

Abstract
Objective.
To evaluate correlations between rucaparib exposure and selected efficacy and safety endpoints in patients with recurrent ovarian carcinoma using pooled data from Study 10 and ARIEL2.
Methods.
Efficacy analyses were limited to patients with carcinomas harboring a deleterious BRCA1 or BRCA2 mutation who had received ≥2 prior lines of chemotherapy. Safety was evaluated in all patients who received ≥1 rucaparib dose. Steady-state daily area under the concentration-time curve (AUCss) and maximum concentration (Cmax,ss) for rucaparib were calculated for each patient and averaged by actual dose received over time (AUCavg,ss and Cmax,avg,ss) using a previously developed population pharmacokinetic model.
Results.
Rucaparib exposure was dose-proportional and not associated with baseline patient weight. In the exposure-efficacy analyses (n = 121), AUCavg,ss was positively associated with independent radiology review-assessed RECIST response in the subgroup of patients with platinum-sensitive recurrent disease (n = 75, p = 0.017). In the exposure-safety analyses (n = 393, 40 mg once daily to 840 mg twice daily [BID] starting doses), most patients received a 600 mg BID rucaparib starting dose, with 27% and 21% receiving 1 or ≥2 dose reductions, respectively. Cmax,ss was significantly correlated with grade ≥2 serum creatinine increase, grade ≥3 alanine transaminase/aspartate transaminase increase, platelet decrease, fatigue/asthenia, and maximal hemoglobin decrease (p < 0.05).
Conclusion.
The exposure-response analyses provide support for the approved starting dose of rucaparib 600 mg BID for maximum clinical benefit with subsequent dose modification only following the occurrence of a treatment-emergent adverse event in patients with BRCA-mutated recurrent ovarian carcinoma.
Keywords: Efficacy, Exposure, Ovarian carcinoma, Pharmacokinetics, Rucaparib, Safety
1. Introduction
Ovarian cancer is the fifth leading cause of cancer-related death in women in the United States and the European Union and the eighth leading cause of cancer-related death in women worldwide [1]. Approximately half of all high-grade ovarian carcinomas (HGOCs), including fallopian tube and primary peritoneal carcinomas, have homologous recombination deficiency (HRD), with 18% estimated to harbor a germline BRCA1 or BRCA2 (BRCA) mutation, 7% a somatic BRCA mutation, and 20% a mutation in, or epigenetic silencing of, another homologous recombination gene [2,3].
Rucaparib is a potent, oral small molecule inhibitor of poly(ADP-ribose) polymerase (PARP) enzymes, including PARP1, PARP2, and PARP3. In cancer cells with HRD, defects in the homologous recombination repair pathway combine with rucaparib-induced enzymatic inhibition of PARP proteins in an interaction known as synthetic lethality, resulting in the accumulation of DNA damage, stalled replication forks, and cell death [4–6]. The efficacy and safety of rucaparib in clinical studies of treatment, including Study 10 (NCT01482715) and ARIEL2 (NCT01891344), as well as maintenance therapy in ARIEL3 (NCT01968213), have resulted in the approval of rucaparib in the United States and Europe for treatment and maintenance therapy of patients with recurrent HGOC [7,8]. The approved starting dose of rucaparib is 600 mg twice daily (BID) for all patients to allow for maximum clinical benefit, with dose modifications as needed to manage treatment-related adverse effects. In this study, we used a previously published population pharmacokinetic (PK) model [9] and pooled data from Study 10 and ARIEL2 to evaluate the correlations of rucaparib exposure with efficacy and safety and better understand the covariates influencing rucaparib PK variability in the treatment setting for patients with recurrent ovarian carcinoma.
2. Methods
In Study 10 Part 1, patients with solid tumors received oral rucaparib 40–500 mg once daily (QD) or 240–840 mg BID. In Study 10 Part 2A and ARIEL2 Parts 1 and 2, patients with relapsed HGOC or fallopian tube or primary peritoneal carcinoma received rucaparib 600 mg BID. A population PK model [9] was developed based on the data from Study A4991014 (NCT01009190), Study 10, and ARIEL2 (Table S1). Exposure variables were calculated from individual PK parameter estimates from the population PK analysis, nominal dose and frequency, and actual dosing information. Steady-state daily (0–24 h) area under the concentration-time curve (AUCss) and maximum concentration (Cmax, ss) for rucaparib were calculated for each patient, and these PK parameters were averaged by the actual dose received over time (AUCavg,ss and Cmax,avg,ss). For the exposure-efficacy analysis, AUCavg,ss up to the confirmed response (or the last dose if there was no confirmed response) was used. For the exposure-safety analysis, Cmax,avg,ss up to the last dose was used.
2.1. Exposure-efficacy analysis population
The US Food and Drug Administration approved rucaparib for the treatment of patients with ovarian carcinoma associated with deleterious germline and/or somatic BRCA mutation who have been treated with 2 or more chemotherapies based on integrated analysis of data from Study 10 and ARIEL2 [7]. We sought to better characterize the relationship between exposure and efficacy in this population, and thus the exposure-efficacy analysis population was limited to patients with ovarian carcinoma associated with a deleterious germline or somatic BRCA mutation who participated in Study 10 Parts 1 and 2A or ARIEL2 Parts 1 and 2, received at least 1 dose of rucaparib, and underwent at least 1 efficacy assessment. BRCA mutation status was determined based on results from local and central laboratory tests.
2.2. Exposure-efficacy analysis endpoints, models, and covariates
The exposure-efficacy analysis tested 7 endpoints (Table S2): confirmed investigator- and independent radiologic review (IRR)-assessed response according to Response Evaluation Criteria In Solid Tumors, version 1.1 (RECIST), composite of confirmed response by Gynecologic Cancer InterGroup (GCIG) CA-125 criteria or RECIST response, best percent change from baseline in sum of diameters of target lesions (per RECIST) and in serum cancer antigen 125 (CA-125), progression-free survival (PFS), and duration of response (DOR). PFS was analyzed in all patients in the exposure-efficacy analysis population, and DOR was analyzed in patients with a confirmed investigator-assessed RECIST response.
