

SUMMARY
Genetics in model organisms has progressively broken down walls that previously separated different disciplines of biology. One example of this holistic evolution is the recognition of the complex relationship that exists between the control of bone mass (bone remodeling) and energy metabolism in mammals. Numerous hormones orchestrate this crosstalk. In particular, the study of the leptin-mediated regulation of bone mass has not only revealed the existence of a central control of bone mass but has also led to the realization that sympathetic innervation is a major regulator of bone remodeling. This happened at a time when the use of drugs aiming at treating osteoporosis, the most frequent bone disease, has dwindled. This review will highlight the main aspects of the leptin-mediated regulation of bone mass and how this led to the realization that β-blockers, which block the effects of the sympathetic nervous system, may be a viable option to prevent osteoporosis.
INTRODUCTION
Like a machine, any organism, even the simplest, is a functional unit coherent and integrated…. We are far to have in complex organisms elucidated the entire structure of these systems.
Each of these two sentences makes a fundamental point. The first is that elucidating how multiple physiological functions unfold simultaneously in a complex organism, like a mammal, remains a central interrogation of biology. The second statement acknowledges that the study of the fundamental bases of physiological functions in a living organism, i.e., whole-organism physiology, was in 1970, a largely uncharted territory that would remain so until the very end of the 20th century, when technical tools needed to study the genetic bases of whole-organism physiology became available. It is not the least important aspect of this statement that it was made by a molecular biologist who never studied, even remotely, physiology. This appreciation reifies the broad importance of whole-organism physiology by inferring that understanding how organismal homeostasis is achieved may be the final frontier of biology.
It is a tribute to Monod’s remarkable foresight that between 1970 and now, molecular biology and genetics manipulations in model organisms have steadily worked their way toward reaching the goal he described as central to all biological research. Both disciplines have, through numerous and increasingly sophisticated advances, made it possible to modify at will, in an inducible manner, the level of expression of a given gene in one cell type at a time in a living animal, invertebrate, or vertebrate. As a result, it is now possible to define the function of any gene in virtually any cell type in the context of the entire animal, this often being a mammal. This remarkable molecular and genetic accomplishment allowed biology to move beyond prior constraints by studying the functions of a gene in a single-cell type and, in doing so, has revived the study of whole-organism physiology. This revival of whole-organism physiology has affected every aspect of physiology from neuroscience to metabolism (Friedman and Halaas, 1998; Ganz, 2011; Karsenty and Ferron, 2012; Kliewer and Mangelsdorf, 2019). It has also revealed the existence of and often molecularly defined connections between distinct organs and different physiological functions that were not known before and, for most of them, were unexpected and yet may be critically important. It is an under-statement to say that the influence that energy metabolism and bone physiology exert on each other postulated 22 years ago (Ducy et al., 2000) was not then a central theme of research of either of these two fields of physiology. Without the advent of modern genetics, they might have remained independent of each other. A logical consequence of this revival of whole-organism physiology through the genetics in model organisms is that it has also impacted our understanding of the pathogenesis of degenerative diseases. As a result, it has led, as will be illustrated in this review, to the identification of novel adapted therapies for degenerative diseases. In that context, this review article will address how the established coordinated regulation of bone mass accrual and energy metabolism led to a nation-wide clinical trial that attempts to prevent the development of osteoporosis (Ducy et al., 2000; Khosla et al., 2018).
The hypothesis that underlies the work described in this review article is that there should be a coordinated regulation, endocrine in nature, of bone remodeling and energy metabolism. This hypothesis that is implied by the cell biology of bone is verified by clinical medicine (Ducy et al., 2000). Bone renews itself, and bone mass is maintained constant through a process, unique to this tissue that comprises two arms. The first is an active destruction or resorption of the pre-existing mineralized bone extracellular matrix (ECM) by osteoclasts; the second arm, which invariably follows bone resorption, is de novo bone formation by osteoblasts. These cellular events occur daily in alternation in all bones from birth to death. When considered together, they are called bone modeling during childhood and bone remodeling during adulthood. Bone modeling contributes to the longitudinal growth of a child and therefore to its ability to walk. Bone remodeling repairs micro- and macro-damage (fractures)—in other words, bone remodeling was, for the longest time, the only orthopedic surgeon around. Today, bone remodeling is mostly known for what its disruption causes, osteoporosis. In that age-related disease that most often begins following the cessation of gonadal functions, there is an increase in bone resorption that is not compensated by an increase of similar amplitude of bone formation. As a result, bone mass decreases, and the risk of fracture after a minimal impact increases. Even if it is difficult to precisely measure it in vivo (Zoch et al., 2016), destroying the mineralized bone ECM, as occurs during resorption, is energetically exorbitant; the same is true of synthesizing de novo and secreting huge quantities of collagen and other proteins of the bone ECM during bone formation. Indeed, type I collagen is one of the most abundant proteins in the body. Another reason to surmise that the energetic cost of bone remodeling is high is that bone covers a large surface in our body. However, eventually what gave the most credence to the hypothesis that there is a coordinated regulation of bone mass and energy metabolism are clinical observations. A decrease in food intake, i.e., in energy intake, invariably causes an arrest of growth in children and a low bone mass phenotype in adults. In closing, we should underscore one critical aspect of this hypothesis. Since it is the existence of bone and bone remodeling that justifies formulating it, the hormones that allowed the experimental verification of this hypothesis should exert their regulatory functions only in bony vertebrates. To the best of our knowledge, when it comes to the regulation of appetite and energy metabolism, this is the case for leptin.
Why would the crosstalk between energy metabolism and bone physiology be worth writing about in 2022? We can think of two reasons that justify this exercise. A first is that if this reciprocal regulation was initially viewed as surprising and, in any case, limited to a single hormone, leptin, regulating energy metabolism and bone mass accrual, this is not the case anymore. The field now implicates many more hormones besides leptin, including insulin, adiponectin, irisin, osteoglycin, lipocalin 2, and osteocalcin, to name a few. It also embroils other types of regulatory molecules such as transcription factors like ATF4, FoxO1, AP1, and signaling molecules that all contribute to define the different aspects of the crosstalk between bone physiology and energy metabolism (Dirckx et al., 2019; Fulzele et al., 2010; Kajimura et al., 2013; Kim et al., 2018; Lee et al., 2018; Mosialou et al., 2017; Rached et al., 2010; Rowe et al., 2012; Wagner, 2010; Zou et al., 2019). Studying this crosstalk between energy metabolism and bone has now become a mainstream theme of research in bone biology and has attracted the attention of laboratories outside the field and interested in energy metabolism and its neuronal control (Farooqi et al., 2000; Kim et al., 2018). A second reason justifying this review is of a medical nature. In the last 22 years, the cellular and molecular elucidation of the mechanisms, whereby leptin inhibits bone physiology, has begun to be harnessed for therapeutic purposes (Khosla et al., 2018). This eventually has led to a nation-wide clinical trial that aims at preventing the most frequently acquired bone disease: osteoporosis.
NUTRIENT AND BONE CELL DIFFERENTIATION AND FUNCTIONS
We will focus in this review on specific aspects of the crosstalk between bone physiology and energy metabolism currently. For energy metabolism, those will be (1) the identity and fate of nutrients taken up by bone cells, (2) the regulation of whole-body glucose homeostasis, and (3) the hormonal control of appetite and energy expenditure and their impact on bone mass accrual. As for bone physiology, we will focus on the maintenance of bone mass or bone (re)modeling. Even if we will in some places refer to them, the regulation by bone-derived hormones of glucose homeostasis, energy expenditure, and appetite will not be reviewed here (Lee et al., 2007; Mosialou et al., 2017). We should mention that these effects, established by loss- and gain-of-function mutations in mice and primates, are primary ones, i.e., they are caused by cellular events occurring in target cells when these hormones bind to their cognate receptor and trigger a pathway of gene expression (Berger et al., 2019; Khrimian et al., 2017; Lee et al., 2007; Mera et al., 2016; Mosialou et al., 2017; Oury et al., 2011; Wei et al., 2014b).
It has been known for over half a century that osteoblasts could uptake glucose, the main nutrient of most cell types, and that this was correlated in vitro with their activity (Neuman et al., 1979). In addition, glycogen droplets were also identified in osteoblasts 50 years ago (Neuman et al., 1979; Scott and Glimcher, 1971). Since the biological relevance of these observations could not be studied in vivo at the time, these studies were for a long time a mere footnote in bone biology. This has changed in part because of advances in the genetic manipulations of model organisms and, in part, because of our much-improved molecular understanding of how cell differentiation unfolds in the osteoblast, a cell of mesenchymal origin (Akiyama et al., 2005; Ducy et al., 1997, 1999; Nakashima et al., 2002; Yang et al., 2004). Hence, one can now study the role of nutrients in bone cells at specific stages of differentiation. Since overall, more information is currently available about the role of nutrients in osteoblasts than is the case for osteoclasts, we will focus principally on the former cell type.
The analysis of oxygen consumption by mouse osteoblasts cultured in the presence as nutrients of glucose, glutamine, an amino acid and a proxy of proteins, or fatty acids established that glucose is the main nutrient of osteoblasts. Of note, the same is true for osteoclasts (Wei et al., 2015; Kim et al., 2017b). To put the significance of glucose uptake by bone in perspective, bone is one of the biggest consumers of glucose; specifically, it takes up one-fifth of the quantity of glucose taken up by muscle, which is the main consumer of glucose in the mouse (Zoch et al., 2016). Unlike in muscle, however, glucose uptake occurs in an insulin-independent manner in both osteoblasts and osteoclasts. This is congruent with the fact that Glut1, a glucose transporter that allows glucose uptake in an insulin-independent manner, is expressed at two orders of magnitude higher in osteoblasts and osteoclasts than Glut4 that transports glucose in an insulin-dependent manner or any other glucose transporter. Glut1 is also the only glucose transporter so far whose deletion affects glucose uptake in osteoblasts at steady state. That Glut1 transports glucose in an insulin-dependent manner does not exclude that insulin signaling in osteoblasts also controls bone remodeling and bone endocrine functions (Ferron et al., 2010; Fulzele et al., 2010; Lee et al., 2007; Wei et al., 2014a).
The in vivo study of glucose uptake in cells of the osteoblast lineage at various stages of differentiation has shed important light on the regulation of osteoblast differentiation and bone formation. In doing so, it has also provided an explanation for a major paradox of bone biology, which is that the expression of type I collagen genes in prospective bone cells and the synthesis of this huge protein, which accounts for over 90% of the protein content of the bone ECM, precedes the expression in prospective osteoblasts of Runx2, the master gene of osteoblast differentiation (Ducy et al., 1997, 1999). Glut1 expression marks type I collagen-expressing mesenchymal osteoblast progenitors before they express Runx2 (Wei et al., 2015). Glucose uptake through Glut1 is necessary in vivo for the accumulation of the Runx2 protein in prospective osteoblasts and therefore for the differentiation of these cells into mature osteoblasts, for their proliferation, and for type I collagen synthesis, i.e., bone formation by these osteoblasts. Inside the osteoblast, glucose is metabolized mostly through aerobic glycolysis to generate ATP molecules necessary for bone formation. The central role of glucose and aerobic glycolysis in osteoblasts is vividly illustrated by the phenotype of mice lacking the von Hippel-Landau protein, a key component of the hypoxia signaling pathway (Dirckx et al., 2018). These mutant mice exhibit a significant increase in glucose uptake and aerobic glycolysis in osteoblasts that results in a high bone mass phenotype and hypoglycemia, a phenotype related to the endocrine functions of bone, which have been reviewed elsewhere recently (Ducy, 2020).
