

Abstract
N‐terminal sequences are important sites for post‐translational modifications that alter protein localization, activity, and stability. Dipeptidyl peptidase 9 (DPP9) is a serine aminopeptidase with the rare ability to cleave off N‐terminal dipeptides with imino acid proline in the second position. Here, we identify the tumor‐suppressor BRCA2 as a DPP9 substrate and show this interaction to be induced by DNA damage. We present crystallographic structures documenting intracrystalline enzymatic activity of DPP9, with the N‐terminal Met1‐Pro2 of a BRCA21‐40 peptide captured in its active site. Intriguingly, DPP9‐depleted cells are hypersensitive to genotoxic agents and are impaired in the repair of DNA double‐strand breaks by homologous recombination. Mechanistically, DPP9 targets BRCA2 for degradation and promotes the formation of RAD51 foci, the downstream function of BRCA2. N‐terminal truncation mutants of BRCA2 that mimic a DPP9 product phenocopy reduced BRCA2 stability and rescue RAD51 foci formation in DPP9‐deficient cells. Taken together, we present DPP9 as a regulator of BRCA2 stability and propose that by fine‐tuning the cellular concentrations of BRCA2, DPP9 alters the BRCA2 interactome, providing a possible explanation for DPP9's role in cancer.
Keywords: BRCA2, DNA repair, DPP9, N‐degron, proteolysis
Subject Categories: DNA Replication, Recombination & Repair; Post-translational Modifications & Proteolysis
Dipeptidyl peptidase 9 triggers BRCA2 degradation and promotes repair of DNA double strand breaks by homologous recombination.
Introduction
N‐termini sequences are important sites for post‐translational modifications that alter protein localization, activity, and also stability via the N‐degron pathway that targets proteins with a destabilizing amino acid in the first N‐terminal position for proteasomal degradation (Varshavsky, 2019). Regulated degradation of cellular proteins is critical for the removal of damaged or mislocalized proteins, to establish optimal protein stoichiometry and to allow for changes in the cellular proteome in response to different stimuli. Among others, the N‐degron pathway regulates apoptosis (Ditzel et al, 2003; Piatkov et al, 2012; Eldeeb & Fahlman, 2014; Wang et al, 2017; Weaver et al, 2017), pyroptosis (Chui et al, 2019; Xu et al, 2019), glucose homeostasis (Chen et al, 2017), mitochondrial import (Finger et al, 2020), autophagy (Cha‐Molstad et al, 2017), G‐protein signaling (Lee et al, 2005; Park et al, 2015), and B‐cell signaling (Justa‐Schuch et al, 2016). The initiator methionine acts as an N‐Degron only in rare cases (Kim et al, 2014). More frequently, N‐degrons are formed following post‐translational modifications of the substrate such as N‐terminal acetylation (Hwang et al, 2010; Shemorry et al, 2013), N‐arginylation (Wang et al, 2009; Park et al, 2020), or limited proteolysis (Rao et al, 2001; Piatkov et al, 2012, 2014; Brower et al, 2013; Eldeeb & Fahlman, 2014; Weaver et al, 2017; Nguyen et al, 2019). Initial characterization of the N‐degron pathway had shown a highly stabilizing effect of the imino acid proline, both in the first or second N‐terminal position (Bachmair et al, 1986). N‐terminal prolines cannot be arginylated and are only rarely acetylated (Arnesen et al, 2009; Helbig et al, 2010). In yeast, however, gluconeogenic enzymes with N‐terminal prolines are rapidly degraded upon transition to glucose‐rich medium. Ubiquitination of these otherwise highly stable enzymes is carried out by multi‐subunit E3 ubiquitin ligases called the GID/CTLH complexes (Pro/N‐Recognins). These multi‐subunit E3 complexes associate in response to extracellular stimuli (Santt et al, 2008; Chen et al, 2017; Dong et al, 2018; Menssen et al, 2018; Melnykov et al, 2019; Qiao et al, 2020).
Dipeptidyl peptidase DPP9 and its homolog DPP8 are two intracellular serine aminopeptidases with the rare ability to cleave postproline (Xaa‐Pro/Ala↓Zaa) (Geiss‐Friedlander et al, 2009; Zhang et al, 2015). DPP9 is more abundant than DPP8 and its depletion leads to a large reduction in the capacity of cells to process proline‐containing peptides (Geiss‐Friedlander et al, 2009). Two isoforms of DPP9 are expressed in cells, DPP9‐S that localizes to the cytoplasm (Ajami et al, 2004) and DPP9‐L that contains an N‐terminal nuclear localization signal targeting the protease to the nucleus (Justa‐Schuch et al, 2014). DPP9 plays a role in neonatal survival (Gall et al, 2013) and in the immune response (Geiss‐Friedlander et al, 2009; Justa‐Schuch et al, 2016; Okondo et al, 2017, 2018; Johnson et al, 2018; de Vasconcelos et al, 2019). Deregulation of DPP9 is connected with tumorigenicity (Spagnuolo et al, 2013; Smebye et al, 2017; Tang et al, 2017; Saso et al, 2020), albeit the underlying mechanisms are poorly understood. Recently, we showed that DPP9 acts upstream to the N‐degron pathway by processing the precursor of the mitochondria protein AK2 to prevent its accumulation in the cytosol (Finger et al, 2020). Another protein that is targeted by DPP9 to the N‐degron pathway is the tyrosine kinase Syk, which is central for B‐cell receptor‐mediated signaling. The interaction between DPP9 and Syk requires FLNA that acts as a scaffold linking DPP9 to Syk (Justa‐Schuch et al, 2016). Here we asked whether additional FLNA interaction partners are targeted by DPP9 for proteasome‐degradation. Thus, we inspected the N‐terminus of known FLNA‐binding proteins for N‐terminal prolines, which are predicted to localize to disordered regions. Analysis of 90 different proteins known to interact with FLNA (Savoy & Ghosh, 2013) highlighted the human breast cancer‐associated protein 2 (BRCA2) (Yuan & Shen, 2001; Mondal et al, 2012; Yue et al, 2012). The N‐terminal sequence of BRCA2 is intrinsically disordered (Le et al, 2020; Sidhu et al, 2020) and includes a classical DPP9 cleavage site. The alignment of BRCA2 protein sequences from 108 placental mammals reveals high conservation of the Met1‐Pro2‐Ile3/Val3 sequence in the N‐terminus of BRCA2 (Fig 1A).