Endpoints with continuous data (i.e. change from baseline in sum of diameters of target lesions) were modeled using linear regression (Table S2). Endpoints with binary data (i.e. investigator-assessed response, IRR-assessed response, and confirmed response by GCIG CA-125 criteria or RECIST) were modeled using linear logistic regression. Endpoints with time-to-event data (i.e. PFS and DOR) were modeled using Cox regression.
Efficacy covariates included age (<65 vs ≥65 years), race (white vs non-white), region (North America vs Europe/other), mutation origin (germline vs somatic vs indeterminate germline or somatic BRCA mutation), BRCA gene (BRCA1 vs BRCA2), number of prior chemotherapies (2 vs ≥3), progression-free interval (PFI) following last platinum-based therapy (<6 months vs ≥6–≤12 months vs >12 months), Eastern Cooperative Oncology Group (ECOG) performance status (PS; 0 vs 1), and baseline sum of diameters of target lesions (Table S2).
In all covariate models, exposure was included in the model regardless of significance. Logistic and linear regression models tested covariates.
Cox regression models were centered at the median exposure in all ovarian cancer patients receiving 600 mg BID rucaparib; covariates were tested on the reference hazard at that median exposure. Covariates were tested in a stepwise forward search with a p < 0.05 level of significance. Only covariates that were significant in a univariate search were included in multivariate testing.
2.3. Exposure-safety analysis population
To comprehensively evaluate the safety of rucaparib in patients with ovarian carcinoma, the exposure-safety analysis population included patients with ovarian cancer in Study 10 Parts 1 and 2A or ARIEL2 Parts 1 and 2 who received at least 1 dose of rucaparib, regardless of BRCA mutation status.
2.4. Exposure-safety analysis endpoints, models, and covariates
The exposure-safety analysis tested treatment-emergent hematologic laboratory variables (grade ≥3 neutrophil decrease, grade ≥3 platelet decrease, grade ≥3 lymphocyte decrease, grade ≥3 hemoglobin decrease, maximum hemoglobin reduction from baseline), nonhematologic laboratory variables (grade ≥3 alanine aminotransferase [ALT] increase, grade ≥3 aspartate aminotransferase [AST] increase, grade ≥2 bilirubin increase, grade ≥2 creatinine increase), and other qualitative adverse events (grade ≥3 fatigue/asthenia, grade ≥3 nausea) (Table S2). All laboratory endpoints were based on standard Common Terminology Criteria for Adverse Events version 4.03 grading [10].
Maximum change from baseline in hemoglobin, a continuous endpoint, was modeled using linear regression (Table S2). The other endpoints were binary and modeled using linear logistic regression.
Safety covariates included age (<65 vs ≥65 years), race (white vs non-white), germline BRCA status (germline BRCA mutation vs BRCA wild type/unknown), number of prior chemotherapies (2 vs ≥3), ECOG PS (0 vs 1), and baseline albumin (Table S2).
As with the efficacy models, in all covariate models the exposure was included in the model regardless of significance. Logistic and linear regression models tested covariates in a stepwise forward search with a p < 0.05 level of significance. Only covariates that were significant in a univariate search were included in multivariate testing.
2.5. Concentration-QT analysis
Concentration-QT prolongation analysis was conducted using data from patients in Study 10 Part 1. QT interval was obtained from triplicate 12-lead electrocardiogram (ECG) recordings and corrected for heart rate by Fridericia’s method (QTcF). Per study design, triplicate ECG data were collected from dose-escalation cohorts (3 patients per dose level based on 3 + 3 design) at predose, and 2 and 7 h postdose on day 1 and day 15 of cycle 1 following continuous rucaparib treatment at escalating dose levels (40 mg QD to 500 mg QD, then further escalated from 240 mg BID to 840 mg BID). PK samples were collected before and after each postdose ECG measurement (1.5 and 2.5 h PK for 2 h ECG; 6 and 8 h PK for 7 h ECG) to avoid any effect of PK sampling on QT interval. The PK sample concentration at each ECG measurement was calculated by linear interpolation. A linear mixed-effects model was conducted with the calculated PK sample concentration and QTcF change from baseline (ΔQTcF). The expected ΔQTcF and confidence intervals following 600 mg rucaparib BID were calculated at the median Cmax,ss and at the upper 95th percentile of Cmax,ss based on the final model.
3. Results
3.1. Model-predicted steady-state exposure
Comprehensive PK data collected in 375 patients from Study 10 and ARIEL2 allowed us to extend upon and confirm earlier findings that the model-estimated steady-state rucaparib exposure, as measured by Cmax, avg,ss and AUCavg,ss, is dose-proportional [11].
Other studies of maintenance treatment with the PARP inhibitor niraparib in the setting of recurrent ovarian carcinoma have suggested that baseline patient body weight could guide dose selection [12]. Given these results, we evaluated whether patient weight may be associated with rucaparib exposure. Because baseline creatinine clearance was a significant covariate on rucaparib clearance [11], we accounted for the effect of baseline creatinine clearance on rucaparib clearance and analyzed the association between body weight and rucaparib exposure stratified by renal function. We found no statistically significant association between the model-predicted rucaparib exposures (Cmax,avg,ss and AUCavg,ss) and baseline body weight in patients with normal, mild or moderate renal insufficiency (Fig. 1).
Fig. 1.

Model-predicted Cmax,avg,ss (A), p > 0.05, and AUCavg,ss (B), p > 0.05 at 600 mg twice daily vs baseline weight stratified by renal function to account for the effect of baseline creatinine clearance on rucaparib apparent clearance. The points are individual model-predicted estimates of the Cmax,avg.ss or AUCavg,ss following a dose at steady state vs baseline weight. The solid blue line is a local regression smooth showing the trend of the relationship. The shaded blue region represents 5th to 95th percentile confidence interval around the trend. AUCavg,ss, average daily area under the concentration-time curve at steady state; Cmax,avg,ss, average maximum concentration at steady state.