If these observations underscore the paramount importance of glucose as a nutrient for osteoblast differentiation and function (Li et al., 2016; Wei et al., 2015), glucose is by no means the only nutrient used by osteoblasts. As one would expect, amino acids, proteins, and lipids are also critical for osteoblast differentiation and bone formation (Elefteriou et al., 2006; Karner et al., 2015; Shen et al., 2021). However, this aspect of the multilayered relationship between bone remodeling and energy metabolism will not be developed here as it has not yet been harnessed for therapeutic purposes.
INFLUENCE OF HORMONES AFFECTING GLUCOSE HOMEOSTASIS ON BONE REMODELING
The preeminence of glucose as a nutrient for osteoblasts explains why the influence that insulin, the major endocrine regulator of glucose homeostasis, has on bone functions was the first to be studied. This work was also prompted by another observation: the rather mild dysregulation of glucose homeostasis in mice that lack the insulin receptor in classical insulin target organs such as muscle and white adipose tissue when fed a normal diet was an incentive to look for other insulin target organs that could also contribute to glucose homoestasis (Blüher et al., 2002; Brüning et al., 1998). The role of osteoblasts in whole-body glucose homeostasis was defined through the generation of mice lacking the insulin receptor exclusively in cells of the osteoblast lineage at various stages of differentiation (Ferron et al., 2010; Fulzele et al., 2010). This analysis performed independently in two different laboratories showed the profound influence that insulin has on bone remodeling and, beyond that, on its own secretion and whole-body glucose homeostasis. On one hand, insulin signaling in cells of the osteoblast lineage is needed for osteoblast differentiation and proliferation and, as a result, for bone formation because it favors Runx2 expression. On the other hand, insulin signaling in osteoblasts favors osteoclast differentiation and bone resorption by inhibiting the expression of osteoprotegerin, a decoy receptor for the osteoclast differentiation factor, Rankl. The result of this complex influence of insulin signaling in osteoblasts is to favor bone mass accrual in adult animals. However, the ability of insulin signaling in osteoblasts to favor osteoclast differentiation has another important consequence: it increases the release in the general circulation of osteocalcin, an insulin secretagogue. Illustrating the importance of this latter function of insulin signaling in osteoblasts, increasing or decreasing insulin signaling in osteoblasts improves or worsens whole-body glucose intolerance and insulin resistance in mice fed a high-fat diet (HFD) (Wei et al., 2014a). This is caused, in part, by the insulin resistance that develops in bone of wild-type (WT) mice fed an HFD. We should mention here that these observations have not been extended to type 2 or type 1 diabetic patients yet. Although type 1 diabetic patients often have low bone mass, type 2 diabetic patients often have a high bone mass that may be due to their obesity and the leptin resistance that ensues (Hofbauer et al., 2007; Myers et al., 2008).
Together, the analysis of mice lacking one or two copies of the insulin receptor in osteoblasts and that one of the mice with instead an increased expression of the insulin receptor in osteoblasts allow us to make several points that are relevant to the topic of this review article (Ferron et al., 2010; Fulzele et al., 2010; Wei et al., 2014a). The first is that insulin signaling in osteoblasts is a significant determinant of bone mass accrual. The second is that through its signaling in osteoblasts, insulin favors its own secretion. These two points epitomize how interwoven glucose homeostasis and bone physiology are. The third point that supports the first two is that insulin signaling in osteoblasts is a significant contributor to the whole-body glucose intolerance and insulin resistance seen in WT mice fed an HFD.
In addition to insulin, several other hormones contribute to glucose homeostasis. Several of them called “incretins” are secreted by intestinal cells in response to a meal and, directly or indirectly, favor insulin secretion in a glucose-dependent manner. This family of hormones includes glucose-dependent insulinotropic polypeptide (GIP), glucagon-like polypeptide 1 (GLP-1), and glucagon-like polypeptide 2 (GLP-2); these hormones signal through GPCR5 in their target organs (Iepsen et al., 2015; Xie et al., 2005). Although the effects on bone cells of the incretin vary depending on whether animals or humans are hyperglycemic or not, schematically, GLP-1 can favor osteoblast differentiation and proliferation, whereas GIP can decrease bone resorption (Iepsen et al., 2015; Zhong et al., 2007). The exact mechanism, i.e., direct or indirect action on bone cells, whereby these hormones affect bone mass accrual has not been established yet with complete certainty. Again, emphasizing the potential involvement of bone on glucose homeostasis, it has been proposed that one mechanism used by osteocalcin to promote insulin secretion is by favoring the secretion of GLP-1 by the gut (Mizokami et al., 2013, 2014, 2020). FGF21, a hormone that regulates energy metabolism and is secreted by the liver, also exerts an influence on bone remodeling by increasing the differentiation of osteoclasts (Wang et al., 2015). Another more recently identified hormone that affects energy metabolism (thermogenesis and browning of white fat) is irisin, a cleavage product of FNDC5, a membrane protein. Irisin was initially characterized as being secreted by muscle during exercise, stimulating UCP1 expression in white adipocytes that could result in improvement in body weight and glucose homeostasis in mice fed an HFD. Subsequently, it was shown that irisin also exerts anabolic effects on the skeleton through several mechanisms (Kim et al., 2018). In cell culture, irisin favors osteoblast differentiation and proliferation as well as aerobic glycolysis in osteoblasts. Irisin also promotes osteoclast proliferation and, in one study, bone resorption (Estell et al., 2020). In vivo, the analysis of irisin-null mice indicates that irisin may increase the survival of a population of osteoblasts embedded in the bone ECM, the osteocytes, and favors the osteolysis that osteocytes otherwise perform following ovariectomy or lactation.
COORDINATED REGULATION OF APPETITE AND BONE REMODELING AND ITS THERAPEUTIC IMPLICATIONS
The growing body of work summarized above established the importance of the functional connection between bone remodeling and glucose homeostasis. However, the notion that a crosstalk between hormones regulating various aspects of energy metabolism and bone remodeling may exist began with the hypothesis that adipocyte-derived hormones regulate bone mass accrual (Ducy et al., 2000). It is the study of this aspect of the crosstalk between energy metabolism and bone physiology that has allowed investigators to propose and test a preventative treatment for osteoporosis. For the purpose of this review, which is centered on the treatment of osteoporosis, we may not cite all the molecular features of this regulation, those have been reviewed in detail elsewhere (Karsenty and Ferron, 2012).
The proposal that there may be a reciprocal regulation of energy metabolism and bone physiology was catalyzed by an apparently unrelated finding. Mice lacking an osteoblast-specific secreted protein, osteocalcin, exhibited as their most overt phenotype, albeit not initially simply because it was not understood, an increase in their abdominal fat mass (Ducy et al., 1996). Paradoxically and for a variety of reasons, this observation that suggested that bone might be an endocrine organ regulating fat mass was not directly tested initially. Instead, it triggered the study of the influence of adipocyte-derived hormones and altogether of white adipocytes on bone remodeling, a work that had a long-lasting and possibly clinical influence on bone biology. This body of work is based exclusively on the analysis of genetic mouse models of loss of function and, in some cases, on even more important human genetics studies (Ducy et al., 2000; Elefteriou et al., 2004; Pogoda et al., 2006). Results of these physiological and genetic analyses of loss-of-function models are at variance with the results obtained in various pharmacological experiments (Reseland et al., 2001). Although these pharmacological studies have their own merits, given that it was the analysis of loss-of-function models that uncovered the existence of the sympathetic regulation of bone mass and its potential therapeutic application, it is those analyses that will be summarized in this review.
In a nutshell, these studies showed that leptin and, to a lesser extent, adiponectin inhibit bone mass accrual. Specifically, mice lacking leptin (ob/ob) or its receptor (db/db) or adiponectin exhibit a high bone mass phenotype (Ducy et al., 2000; Kajimura et al., 2013). This phenotype is significantly more severe in leptin signaling-deficient than in adiponectin-deficient mice. As one would expect in view of these findings, mice lacking all adipocytes—the so-called fat-free mice, mice deprived of adipocytes—develop a high bone mass phenotype as well (Ducy et al., 2000). A mechanistic link between the high bone mass observed in fat-free mice and the one observed in mice lacking leptin or adiponectin was established by showing that transplantation of WT adipocytes corrected the high bone mass phenotype of the fat-free mice they studied. On the other hand, transplantation of leptin-deficient or adiponectin-deficient or double-deficient adipocytes could not correct this phenotype (Zou et al., 2019).
Not only is leptin regulation of bone mass of greater magnitude than the one exerted by adiponectin, but it has two features that separate it from any other known hormonal regulation of bone mass accrual, and they also explain why this regulation has been so intensively studied. The leptin inhibition of bone mass accrual that has been observed in mice, and subsequently in rats, sheep, and humans, is distinct from the regulation of bone mass by any other hormones since it develops in the absence of leptin signaling and does so in the face of an equally severe hypogonadism, i.e., a condition that in every other known circumstance leads to osteoporosis in all mammals (Elefteriou et al., 2004; Pogoda et al., 2006; Vaira et al., 2012). This occurs because in the absence of leptin signaling, there is an increase in osteoblast numbers and in bone formation parameters that far overcome the increase in osteoclast numbers and in bone resorption that gonadal failure induces. This unique feature of leptin biology immediately made leptin regulation of bone mass potentially important from a therapeutic point of view.
The leptin regulation of bone mass is original for a second reason. To affect bone formation and bone resorption, leptin, as it does for the control of appetite, signals in the brain to initiate what is now referred to as the central control of bone mass. As shown in Figure 1, leptin uses, in part, different mediators to regulate appetite and bone mass accrual. The road map of leptin signaling in the brain, as far as the regulation of bone mass is concerned, starts with the signaling of leptin in serotonergic neurons of the dorsal raphe that control both bone mass and appetite (Elefteriou et al., 2005; Nectow et al., 2017; Ortuño et al., 2016, 2021; Takeda et al., 2002; Yadav et al., 2009, 2011). Genetic analysis as well as anterograde and retrograde labeling showed that serotonin signals in neurons of the ventromedial hypothalamic nuclei and recruits two distinct mediators to inhibit bone mass accrual, the neuropeptide cocaine and amphetamine regulated transcript (CART) and the sympathetic nervous system (Figure 1; Elefteriou et al., 2005; Takeda et al., 2002). The hypothesis that the sympathetic tone may mediate the leptin-dependent regulation of bone mass was also suggested by a clinical observation: patients with reflex sympathetic activity, a disease characterized by high sympathetic activity, develop a severe osteoporosis that can be treated by β-blockers (Schwartzman, 2000). Neither CART nor the sympathetic tone is implicated in the leptin regulation of appetite or energy expenditure in mice fed a normal chow diet. That leptin used distinct signaling mechanisms to affect appetite and bone mass is of fundamental importance when considering the possibility of harnessing the leptin-dependent regulation of bone mass for therapeutic purposes.
Figure 1. Leptin signaling in the central nervous system recruits the sympathetic nervous system to inhibit bone mass accrual.
