL levels (6.8% vs 19%).
Table 1.
Prevalence of Risk Factors in the Cooperative Health Research in the Region of Augsburg Surveys (Germany, 1994–2001) and the Pathobiological Determinants of Atherosclerosis in Youth Study (United States, 1987–1990)
| Study Group | Active Smoking, % | Hypertension, % |
Unfavorable HDL
(<0.91 mmol/L) a , % |
Unfavorable Non-HDL
( 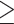 4.14 mmol/L)
a
, %
4.14 mmol/L)
a
, %
|
|---|---|---|---|---|
| KORA | 24 | 39 | 7.9 | 57 |
| KORA, age <35 yearsb | 33 | 11 | 6.8 | 34 |
| PDAY | 44 | 16 | 19 | 28 |
Abbreviation: HDL, high-density lipoprotein; KORA, Cooperative Health Research in the Region of Augsburg; PDAY, Pathobiological Determinants of Atherosclerosis in Youth.
a Thresholds for lipid levels were adapted from McGill et al. (14).
b For direct comparison, KORA results are also shown restricted to ages below 35 years, the maximum age in the PDAY study.
Model results
Smoking.
Risk factors were successively included in the model, starting with smoking. Different model variants were fitted; for each variant a specific model parameter was assumed to change during the active smoking period. Resulting deviances are shown in Table 2 for variants with smoking-dependent transition rates 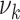 (
(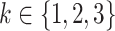 ) and
) and 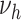 . Based on AIC, the only valid models were those with a change in the late disease stages, such as formation (
. Based on AIC, the only valid models were those with a change in the late disease stages, such as formation (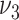 ) of complicated lesions and occlusion at the site of the plaque (
) of complicated lesions and occlusion at the site of the plaque (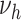 ). The differences in the deviance can be understood by the fact that in the process-oriented model, the age-trend of the risk is directly related to the parameter affected by smoking. For an effect on a late stage of an already existing disease, the impact on risk is fast. On the other hand, the increase in risk may be lagged by decades after an increase in early fatty streaks. This is illustrated in Web Figure 1.
). The differences in the deviance can be understood by the fact that in the process-oriented model, the age-trend of the risk is directly related to the parameter affected by smoking. For an effect on a late stage of an already existing disease, the impact on risk is fast. On the other hand, the increase in risk may be lagged by decades after an increase in early fatty streaks. This is illustrated in Web Figure 1.
Table 2.
Total Deviance Improvements for Various Model Variants Compared With a Model Without Risk Factors Included
| Men | Women | |||||
|---|---|---|---|---|---|---|
| Parameter a | + Smoking b | + Blood Pressure b , c | + Lipid Levels b , c | + Smoking b | + Blood Pressure b , c | + Lipid Levels b , c |
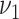
|
13.4 | 65.7 | 86.2 | 1.3 | 61.8 | 81.6 |
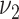
|
5.7 | 57.1 | 81.1 | 11.5 | 45.9 | 77.8 |
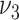
|
41.5 | 68.0d | 88.8 | 25.0 | 72.9d | 82.8 |
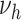
|
38.7d | 60.7 | 88.3 | 30.9d | 66.2 | 83.5 |
a See Figure 1 for definition of the parameters.
b In each row, the deviance improvement is shown for a model with the risk factor acting only on the specific model parameter.
c For each column from left to right, an additional risk factor was taken into account in the model, building on the preferred model of the previous column.
d Preferred model.