3.2. Exposure-efficacy relationships
Of 121 patients in the exposure-efficacy analysis dataset, PK concentrations were available from 117 patients for whom steady-state exposures were estimated (Table 1). Cmax,avg,ss (standard deviation [SD]) was 1712 (756) ng/mL (range, 166–4413 ng/mL) and AUCavg,ss (SD) was 37,145 (17,140) ng/mL × h (range, 2328–99,524). A majority of patients (110/121; 90.9%) were treated with a starting dose of rucaparib 600 mg BID. Ten patients were treated at doses below 600 mg BID and 1 patient received 840 mg BID in the phase I dose-escalation part of Study 10 (Part 1). The number of patients varied for different efficacy endpoints: 114 patients were evaluated for the endpoint of investigator-assessed response and 102 for the endpoint of IRR-assessed response. Most patients were younger than 65 years old (81/121; 66.9%), white (98/121; 81.0%), and from North America (65/121; 53.7%). All had HGOC with a BRCA1 (77/121; 63.6%) or BRCA2 (44/121; 36.4%) mutation, the majority of which were germline mutations (102/121; 84.3%). Most patients had received 3 or more prior chemotherapies (76/121; 62.8%).
Table 1.
Exposure summary in the exposure-efficacy and exposure-safety analyses.a
| Variable | Exposure-efficacy analysis | Exposure-safety analysis |
|---|---|---|
|
| ||
| Patients (n) | 121 | 393 |
| PKs (n)b | 117 | 375 |
| Cmax,avg,ss, mean ng/mL | 1712 | 1839 |
| SD | 756 | 732 |
| Range | 166–4413 | 75–6254 |
| AUCavg,ss, mean ng/mL × h | 37,145 | 40,200 |
| SD | 17,140 | 16,767 |
| Range | 2328–99,524 | 690–141,157 |
AUCavg,ss, average daily area under the concentration-time curve at steady-state; Cmax,avg,ss, average maximum concentration at steady state; PK, pharmacokinetic; SD, standard deviation.
Overall exposure estimates over the full duration of treatment.
Number of patients with at least 1 evaluable PK concentration.
Patients with a confirmed IRR-assessed partial or complete RECIST response showed statistically higher exposures than patients without a confirmed response (Fig. 2A; p = 0.029). We also observed a trend of increasing response rate with increasing rucaparib exposure in a linear logistic regression analysis of IRR-assessed RECIST response (n = 102), although in that analysis the exposure-response relationship was not found to be statistically significant (Fig. 2B). We then further assessed the relationship between steady-state exposure and IRR-assessed RECIST response based on patients’ platinum sensitivity status. In the patients with PFI <6 months following their last platinum-based chemotherapy (platinum-resistant or refractory), the observed response rate was 11.1% (3/27), while the response rates in patients with PFI 6–12 months (partially platinum-sensitive) and ≥12 months (platinum-sensitive) were 53.8% (28/52) and 56.5% (13/23) respectively. No correlation between AUCavg,ss and IRR-assessed RECIST response was detectable among patients with platinum-refractory/resistant ovarian cancer (PFI <6 months) (Fig. 2C); however, a significant correlation (p = 0.017) was observed for patients with platinum-sensitive disease (PFI ≥6 months; Fig. 2D).
Fig. 2.
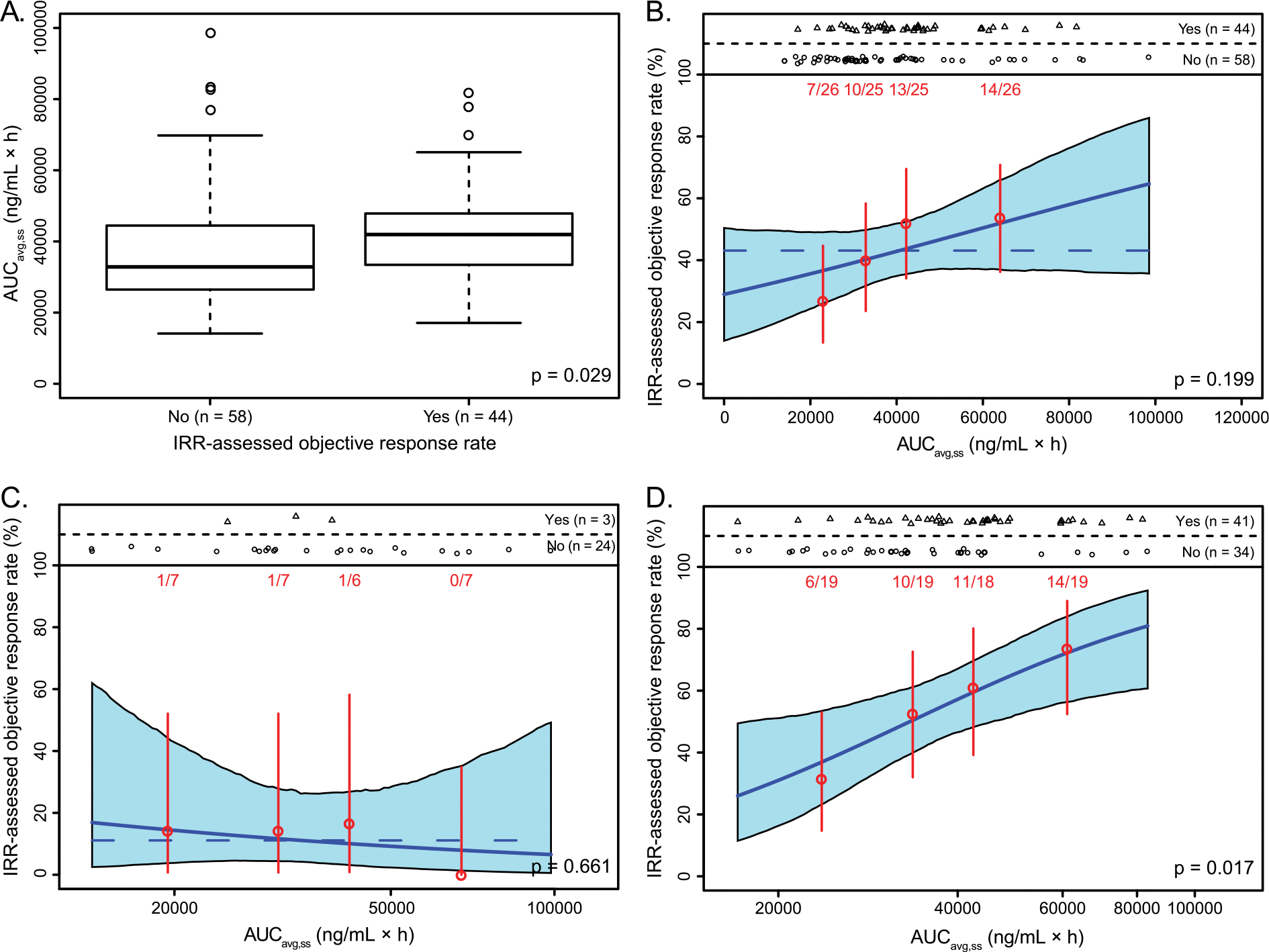
Box plots show the exposure distributions, stratified by response (A). Exposure-response relationships for IRR-assessed RECIST response in patients with a BRCA mutation were tested for overall patients (B), for patients with PFI <6 months (C), and for patients with PFI ≥6 months (D). Red points represent the mean exposure and event rates in patients stratified by exposure quartile. Vertical red bars represent the 5th to 95th percentile confidence intervals on the event rate. Event numbers (patients with event/total patients) are displayed above each vertical bar. In each plot, the solid blue line is the logistic regression model fit for that patient subset. The shaded blue region represents 5th to 95th percentile confidence interval on the predicted event rate. The horizontal blue dashed line shows the average response rate when the p value is >0.05. AUCavg,ss, average daily area under the concentration-time curve at steady state up until the relevant event; BRCA, BRCA1 or BRCA2; IRR, independent radiologic review; PFI, progression-free interval; RECIST, Response Evaluation Criteria In Solid Tumors version 1.1.
