

Abstract

Chlorfortunones A (1) and B (2), two novel sesquiterpenoid dimers, were isolated from the roots of Chloranthus fortunei. Their structures were elucidated by spectroscopic analysis and X-ray diffraction analysis. Compounds 1 and 2 represent a new type of sesquiterpenoid dimer possessing an unprecedented 3/5/6/6/6/5 hexacyclic system with a unique dispiro[4,2,5,2]pentadecane-6,10,14-trien moiety. A plausible biosynthetic pathway of 1 and 2 was proposed. Compound 1 showed transforming growth factor (TGF)-β inhibitory activity in MDA-MB-231 cells.
1. Introduction
Sesquiterpenoid dimers (SDs), the characteristic secondary metabolites of genus Chloranthus, possess more fascinating structures and exhibited more potent biological activities than their monomeric precursors in previous research.1−5 These compounds have attracted considerable attention from the scientific communities of natural products and organic synthesis. These SDs are biosynthesized by the Diels–Alder cycloaddition or free-radical coupling reaction of two sesquiterpenoid monomers, in which a new single C–C bond, 4-membered ring, 6-membered ring, or 12-membered ring was formed.3,6Chloranthus fortunei is widely distributed in provinces south of the Yangtze River in China.7 It has long been used as a traditional Chinese medicine to treat blood stasis, inflammatory swelling, drainage, and detoxification.7,8 In previous phytochemical studies, 26 dilindenane-type and lindeane-eudesmane-type SDs had been isolated from C. fortunei.1,7−10 In this paper, we reported two novel SDs, chlorfortunones A (1) and B (2), isolated from the roots of C. fortunei (Figure 1). They are a new class of SDs formed by the [4 + 2] cycloaddition of lindenane- and acrane-type sesquiterpenoid monomers and possess an unprecedented 3/5/6/6/6/5 hexacyclic system with a unique dispiro[4,2,5,2]pentadecane-6,10,14-trien moiety.
Figure 1.

Structures of compounds 1 and 2.
2. Results and Discussion
Chlorfortunone A (1), a colorless crystal ([α]D25 −158.6), was assigned its molecular formula C31H38O5 by the HRESIMS ion at m/z 491.2799 [M + H]+ (calcd for 491.2792), suggesting 13 degrees of unsaturation. The 1H NMR (Table 1) of 1 displayed one typical methylene of 1,2-disubstituted cyclopropane moiety at δH 0.20 and 0.91, two singlet methyls [δH 1.04 (s, H-14) and 1.99 (s, H-13)], three doublet methyls [δH 0.81 (d, J = 7.1 Hz, H-14′), 0.83 (d, J = 6.7 Hz, H-12′), and 0.97 (d, J = 6.7 Hz, H-13′)], one methoxy group, and four olefinic protons. The 13C NMR (Table 1) data showed 31 carbon signals, which were classified by DEPT and HSQC data as 5 methyls, 4 methylenes, 11 methines (including one oxygenated and four olefinic), and 10 quaternary carbons. Detailed analysis of its one-dimensional (1D) and two-dimensional (2D) NMR data allowed for the establishment of units A and B substructures for 1. Unit A (Figure 2, in red) was deduced to be a lindenane-type sesquiterpenoid. The 1H–1H COSY correlations (Figure 2) of H-1/H-2/H-3 confirmed the typical 1,2-disubstituted cyclopropane moiety.5 The HMBC cross-peaks (Figure 2) from H-14 to C-1, C-5, C-9, and C-10, from H-15 to C-5, from H-3 to C-5, from H-6 to C-4, C-8, and C-11, and from H-9 to C-7 were structural characteristics of lindenane-type sesquiterpenoid and indicated a double bond at C-4 and C-5, one carbonyl group at C-8, and one hydroxy group at C-9. In addition, a senecioyl moiety connected to C-7 was suggested by HMBC cross-peaks from H-6 to C-11 and from H-13 to C-7 and C-12.11
Table 1. 1H NMR (600 MHz) and 13C NMR (150 MHz) Spectroscopic Data for Compounds 1 and 2 (δ in ppm, CDCl3).