The model fit implies that smoking affects mainly a late disease stage. Therefore, there should be no substantial difference in the extent of fatty streaks and raised lesions between smokers and nonsmokers. Indeed, no such difference was observed in actual data (12). This is illustrated in Figure 3A
and 3E. The figure refers only to men, but the same conclusions could be drawn for women. Other model variants are excluded. Variants of the process-oriented model with smoking associated with first lesion formation 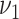 (light gray bars) show substantial differences to the actual extent of fatty streaks, and variants with smoking associated with the transition rate to raised lesions
(light gray bars) show substantial differences to the actual extent of fatty streaks, and variants with smoking associated with the transition rate to raised lesions 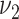 (medium gray bars) substantial differences to the actual extent of raised lesions. Therefore, these variants are excluded by poor model fit to the epidemiologic data and, independently, as they predict a substantial association between smoking and the extent of early lesions. Model variants
(medium gray bars) substantial differences to the actual extent of raised lesions. Therefore, these variants are excluded by poor model fit to the epidemiologic data and, independently, as they predict a substantial association between smoking and the extent of early lesions. Model variants 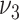 and
and 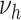 differ only in their prediction on complicated lesions. However, there is only limited information on complicated lesions in the PDAY study, which is thus not shown in Figure 3. Therefore, we evaluated smoking associated with
differ only in their prediction on complicated lesions. However, there is only limited information on complicated lesions in the PDAY study, which is thus not shown in Figure 3. Therefore, we evaluated smoking associated with 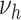 , corresponding to an effect of smoking on thrombosis.
, corresponding to an effect of smoking on thrombosis.
Figure 3.

Observed and modeled extent of fatty streaks and raised lesions in men by age and risk factor levels. Horizontal lines show observations: Adjusted estimates (solid lines) and standard errors (dotted lines) from the US Pathobiological Determinants of Atherosclerosis in Youth (PDAY) autopsy study for smoking, cholesterol (12) and hypertension (13). The bars show the corresponding results from different model variants. In each variant, one model parameter is associated with the relevant risk factor. Groupings correspond to age categories in years, either ages 15–24 and 25–34 or ages 20–24, 25–29, and 30–34. The panels show the relationships of: A) smoking to fatty streak area; B) hypertension to fatty streak area; C) high-density lipoprotein (HDL) cholesterol to fatty streak area; D) non-HDL cholesterol to fatty streak area; E) smoking to raised lesion area; F) hypertension to raised lesion area; G) HDL cholesterol to raised lesion area; H) non-HDL cholesterol to raised lesion area. For definition of the parameters see Figure 1. Fatty streaks and raised lesions do not depend on risk factor levels for model variants 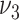 and
and 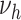 . Thus variant
. Thus variant 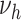 is not shown. Of note, the presented PDAY results were not used for model calibration, which is based solely on myocardial incidence data (9) and risk factor–agnostic lesion data (10), restricted to White ethnicity. NT, normotensive; HT, hypertensive; L, lower tertile; M, mid tertile; H, upper tertile.
is not shown. Of note, the presented PDAY results were not used for model calibration, which is based solely on myocardial incidence data (9) and risk factor–agnostic lesion data (10), restricted to White ethnicity. NT, normotensive; HT, hypertensive; L, lower tertile; M, mid tertile; H, upper tertile.
According to our best estimates, 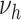 was 4.5-fold increased for male and even 11-fold increased for female smokers compared with nonsmokers. As
was 4.5-fold increased for male and even 11-fold increased for female smokers compared with nonsmokers. As 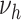 parameterizes the very last process, this fold increase directly translates to a fold increase in the individual hazard, which is illustrated with a dotted line in the left panel of Figure 4. Despite this large increase in individual risk, a lower HR is observed at the population level; see the dashed line in Figure 4. Within the group of smokers, more infarctions occur, leading to a stronger selection of individuals without extensive complicated lesions. This leads to an attenuation of the marginal hazard, and this attenuation is stronger in smokers and increases with age.
parameterizes the very last process, this fold increase directly translates to a fold increase in the individual hazard, which is illustrated with a dotted line in the left panel of Figure 4. Despite this large increase in individual risk, a lower HR is observed at the population level; see the dashed line in Figure 4. Within the group of smokers, more infarctions occur, leading to a stronger selection of individuals without extensive complicated lesions. This leads to an attenuation of the marginal hazard, and this attenuation is stronger in smokers and increases with age.
Figure 4.