The exposure in patients with a confirmed investigator-assessed partial or complete RECIST response was similar to the exposure in patients without a confirmed response (Fig. S1A; p = 0.606). When the exposure-response relationship analysis was performed for investigator-assessed RECIST response (n = 114), no apparent correlation or trends were observed over the range of exposures (Fig. S1B). Further, no significant correlations were seen for the other tested efficacy endpoints with continuous data (i.e. change from baseline in sum of diameters of target lesions, change in CA-125, PFS or DOR) or with binary data (i.e. confirmed response by GCIG CA-125 criteria or RECIST; Table S3).
3.3. Exposure-safety relationships
Of 393 patients in the exposure-safety analysis dataset, PK parameters were determined in 375 patients (Table 1). Cmax,avg,ss (SD) was 1839 (732) ng/mL (range, 75–6254 ng/mL) and AUCavg,ss (SD) was 40,200 (16,767) ng/mL × h (range, 690–141,157). The starting doses of rucaparib ranged from 40 mg QD up to 840 mg BID, but most patients (378/393; 96.2%) received a starting dose of 600 mg BID, with 27% and 21% of patients requiring 1 and ≥2 dose reductions, respectively.
Increased incidence of adverse events with increased exposure was observed for selected safety endpoints (Table 2). Among nonhematologic endpoints evaluated, the exposure-response relationship was statistically significant for grade ≥3 ALT increase (p = 0.033, incidence rate 13.3%) and grade ≥3 AST increase (p = 0.027, incidence rate 4.0%), but not for grade ≥2 bilirubin increase (p = 0.723, incidence rate 6.4%). In addition, a statistically significant correlation was identified for grade ≥2 creatinine increase (p < 0.001, incidence rate 28.8%; Fig. 3). Among hematologic endpoints evaluated, correlations with grade ≥3 platelet decrease and maximum hemoglobin reduction were observed (Table 2). Although additional assessment suggested albumin as a significant covariate for maximum hemoglobin reduction and grade ≥3 nausea, and age (≥65 years) as a significant covariate for grade ≥2 creatinine increase (Table 2), none were considered clinically significant, as these relationships are typically observed in patients regardless of treatment [13–15].
Table 2.
Exposure-safety relationship with covariates.
| Exposure metric | Endpoint |
p value for exposure |
Significant covariates (p < 0.05)b | |
|---|---|---|---|---|
| No covariate | Adjusted for covariatesa | |||
|
| ||||
| Exposure-safety with nonhematologic variables | ||||
| Cmax,avg,ss | ALT increased, grade ≥3 | 0.033 | 0.025 | ECOG PS |
| Cmax,avg,ss | AST increased, grade ≥3 | 0.027 | NA | NA |
| Cmax,avg,ss | Bilirubin increased, grade ≥2 | 0.723 | NA | NA |
| Cmax,avg,ss | Creatinine increased, grade ≥2 | <0.001 | <0.001 | Age ≥65 years |
| Cmax,avg,ss | Fatigue/asthenia, grade ≥3 | 0.029 | NA | NA |
| Cmax,avg,ss | Nausea, grade ≥3 | 0.101 | 0.245 | Albumin |
| Exposure-safety with hematologic variables | ||||
| Cmax,avg,ss | Neutrophils count decreased, grade ≥3 | 0.061 | NA | NA |
| Cmax,avg,ss | Lymphocytes count decreased, grade ≥3 | 0.548 | NA | NA |
| Cmax,avg,ss | Platelet count decreased, grade ≥3 | 0.040 | NA | NA |
| Cmax,avg,ss | Hemoglobin decreased, grade ≥3 | 0.067 | NA | NA |
| Cmax,avg,ss | Hemoglobin, CFB | 0.001 | <0.001 | Albumin |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CFB, change from baseline; Cmax,avg,ss, average maximum concentration at steady state; ECOG, Eastern Cooperative Oncology Group; NA, not applicable (no significant covariates identified); PS, performance status.
The model includes significant covariates as listed.
The p value is the significance level for the exposure variable.
Fig. 3.