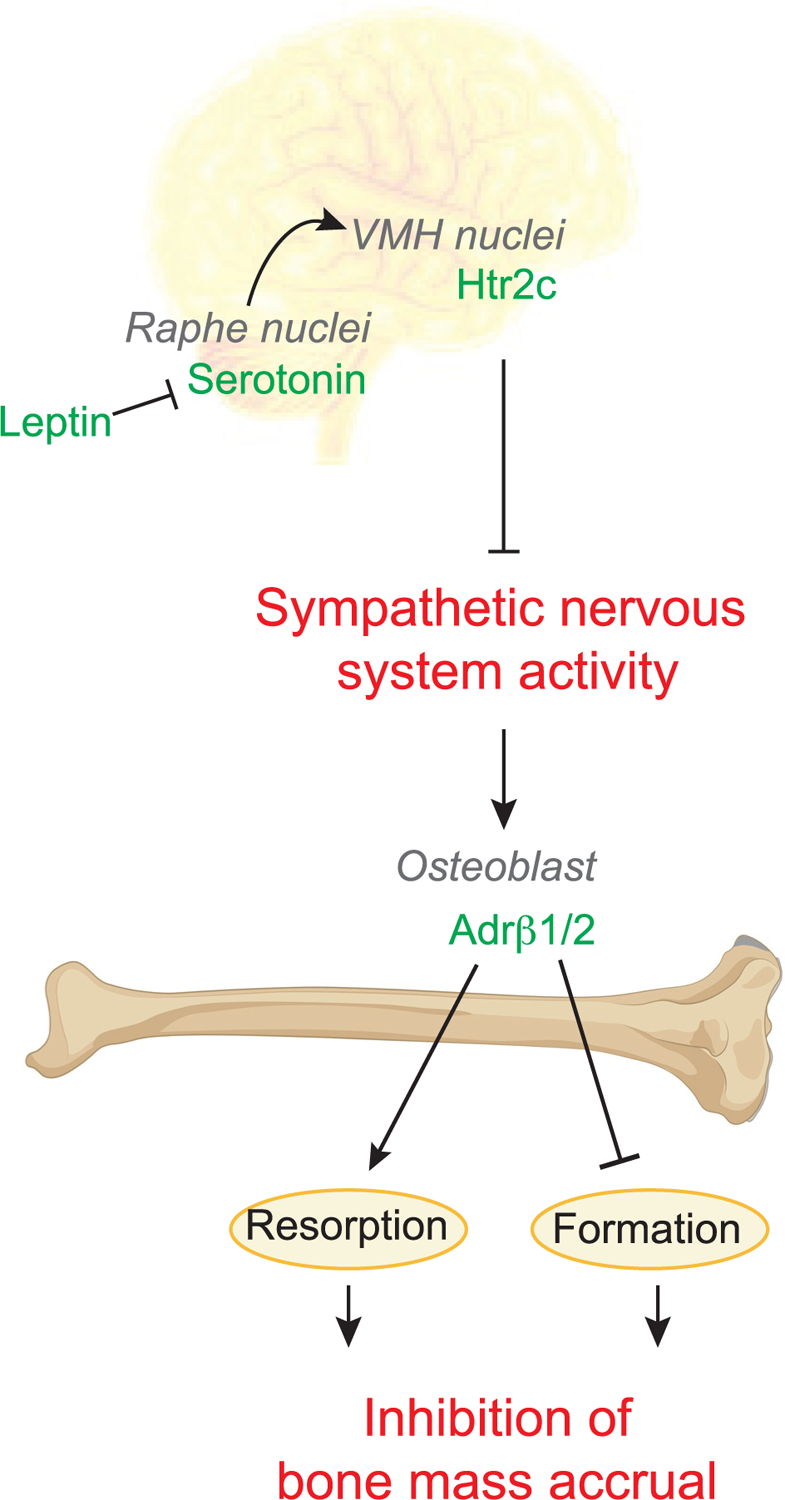
Leptin, a hormone responsible for enforcing energy balance, is a powerful regulator of bone mass. Leptin signals in the dorsal raphe of the brainstem to inhibit serotonin synthesis by Tph2. Serotonergic neurons from the dorsal raphe synapse at several nuclei in the hypothalamus including the VMH. Htr2c signaling in the VMH inhibits sympathetic nervous system activity in the skeleton, thereby inhibiting bone mass accrual. Within the skeleton, the sympathetic nervous system releases norepinephrine, which signals on osteoblasts to stimulate bone resorption and inhibits bone formation. In mice, norepinephrine signals through Adrb2, but in humans, sympathetic norepinephrine signals through both Adrb1 and Adrb2. The effects of norepinephrine signaling on bone mass accrual are conserved between mice and humans. Altogether, this series of findings made several points. First, an energy metabolism hormone does regulate bone mass accrual, a process that consumes a great deal of energy. Second, the central nervous system controls bone mass. Third, the sympathetic nervous system is a powerful inhibitor of bone mass accrual.
The realization that leptin regulates bone mass through the central nervous system opened a floodgate in the study of the regulation of bone mass (Hu et al., 2020). Rather quickly, this was followed by the demonstration that other molecules signaling in the brain, whether they are neuropeptides such as NPY or Neuromedin U, receptors for hormones like Mc4r, or transcription factors like delta FosB, originally thought of as acting within bone cells were all shown to regulate bone mass through their central actions (Ahn et al., 2006; Baldock et al., 2009; Elefteriou et al., 2003; Herber et al., 2019; Rowe et al., 2012; Sabatakos et al., 2000; Sato et al., 2007). Furthermore, and independently of the leptin regulation of bone mass, additional studies linked the sympathetic control of bone mass to the bone loss induced by neuroleptics (Motyl et al., 2015). Figure 2 summarizes our current understanding of the regulation of bone remodeling by factors important in regulating energy metabolism.
Figure 2. Energy metabolism endocrine systems calibrate the balance between bone quality and energy conservation.
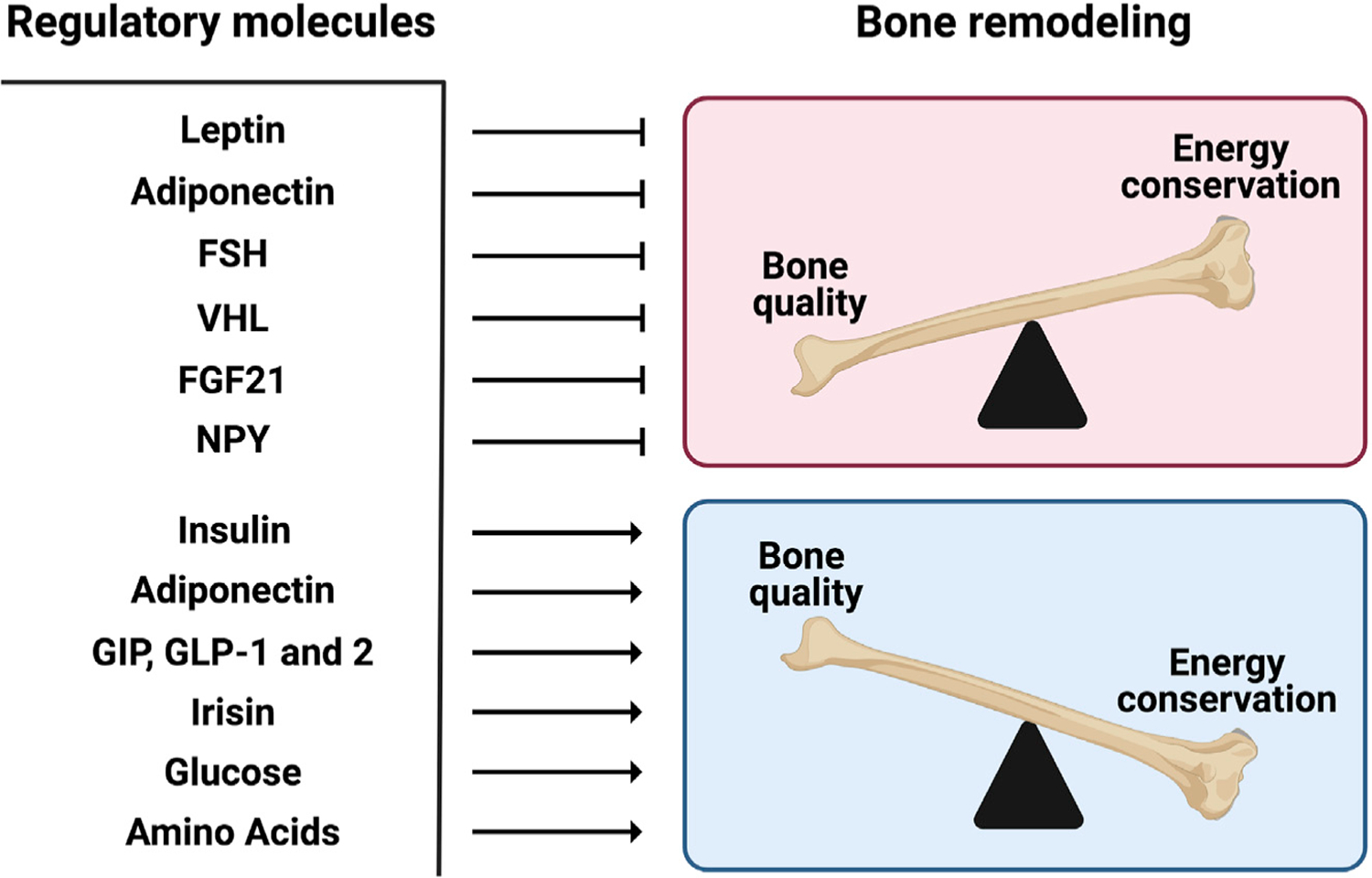
Bone remodeling is the lifelong creation and destruction of bone tissue that preserves bone quality. Because bone is one of the largest organs in the body and because bone remodeling is one of the only healthy biological processes that requires the active destruction of tissue, bone remodeling is one of the largest consumers of energy in bony vertebrates. For these reasons and others, it was proposed that there would be a coordinated regulation of organismal energy metabolism and bone remodeling. This concept stimulated the discovery that leptin inhibits bone formation and stimulates bone resorption by signaling through the brain to activate the sympathetic nervous system, which then signals through the β-adrenergic receptor in osteoblasts. Over the subsequent two decades, a host of energy metabolism hormones and metabolites, listed in this figure, have been shown to influence bone remodeling and, as hypothesized, help calibrate the balance between bone quality and energy conservation.
The molecular pathway whereby the sympathetic nervous system inhibits bone mass accrual was deciphered in the mouse. Only one adrenergic receptor is expressed in osteoblasts, the β2-adrenergic receptor or Adrβ2. As will be presented below, the picture appears to be different in human osteoblasts since they harbor on their surface not only Adrβ2 but also Adrβ1 (Khosla et al., 2018). Sympathetic signaling in mouse osteoblasts through Adrβ2 exerts two functions. It inhibits osteoblast proliferation and bone formation and favors the expression of the osteoclast differentiation factor, Rankl (Elefteriou et al., 2005; Fu et al., 2005; Hu et al., 2020; Takeda et al., 2002). As a result, sympathetic signaling in osteoblasts favors osteoclast differentiation and bone resorption. In agreement with this dual mechanism of action of the sympathetic nervous system in bone, inhibition of bone formation and promotion of bone resorption, mice lacking Adrβ2 in all cells or in osteoblasts only develop a high bone mass phenotype caused by the conjunction of an increase in bone formation and a decrease in bone resorption (Kajimura et al., 2011). In full support of the notion that the sympathetic tone is a mediator of the leptin inhibition of bone mass accrual, the high bone mass phenotype observed in Adrβ2−/− mice cannot be corrected by the delivery of leptin in their brains (Kajimura et al., 2011; Takeda et al., 2002).