Hazard ratios (HRs) for certain levels of risk factors in men compared with a life-long nonsmoker with age-related mean blood pressure and lipid levels. Dotted and dashed lines correspond to results of the preferred process-oriented model variant. The dotted lines represent the view of an average individual with enhanced risk factor. The dashed lines represent marginal HRs, which compare groups with different risk factor levels. Only the marginal HRs can be estimated with traditional descriptive analysis. Marginal HRs decrease with age as the most susceptible individuals in the high-risk group are most likely to suffer myocardial infarction, thus successively dropping out. For comparison, the result of a traditional descriptive model is shown, together with 95% confidence intervals. A) HR for smoking initiation at age 18 years. B) HR for a blood pressure increase by 10 mm Hg. C) HR for an increase in non–high-density lipoprotein cholesterol by 1 mmol/L.
In traditional descriptive analyses, selection effects are not accounted for, and only marginal HRs can be derived. Assuming age-independence, the descriptive model, equation 1 in the Web Appendix 1, yielded HRs of 2.3 for men and 3.1 for women. The age dependence was analyzed by a spline and is illustrated with solid lines in Figure 4A. As expected, it roughly agrees with the modeled marginal HR.
Therefore, while it is known that population-based marginal HRs underestimate individual folds in risk (16), our analysis shows that the underestimation of the effect of smoking can be quite strong.
Blood pressure.
Next, blood pressure is included in the model in addition to smoking. One at a time, logarithms of 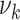 (
(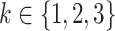 ) and
) and 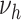 were tested for linear dependence on systolic blood pressure and results are again shown in Table 2. For both, men and women, the best fit was achieved for blood pressure affecting the transition rate to complicated lesions,
were tested for linear dependence on systolic blood pressure and results are again shown in Table 2. For both, men and women, the best fit was achieved for blood pressure affecting the transition rate to complicated lesions, 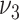 , with deviance improvements of 29.3 for men and 42.0 for women.
, with deviance improvements of 29.3 for men and 42.0 for women.
Comparison with the actual subclinical data is depicted in Figure 3B and 3F. In the PDAY study (13) no association of hypertension with the extent of fatty streaks was observed in men, and in women hypertension was even associated with a reduction in fatty streaks (not shown). However, hypertension was associated with a significant increase of raised lesions for both sexes, for ages above 25 years. Clearly, the model variant with blood pressure associated only with 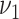 (light gray) is in conflict with these observations. The model variant with blood pressure affecting
(light gray) is in conflict with these observations. The model variant with blood pressure affecting 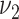 , the transition rate to raised lesions, can be expected to qualitatively lead to the observed behavior: increase of the extent of raised lesions, and even some decrease of fatty streaks by a faster transition to raised lesions. However, transition to and growth of raised lesions are more rapid in the model compared with other transition and growth rates. This made necessary a strong increase of
, the transition rate to raised lesions, can be expected to qualitatively lead to the observed behavior: increase of the extent of raised lesions, and even some decrease of fatty streaks by a faster transition to raised lesions. However, transition to and growth of raised lesions are more rapid in the model compared with other transition and growth rates. This made necessary a strong increase of 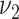 if this was the only mechanism leading to the observed higher risk in hypertensive patients, associated with an unrealistically large extent of raised lesions (medium gray). In summary, none of the tested mechanisms alone can account at the same time for the observed relations of lesion extent and risk with blood pressure. Association of risk factors with several parameters, however, is beyond the scope of the present study. Here we evaluated the variant with affected
if this was the only mechanism leading to the observed higher risk in hypertensive patients, associated with an unrealistically large extent of raised lesions (medium gray). In summary, none of the tested mechanisms alone can account at the same time for the observed relations of lesion extent and risk with blood pressure. Association of risk factors with several parameters, however, is beyond the scope of the present study. Here we evaluated the variant with affected 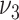 as it led to the best description of risk.
as it led to the best description of risk.
Similar to the HRs for smoking, the marginal HR for increased blood pressure decreases with age (Figure 4B), while the mean conditional HR is practically stable. The descriptive model does barely show a decrease with age but is consistent with the marginal HR derived from the process-oriented model.
Lipid levels.
For simplicity, HDL and non-HDL cholesterol were assumed to affect the same process of disease development. As before, fits were performed modeling the logarithms of the parameters 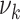 (
(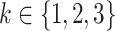 ),
), 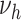 as a linear function of HDL and non-HDL cholesterol levels.
as a linear function of HDL and non-HDL cholesterol levels.