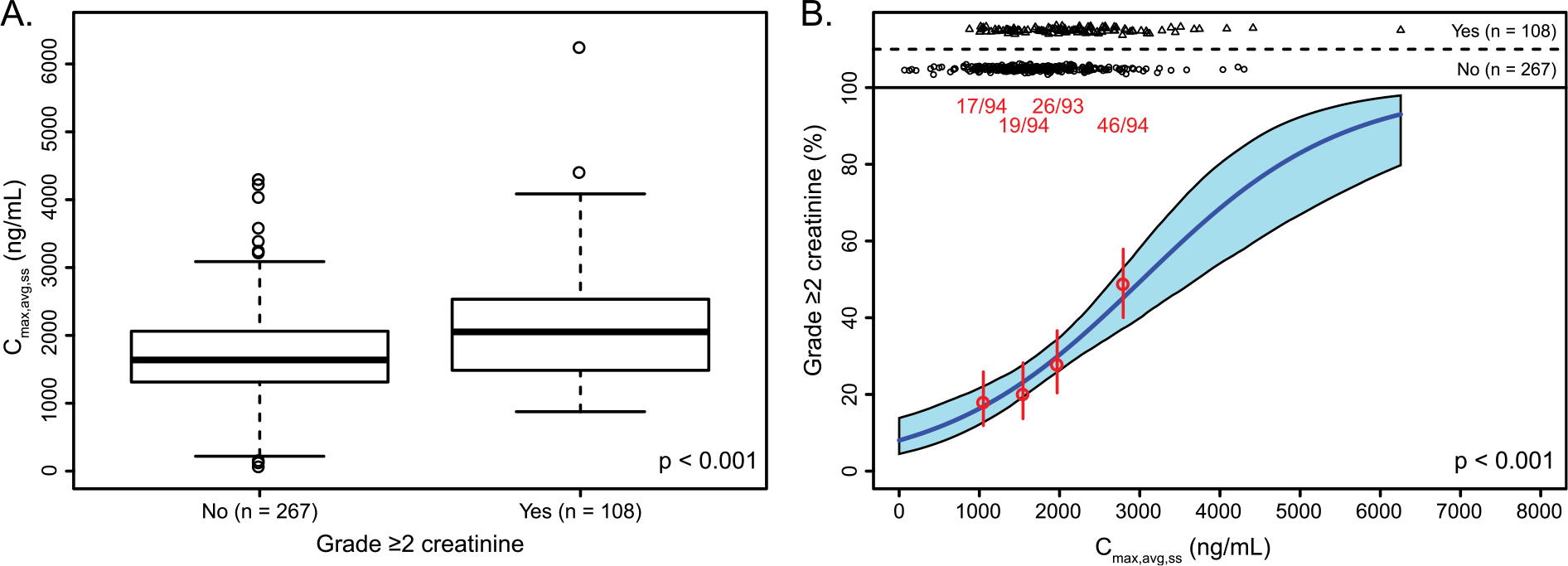
Exposure-safety relationships for creatinine. (A) Box plots show the maximum concentration of rucaparib, stratified by grade ≥2 creatinine increase. (B) Red points represent the mean exposure and event rates in patients stratified by exposure quartile. Vertical red bars represent the 5th to 95th percentile confidence intervals on the event rate. Event numbers (patients with event/total patients) are displayed above each vertical bar. Regression analyses were conducted for patients with grade ≥2 creatinine increase. The solid blue line is the logistic regression model fit for that patient subset. The shaded blue region represents 5th to 95th percentile confidence interval on the predicted event rate. Cmax,avg,ss, average maximum concentration at steady state.
The concentration-QT analysis included 54 patients with a total of 522 paired ECG-concentration data points. A linear mixed-effects model was used to describe the moderate increase in ΔQTcF at increasing rucaparib concentrations. Based on the population PK model developed with data from Study 10 and ARIEL2, the median and 95th percentile Cmax,ss following 600 mg BID was 2079 ng/mL and 3627 ng/mL, respectively. At the predicted median steady-state Cmax,ss the projected ΔQTcF was 11.5 ms (90% confidence interval CI 8.8–14.2 ms); at the 95th percentile of Cmax,ss the projected ΔQTcF was 17.2 ms (90% confidence interval 12.6–21.7 ms). The observed data, model fitting, and prediction are presented in Fig. 4.
Fig. 4.
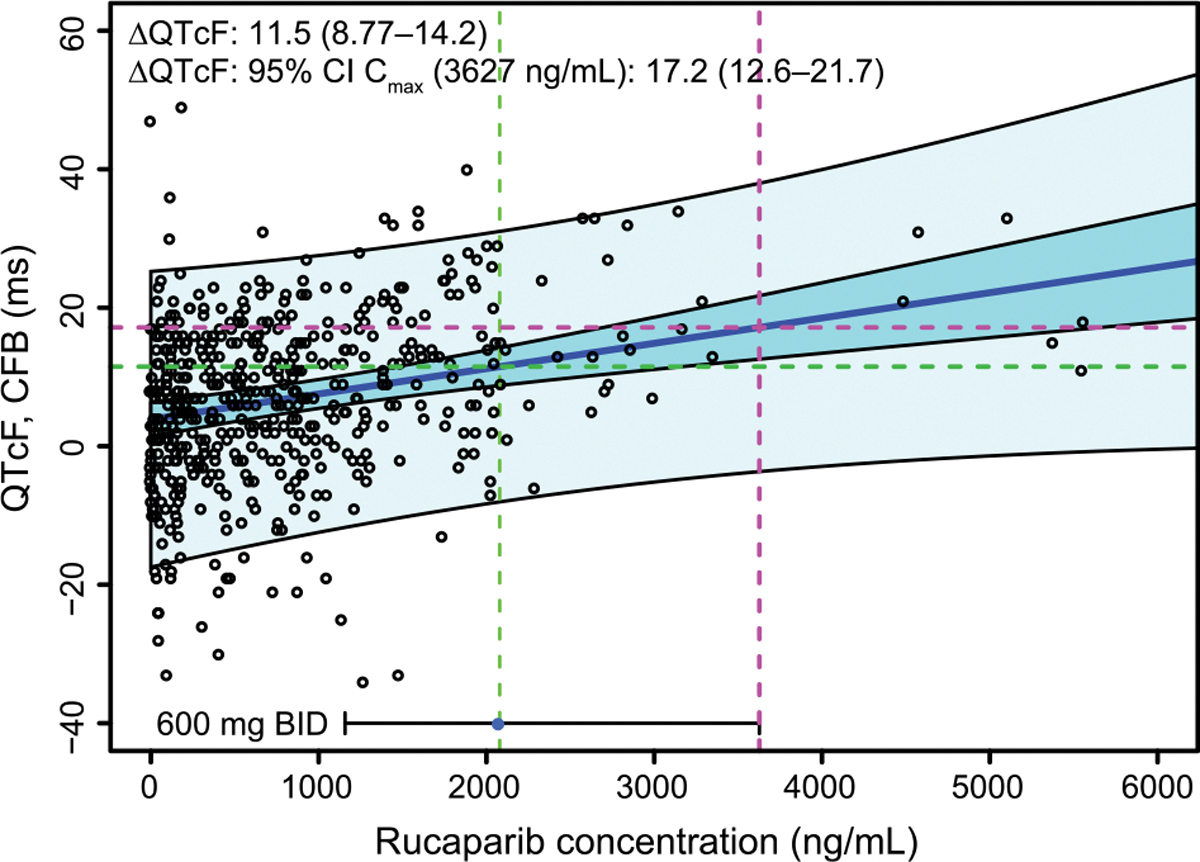
QTcF analysis based on data from Study 10 dose escalation. The points are triplicate mean ΔQTcF vs time-matched concentrations. The solid blue line is the line of best fit for mean ΔQTcF vs concentration using a linear mixed-effects model. The dark blue shaded band is the 5th to 95th percentile confidence interval in the mean fit. The light blue shaded band is the 5th to 95th percentile prediction interval of the data. The horizontal bar at the bottom of the plot is the 5th to 95th percentile range of Cmax,avg,ss data following 600 mg rucaparib BID from patients with high-grade ovarian cancer included in the exposure-safety analysis. The point is the median Cmax, and the vertical dashed green and magenta lines indicate the median and 95th percentile concentrations, respectively. The projected ΔQTcF at the median and the 95th percentile Cmax (labeled as ‘95% CI’ in the figure) are indicated by the green and magenta horizontal dashed lines, respectively. ΔQTcF, QTcF change from baseline; CFB, change from baseline; CI, confidence interval; Cmax, maximum concentration; QT, time from the start of the Q wave to the end of the T wave on an electrocardiogram; QTcF, QT corrected according to Fridericia’s formula.