|
1 |
2 |
|||
|---|---|---|---|---|
| no. | δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) |
| 1 | 25.4 CH | 1.96 m | 25.3 CH | 1.95 m |
| 2 | 15.4 CH2 | 0.20 q (4.0) | 15.3 CH2 | 0.19 q (4.0) |
| 0.91 dt (11.6, 4.1) | 0.89 dt (11.6, 4.1) | |||
| 3 | 23.6 CH | 1.68 m | 23.6 CH | 1.67 m |
| 4 | 142.8 C | 143.0 C | ||
| 5 | 132.1 C | 132.2 C | ||
| 6 | 43.5 CH | 3.43 t (3.2) | 43.8 CH | 3.42 t (3.2) |
| 7 | 133.9 C | 143.6 C | ||
| 8 | 201.8 C | 201.9 C | ||
| 9 | 80.7 CH | 3.72 overlap | 80.9 CH | 3.77 overlap |
| 10 | 51.4 C | 51.3 C | ||
| 11 | 143.3 C | 134.2 C | ||
| 12 | 171.5 C | 171.5 C | ||
| 13 | 19.5 CH3 | 1.99 s | 19.6 CH3 | 1.99 s |
| 14 | 14.7 CH3 | 1.04 s | 14.7 CH3 | 1.03 s |
| 15 | 22.9 CH2 | 2.21 m | 22.8 CH2 | 2.22 m |
| 2.37 m | 2.29 m | |||
| 1′ | 51.9 CH | 1.82 m | 55.1 CH | 1.72 m |
| 2′ | 38.0 CH2 | 2.03 m | 36.5 CH2 | 1.72 m |
| 2.38 m | 2.02 m | |||
| 3′ | 217.7 C | 77.0 CH | 3.84 m | |
| 4′ | 57.2 CH | 2.09 dd (13.8, 7.0) | 54.4 CH | 1.56 m |
| 5′ | 50.7 C | 53.0 C | ||
| 6′ | 133.4 CH | 5.49 dd (10.1, 2.3) | 137.9 CH | 5.38 dd (10.3, 2.2) |
| 7′ | 133.8 CH | 5.64 dd (10.1, 2.3) | 135.8 CH | 5.68 dd (10.4, 2.2) |
| 8′ | 42.5 C | 42.7 C | ||
| 9′ | 128.6 CH | 5.74 dd (10.7, 2.2) | 125.3 CH | 5.55 dd (10.3, 2.2) |
| 10′ | 128.3 CH | 5.24 dd (10.7, 2.3) | 126.1 CH | 5.33 dd (10.4, 2.1) |
| 11′ | 28.1 CH | 1.82 m | 28.6 CH | 1.49 m |
| 12′ | 20.7 CH3 | 0.83 d (6.7) | 21.5 CH3 | 0.83 d (4.6) |
| 13′ | 23.8 CH3 | 0.97 d (6.7) | 23.9 CH3 | 0.78 d (6.5) |
| 14′ | 8.4 CH3 | 0.81 d (7.1) | 13.0 CH3 | 0.82 d (4.8) |
| 15′ | 36.9 CH2 | 1.69 m | 38.2 CH2 | 1.58 m |
| 1.89 m | 1.87 m | |||
| OCH3 | 52.2 CH3 | 3.72 s | 52.0 CH3 | 3.73 s |
Figure 2.

Key 1H–1H COSY, HMBC, and NOESY correlations of compounds 1 and 2.
The remaining data of unit B was similar to those of Shizuka-acoradienol, an acrane-type sesquiterpenoid isolated from C. japonicus.12 The 1H–1H COSY correlations of unit B (Figure 2, in blue) revealed four structural fragments, as depicted with bold bonds (Figure 2), which were then connected by the HMBC correlations (Figure 2) to furnish its acrane framework. The HMBC cross-peaks from H-14′ to C-3′ and C-5′, from H-15′ to C-7′ and C-9′, from H-1′ to C-6′ and C-10′, from H-4′ to C-2′, C-6′, and C-10′, and from H-2′ to C-5′ verified the structure of unit B as 4-isopropyl-1,8-dimethylspiro[4.5]deca-6,9-dien-2-one.