Resulting deviances are again presented in Table 2. The total deviance was reduced by up to 21 points for men and 11 for women but no clear pattern emerged from this analysis.
Comparison with the actual subclinical data is depicted in Figure 3C, 3D, 3G, and 3H. In the PDAY study, associations have been observed between HDL and non-HDL cholesterol to the extent of fatty streaks and raised lesions (12). Clearly, the model simulations with cholesterol-dependent rate of first lesion initiation 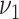 mirrors this observation best.
mirrors this observation best.
As can be seen in Figure 4C, the difference between marginal and mean conditional HR is minor. Reasons include the smaller effect size and the modification of an early disease process.
All risk factors.
Best estimates of all parameters are shown in Table 3 together with modeled risk factors dependencies. For example, for men 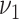 was estimated to decrease by 26% for each mmol/L increase of HDL and to increase by 23% for each mmol/l increase of non-HDL cholesterol. Best estimates for risk factor–independent model parameters are mostly similar to earlier results with the process-oriented model (8). Only for
was estimated to decrease by 26% for each mmol/L increase of HDL and to increase by 23% for each mmol/l increase of non-HDL cholesterol. Best estimates for risk factor–independent model parameters are mostly similar to earlier results with the process-oriented model (8). Only for 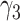 for men, the present best estimate
for men, the present best estimate 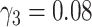 does not agree with earlier results,
does not agree with earlier results, 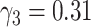 (95% confidence interval: 0.11, [no upper bound could be determined]). With the process-oriented model, inclusion of risk factors improved goodness-of-fit to the epidemiologic data as much as or even more than with a descriptive model, see Web Table 2.
(95% confidence interval: 0.11, [no upper bound could be determined]). With the process-oriented model, inclusion of risk factors improved goodness-of-fit to the epidemiologic data as much as or even more than with a descriptive model, see Web Table 2.
Table 3.
Maximum Likelihood Estimates From the Likelihood Profile for the Final Fit
| Men | Women | |||
|---|---|---|---|---|
| Parameter a | Estimate | CI | Estimate | CI |
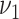 b
, 1.4 mmol/l HDL, 4.0 mmol/l non-HDL
b
, 1.4 mmol/l HDL, 4.0 mmol/l non-HDL |
0.040 | 0.034, 0.046 | 0.054 | 0.035, 0.081 |
| Factorc for HDL cholesterol | 0.74 | 0.42, 1.14 | 0.62 | 0.35, 1.03 |
| Factorc for non-HDL cholesterol | 1.23 | 1.11, 1.43 | 1.29 | 0.97, 1.63 |
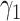 b
b
|
0.11 | 0.10, 0.13 | 0.11 | 0.08, 0.13 |
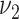 b
b
|
1.5 | 1.1, 2.0 | 0.62 | 0.32, 1.5 |
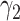 b
b
|
0.32 | 0.22, 0.49 | 0.57 | 0.16, 1.0d |
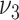 b
, 130 mm Hg
b
, 130 mm Hg |
0.051 | 0.025, 0.10 | 0.023 | 0.014, 0.11 |
| Factore for SBP | 1.22 | 1.12, 1.36 | 1.37 | 1.23, 1.58 |
| SBP reduction by treatmentf | 2.0d | 0.9, 2.0d | 2.0d | 1.4, 2.0d |
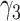 b
b
|
0.082 | 0.02, 1.0d | 0.15 | 0.06, 0.25 |
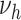 b
, nonsmoking
b
, nonsmoking |
0.30 | 0.04, 1.3 | 0.15 | 0.06, 0.40 |
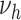 b
, during smoking period
b
, during smoking period |
1.6 | 0.09, 5.6 | 3.4 | 0.34, 21 |
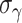 g
g
|
0.51 | 0.36, 0.67 | 0.57 | 0.36, 1.2 |
Abbreviations: CI, confidence interval; HDL, high-density lipoprotein, SBP, systolic blood pressure.
a See Figure 1 for definition of the parameters.
b Rates are given per year.
c Relative increase per mmol/L.
d Maximum value permitted in the fit.
e Relative increase per 10 mm Hg.
f As multiple of the standard deviation of the age-specific distribution of SBP in the study group.
g Individual growth rates are assumed to be normally distributed with mean 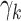 (with
(with 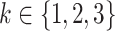 and standard deviation
and standard deviation 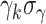 ).