4. Discussion
An exploratory exposure-efficacy analysis was conducted using a previously developed population PK model and the pooled clinical efficacy data from patients with ovarian carcinoma and a BRCA mutation in Study 10 and ARIEL2. In this analysis, the relationship between AUCavg,ss and IRR-assessed RECIST response was found to be statistically significant in the subgroup of patients with platinum-sensitive recurrent disease, suggesting a clinical benefit for maximizing the rucaparib dose in these patients. Most patients (91%) in the efficacy population received a starting dose of 600 mg BID rucaparib, which may have reduced the chance of finding significant correlations for the other tested efficacy endpoints (i.e. maximum percent change in target lesions, change in CA-125, PFS, or DOR). However, these endpoint variables, which are assessed on a continuum, may be considered less clinically robust compared with the binary endpoint of RECIST response for assessing drug activity or lack thereof in a small patient cohort. It is also important to note the lack of a significant correlation when evaluating exposure-efficacy using investigator-assessed objective response, which was likely attributable, in part, to discordance between local and central radiology review, as has previously been reported in assessments of RECIST response [16,17]. Despite these limitations, the wide range in the observed AUCavg,ss levels (range, 2328–99,524 ng/mL × h) still allowed detection of a statistically significant correlation between rucaparib exposure and IRR-assessed RECIST response. Although this significant correlation was observed in a relatively small group of patients (n = 75), this exposure-efficacy correlation is the first to our knowledge that has been identified among PARP inhibitors that are approved for the treatment of patients with ovarian cancer.
In this analysis, we also demonstrate that body weight shows no significant effect on rucaparib PK, suggesting that body weight–normalized dosing is not required to optimize the overall benefit-risk profile. In the frontline maintenance PRIMA trial (NCT02655016), an individualized starting dose for niraparib (200 vs 300 mg daily; selected based on baseline body weight and platelet counts) was introduced, after approximately 65% of patients had been enrolled, in an effort to improve the tolerability of the treatment [18]. While results have been reported for patients in PRIMA who received an individualized vs a fixed starting dose, the efficacy data for patients receiving lower vs higher doses have not yet been reported, which is an important consideration for evaluating if patients are underexposed. In our view, the advantages of selecting an individualized dose based on baseline body weight and platelets remain inconclusive.
The current study adds to our understanding of the effects of other clinical covariates and baseline patient characteristics on rucaparib PK. In the previously published analysis of rucaparib PK, the efficacy and safety covariates tested included effect of food, renal function, hepatic function, cytochrome P450 (CYP1A2 and CYP2D6) phenotypes, and concomitant photon-pump inhibitor medication [9]. The previous population PK analysis showed that baseline renal function had a moderate effect on rucaparib elimination, with 33% higher AUCss observed in patients with moderate renal impairment than in patients with normal renal function [9]. However, mean rucaparib exposure (AUCss) largely overlapped between the subgroups of patients with normal, mildly impaired, and moderately impaired renal function, suggesting that no starting dose adjustment is required in patients with mild or moderate renal impairment [7–9]. The correlation observed between rucaparib exposure and increased risk of grade ≥2 creatinine elevation was likely due to the inhibition of renal transporters (e.g. MATE1 and MATE2-K) by rucaparib rather than direct impact on renal function [19]. Although a relationship was observed between age (≥65 years) and increased grade ≥2 creatinine, this may be attributable to a trend towards declining renal function and physical condition in older adults [15]. Similarly, the clinical significance of albumin as a covariate for maximum hemoglobin reduction and grade ≥3 nausea may be explained by the fact that low albumin has generally been found to be associated with nausea, fatigue, and lower hemoglobin concentration in patients who are not receiving anticancer treatment [13,14]. A significant exposure-response relationship was observed for grade ≥3 ALT or AST increase; however, consistent with previous reports [20], these increases were transient. Nevertheless, increased incidence and severity of adverse effects in patients with higher rucaparib exposures underscores the importance of routine safety monitoring.
Concentration-QT prolongation analysis showed an increase in ΔQTcF as rucaparib concentrations approached Cmax,ss in patients receiving 600 mg rucaparib BID. The upper limit of the confidence interval for Cmax,ss was between 10 and 20 ms, indicating mild-to-moderate QT prolongation at the recommended clinical dose of rucaparib [21]. Consistent with results reported with other PARP inhibitors (e.g. olaparib, niraparib, veliparib [22–24]), our analysis suggested that clinically significant QTcF prolongation from baseline (>20 ms) is unlikely following continuous dosing with 600 mg rucaparib BID [25,26].