The functional groups and ring systems in the two monomeric sesquiterpenoids took 12 out of the 13 total degrees of unsaturation, and the remaining one thus required the existence of an additional ring. The presence of a new six-membered ring between units A and B was supported by the HMBC correlations from H-6 to C-7′ and C-9′, from H-15 to C-8′, from H-15′ to C-6, combined with the 1H–1H COSY correlations of H-15/H-15′, which was connected between C-15 and C-15′ and between C-6 and spiro carbon C-8′ (δC 42.5). The planar structure of 1 was established to be a heterodimeric framework of lindenane- and acrane-type SDs, which possesses a unique 3/5/6/6/6/5 hexacyclic system with dispiro[4,2,5,2]pentadecane-6,10,14-trien moiety. The relative configuration of 1 was fixed by NOESY data (Figure 2). The NOESY correlations of H-1/H-9, H-14/H-2, and H-14/H-6 indicated that the cyclopropane moiety, H-6, H-14, and OH-9 were β-orientation, which are the same relative configuration as those of lindenane-type sesquiterpenoids.13 The NOESY cross-peaks of H-6/H-7′, H-6′/H-1′, and H-1′/H-4′ indicated that these protons were on the same side and were β-oriented, while the NOESY correlations of H-9/H-9′ and H-14′/H-10′ suggested that these protons were α-orientation.
To prove the absolute configuration of 1, the single crystal with good quality of 1 was obtained in a methanol/water (10:1) system and subjected to an X-ray diffraction experiment with Cu Kα radiation. The crystal data [Figure 3; CCDC: 2172881, Flack = 0.01(8)] not only confirmed our deduction about the planar and relative structure of 1 but also unambiguously gave its absolute configuration as 1R,3S,6R,9R,10S,1′S,4′S,5′S,8′R.
Figure 3.

X-ray ORTEP drawing of compound 1.
Chlorfortunone B (2) ([α]D25 −118.0), an amorphous powder, afforded the molecular formula C31H40O5 by HRESIMS ion at m/z 515.2782 [M + Na]+ (calcd for 515.2768), indicating 12 degrees of unsaturation. The similar NMR data (Table 1) revealed that 2 is the analogue of 1. Comparison of the 1H and 13C data of 2 with those of 1 revealed a hydroxy group at C-3′ (δH 3.84, δC 77.0) in 2 instead of those of a carbonyl group (δC 217.7) in 1. The planar structure of 2 was assigned by 1H–1H COSY, HSQC, and HMBC spectroscopic data analysis (Figure 2). The H-3′/H-10′ and H-14′ indicated that the OH-3′ was β-orientation. The configurations of the remaining chiral carbons in 2 are the same as those of 1 suggested by the NOESY correlations (Figure 2) and their similar ECD spectra (Figure 4). Thus, the structure of 2 was determined as shown.
Figure 4.

ECD spectra of compounds 1 and 2 in MeOH.
A plausible biosynthetic pathway for 1 and 2 was proposed (Scheme 1). According to their structures, chloranthalactone A and Shizuka-acoradienol existing in Chloranthaceae plants would be considered as the precursors of 1 and 2.12,14 The intermediate iv with the required conjugated Δ15(4),5(6) system for the Diels–Alder [4 + 2] cycloaddition should be generated by a series of reactions including the olefination of allylic position at C-5/C-6, stereoselective epoxidation of the active Δ8,9 olefinic bond, epoxide cleavage, and lactone opening of chloranthalactone A.14 A Diels–Alder [4 + 2] cycloaddition between iv and Shizuka-acoradien (red bond) led to the formation of spiro[5.5]undecane-4,8-dien skeleton of v. Furthermore, the v would be dehydrated at C-9′ and oxidized at C-3′ to afford 2. Compound 1 should be formed by further oxidization at C-3′ of 2.
Scheme 1. Hypothetic Biosynthetic Pathway for 1 and 2.