).
DISCUSSION
In this study, we have extended a process-oriented mathematical model of atherosclerosis development and progression to include individual risk factor levels. We have incorporated data from a well-characterized population-based cohort as well as subclinical coronary artery data from a younger autopsy cohort. The model performed well, compared with traditional descriptive models, and allowed for investigating the biological role of individual risk factors in the pathway of atherosclerosis progression. We thus conclude that the newly proposed statistical model is a helpful addition to traditional cardiovascular disease (CVD) risk models and can contribute to a more thorough understanding and individualized risk assessment.
Regarding the biological pathways of risk factor effects, our results corroborate and extend the current literature. Effects of smoking on atherosclerosis progression are present at all phases, starting already at endothelial dysfunction (17) and continuing through promotion of unstable plaques by inducing oxidative stress-related activation of extracellular matrix proteins (18). However, evidence points to a major thrombogenic effect of smoking (19): Cigarette smoke induces expression of tissue factor in endothelial cells, thus activating the coagulation cascade and increasing the risk for vascular clotting (20). Smokers have an elevated immature platelet fraction, which promotes thrombus formation (21). Furthermore, smoking stimulates platelet activation and aggregation and is associated with increased fibrinogen and decreased fibrinolytic capacity, predisposing for formation and propagation of thrombi (22, 23). Our model is consistent with such a thrombogenic effect by providing strong statistical evidence that the major impact of smoking indeed lies within the last stages of disease. Smoking was associated with HRs of about 2.3 for men and 3.1 for women (descriptive analysis). Findings from the population-based Tromsø study showed incidence rate ratios for MI of 2.2 for men and 2.1 for women for daily smokers (24). A large study from the United States, based on the National Health Interview Survey, found HRs for death of ischemic heart disease of 3.2 for men and 3.5 for women (25). In a large case-control study within INTERHEART, current smoking was associated with nonfatal MI with an odds ratio of 2.95. Of note, this was already reduced for former smokers to 1.87 1–3 years after quitting (26). A meta-analysis calculated the relative risk of smoking for MI to be 2.88, which rapidly decreased within a few years after cessation to finally yield 1.17 (27). Thus, our findings are consistent with the literature. The model sheds some light on the rapid and almost complete decrease of CVD risk right after smoking cessation: Acute risk of plaque rupture and thrombus formation is reduced immediately and other biological effects on lesions are minor.
Hypertension contributes to atherosclerotic progression by inducing hemodynamic instability and a proinflammatory environment. The contribution of hypertension to atherosclerosis development is multifactorial and covers multiple phases, from initial endothelial dysfunction to coronary vasospasm and vessel constriction (28). Our findings indicate that hypertension is mainly involved in the formation of unstable lesions. This could be due to modulation of blood flow mechanics, as local flow changes induce lower wall shear stress, increasing the risk of transformation from stable to unstable plaques (29, 30). Moreover, hypertension induces up-regulation of matrix-metalloproteinases, such as MMP-9, which catalyze the transition from stable lesions to advanced, complex lesions with necrotic cores (31). A recent animal study showed that prolonged exposure to hypertension caused complex lesions, including intraplaque hemorrhage, in rabbits with both hypertension and hypercholesterolemia; however, no rupture or aneurysms occurred in the aorta (32). This is in line with our results, which exclude a major association of hypertension with 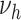 . On a side note, the effect of hypertension on coronary artery disease and MI also includes the pathway of vascular remodeling, left ventricular hypertrophy, and reduced coronary flow reserve; however, this pathway is not covered by our presented model.
. On a side note, the effect of hypertension on coronary artery disease and MI also includes the pathway of vascular remodeling, left ventricular hypertrophy, and reduced coronary flow reserve; however, this pathway is not covered by our presented model.