A confirmation of the importance of this sympathetic regulation of bone mass and a first step toward translating these findings into a treatment for a osteoporosis in humans was the demonstration that propranolol, a β-blocker that can inhibit signaling through Adrβ2 and to a lesser extent through Adrβ1, could prevent the bone loss observed in ovariectomized WT mice (Takeda et al., 2002). This initial observation was cleverly refined by Bonnet et al., who used ovariectomized WT rats as an experimental model. These investigators showed that at low doses (0.1–1 mg/kg/day), propranolol was acting only through Adrβ2 and affecting bone mass without affecting heart rate or respiration, whereas increasing the dose of propranolol and allowing this drug to block signaling through both Adrβ1 and Adrβ2 did not increase the beneficial effect on bone mass but affected heart rate and respiration (Bonnet et al., 2006). These results were consistent with the observation that even heterozygous Adrβ2-deficient mice had a high bone mass phenotype (Takeda et al., 2002). This illustrates the unanticipated importance of the sympathetic tone for the regulation of bone remodeling (Takeda et al., 2002). This immediately suggested the testable possibility that this regulation could be harnessed for therapeutic purposes—specifically, for the prevention of osteoporosis.
THE PROBLEM OF PRIMARY PREVENTION OF OSTEOPOROSIS
Concomitant with the description of the central control of bone mass and its peripheral mediation by the sympathetic nervous system, a growing “crisis” in the treatment and prevention of osteoporosis was developing in front of our eyes (Khosla et al., 2017; Khosla and Shane, 2016). Before describing the nature and magnitude of this crisis, the existence of which led to the current search for a new treatment for the disease, we will provide a brief overview of the current definition and management of osteoporosis and then delineate why the fundamental observations described above have the potential to significantly impact the management of this devastating disease.
Osteoporosis, the most frequent bone degenerative disease, is currently defined clinically by the measurement of bone mineral density (BMD) or by the occurrence of adulthood hip or vertebral fracture in the absence of major trauma (e.g., motor vehicle accident or fall from greater than standing height) (Cosman et al., 2014). The BMD criteria are based on T-scores (standard deviation [SD] decrease as compared to a young adult reference population of the same sex): a T-score of 2.5 SD or more below that of the mean level for a young adult reference population at the lumbar spine or hip is diagnostic of osteoporosis and a T-score between −1.0 and −2.5 is defined as “osteopenia,” i.e., low bone mass, representing an at-risk population (Cosman et al., 2014). Osteoporosis is overall an underestimated public health problem. For example, it has been estimated that the number of women who will experience a fracture in 1 year due to osteoporosis in fact exceeds the combined number of women who will experience incident breast cancer, myocardial infarction, or stroke across all ethnic groups (Cauley et al., 2008).
As explained in the first part of this review article, bone is constantly being remodeled during adult life to maintain structural integrity—specifically, to repair microcracks in the skeleton that occur with normal activities (Riggs et al., 2002). At the cell biological level, bone loss occurs following menopause in women or with aging in both sexes due to an imbalance favoring bone resorption by osteoclasts over bone formation by osteoblasts (Riggs et al., 2002). This increase in bone resorption can be monitored by biomarkers, e.g., serum C-terminal telopeptide of type I collagen (CTX) and tartrate resistant acid phosphatase (TRAP)5b, whereas biomarkers of bone formation are typically serum amino-terminal propeptide of type I collagen (PINP) and total osteocalcin (Naylor and Eastell, 2012).
It is this cellular basis for osteopenia and osteoporosis that explains why there are currently two types of drugs used to treat osteoporosis: those that inhibit bone resorption (antiresorptive agents) and those that stimulate bone formation (anabolic agents). The former include four bisphosphonates (alendronate, risedronate, ibandronate, and zoledronate) and an anti-RANKL antibody (denosumab), and the latter include derivatives of parathyroid hormone (PTH; teriparatide and abaloparatide) and an antibody against sclerostin (romozosumab) (Khosla and Hofbauer, 2017). Paradoxically, despite the availability of these drugs, many, if not most, patients with osteoporosis are not currently receiving appropriate treatment, even following an event as devastating as a hip fracture (Kim et al., 2016).
A major reason for the fact that so many osteoporotic patients are not adequately treated is the fear, on the part of patients and some physicians, of rare side effects related to bisphosphonates and denosumab. These include the uncommon occurrence of atypical femur fractures (AFFs) and osteonecrosis of the jaw (ONJ) (Khosla and Hofbauer, 2017). Although the absolute risk of AFFs ranges from 3.2 to 50 cases per 100,000 person years (Shane et al., 2014) and the estimated incidence of ONJ is <0.01% in bisphosphonate-treated patients (Khosla et al., 2007), fear of these rare side effects in the general population and even among physicians has led to strong reluctance on the part of many patients to accept appropriate treatment for reducing their fracture risk. The anabolic drugs are not associated with these risks. However, like derivatives of PTH, they require refrigeration at 4°C, which is cumbersome when traveling, and they involve subcutaneous injections rather than oral therapy, which are extremely expensive and necessitate follow-up treatment with a bisphosphonate or denosumab to maintain the gains in bone mass. Both factors have limited their broad use (Khosla and Hofbauer, 2017). Thus, there is a clear need for new approaches to reduce the burden of fractures, with perhaps the best one being primary prevention. Figure 3 provides a summary of current options for the treatment of osteoporosis and potential risks associated with each therapy.
Figure 3. Current clinical tools in the treatment of osteoporosis.
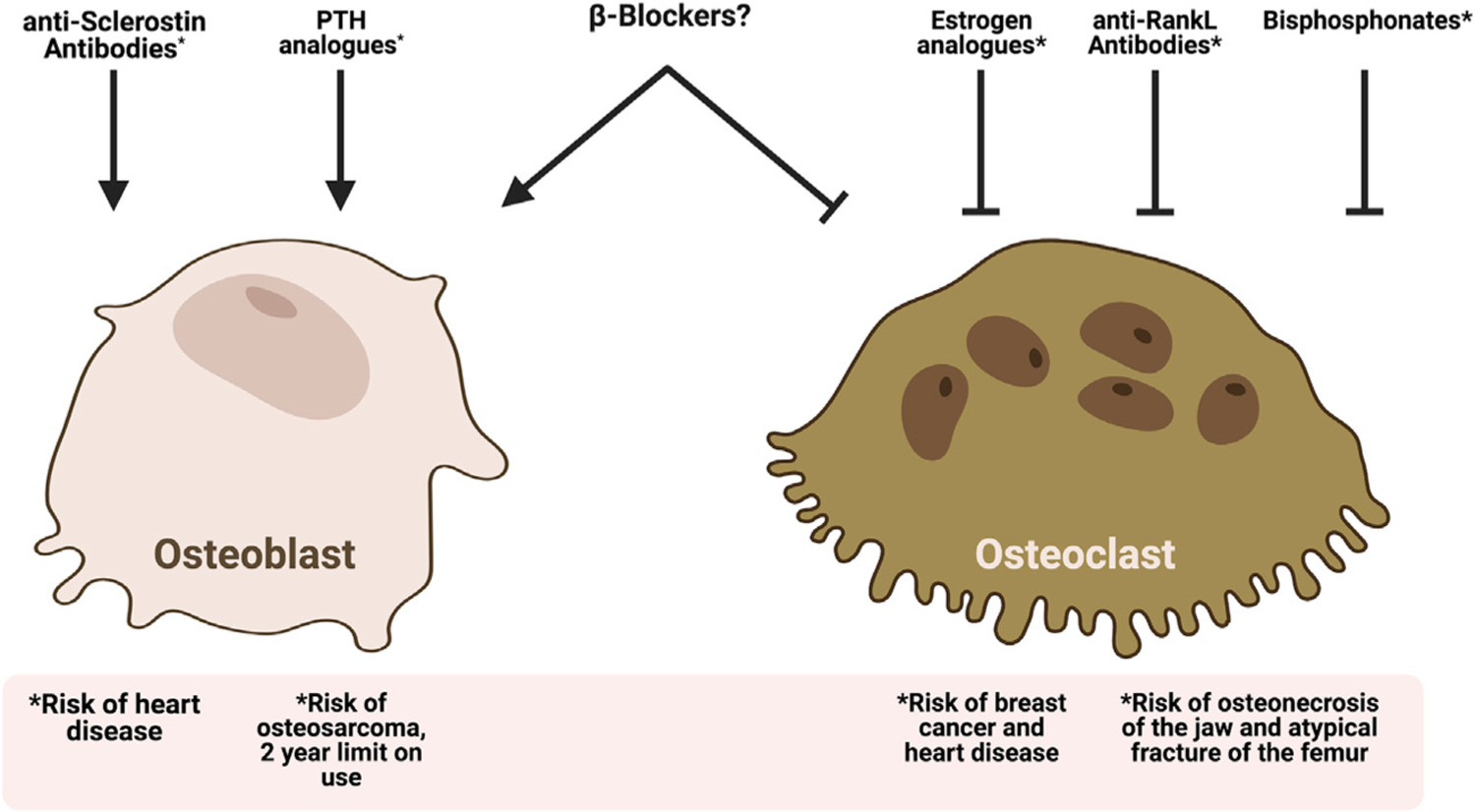
There are several classes of drugs listed in this figure that can improve outcomes in osteoporosis. Each of these drug classes either inhibits bone resorption by osteoclasts or stimulates osteoblastic bone formation to improve bone mass and quality in osteoporotic patients. In addition to acting on only one arm of bone remodeling, each of these drugs has significant adverse side effects or limits on duration of use as represented in the figure. *The adverse side effects caused by each osteoporosis treatment.
In terms of the effects of osteoporosis drugs on glucose homeostasis, the RANKL inhibitor, denosumab, did not have an effect on glucose and insulin levels in otherwise healthy, osteoporotic women (Ala et al., 2020). However, an analysis of the FREEDOM Trial did show a significant reduction in fasting plasma glucose in denosumaβ-treated women with diabetes or prediabetes not being pharmacologically treated for diabetes (Napoli et al., 2018). More recently, it was found that patients with type 2 diabetes treated with denosumab had a significant reduction in DPP4 and increase in GLP1 levels, leading to a greater improvement of HbA1c than subjects treated with bisphosphonates or calcium and vitamin D supplementation (Weivoda et al., 2020; Anastasilakis et al., 2021). Although preliminary, these studies agree with regulatory events identified in mouse models (Ferron et al., 2010).
It is also important to emphasize that the drugs shown in Figure 3 are generally only used for the treatment of established osteoporosis. A major problem in terms of primary prevention of osteoporosis is that although estrogen had been used in the past in women with osteopenia to prevent the development of osteoporosis, following the results of the Women’s Health Initiative demonstrating increased risks of cardiovascular events and breast cancer with long-term estrogen therapy (Rossouw et al., 2002), use of estrogen for this indication has largely been discontinued. In other words, there is currently no clear viable pharmacological option for postmenopausal women (or men) who have low bone mass or osteopenia and continue to lose bone over time. Given the large population these individuals represent, estimated to include >40 million Americans (Cosman et al., 2014), an effective and inexpensive intervention, particularly one that could decrease bone resorption and at the same time increase bone formation, could have an enormous clinical impact. Highlighting the importance of basic biology to inform the clinic and define novel and adapted therapies for degenerative diseases, the work presented above raised the question as to whether β-blockers could be used to prevent osteoporosis, translating the fundamental observations noted above into clinical practice.