Transforming growth factor-β (TGF-β) is the key regulator of cancer metastasis and fibrosis diseases.15 As our group′s ongoing research on natural TGF-β inhibitors,16−18 compounds 1 and 2 were tested for in vitro TGF-β inhibitory activity. The results showed that only compound 1 significantly downregulated the TGF-β-induced p-Smad2 expression in a concentration-dependent manner without any impact on the expression of Smad2/3 protein in MDA-MB-231 cells (Figure 5A). The immunofluorescence assay (Figure 5B) exhibited that 25 μM of 1 significantly downregulated the expression of vimentin, a Smad-regulated EMT-marker. This data suggested that 1 is a potential natural TGF-β inhibitor.
Figure 5.
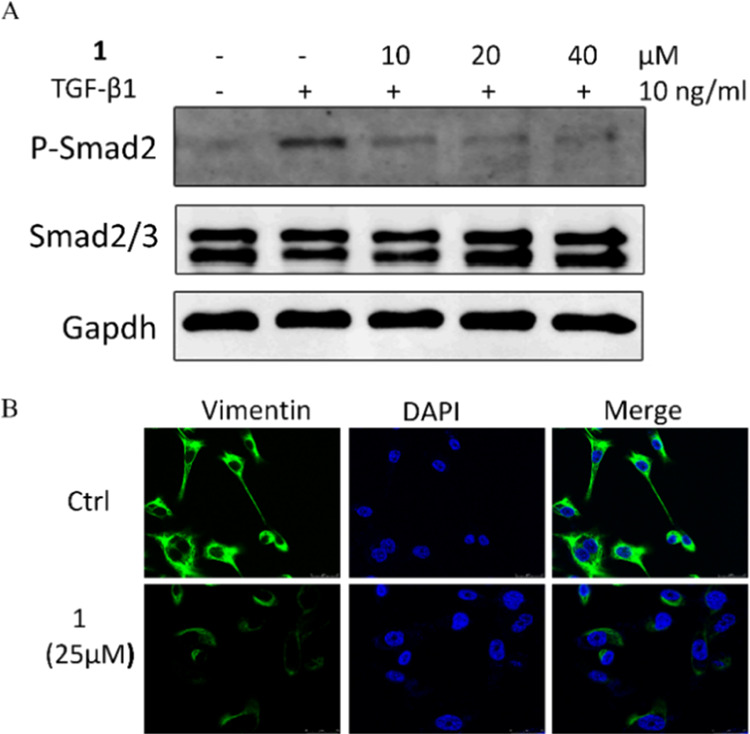
Compound 1 downregulated TGF-β-induced Smad2 phosphorylation (A) and the expression of vimentin (B) in MDA-MB-231 cells.
In summary, two novel SDs, chlorfortunones A (1) and B (2), were isolated from the roots of C. fortunei. Compounds 1 and 2 possess an unprecedented 3/5/6/6/6/5 hexacyclic skeleton containing a unique dispiro[4,2,5,2]pentadecane-6,10,14-trien moiety. They are the first example of SDs fused by the lindenane- and acrane-type sesquiterpenoid monomers in nature. Bioassay results showed that 1 is a potential natural TGF-β inhibitor. These findings further demonstrated the diversity of chemical structures and bioactivities of natural SDs.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured on a Rudolph Research Autopol I automatic polarimeter. Ultraviolet (UV) spectra were recorded on a Jasco J-1500 circular dichroism spectrometer. IR spectra were carried on an Agilent Cary 660 series FT-IR spectrometer. 1D and 2D NMR spectra were performed on a Bruker Ascend-600 spectrometer. Chemical shifts are given as δ values concerning tetramethylsilane (TMS) as the internal standard. Silica gel (40–63 mesh) was used for column chromatography (CC). HRESIMS data were determined by an Agilent-6230 Q-TOF UHPLC/MS spectrometer. Preparative medium-pressure liquid chromatography (MPLC) was carried out on an Unips 40–300 gel (10 μm, 460 × 49 mm). Semipreparative HPLC separations were performed on a Vision HT C18 HL column and a Waters BEH phenyl column with an Agilent 1200 liquid chromatography instrument.