5. Conclusion
The lack of clinically relevant safety covariates in our analysis supports not modifying the starting dose for rucaparib based on baseline covariates. Furthermore, the significant correlation observed between exposure and IRR-assessed RECIST response supports maximizing the rucaparib dose for improved clinical benefit. Given these analyses, the rucaparib starting dose should not be modified based on baseline characteristics, including weight. Alternatively, tolerability to therapy should guide a clinician’s decision to modify the dose. Overall, the exposure-response analyses presented here support the approved starting dose of rucaparib 600 mg BID with subsequent dose modification following the occurrence of a treatment-emergent adverse event in patients with BRCA-mutated recurrent ovarian carcinoma.
Supplementary Material
{kind=link}
HIGHLIGHTS.
Rucaparib exposure and efficacy/safety relationship were studied using pooled Study 10 and ARIEL2 data.
Rucaparib exposure and response were positively correlated in patients with platinum-sensitive recurrent ovarian carcinoma.
We found no significant association between the model-predicted rucaparib exposure and baseline body weight.
Our analyses support starting rucaparib 600 mg twice daily with subsequent dose modification based on adverse events.
Acknowledgments
Support for medical writing and copyediting was paid for by Clovis Oncology, Inc., and provided by Shelly Lim and Frederique H. Evans of Ashfield MedComms, an Ashfield Health Company.
Funding
The current analyses were funded by Clovis Oncology, Inc., and were designed by the sponsor.
Footnotes
Declaration of competing interest
G.E. Konecny has served on speaker bureaus for Clovis Oncology, GSK and AstraZeneca and received research funding from Lilly and Merck. A.M. Oza reports grants from AstraZeneca; has served on steering committee for Clovis Oncology; and has served as a principal investigator of an investigator initiated clinical trial from GSK. A.V. Tinker received grants and honoraria from AstraZeneca. A. Oaknin reports grants from Clovis Oncology, Abbie Deutchland, Ability Pharma, Advaxis, Aeterna Zentaris, Amgen SA, Aprea Therapeutics AB, BMS, Bristrol Meyers Squibb, Eisai, F. Hoffmann - La Roche Ltd., Regeneron Pharmaceuticals, Immunogen, Merck Sharp & Dohme de España SA, Millennium Pharmaceutials, PharmaMar, and Tesaro; received personal fees from Clovis Oncology, AstraZeneca, Deciphera Pharmarceutial, Genmab, GSK, Immunogen, Mersana Therapeutic, Pharma Mar, Roche, and Tesaro; and received support for travel or accommodation from Clovis Oncology, AstraZeneca, PharmaMar, and Roche. R. Shapira-Frommer has no conflicts to report. I. Ray-Coquard has reports grants from BMS, GSK, and Roche; received personal fees from Clovis Oncology, AstraZeneca, BMS, Deciphera Pharmaceuticals, GSK, and Roche; and received support for travel from AstraZeneca. C. Aghajanian has served on steering committees for AbbVie and Genentech; has served on advisory boards for and received honoraria from Clovis Oncology, AbbVie, AstraZeneca, AstraZeneca/Merck, Eisai/Merck, Immunogen, Mersana Therapeutics, Roche/Genentech, and Tesaro; has served as a National Coordinating Investigator to AstraZeneca; and reports grants from Clovis Oncology, AbbVie, AstraZeneca, and Genentech. R.L. Coleman reports grants from Clovis Oncology, AstraZeneca, Genmab, Janssen, Merck, and Roche/Genentech; and has served as a consultant to Clovis Oncology, Agenus, AstraZeneca, Genmab, GSK, Janssen, OncoQuest, Regeneron Pharmaceuticals, and Roche/Genentech. D.M. O’Malley reports grants, personal fees, and other from Clovis during the conduct of the study; has served on advisory boards for Agenus, AstraZeneca, Eisai, Genentech/Roche, Immunogen, Iovance Biotherapeutics, Janssen/Johnson & Johnson, Merck, Mersana, Myriad, Novartis Pharmaceuticals, Novocure, Regeneron Pharmaceuticals, SeaGen, Tarveda, and Tesaro/GSK; has served on steering committees for Amgen; has served as a consultant to AbbVie, Agenus, Ambry, AstraZeneca, Eisai, Genentech/Roche, GOG Foundation, Immunogen, Iovance Biotherapeutics, Merck, Mersana, Novartis Pharmaceuticals, Novocure, Seagen, and Tesaro/GSK; and his institution has received research support from AbbVie, Agenus, Ajinomoto, Amgen, Array BioPharma, AstraZeneca, Bristol-Myers Squibb, Cerulean Pharma, Eisai, EMD Serono, Ergomed Clinical Research, Genmab, Genentech/Roche, GOG Foundation, ImmunoGen, INC Research, inVentiv Health Clinical, Iovance Biotherapeutics, Janssen/Johnson & Johnson, Ludwig Institute for Cancer Research, Merck, Mersana, New Mexico Cancer Care Alliance, Novocure, PRA International, Regeneron Pharmaceuticals, Seagen, Serono, Stemcentrx, Tesaro/GSK, TRACON Pharmaceuticals, VentiRx, and Yale University. A. Leary reports grants from Clovis Oncology, Ability Pharma, Agenus, AstraZeneca, GSK, Inivata, Iovance Biotherapeutics, MSD, Roche, Sanofi, and Tesaro; and received personal fees from Clovis Oncology, Ability Pharma, AstraZeneca, Biocad, GSK, MSD, Tesaro, and Zentalis. L.m. Chen has no conflicts to report. D. Provencher has served on advisory boards for Clovis Oncology, AstraZeneca, GlaxoSmithKline, Roche-Genentech, and Tesaro. L. Ma has no conflicts to report. J.D. Brenton has served as a leader to Tailor Bio; owns stock and other ownership interests in Inivata Ltd. and Tailor Bio; has received honoraria from GSK; has served as a consultant or on advisory boards for AstraZeneca; his institution has received research funding from Aprea; has patents, royalties, other intellectual property for TAm-Seq v2 method for ctDNA estimation and for enhanced detection of target DNA by fragment size analysis; and received support for travel, accommodation, and expenses from GSK. C. Castro received personal fees from Qiagen. M. Green is an employee of Certara and was a paid contractor to Clovis Oncology in connection with the analysis and development of this manuscript. A. D. Simmons, J. Beltman, T. Harding, K. K. Lin, S. Goble, L. Maloney, and J.J. Xiao are employees of Clovis Oncology and may own stock or have stock options in that company. R.S. Kristeleit reports grants from MSD; reports personal fees from Clovis Oncology, Eisai, GSK, MSD, and Roche; and received non-financial support from GSK. I.A. McNeish has served on advisory boards for Clovis Oncology. E.M. Swisher has no conflicts to report.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygyno.2021.03.015.