OBSERVATIONAL EVIDENCE LINKING β-BLOCKER USE TO BMD AND FRACTURE RISK
As mentioned above, the evidence regarding the efficacy of β-blockers in the treatment of osteoporosis gathered in animal models raised the legitimate question of their clinical relevance. This novel question in the management of osteoporosis was first addressed through multiple observational studies examining the relationship between β-blocker use and BMD as well as fracture risk. For instance, and to cite only a few of these studies, use of β-blockers was associated with a 23% reduction in fracture risk in the UK General Practice Research Database (Schlienger et al., 2004); several more recent meta-analyses consistently found protective effects of β-blocker use on BMD and fracture risk (Toulis et al., 2014; Wiens et al., 2006; Yang et al., 2011, 2012). Importantly, the most recent of these analyses that pooled results of 16 other studies involving as many as 1,644,570 individuals found that the risk of any fracture was significantly reduced in those receiving β-blockers as compared to controls (random effects pooled effect size of 0.86, 95% confidence interval 0.78–0.93), with similar protective effects in women and men (Toulis et al., 2014). Unexpectedly, given the evidence noted above for a predominant role for β2-adrenergic receptors in regulating bone metabolism in rodents (Kajimura et al., 2011; Takeda et al., 2002), the analysis of Toulis et al. (2014), as well as an earlier meta-analysis (Yang et al., 2012), found that it was predominantly β1-selective blockers that were associated with higher lumbar spine and femur neck BMD and reduced fracture risk in humans.
Studies in patients with pheochromocytomas, which are catecholamine-producing neuroendocrine tumors that result in generalized stimulation of the β-adrenergic receptors (Mercado-Asis et al., 2018), also support the hypothesis that the sympathetic tone, acting through β-adrenergic receptors, regulates bone metabolism. Thus, bone resorption (serum CTX) and bone formation (serum PINP) marker changes were examined following adrenalectomy in 21 patients with pheochromocytomas. Although this study was confounded by the fact that 14 of the 21 patients were treated with β-blockers preoperatively, removal of the pheochromocytoma was associated with a decrease in serum CTX but no change in serum PINP and, hence, a positive effect on bone balance (an increase in the PINP/CTX ratio) (Veldhuis-Vlug et al., 2012). The finding that bone resorption decreased without a decrease in bone formation is entirely consistent with the animal findings indicating that β-adrenergic receptor activation results in an increase of bone resorption and a decrease of bone formation (Elefteriou et al., 2005; Kondo et al., 2001; Takeda et al., 2002). Consistent with these findings, patients with pheochromocytoma had reduced spine BMD and increased CTX levels as compared to control subjects (Kim et al., 2017a).
To further address this issue, sympathetic activity (measured directly using sensitive microneurography at the peroneal nerve) was related to bone microarchitecture (assessed by high resolution-peripheral quantitative computed tomography [HR-pQCT]) and bone turnover biomarkers in 23 women (10 premenopausal, 13 postmenopausal) aged between 20 and 72 years (Farr et al., 2012). In this study, sympathetic activity (bursts/100 heart beats) was 2.4-fold higher (p < 0.001) in postmenopausal compared with premenopausal women. Furthermore, in the two groups combined, sympathetic activity was inversely correlated with trabecular bone volume fraction (BV/TV; age-adjusted Spearman correlation, r = −0.55, p < 0.01) and thickness (TbTh; r = −0.59, p < 0.01) and positively correlated with trabecular separation (TbSp; r = +0.45, p < 0.05) (Farr et al., 2012). Sympathetic activity was also negatively correlated with serum PINP levels in postmenopausal women (r = −0.65, p < 0.05). Although preliminary and limited to a small number of individuals, these findings are of great importance as they represented the first demonstration in humans of a relationship between directly measured sympathetic activity and bone microstructure and turnover. These findings were recently extended to evaluate the relationship of β-blocker use to bone microarchitecture in a population-based sample (Khosla et al., 2018). From a previously described population-based cohort (Khosla et al., 2006a, 2006b), 67 individuals over age 50 years were identified who had used β-blockers for at least 1 year over the past 5 years; of these, 63 were on β1-selective blockers (atenolol or metoprolol). As there was an insufficient number of individuals on β-non-selective blockers (propranolol; n = 4) and given the observations made by Toulis et al. (2014) and Yang et al. (2012), the analysis was focused on the β1-selective blocker users (atenolol or metoprolol), which were more numerous (n = 63). The findings obtained were remarkably consistent with previous data as they demonstrated that trabecular microarchitectural parameters assessed by HR-pQCT were significantly better in the β1-selective blocker users as compared to nonusers, with similar trends for the cortical parameters.
DIRECT INTERVENTIONAL EVIDENCE LINKING β-BLOCKER USE TO BONE METABOLISM IN HUMANS
The body of work presented above was a strong incentive to establish causality between sympathetic activity and bone metabolism in a “proof-of-concept” interventional study (Khosla et al., 2018). To do so, we recruited 165 postmenopausal women and randomized them to one of five treatment groups for 20 weeks: (1) placebo, (2) 20 mg BID propranolol (β-adrenergic receptor non-selective), (3) 40 mg BID propranolol, (4) 50 mg/day atenolol (β1-adrenergic receptor selective), and (5) 5 mg/day nebivolol (highly β1-adrenergic receptor selective). In total, 155 women received the allocated intervention and 129 completed the full 20 weeks of the study. The propranolol doses were chosen based on animal data noted above showing that lower doses of propranolol had greater skeletal efficacy than higher doses (Bonnet et al., 2006; Sato et al., 2010). In contrast, a previous human study that had used a rather high dose, 160 mg/day, found no significant effects of β-blockers on bone turnover (Reid et al., 2005). In view of these observations, we chose to use 50% and 25% of the previously ineffective 160 mg/day dose (Reid et al., 2005); these doses span the range of propranolol dosing for cardiovascular effects. Moreover, as no clinically available β2-adrenergic receptor selective antagonists exist, we took advantage of the β1-adrenergic receptor selectivity gradient of these drugs (propranolol [non-selective] ≪ atenolol [relatively β1-selective/some β2-adrenergic receptor antagonism] < nebivolol [highly β1-adrenergic receptor selective]) (Ladage et al., 2013; Nuttall et al., 2003) to define the β-adrenergic receptor selectivity for the sympathetic regulation of bone remodeling in humans.
Figure 4 shows the changes over 20 weeks in the DXA BMD at the ultradistal and distal radius sites observed in this pilot study. Despite the relatively short duration of the intervention (only 20 weeks, as compared to the typical trial for osteoporosis that involves treatment for 2–3 years), ultradistal radius BMD increased significantly following treatment with atenolol (3.6% relative to placebo) and nebivolol (2.9% relative to placebo) (Figure 4A). Although it did not reach statistical significance, a similar pattern of changes was seen at the distal radius (Figure 4B). As anticipated, because of the relatively small number of subjects (~30 per group) and short duration of the study, changes in lumbar spine or femur BMD were not significant (Khosla et al., 2018). This is most likely a function of dose or duration of treatment and does not appear to be due to selective effects of atenolol or nebivolol on peripheral versus central skeletal sites, since the previous observational studies described above (Yang et al., 2011) have found higher BMD by DXA at central sites (lumbar spine and femur neck) in β1-selective blocker users as compared to nonusers.
Figure 4. Effects of β-blockers on BMD and bone turnover markers in humans.
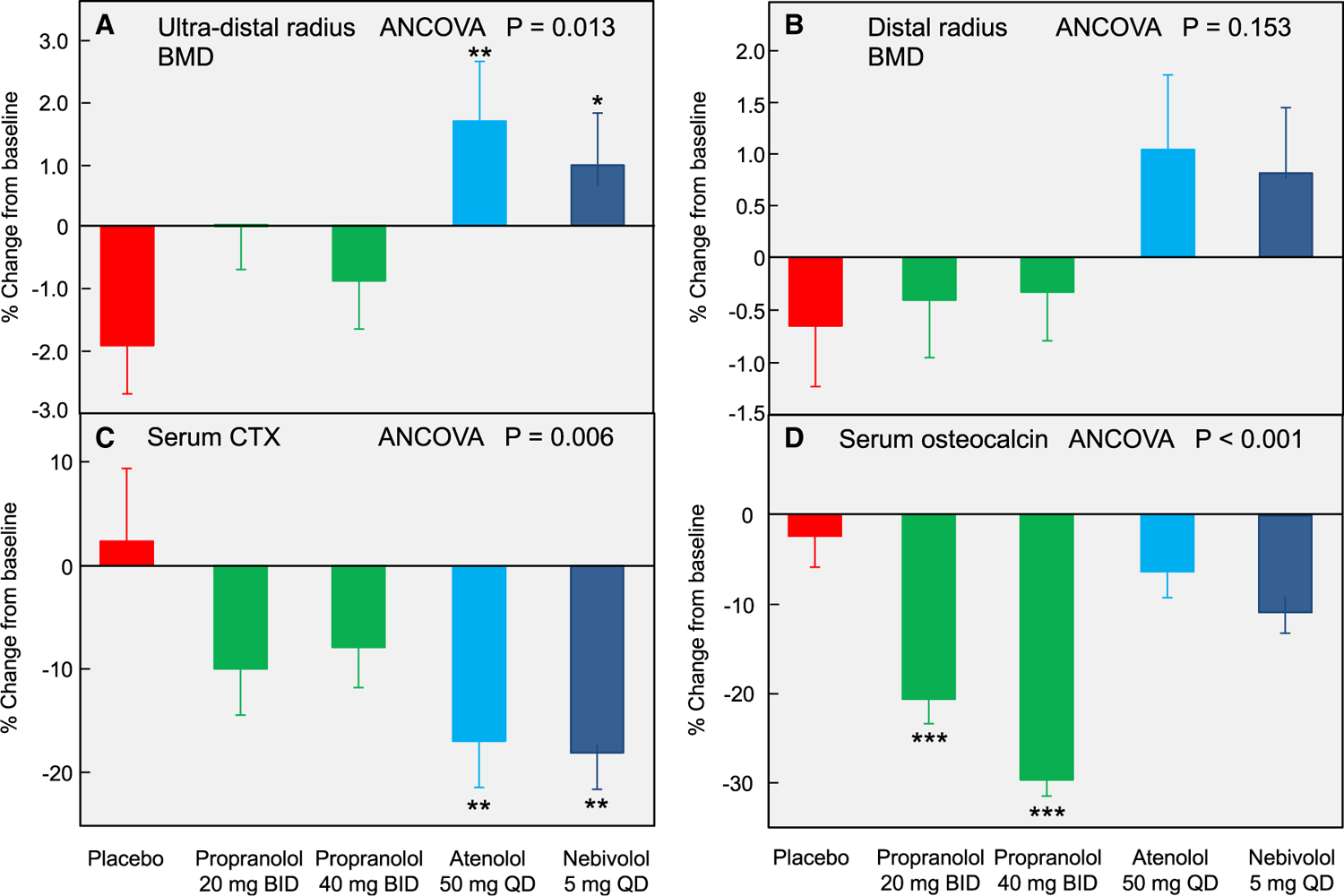
Percent change from baseline following 20 weeks of treatment in (A) ultradistal radius and (B) distal radius BMD, as well as serum levels of (C) CTX and (D) total osteocalcin; ANCOVA p values are indicated, and when these were <0.05, individual groups were compared with placebo using the Dunnett’s two-tailed t test; *p < 0.05; **p < 0.01; ***p < 0.001. Data are mean ± SEM. Adapted from Khosla et al. (2018).