The initial process in plaque formation is the recruitment of oxidized low-density lipoprotein in the subendothelial matrix, thus initiating fatty streak formation (33). According to our model, this increased fatty streak formation is sufficient to explain the association of lipids with MI risk. However, lipid profile is also related to plaque composition and stability (34), and lowering lipid levels leads to reductions in atheroma volumes (35). The relevance of these associations with regard to MI risk remains unclear.
Results have to be interpreted within the context of the model’s limitations. Unavoidably, the applied model represents a strong simplification of actual biological processes. The model was fitted against subclinical data for persons up to age 35 years, and against MI incidence data. Complicated lesions mostly arise after this age, and their development is thus inferred indirectly by the age-dependence of MI risk. The estimated effects of risk factors on single model parameters (see Table 3) are likely overestimated, as effects on growth parameters and other, subordinate effects are likely to contribute to risk but were not taken into account. Likewise, possible interactions between risk factors were not investigated here. For example, in addition to the direct effect of smoking on risk, smoking may also increase blood pressure and affect lipid levels. The total harm of smoking may thus be larger than the association presented. Furthermore, data of the present analysis originated from 2 different studies, potentially leading to increased variability and uncertainty in the model simulations. A single study covering both subclinical and epidemiologic data would be desirable; however, to our knowledge no such study is available to date.
To summarize, we have shown that process-oriented mathematical modeling of atherosclerosis development and progression provides novel insights into risk estimation. The proposed model has substantial potential for epidemiologic application. First, a strength of the presented approach is the inference on pathological mechanisms from the combination of population-based epidemiologic and subclinical coronary artery data in a process-oriented model. In order to administer effective treatment (e.g., lipid-lowering or antihypertensive medication), it is important to know which main biological process, and corresponding time point in the chronological sequence of atherosclerosis, is addressed. This helps with more accurate CVD risk assessment before and after treatment initiation, and can thus guide further interventions. Moreover, this information can be used to improve risk communication to the individual patient. Our model results showed this most strikingly for the effect of smoking, where the immediate benefits of smoking cessation can be communicated quite concretely and can thus potentially increase adherence to intervention. Second, model results may be more reliable compared with the usual analysis of incidence data only, which is usually based on simplistic statistical assumptions without considering underlying biological processes. For example, this might be relevant for extrapolation of risk to young ages. Our approach adds an estimate of heterogeneity of individual risk, which is not accessible by incidence data alone. In a heterogeneous population, those individuals with higher risk tend to suffer MI earlier. This selection effect biases comparisons of risks in different groups (16), such as smokers versus nonsmokers. Indeed, our model revealed that individual risk of smoking-induced MI is strongly underestimated if heterogeneity is not taken into account. Therefore, use of the process-oriented model will help with more individualized CVD risk prediction and identification of those individual risk factors for which intervention might be most promising. Third, from a technical point of view, the model is suitable for extension to more complex risk factor effects, other risk factors, and additional data, such as imaging data. Taken together, the process-oriented model in atherosclerosis research is a promising tool to improve individualized CVD risk assessment and thus promote CVD prevention.
Supplementary Material
ACKNOWLEDGMENTS
Author affiliations: Institute of Radiation Medicine, Helmholtz Zentrum München German Research Center for Environmental Health (GmbH), Neuherberg, Germany (Cristoforo Simonetto, Jan Christian Kaiser); Institute of Epidemiology, Helmholtz Zentrum München German Research Center for Environmental Health (GmbH), Neuherberg, Germany (Margit Heier, Annette Peters, Susanne Rospleszcz); KORA Study Centre, University Hospital of Augsburg, Augsburg, Germany (Margit Heier); Institute for Medical Information Processing, Biometry and Epidemiology, Ludwig-Maximilians-Universität München, Munich, Germany (Annette Peters, Susanne Rospleszcz); and German Center for Cardiovascular Disease (DZHK), Partner Site Munich Heart Alliance, Munich, Germany (Annette Peters, Susanne Rospleszcz).
J.C.K and S.R. contributed equally to this work.
This project has received funding from the Euratom research and training programme 2014–2018 under grant agreement No. 755523 (MEDIRAD).