3.2. Collection of Sample
The roots of C. fortunei were collected from the Bozhou market of Anhui Province, China, in May 2019 and identified by Prof. G.-Y. Zhu. The voucher specimen (No. 20190901) was deposited at the Science Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology.
3.3. Extraction and Isolation
The roots of C. fortunei (15.0 kg) were ground to powder and extracted with 85% EtOH (5 × 12 L) at room temperature. After evaporation of the solvent under reduced pressure, the residue (1.8 kg) was dissolved in 10 L water and partitioned with EtOAc (3 × 2.5 L). After solvent removal, the EtOAc fraction (858.2 g) was subjected to silica gel (40–63 mesh) column chromatography and subsequently eluted with a gradient of increasing acetone (0–100%) in EtOAc to afford 16 fractions (Fr. A–P). Fr. E was subjected to MPLC on a Unips 40–300 gel column eluted with MeCN–H2O (90:10 to 100:0, v/v) to provide eight subfractions (Fr. E1–E8). Fr. E7 was subjected to a Vision HT C18 HL column and eluted with MeCN–H2O (80:20, v/v) to get eight subfractions (Fr. E7a–E7j). Fr. F7d was fractioned on a Vision HT C18 HL column using MeCN–H2O (77:23, v/v) to afford five subfractions (Fr. E7d1–E7d5). Fr. E7d3 was purified by an Xterra C18 OBD column and semipreparative HPLC with MeOH–H2O (79:21, v/v) to yield compound 1 (4 mg, tR = 21 min). Fr. F was separated with MPLC using a Unips 40–300 gel column eluted with MeCN–H2O (30:70 to 100:0, v/v) to afford 12 fractions (Fr. F1–F8). Fr. F3 was fractioned on a Waters BEH phenyl column with MeCN–H2O (57:43, v/v) to get 14 fractions (Fr. F3a-F3n). Fr. F3f was purified by a Sunfire C18 OBD column and semipreparative HPLC with MeCN–H2O (61:39, v/v) to yield compound 2 (3.7 mg, tR = 25 min).
Chlorfortunone A (1)
Colorless crystals; mp. 189 °C; [α]D25 −158.6 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 216 (4.00), 270 (4.11) nm; IR (KBr) νmax 3457, 2931, 2870, 1735, 1604, 1450, 1172, 1087 cm–1; 1H and 13C NMR data; see Table 1; HRESIMS m/z 491.2799 [M + H]+ (calcd for 491.2792).
Chlorfortunone B (2)
Amorphous powder; [α]D25 −118.0 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 216 (4.00), 270 (4.11) nm; IR (KBr) νmax 1728, 1612, 1535, 1450, 1273, 1219, 1033, 995 cm–1; 1H and 13C NMR data; see Table 1; HRESIMS m/z 515.2782 [M + Na]+ (calcd for 515.2768).
3.4. X-ray Diffraction Crystallographic Data of Compound 1
After many attempts with various solvent systems, we successfully obtained the colorless orthorhombic crystals of compound 1 by culturing the compound in a mixture of methanol/water (10:1) at room temperature for several days. The intensity data were collected on a diffractometer Rigaku Oxford Diffraction SuperNova, Dual, Cu at zero, equipped with an AtlasS2 CCD using Cu Kα radiation. The crystal of 1 was kept at 150.00 (10) K during data collection. The crystallographic data of 1 have been deposited in the Cambridge Crystallographic Data Center database (CCDC 2172881).
Crystal Data for Compound 1 (CCDC 2172881)
C31H38O5 (M = 490.61 g/mol), orthorhombic, space group P212121 (no. 19), a = 6.57981(11) Å, b = 12.10779(19) Å, c = 32.5315(5) Å, V = 2591.69(7) Å, Z = 4, T = 150.00(10) K, μ (Cu Kα) = 0.669 mm–1, Dcalc = 1.257 g/cm3, 13747 reflections measured (5.434° ≤ 2Θ ≤ 147.772°), 5087 unique (Rint = 0.0305, Rsigma = 0.0293), which were used in all calculations. The final R1 was 0.0332 (I > 2σ(I)), and wR2 was 0.0873 (all data).