Figures 4C and 4D show the changes in the bone turnover markers in the 5 groups following 20 weeks of the interventions. In terms of the bone resorption markers, serum CTX (Figure 4A) decreased significantly following treatment with atenolol (19.5% relative to placebo) and nebivolol (20.6% relative to placebo), but not following treatment with either dose of propranolol. Serum TRAP5b levels were reduced following treatment with atenolol and nebivolol (by 13.6% and 15.0%, respectively, relative to placebo). Propranolol at 20 mg BID did significantly reduce serum TRAP5b levels and nearly significantly with the 40 mg BID dose (p = 0.066). With regard to the bone formation markers, serum PINP levels did not change relative to placebo with either dose of propranolol but decreased significantly following treatment with atenolol and nebivolol, likely due to the known “coupling” between bone resorption and bone formation (Khosla, 2012). The changes in another bone formation marker, serum total osteocalcin levels, are shown in Figure 4D. Relative to placebo, neither atenolol nor nebivolol significantly reduced serum osteocalcin levels; by contrast, both doses of propranolol markedly decreased serum osteocalcin. Collectively, these data demonstrate that atenolol and nebivolol, but not propranolol, consistently reduce bone resorption; although serum PINP was reduced with both β1-selective blockers, the lack of change in osteocalcin, particularly with atenolol, raises the possibility of reductions in bone resorption with a maintenance of bone formation by these agents, which would overcome the coupling effects of bone resorption and bone formation. We should note, however, that these human studies do not establish whether the effects of β-blockers on bone are mediated centrally, peripherally, or both, and additional studies are needed to address this issue.
Based on these findings, we are now conducting an NIH-funded multisite phase 3 double-blinded randomized placebo-controlled trial (atenolol for the prevention of osteoporosis [APO], NCT04905277) to test the efficacy, over 2 years, of atenolol in preventing bone loss in postmenopausal women. If results of this prospective study confirm and extend the ones observed in the pilot study presented above, then β-blockers could fill a crucial clinical need in the primary prevention of osteoporosis, as depicted in Figure 5. As noted earlier, this clinical need was previously filled, in large part, by estrogen replacement therapy. However, estrogen is now generally only used for the short-term relief of vasomotor or genitourinary symptoms due to concerns regarding risks of coronary heart disease, stroke, venous thromboembolism, and dementia with long-term estrogen therapy (NAMS, 2018). By contrast, β-blockers lack these potential risks and are generally well tolerated by most patients; indeed, an estimated 60 million or more prescriptions for these agents are written annually (Cruickshank, 2017). As such, atenolol (or other β-blockers) would be preferred alternatives to estrogen for the primary prevention of osteoporosis. Moreover, a positive finding from this definitive trial would represent a remarkable straight line from the fundamental bone biology observations on the sympathetic regulation of bone mass made in rodents to observational and then interventional studies in humans. This could lead eventually to a radical change in clinical practice.
Figure 5. Potential clinical niche for a β-blocker for the primary prevention of osteoporosis.
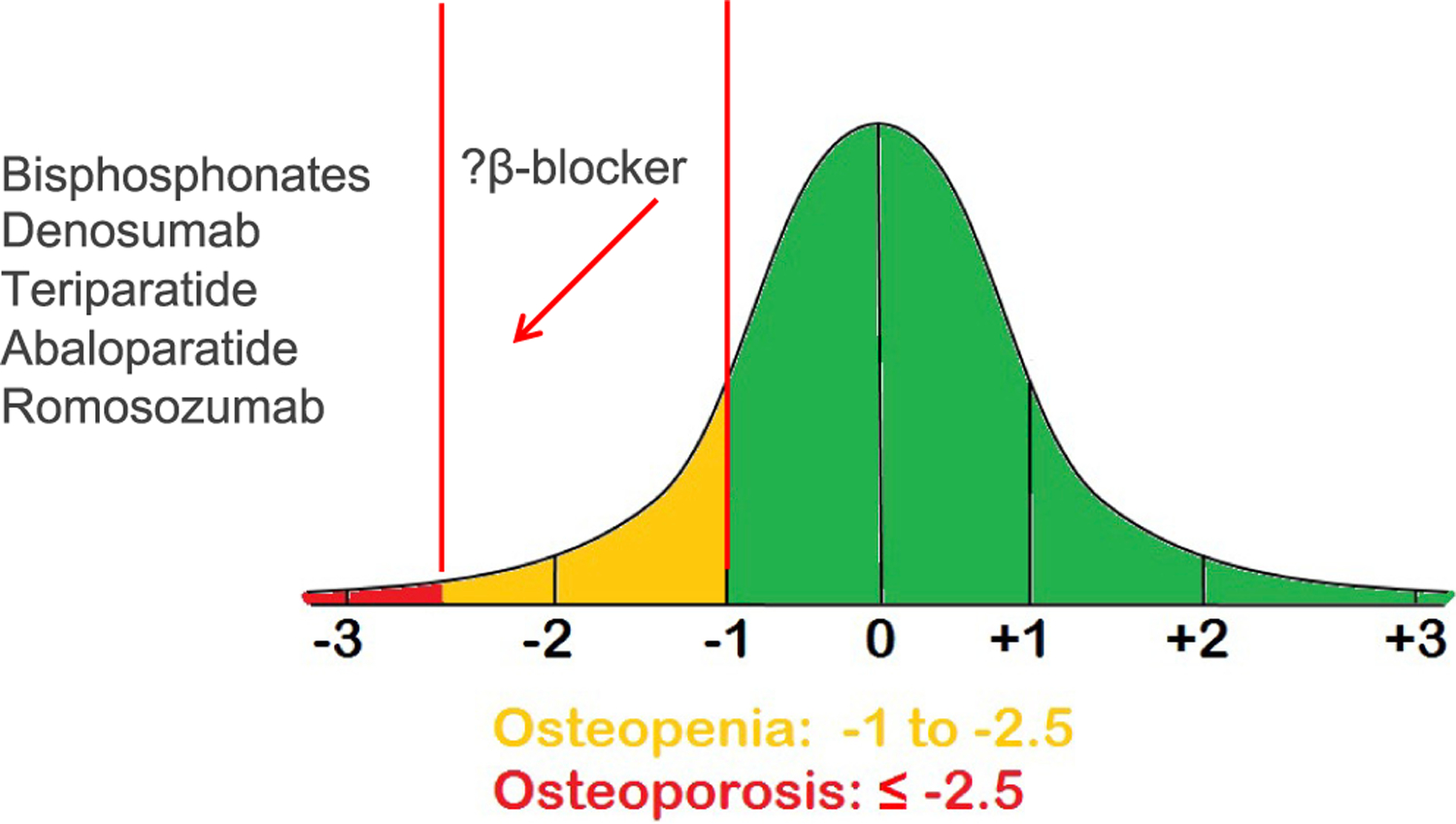
Although the established osteoporosis drugs (bisphosphonates, denosumab, teriparatide, abaloparatide, and Romosozumab) are prescribed for the treatment of established osteoporosis, following the results of the Women’s Health Initiative, there is virtually no viable option for the primary prevention of osteoporosis. β-blockers could potentially fill this niche.
SUMMARY AND PERSPECTIVES
We have summarized here the journey of a fundamental observation in bone biology that grew out of extensive mechanistic studies examining the relationships between energy metabolism and bone physiology. Specifically, this comprises the sympathetic control of bone mass accrual in rodents and the confirmation of its existence in humans through observational studies followed by its subsequent validation through a direct interventional clinical study leading, in turn, to a definitive, multisite clinical trial. From a clinical point of view, the high bone mass observed in mice, rats, and humans deprived of leptin signaling because it developed in the face of an absence of any gonadal functions is more than a medical exception; it was an opportunity to seize. The hope was that the identification of a cellular and molecular mechanism whereby leptin signaling inhibits bone mass accrual could be harnessed to reproduce this high bone mass in the face of an absence of gonadal functions to prevent if not to treat osteoporosis. To be clear, neither the complexities of the mechanisms whereby leptin regulates bone mass accrual nor the entire lexicon underlying the crosstalk between energy metabolism and bone physiology have been identified and deciphered.