The informed consent given by Cooperative Health Research in the Region of Augsburg (KORA) study participants does not cover data posting in public databases. However, data are available upon request from KORA-gen (http://epi.helmholtz-muenchen.de/kora-gen/) by means of a project agreement. Requests should be sent to kora.passt@helmholtz-muenchen.de and are subject to approval by the KORA Board. The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study data used in this work are available as published.
Special thanks go to Dr. Noemi Castelletti for fruitful discussions, proofreading of the manuscript, and help in the preparation of graphics. The KORA study was initiated and financed by the Helmholtz Zentrum München–German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ.
Conflict of interest: none declared.
REFERENCES
- 1. Moolgavkar SH, Knudson AG. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 1981;66(6):1037–1052. [DOI] [PubMed] [Google Scholar]
- 2. Luebeck EG, Moolgavkar SH. Multistage carcinogenesis and the incidence of colorectal cancer. Proc Natl Acad Sci U S A. 2002;99(23):15095–15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meza R, Hazelton WD, Colditz GA, et al. Analysis of lung cancer incidence in the Nurses’ Health and the Health Professionals’ Follow-up Studies using a multistage carcinogenesis model. Cancer Causes Control. 2008;19(3):317–328. [DOI] [PubMed] [Google Scholar]
- 4. Peto R. Epidemiology, multistage models, and short-term mutagenicity tests. Int J Epidemiol. 2016;45(3):621–637. [DOI] [PubMed] [Google Scholar]
- 5. Simonetto C, Azizova TV, Barjaktarovic Z, et al. A mechanistic model for atherosclerosis and its application to the cohort of Mayak workers. PLoS One. 2017;12(4):e0175386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rühm W, Eidemüller M, Kaiser JC. Biologically-based mechanistic models of radiation-related carcinogenesis applied to epidemiological data. Int J Radiat Biol. 2017;93(10):1093–1117. [DOI] [PubMed] [Google Scholar]
- 7. Richardson DB. Multistage modeling of leukemia in benzene workers: a simple approach to fitting the 2-stage clonal expansion model. Am J Epidemiol. 2009;169(1):78–85. [DOI] [PubMed] [Google Scholar]
- 8. Simonetto C, Rospleszcz S, Heier M, et al. Simulating the dynamics of atherosclerosis to the incidence of myocardial infarction, applied to the KORA population. Stat Med. 2021;40(14):3299–3312. [DOI] [PubMed] [Google Scholar]
- 9. Löwel H, Döring A, Schneider A, et al. The MONICA Augsburg surveys—basis for prospective cohort studies. Gesundheitswesen. 2005;67(suppl 1):S13–S18. [DOI] [PubMed] [Google Scholar]
- 10. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Natural history of aortic and coronary atherosclerotic lesions in youth. Findings from the PDAY study. Arterioscler Thromb. 1993;13(9):1291–1298. [DOI] [PubMed] [Google Scholar]
- 11. McGill HC, McMahan CA. Determinants of atherosclerosis in the young. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Am J Cardiol. 1998;82(10B):30T–36T. [DOI] [PubMed] [Google Scholar]
- 12. McGill HC, McMahan CA, Malcom GT, et al. Effects of serum lipoproteins and smoking on atherosclerosis in young men and women. The PDAY Research Group. Pathobiological Determinants of Atherosclerosis in Youth. Arterioscler Thromb Vasc Biol. 1997;17(1):95–106. [DOI] [PubMed] [Google Scholar]
- 13. McGill HC, McMahan CA, Tracy RE, et al. Relation of a postmortem renal index of hypertension to atherosclerosis and coronary artery size in young men and women. Arterioscler Thromb Vasc Biol. 1998;18(7):1108–1118. [DOI] [PubMed] [Google Scholar]
- 14. McGill HC, McMahan CA, Zieske AW, et al. Associations of coronary heart disease risk factors with the intermediate lesion of atherosclerosis in youth. The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Circulation. 2000;102:374–379. [DOI] [PubMed] [Google Scholar]