References
- [1].Global Cancer Observatory, Cancer Today, International Agency for Research on Cancer, Lyon, France, 2018. https://gco.iarc.fr/today Accessed March 4, 2019. [Google Scholar]
- [2].Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. , Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas, Clin. Cancer Res. 20 (2014) 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].The Cancer Genome Atlas (TCGA) Research Network, Integrated genomic analyses of ovarian carcinoma, Nature 474 (2011) 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S, et al. , Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2, J. Natl. Cancer Inst. 103 (2011) 334–346. [DOI] [PubMed] [Google Scholar]
- [5].Nguyen M, Simmons AD, Harding TC, Preclinical assessment of the PARP inhibitor rucaparib in homologous recombination deficient prostate cancer models, Cancer Res. 77 (2017) Abst 2476. [Google Scholar]
- [6].Robillard L, Nguyen M, Harding TC, Simmons AD, In vitro and in vivo assessment of the mechanism of action of the PARP inhibitor rucaparib, Cancer Res. 77 (2017) Abst 2475. [Google Scholar]
- [7].Rubraca (rucaparib) tablets [prescribing information]., Clovis Oncology, Inc., Boulder, CO, 2020. [Google Scholar]
- [8].Rubraca (Rucaparib) Tablets [Summary Of Product Characteristics]., Clovis Oncology Ireland Ltd., Swords, Ireland, 2019. [Google Scholar]
- [9].Xiao JJ, Green M, Ma SC, Goble S, Giordano H, Maloney L, et al. , Population pharmacokinetics (PK) of rucaparib (CO-338) in patients with advanced ovarian cancer (AOC) or other solid tumors, Clin. Pharmacol. Ther. 101 (2017) Abst PII-144. [DOI] [PubMed] [Google Scholar]
- [10].US Department of Health and Human Services, Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0, 2010.
- [11].Shapiro GI, Kristeleit RS, Burris HA, LoRusso P, Patel MR, Drew Y, et al. , Pharmacokinetic study of rucaparib in patients with advanced solid tumors, Clin. Pharmacol. Drug Dev. 8 (2019) 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Berek JS, Matulonis UA, Peen U, Ghatage P, Mahner S, Redondo A, et al. , Safety and dose modification for patients receiving niraparib, Ann. Oncol. 29 (2018) 1784–1792. [DOI] [PubMed] [Google Scholar]
- [13].Agarwal R, Davis JL, Smith L, Serum albumin is strongly associated with erythropoietin sensitivity in hemodialysis patients, Clin. J. Am. Soc. Nephrol. 3 (2008) 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gounden V, Vashisht R, Jialal I, Hypoalbuminemia, StatPearls, Treasure Island (FL), 2020. [PubMed] [Google Scholar]
- [15].Glassock RJ, Rule AD, The implications of anatomical and functional changes of the aging kidney: with an emphasis on the glomeruli, Kidney Int. 82 (2012) 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tang PA, Pond GR, Chen EX, Influence of an independent review committee on assessment of response rate and progression-free survival in phase III clinical trials, Ann. Oncol. 21 (2010) 19–26. [DOI] [PubMed] [Google Scholar]
- [17].Zhang J, Zhang Y, Tang S, Liang H, Chen D, Jiang L, et al. , Evaluation bias in objective response rate and disease control rate between blinded independent central review and local assessment: a study-level pooled analysis of phase III randomized control trials in the past seven years, Ann Transl. Med. 5 (2017) 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mirza MR, Martin AG, Graybill W, O’Malley DM, Gaba L, Yap OW, et al. , Evaluation of an individualized starting-dose of niraparib in the PRIMA/ENGOT-OV26/GOG-3012 study, J. Clin. Oncol. 38 (2020) Abst 6050. [Google Scholar]
- [19].Zibetti Dal Molin G, Westin SN, Msaouel P, Gomes LM, Dickens A, Coleman RL, Discrepancy in calculated and measured glomerular filtration rates in patients treated with PARP inhibitors, Int. J. Gynecol. Cancer 30 (2020) 89–93. [DOI] [PubMed] [Google Scholar]
- [20].Oza AM, Tinker AV, Oaknin A, Shapira-Frommer R, McNeish IA, Swisher EM, et al. , Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: integrated analysis of data from Study 10 and ARIEL2, Gynecol. Oncol. 147 (2017) 267–275. [DOI] [PubMed] [Google Scholar]
- [21].Sarapa N, Britto MR, Challenges of characterizing proarrhythmic risk due to QTc prolongation induced by nonadjuvant anticancer agents, Expert Opin. Drug Saf. 7 (2008) 305–318. [DOI] [PubMed] [Google Scholar]
- [22].Moore K, Chan JK, Secord AA, Patel MR, Callahan T, Guo W, et al. , Effect of niraparib on cardiac repolarization in patients with platinum-sensitive, recurrent epithelial ovarian, fallopian tube, and primary peritoneal cancer, Cancer Chemother. Pharmacol. 83 (2019) 717–726. [DOI] [PubMed] [Google Scholar]
- [23].Munasinghe W, Stodtmann S, Tolcher A, Calvo E, Gordon M, Jalving M, et al. , Effect of veliparib (ABT-888) on cardiac repolarization in patients with advanced solid tumors: a randomized, placebo-controlled crossover study, Cancer Chemother. Pharmacol. 78 (2016) 1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Swaisland H, Plummer R, So K, Garnett S, Bannister W, Fabre MA, et al. , Olaparib does not cause clinically relevant QT/QTc interval prolongation in patients with advanced solid tumours: results from two phase I studies, Cancer Chemother. Pharmacol. 78 (2016) 775–784. [DOI] [PubMed] [Google Scholar]
- [25].U.S. Food, Drug Administration, Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-antiarrhythmic Drugs, October 2012.
- [26].Sarapa N, Britto MR, Challenges of characterizing proarrhythmic risk due to QTc prolongation induced by nonadjuvant anticancer agents, Expert Opin. Drug Saf. 7 (2008) 305–318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.