Nevertheless, and beyond the intricacies of the road map of leptin in the brain, the identification of the sympathetic nervous system as a peripheral mediator of leptin regulation of bone mass could not have been timelier. This is because it involves a pathway that has been so thoroughly studied and used pharmacologically, and it emerged at a time when the pharmacology of osteoporosis was entering a crisis. This finding appeared as a unique therapeutic opportunity. What is now at stake is nothing less than the repurposing of a class of drugs already on the market for decades and for which the safety has been tested to prevent osteoporosis altogether. Of course, the identification of β-blockers as a viable option for the prevention of osteoporosis comes with its own set of questions, but again, those can be addressed quickly because the drugs are already approved for other indications. Should β-blockers be used as a curative or a preventative treatment? If it is curative, how do β-blockers compare with other therapeutic options in terms of efficacy, safety, and cost? Should these agents ideally target in humans Adrβ1 only or both Adrβ1 and Adrβ2? What will be the best dose and for how long should they be used for? Should they be used alone or in combination with other drugs? If yes, which one? On the other hand, if β-blockers emerge as a true preventative option for osteoporosis, could long-acting compounds be designed, could they be targeted better to the skeleton to minimize nonskeletal (e.g., cardiovascular) effects, could they change the face of clinical medicine when it comes to the care of degenerative bone diseases, and could we hope that at least in developed countries it could lead to the prevention of this crippling disease that is also, we should not forget, a heavy burden in terms of public health? These important questions notwithstanding, this evolving story does represent a true effort at translating unexpected fundamental biology of bone to the clinic.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AR073180-03, R01 DE027887-04, and P01 AG032959-12 (G.K.) and P01 AG063413 and R01 AG065154 (S.K.). The authors thank Dr. J. Berger for numerous suggestions. We apologize to those whose work was not cited due to space constraints.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahn JD, Dubern B, Lubrano-Berthelier C, Clement K, and Karsenty G (2006). Cart overexpression is the only identifiable cause of high bone mass in melanocortin 4 receptor deficiency. Endocrinology 147, 3196–3202. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, et al. (2005). Osteochondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 102, 14665–14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ala M, Jafari RM, and Dehpour AR (2020). Diabetes mellitus and osteoporosis correlation: challenges and hopes. Curr. Diabetes Rev 16, 984–1001. [DOI] [PubMed] [Google Scholar]
- Anastasilakis AD, Tsourdi E, Tabacco G, Naciu AM, Napoli N, Vescini F, and Palermo A (2021). The impact of antiosteoporotic drugs on glucose metabolism and fracture risk in diabetes: good or bad news? J. Clin. Med 10, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldock PA, Lee NJ, Driessler F, Lin S, Allison S, Stehrer B, Lin EJ, Zhang L, Enriquez RF, Wong IP, et al. (2009). Neuropeptide Y knockout mice reveal a central role of NPY in the coordination of bone mass to body weight. PLoS One 4, e8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JM, Singh P, Khrimian L, Morgan DA, Chowdhury S, Arteaga-Solis E, Horvath TL, Domingos AI, Marsland AL, Yadav VK, et al. (2019). Mediation of the acute stress response by the skeleton. Cell Metab. 30, 890–902.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blüher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, and Kahn CR (2002). Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell 3, 25–38. [DOI] [PubMed] [Google Scholar]
- Bonnet N, Laroche N, Vico L, Dolleans E, Benhamou CL, and Courteix D (2006). Dose effects of propranolol on cancellous and cortical bone in ovariectomized adult rats. J. Pharmacol. Exp. Ther 318, 1118–1127. [DOI] [PubMed] [Google Scholar]
- Brüning JC, Michael MD, Winnay JN, Hayashi T, Hörsch D, Accili D, Goodyear LJ, and Kahn CR (1998). A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569. [DOI] [PubMed] [Google Scholar]
- Cauley JA, Wampler NS, Barnhart JM, Wu L, Allison M, Chen Z, Hendrix S, Robbins J, and Jackson RD; Women’s Health Initiative Observational Study (2008). Incidence of fractures compared to cardiovascular disease and breast cancer: the Women’s Health Initiative Observational Study. Osteoporos. Int 19, 1717–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, and Lindsay R; National Osteoporosis Foundation (2014). Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos. Int 25, 2359–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank JM (2017). The role of beta-blockers in the treatment of hyper-tension. Adv. Exp. Med. Biol 956, 149–166. [DOI] [PubMed] [Google Scholar]
- Dirckx N, Tower RJ, Mercken EM, Vangoitsenhoven R, Moreau-Triby C, Breugelmans T, Nefyodova E, Cardoen R, Mathieu C, Van der Schueren B, et al. (2018). Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. J. Clin. Invest 128, 1087–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirckx N, Moorer MC, Clemens TL, and Riddle RC (2019). The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol 15, 651–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P (2020). Bone regulation of insulin secretion and glucose homeostasis. Endocrinology 161, bqaa149. [DOI] [PubMed] [Google Scholar]
- Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, et al. (1996). Increased bone formation in osteocalcin-deficient mice. Nature 382, 448–452. [DOI] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, and Karsenty G (1997). Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89, 747–754. [DOI] [PubMed] [Google Scholar]
- Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, and Karsenty G (1999). A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 13, 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, and Karsenty G (2000). Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100, 197–207. [DOI] [PubMed] [Google Scholar]
- Elefteriou F, Takeda S, Liu X, Armstrong D, and Karsenty G (2003). Monosodium glutamate-sensitive hypothalamic neurons contribute to the control of bone mass. Endocrinology 144, 3842–3847. [DOI] [PubMed] [Google Scholar]
- Elefteriou F, Takeda S, Ebihara K, Magre J, Patano N, Kim CA, Ogawa Y, Liu X, Ware SM, Craigen WJ, et al. (2004). Serum leptin level is a regulator of bone mass. Proc. Natl. Acad. Sci. USA 101, 3258–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, Kondo H, Richards WG, Bannon TW, Noda M, et al. (2005). Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434, 514–520. [DOI] [PubMed] [Google Scholar]
- Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, and Karsenty G (2006). ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 4, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estell EG, Le PT, Vegting Y, Kim H, Wrann C, Bouxsein ML, Nagano K, Baron R, Spiegelman BM, and Rosen CJ (2020). Irisin directly stimulates osteoclastogenesis and bone resorption in vitro and in vivo. eLife 9, e58172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, and O’Rahilly S (2000). Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Invest 106, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr JN, Charkoudian N, Barnes JN, Monroe DG, McCready LK, Atkinson EJ, Amin S, Melton LJ 3rd, Joyner MJ, and Khosla S (2012). Relationship of sympathetic activity to bone microstructure, turnover, and plasma osteopontin levels in women. J. Clin. Endocrinol. Metab 97, 4219–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A, Ducy P, and Karsenty G (2010). Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM, and Halaas JL (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763–770. [DOI] [PubMed] [Google Scholar]
- Fu L, Patel MS, Bradley A, Wagner EF, and Karsenty G (2005). The molecular clock mediates leptin-regulated bone formation. Cell 122, 803–815. [DOI] [PubMed] [Google Scholar]
- Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, Faugere M-C, Aja S, Hussain MA, Brüning JC, et al. (2010). Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T (2011). Hepcidin and iron regulation, 10 years later. Blood 117, 4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber CB, Krause WC, Wang L, Bayrer JR, Li A, Schmitz M, Fields A, Ford B, Zhang Z, Reid MS, et al. (2019). Estrogen signaling in arcuate Kiss1 neurons suppresses a sex-dependent female circuit promoting dense strong bones. Nat. Commun 10, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer LC, Brueck CC, Singh SK, and Dobnig H (2007). Osteoporosis in patients with diabetes mellitus. J. Bone Miner. Res 22, 1317–1328. [DOI] [PubMed] [Google Scholar]
- Hu B, Lv X, Chen H, Xue P, Gao B, Wang X, Zhen G, Crane JL, Pan D, Liu S, et al. (2020). Sensory nerves regulate mesenchymal stromal cell lineage commitment by tuning sympathetic tones. J. Clin. Invest 130, 3483–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iepsen EW, Lundgren JR, Hartmann B, Pedersen O, Hansen T, Jørgensen NR, Jensen JE, Holst JJ, Madsbad S, and Torekov SS (2015). GLP-1 receptor agonist treatment increases bone formation and prevents bone loss in weight-reduced obese women. J. Clin. Endocrinol. Metab 100, 2909–2917. [DOI] [PubMed] [Google Scholar]
- Kajimura D, Hinoi E, Ferron M, Kode A, Riley KJ, Zhou B, Guo XE, and Karsenty G (2011). Genetic determination of the cellular basis of the sympathetic regulation of bone mass accrual. J. Exp. Med 208, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura D, Lee HW, Riley KJ, Arteaga-Solis E, Ferron M, Zhou B, Clarke CJ, Hannun YA, DePinho RA, Guo XE, et al. (2013). Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1. Cell Metab. 17, 901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner CM, Esen E, Okunade AL, Patterson BW, and Long F (2015). Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J. Clin. Invest 125, 551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G, and Ferron M (2012). The contribution of bone to whole-organism physiology. Nature 481, 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S (2012). Odanacatib: location and timing are everying. J. Bone Miner. Res 27, 506–508. [DOI] [PubMed] [Google Scholar]
- Khosla S, and Hofbauer LC (2017). Osteoporosis treatment: recent developments and ongoing challenges. Lancet Diabetes Endocrinol. 5, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S, and Shane E (2016). A crisis in the treatment of osteoporosis. J. Bone Miner. Res 31, 1485–1487. [DOI] [PubMed] [Google Scholar]
- Khosla S, Melton LJ 3rd, Achenbach SJ, Oberg AL, and Riggs BL (2006a). Hormonal and biochemical determinants of trabecular microstructure at the ultradistal radius in women and men. J. Clin. Endocrinol. Metab 91, 885–891. [DOI] [PubMed] [Google Scholar]
- Khosla S, Riggs BL, Atkinson EJ, Oberg AL, McDaniel LJ, Holets M, Peterson JM, and Melton LJ 3rd (2006b). Effects of sex and age on bone microstructure at the ultradistal radius: a population-based noninvasive in vivo assessment. J. Bone Miner. Res 21, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S, Burr D, Cauley J, Dempster DW, Ebeling PR, Felsenberg D, Gagel RF, Gilsanz V, Guise T, Koka S, et al. (2007). Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J. Bone Miner. Res 22, 1479–1491. [DOI] [PubMed] [Google Scholar]
- Khosla S, Cauley JA, Compston J, Kiel DP, Rosen C, Saag KG, and Shane E (2017). Addressing the crisis in the treatment of osteoporosis: a path forward. J. Bone Miner. Res 32, 424–430. [DOI] [PubMed] [Google Scholar]
- Khosla S, Drake MT, Volkman TL, Thicke BS, Achenbach SJ, Atkinson EJ, Joyner MJ, Rosen CJ, Monroe DG, and Farr JN (2018). Sympathetic β 1-adrenergic signaling contributes to regulation of human bone metabolism. J. Clin. Invest 128, 4832–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrimian L, Obri A, Ramos-Brossier M, Rousseaud A, Moriceau S, Nicot AS, Mera P, Kosmidis S, Karnavas T, Saudou F, et al. (2017). Gpr158 mediates osteocalcin’s regulation of cognition. J. Exp. Med 214, 2859–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Kim DH, Mogun H, Eddings W, Polinski JM, Franklin JM, and Solomon DH (2016). Impact of the U.S. Food and Drug Administration’s safety-related announcements on the use of bisphosphonates after hip fracture. J. Bone Miner. Res 31, 1536–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Kwak MK, Ahn SH, Kim H, Lee SH, Song KH, Suh S, Kim JH, and Koh JM (2017a). Lower bone mass and higher bone resorption in pheochromocytoma: importance of sympathetic activity on human bone. J. Clin. Endocrinol. Metab 102, 2711–2718. [DOI] [PubMed] [Google Scholar]
- Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE, Kushwaha P, Ryan KA, Goh BC, Scafidi S, Pickett JE, et al. (2017b). Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex-and diet-dependent manner. JCI Insight 2, e92704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, Zhou C, Chou J, Parkman V-JA, and Novick SJ (2018). Irisin mediates effects on bone and fat via aV integrin receptors. Cell 175, 1756–1768.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, and Mangelsdorf DJ (2019). A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metab. 29, 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo A, Mogi M, Koshihara Y, and Togari A (2001). Signal transduction system for interleukin-6 and interleukin-11 synthesis stimulated by epinephrine in human osteoblasts and human osteogenic sarcoma cells. Biochem. Pharmacol 61, 319–326. [DOI] [PubMed] [Google Scholar]
- Ladage D, Schwinger RH, and Brixius K (2013). Cardio-selective beta-blocker: pharmacological evidence and their influence on exercise capacity. Cardiovasc. Ther 31, 76–83. [DOI] [PubMed] [Google Scholar]
- Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, et al. (2007). Endocrine regulation of energy metabolism by the skeleton. Cell 130, 456–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NJ, Ali N, Zhang L, Qi Y, Clarke I, Enriquez RF, Brzozowska M, Lee IC, Rogers MJ, Laybutt DR, et al. (2018). Osteoglycin, a novel coordinator of bone and glucose homeostasis. Mol. Metab 13, 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Frey JL, Wong GW, Faugere MC, Wolfgang MJ, Kim JK, Riddle RC, and Clemens TL (2016). Glucose transporter-4 facilitates insulin-stimulated glucose uptake in osteoblasts. Endocrinology 157, 4094–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galán-Díez M, Lacampagne A, Mitchell SJ, Mattison JA, Chen Y, et al. (2016). Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab. 23, 1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado-Asis LB, Wolf KI, Jochmanova I, and Taïeb D (2018). Pheochromocytoma: a genetic and diagnostic update. Endocr. Pract 24, 78–90. [DOI] [PubMed] [Google Scholar]
- Mizokami A, Yasutake Y, Gao J, Matsuda M, Takahashi I, Takeuchi H, and Hirata M (2013). Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS One 8, e57375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizokami A, Yasutake Y, Higashi S, Kawakubo-Yasukochi T, Chishaki S, Takahashi I, Takeuchi H, and Hirata M (2014). Oral administration of osteocalcin improves glucose utilization by stimulating glucagon-like peptide-1 secretion. Bone 69, 68–79. [DOI] [PubMed] [Google Scholar]
- Mizokami A, Mukai S, Gao J, Kawakubo-Yasukochi T, Otani T, Takeuchi H, Jimi E, and Hirata M (2020). GLP-1 signaling is required for improvement of glucose tolerance by osteocalcin. J. Endocrinol 244, 285–296. [DOI] [PubMed] [Google Scholar]
- Monod J (1970). Le Hasard et la nécessité: essai sur la philosophie naturelle de la biologie naturelle (Le Seuil).
- Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z, Huang Y, Zong H, Friedman RA, Barasch J, et al. (2017). MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl KJ, DeMambro VE, Barlow D, Olshan D, Nagano K, Baron R, Rosen CJ, and Houseknecht KL (2015). Propranolol attenuates risperi-done-induced trabecular bone loss in female mice. Endocrinology 156, 2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, and Münzberg H (2008). Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol 70, 537–556. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, and de Crombrugghe B (2002). The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29. [DOI] [PubMed] [Google Scholar]
- NAMS (2018). The 2017 hormone therapy position statement of the North American Menopause Society. Menopause 25, 1362–1387. [DOI] [PubMed] [Google Scholar]
- Napoli N, Pannacciulli N, Vittinghoff E, Crittenden D, Yun J, Wang A, Wagman R, and Schwartz AV (2018). Effect of denosumab on fasting glucose in women with diabetes or prediabetes from the FREEDOM Trial. Diabetes Metab. Res. Rev 34, e2991. [DOI] [PubMed] [Google Scholar]
- Naylor K, and Eastell R (2012). Bone turnover markers: use in osteoporosis. Nat. Rev. Rheumatol 8, 379–389. [DOI] [PubMed] [Google Scholar]
- Nectow AR, Schneeberger M, Zhang H, Field BC, Renier N, Azevedo E, Patel B, Liang Y, Mitra S, and Tessier-Lavigne M (2017). Identification of a brainstem circuit controlling feeding. Cell 170, 429–442.e11. [DOI] [PubMed] [Google Scholar]
- Neuman WF, Neuman MW, and Myers CR (1979). Blood: bone disequilibrium. III. Linkage between cell energetics and Ca fluxes. Am. J. Physiol 236, C244–C248. [DOI] [PubMed] [Google Scholar]
- Nuttall SL, Routledge HC, and Kendall MJ (2003). A comparison of the beta1-selectivity of three beta1-selective beta-blockers. J. Clin. Pharm. Ther 28, 179–186. [DOI] [PubMed] [Google Scholar]
- Ortuño MJ, Robinson ST, Subramanyam P, Paone R, Huang YY, Guo XE, Colecraft HM, Mann JJ, and Ducy P (2016). Serotonin-reuptake inhibitors act centrally to cause bone loss in mice by counteracting a local anti-resorptive effect. Nat. Med 22, 1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortuño MJ, Schneeberger M, Ilanges A, Marchildon F, Pellegrino K, Friedman JM, and Ducy P (2021). Melanocortin 4 receptor stimulation prevents antidepressant-associated weight gain in mice caused by long-term fluoxetine exposure. J. Clin. Invest 131, e151976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, Hermo L, Suarez S, Roth BL, Ducy P, et al. (2011). Endocrine regulation of male fertility by the skeleton. Cell 144, 796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoda P, Egermann M, Schnell JC, Priemel M, Schilling AF, Alini M, Schinke T, Rueger JM, Schneider E, Clarke I, et al. (2006). Leptin inhibits bone formation not only in rodents, but also in sheep. J. Bone Miner. Res 21, 1591–1599. [DOI] [PubMed] [Google Scholar]
- Rached MT, Kode A, Xu L, Yoshikawa Y, Paik JH, DePinho RA, and Kousteni S (2010). FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 11, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid IR, Lucas J, Wattie D, Horne A, Bolland M, Gamble GD, Davidson JS, and Grey AB (2005). Effects of a beta-blocker on bone turnover in normal postmenopausal women: a randomized controlled trial. J. Clin. Endocrinol. Metab 90, 5212–5216. [DOI] [PubMed] [Google Scholar]
- Reseland JE, Syversen U, Bakke I, Qvigstad G, Eide LG, Hjertner O, Gordeladze JO, and Drevon CA (2001). Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization. J. Bone Miner. Res 16, 1426–1433. [DOI] [PubMed] [Google Scholar]
- Riggs BL, Khosla S, and Melton LJ 3rd (2002). Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev 23, 279–302. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SAA, Howard BV, Johnson KC, et al. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288, 321–333. [DOI] [PubMed] [Google Scholar]
- Rowe GC, Vialou V, Sato K, Saito H, Yin M, Green TA, Lotinun S, Kveiborg M, Horne WC, Nestler EJ, et al. (2012). Energy expenditure and bone formation share a common sensitivity to AP-1 transcription in the hypothalamus. J. Bone Miner. Res 27, 1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatakos G, Sims NA, Chen J, Aoki K, Kelz MB, Amling M, Bouali Y, Mukhopadhyay K, Ford K, Nestler EJ, et al. (2000). Overexpression of DFosB transcription factor (s) increases bone formation and inhibits adipo-genesis. Nat. Med 6, 985–990. [DOI] [PubMed] [Google Scholar]
- Sato S, Hanada R, Kimura A, Abe T, Matsumoto T, Iwasaki M, Inose H, Ida T, Mieda M, Takeuchi Y, et al. (2007). Central control of bone remodeling by neuromedin U. Nat. Med 13, 1234–1240. [DOI] [PubMed] [Google Scholar]
- Sato T, Arai M, Goto S, and Togari A (2010). Effects of propranolol on bone metabolism in spontaneously hypertensive rats. J. Pharmacol. Exp. Ther 334, 99–105. [DOI] [PubMed] [Google Scholar]
- Schlienger RG, Kraenzlin ME, Jick SS, and Meier CR (2004). Use of beta-blockers and risk of fractures. JAMA 292, 1326–1332. [DOI] [PubMed] [Google Scholar]
- Schwartzman RJ (2000). New treatments for reflex sympathetic dystrophy. N. Engl. J. Med 343, 654–656. [DOI] [PubMed] [Google Scholar]
- Scott BL, and Glimcher MJ (1971). Distribution of glycogen in osteoblasts of the fetal rat. J. Ultrastruct. Res 36, 565–586. [DOI] [PubMed] [Google Scholar]
- Shane E, Burr D, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster DW, et al. (2014). Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone Mineral Research. J. Bone Miner. Res 29, 1–23. [DOI] [PubMed] [Google Scholar]
- Shen L, Sharma D, Yu Y, Long F, and Karner CM (2021). Biphasic regulation of glutamine consumption by WNT during osteoblast differentiation. J. Cell Sci 134, jcs251645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, and Karsenty G (2002). Leptin regulates bone formation via the sympathetic nervous system. Cell 111, 305–317. [DOI] [PubMed] [Google Scholar]
- Toulis KA, Hemming K, Stergianos S, Nirantharakumar K, and Bilezikian JP (2014). Beta-adrenergic receptor antagonists and fracture risk: a meta-analysis of selectivity, gender, and site-specific effects. Osteoporos. Int 25, 121–129. [DOI] [PubMed] [Google Scholar]
- Vaira S, Yang C, McCoy A, Keys K, Xue S, Weinstein EJ, Novack DV, and Cui X (2012). Creation and preliminary characterization of a leptin knockout rat. Endocrinology 153, 5622–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhuis-Vlug AG, El Mahdiui M, Endert E, Heijboer AC, Fliers E, and Bisschop PH (2012). Bone resorption is increased in pheochromocytoma patients and normalizes following adrenalectomy. J. Clin. Endocrinol. Metab 97, E2093–E2097. [DOI] [PubMed] [Google Scholar]
- Wagner EF (2010). Bone development and inflammatory disease is regulated by AP-1 (Fos/Jun). Ann. Rheum. Dis 69, i86–i88. [DOI] [PubMed] [Google Scholar]
- Wang X, Wei W, Krzeszinski JY, Wang Y, and Wan Y (2015). A liver-bone endocrine relay by IGFBP1 promotes osteoclastogenesis and mediates FGF21-induced bone resorption. Cell Metab. 22, 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS, and Karsenty G (2014a). Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J. Clin. Invest 124, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Hanna T, Suda N, Karsenty G, and Ducy P (2014b). Osteocalcin promotes beta-cell proliferation during development and adulthood through Gprc6a. Diabetes 63, 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, Takarada T, Lezaki T, Pessin JE, Hinoi E, et al. (2015). Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell 161, 1576–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weivoda MM, Chew CK, Monroe DG, Farr JN, Atkinson EJ, Geske JR, Eckhardt B, Thicke B, Ruan M, Tweed AJ, et al. (2020). Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat. Commun 11, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens M, Etminan M, Gill SS, and Takkouche B (2006). Effects of antihypertensive drug treatments on fracture outcomes: a meta-analysis of observational studies. J. Intern. Med 260, 350–362. [DOI] [PubMed] [Google Scholar]
- Xie D, Cheng H, Hamrick M, Zhong Q, Ding KH, Correa D, Williams S, Mulloy A, Bollag W, Bollag RJ, et al. (2005). Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone 37, 759–769. [DOI] [PubMed] [Google Scholar]
- Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, Confavreux C, Klemen-hagen KC, Tanaka KF, Gingrich JA, Guo XE, et al. (2009). A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell 138, 976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav VK, Oury F, Tanaka KF, Thomas T, Wang Y, Cremers S, Hen R, Krust A, Chambon P, and Karsenty G (2011). Leptin-dependent serotonin control of appetite: temporal specificity, transcriptional regulation, and therapeutic implications. J. Exp. Med 208, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, et al. (2004). ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry syndrome. Cell 117, 387–398. [DOI] [PubMed] [Google Scholar]
- Yang S, Nguyen ND, Center JR, Eisman JA, and Nguyen TV (2011). Association between beta-blocker use and fracture risk: the Dubbo Osteoporosis Epidemiology Study. Bone 48, 451–455. [DOI] [PubMed] [Google Scholar]
- Yang S, Nguyen ND, Eisman JA, and Nguyen TV (2012). Association between beta-blockers and fracture risk: a Bayesian meta-analysis. Bone 51, 969–974. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Itokawa T, Sridhar S, Ding KH, Xie D, Kang B, Bollag WB, Bollag RJ, Hamrick M, Insogna K, et al. (2007). Effects of glucose-dependent insulinotropic peptide on osteoclast function. Am. J. Physiol. Endocrinol. Metab 292, E543–E548. [DOI] [PubMed] [Google Scholar]
- Zoch ML, Abou DS, Clemens TL, Thorek DL, and Riddle RC (2016). In vivo radiometric analysis of glucose uptake and distribution in mouse bone. Bone Res. 4, 16004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Rohatgi N, Brestoff JR, Zhang Y, Scheller EL, Craft CS, Brodt MD, Migotsky N, Silva MJ, Harris CA, et al. (2019). Congenital lipodys-trophy induces severe osteosclerosis. PLoS Genet. 15, e1008244. [DOI] [PMC free article] [PubMed] [Google Scholar]