- 15. Walsh L. A short review of model selection techniques for radiation epidemiology. Radiat Environ Biophys. 2007;46(3):205–213. [DOI] [PubMed] [Google Scholar]
- 16. Aalen OO. Effects of frailty in survival analysis. Stat Methods Med Res. 1994;3(3):227–243. [DOI] [PubMed] [Google Scholar]
- 17. Zeiher AM, Schaechinger V, Minners J. Long-term cigarette smoking impairs endothelium-dependent coronary arterial vasodilator function. Circulation. 1995;92(5):1094–1100. [DOI] [PubMed] [Google Scholar]
- 18. Ruddy JM, Ikonomidis JS, Jones JA. Multidimensional contribution of matrix metalloproteinases to atherosclerotic plaque vulnerability: multiple mechanisms of inhibition to promote stability. J Vasc Res. 2016;53(1–2):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bøttcher M, Falk E. Pathology of the coronary arteries in smokers and non-smokers. J Cardiovasc Risk. 1999;6(5):299–302. [DOI] [PubMed] [Google Scholar]
- 20. Holy EW, Tanner FC. Tissue factor in cardiovascular disease. In: Cardiovascular Pharmacology—Heart and Circulation. Amsterdam, the Netherlands: Elsevier; 2010:259–292. [DOI] [PubMed] [Google Scholar]
- 21. Nardin M, Verdoia M, Negro F, et al. Impact of active smoking on the immature platelet fraction and its relationship with the extent of coronary artery disease. Eur J Clin Invest. 2020;50(2):e13181. [DOI] [PubMed] [Google Scholar]
- 22. Togna AR, Latina V, Orlando R, et al. Cigarette smoke inhibits adenine nucleotide hydrolysis by human platelets. Platelets. 2008;19(7):537–542. [DOI] [PubMed] [Google Scholar]
- 23. Barua RS, Sy F, Sundararajan S, et al. Effects of cigarette smoke exposure on clot dynamics and fibrin structure. Arterioscler Thromb Vasc Biol. 2010;30(1):75–79. [DOI] [PubMed] [Google Scholar]
- 24. Albrektsen G, Heuch I, Løchen M, et al. Risk of incident myocardial infarction by gender: interactions with serum lipids, blood pressure and smoking. The Tromsø study 1979–2012. Atherosclerosis. 2017;261:52–59. [DOI] [PubMed] [Google Scholar]
- 25. Jha P, Ramasundarahettige C, Landsman V, et al. 21st-century hazards of smoking and benefits of cessation in the United States. N Engl J Med. 2013;368(4):341–350. [DOI] [PubMed] [Google Scholar]
- 26. Teo KK, Ounpuu S, Hawken S, et al. Tobacco use and risk of myocardial infarction in 52 countries in the INTERHEART study: a case-control study. Lancet. 2006;368(9536):647–658. [DOI] [PubMed] [Google Scholar]
- 27. Lightwood JM, Glantz SA. Short-term economic and health benefits of smoking cessation. Circulation. 1997;96(4):1089–1096. [DOI] [PubMed] [Google Scholar]
- 28. Wal AC, Becker AE. Atherosclerotic plaque rupture—pathologic basis of plaque stability and instability. Cardiovasc Res. 1999;41(2):334–344. [DOI] [PubMed] [Google Scholar]
- 29. Vergallo R, Papafaklis MI, Yonetsu T, et al. Endothelial shear stress and coronary plaque characteristics in humans. Circ Cardiovasc Imaging. 2014;7(6):905–911. [DOI] [PubMed] [Google Scholar]
- 30. Brown AJ, Teng Z, Evans PC, et al. Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat Rev Cardiol. 2016;13(4):210–220. [DOI] [PubMed] [Google Scholar]
- 31. Wang M, Kim SH, Monticone RE, et al. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension. 2015;65(4):698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ning B, Chen Y, Waqar AB, et al. Hypertension enhances advanced atherosclerosis and induces cardiac death in watanabe heritable hyperlipidemic rabbits. Am J Pathol. 2018;188(12):2936–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(9):2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang X, Liu X, Xie Z, et al. Small HDL subclass is associated with coronary plaque stability: an optical coherence tomography study in patients with coronary artery disease. J Clin Lipidol. 2019;13(2):326–334.e2. [DOI] [PubMed] [Google Scholar]
- 35. Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078–2087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.