3.5. Cell Lines and Cultures
Human TNBC cell line MDA-MB-231 was provided by the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS, GIBCO, Grand Island, NY) in a 5% CO2-humidified atmosphere at 37 °C.
3.6. Western Blot Analysis
MDA-MB-231 cells were seeded into six-well plates and maintained for 24 h. Cells were treated with 10, 20, and 40 μM compound 1 for 4 h and stimulated with 10 ng/mL TGF-β1(Sigma-Aldrich) for 30 min. Total proteins were lysed in RIPA buffer supplemented with protease inhibitors (Amresco). Protein concentrations were determined by a BCA Protein Assay (Thermo Fisher Scientific). The 30 μg of total protein was loaded onto a 12% SDS-PAGE gel for electrophoretic separation and transferred to poly(vinylidene difluoride) (PVDF) membranes (Millipore, Burlington, MA). Primary antibodies against p-Smad2, Smad2/3, and Gapdh were purchased from Cell Signaling Technology. Western blots were imaged using an LI-COR Odyssey imaging system (Lincoln, NE).
3.7. Immunofluorescence Assay
MDA-MB-231 cells were plated into 35 mm glass-bottom dishes (NEST, Wuxi, China). After a treatment of 25 μM compound 1 for 48 h, cells were fixed with 4% paraformaldehyde and stabilized in 0.1% Triton X-100. After blocking with 3% BSA, cells were incubated with vimentin (1:100 dilution, CST) overnight and incubated with Alexa Fluor 488 antibody IgG (1:200 dilution, Invitrogen, Thermo Fisher Scientific) for 1 h. The cells were observed and photographed with a confocal fluorescence microscope (Leica TCS SP8, Germany).
Acknowledgments
This work was financially supported by grants from the Macao Science and Technology Development Funds (0075/2019/AGJ and 0023/2019/AKP) and GDST-FDCT Projects (2020A050515006).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03927.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhou B.; Liu Q. F.; Dalal S.; Maria B. C.; Yue J. M. Fortunoids A–C, three sesquiterpenoid dimers with different carbon skeletons from Chloranthus fortunei. Org. Lett. 2017, 19, 734–737. 10.1021/acs.orglett.7b00066. [DOI] [PubMed] [Google Scholar]
- Yan H.; Qin X. J.; Li X. H.; Yu Q.; Ni W.; He L.; Liu H. Y. Japonicones A-C: three lindenane sesquiterpenoid dimers with a 12-membered ring core from Chloranthus japonicus. Tetrahedron Lett. 2019, 60, 713–717. 10.1016/j.tetlet.2019.01.040. [DOI] [Google Scholar]
- Ma L. F.; Chen Y. L.; Shan W. G.; Zhan Z. J. Natural disesquiterpenoids: an update. Nat. Prod. Rep. 2020, 37, 999–1030. 10.1039/C9NP00062C. [DOI] [PubMed] [Google Scholar]
- Dong T.; Li C.; Wang X.; Dian L. Y.; Zhang X. G.; Li L.; Chen S.; Cao R.; Li L.; Huang N.; He S. D.; Lei X. G. Ainsliadimer A selectively inhibits IKKα/β by covalently binding a conserved cysteine. Nat. Commun. 2015, 6, 6522 10.1038/ncomms7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A. R.; Song H. C.; An H. M.; Huang Q.; Luo X.; Dong J. Y. Secondary metabolites of plants from the genus Chloranthus: chemistry and biological activities. Chem. Biodiversity 2015, 12, 451–473. 10.1002/cbdv.201300376. [DOI] [PubMed] [Google Scholar]
- Huang H. L.; Xu Y. J.; Liu H. L.; Liu X. Q.; Shang J. N.; Han G. T.; Yao M. J.; Yuan C. S. Eremophilane-type sesquiterpene lactones from Ligularia hodgsonii Hook. Phytochemistry 2011, 72, 514–517. 10.1016/j.phytochem.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Bian X. X.; Zhao X.; Liu S. S.; Wu L.; Yin X. W.; Shen C. P. Sesquiterpene dimers from Chloranthus fortunei and their protection activity against acute lung injury. Fitoterapia 2022, 159, 105191 10.1016/j.fitote.2022.105191. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Wu Y.; Dalal S.; Merino E. F.; Liu Q. F.; Xu C. H.; Yuan T.; Ding J.; Kingston D. G. I.; Cassera M. B.; Yue J. M. Nanomolar antimalarial agents against chloroquine-resistant Plasmodium falciparum from medicinal plants and their structure-activity relationships. J. Nat. Prod. 2017, 80, 96–107. 10.1021/acs.jnatprod.6b00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. C.; Zhang Y. N.; Wang L. L.; Ma S. P.; Liu J. H.; Hu L. H. Lindenane sesquiterpene dimers from Chloranthus fortunei. J. Nat. Prod. 2008, 71, 674–677. 10.1021/np7007544. [DOI] [PubMed] [Google Scholar]
- Wang X. C.; Wang L. L.; Ouyang X. W.; Ma S. P.; Liu J. H.; Hu L. H. Sesquiterpenes and dimers thereof from Chloranthus fortunei. Helv. Chim. Acta. 2009, 92, 313–320. 10.1002/hlca.200800262. [DOI] [Google Scholar]
- Bai B.; Ye S. X.; Yang D. P.; Zhu L. P.; Tang G. H.; Chen Y. Y.; Li Q. G.; Zhao Z. M. Chloraserrtone A, a sesquiterpenoid dimer from Chloranthus serratus. J. Nat. Prod. 2019, 82, 407–411. 10.1021/acs.jnatprod.8b00418. [DOI] [PubMed] [Google Scholar]
- Kawabata J.; Fukushi Y.; Tahara S.; Mizutani J. Structures of novel sesquiterpene alcohols from Chloranthus japonicus (Chloranthaceae). Agric. Biol. Chem. 1984, 48, 713–717. 10.1080/00021369.1984.10866207. [DOI] [Google Scholar]
- Shen C. P.; Luo J. G.; Yang M. H. M.; Kong L. Y. Sesquiterpene dimers from the roots of Chloranthus holostegius with moderate anti-inflammatory activity. Phytochemistry 2017, 137, 117–122. 10.1016/j.phytochem.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Chi J.; Xu W. J.; Wei S. S.; Wang X. B.; Li J. X.; Gao H. L.; Kong L. Y.; Luo J. Chlotrichenes A and B, two lindenane sesquiterpene dimers with highly fused carbon skeletons from Chloranthus holostegius. Org. Lett. 2019, 21, 789–792. 10.1021/acs.orglett.8b04046. [DOI] [PubMed] [Google Scholar]
- Peng D.; Fu M.; Wang M.; Wei Y.; Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104 10.1186/s12943-022-01569-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Fu J.; Shen R. S.; Wu X. J.; Yang J.; Bai L. P.; Jiang Z. H.; Zhu G. Y. Linderanoids A–O, dimeric sesquiterpenoids from the roots of Lindera aggregata (Sims) Kosterm. Phytochemistry 2021, 191, 112924 10.1016/j.phytochem.2021.112924. [DOI] [PubMed] [Google Scholar]
- Chen F. L.; Liu D. L.; Fu J.; Yang J.; Bai L. P.; Zhang W.; Jiang Z. H.; Zhu G. Y. (±)-Atrachinenins A-C, three pairs of caged C-27 meroterpenoids from the rhizomes of Atractylodes chinensis. Chin. J. Chem. 2022, 40, 460–466. 10.1002/cjoc.202100700. [DOI] [Google Scholar]
- Liu X.; Yang J.; Fu J.; Yao X. J.; Wang J. R.; Liu L.; Jiang Z. H.; Zhu G. Y. Aggreganoids A–F, carbon-bridged sesquiterpenoid dimers and trimers from Lindera aggregata. Org. Lett. 2019, 21, 5753–5756. 10.1021/acs.orglett.9b02166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.