Figure 1. DNA damage triggers an interaction between DPP9 and BRCA2.
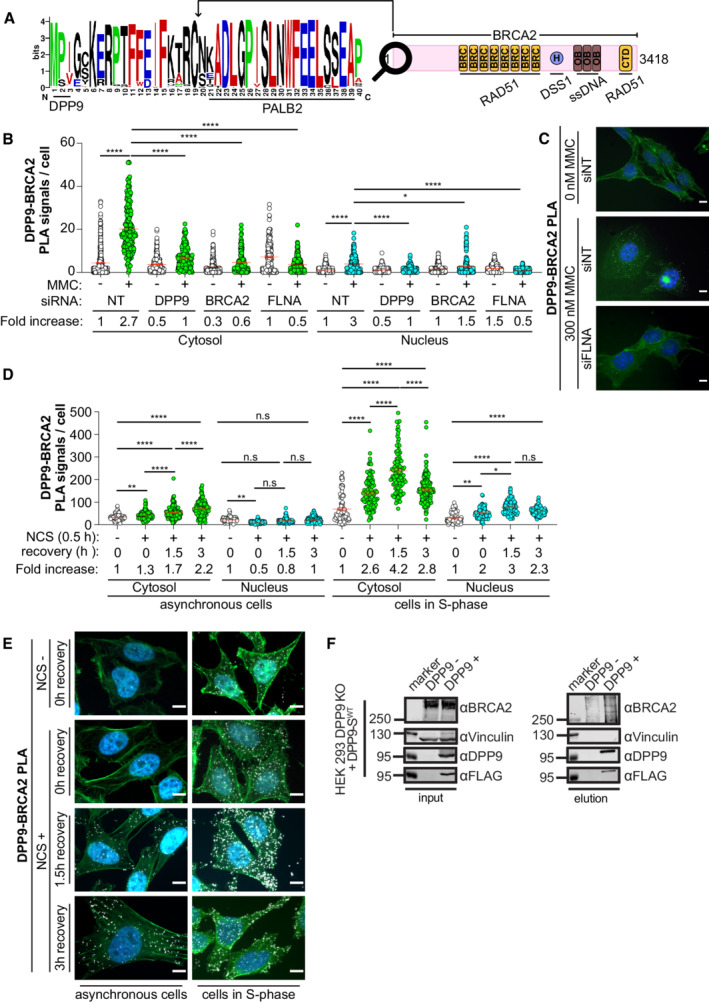
-
ASequence alignment of BRCA2 homologs from 108 placental mammals (Logo plot) shows conservation of the cleavage site for DPP9 at the BRCA2 N‐terminus. The cartoon depicts the full‐length BRCA2 and its conserved domains with the corresponding interaction partners.
-
BQuantification of PLAs between BRCA2 and DPP9 in control HeLa WT cells treated with nontargeting siRNA (siNT) or silenced with the indicated oligos. 300 ng/ml MMC was added for 24 h. Each dot represents the number of PLA events in a single cell, from two to seven biological replicates (siNT‐MMC (n = 6), siNT+MMC (n = 7), siDPP9‐MMC (n = 4), siDPP9+MMC (n = 5), siBRCA2‐MMC (n = 2), siBRCA2+MMC (n = 3), siFLNA‐MMC (n = 5), siFLNA+MMC (n = 6), NgtCntrl‐MMC (n = 3), NgtCntrl+MMC (n = 6)). The number of foci are shown based on their cellular localisation. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (*P ≤ 0.05, ****P ≤ 0.0001).
-
CRepresentative PLA images showing close proximity between endogenous DPP9 and endogenous BRCA2 in HeLa WT cells. Exposure of cells to MMC triggers more PLA events (white). Phalloidin (green) stains actin filaments and DAPI (blue) stains the nucleus. Scale bar 10 μm. Anti‐BRCA2: RRID:AB_2259370, anti‐DPP9: RRID:AB_2889071 (these images, along with the corresponding controls can be found in Appendix Fig S1A and B).
-
DQuantification of PLAs between BRCA2 and DPP9 in asynchronous HeLa WT cells or in cells in S‐Phase. Each dot represents the number of PLA events in a single cell, from three biological replicates. Cells were synchronized with a double thymidine block and released for 3 h before the addition of NCS (250 ng / mL for 30 min). Cells were sampled prior to NCS treatment, after NCS treatment, and with 1.5 and 3 h of recovery after NCS treatment. The number of foci is shown based on their cellular localisation. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (*P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001).
-
ERepresentative PLA images showing close proximity between endogenous DPP9 and endogenous BRCA2 in HeLa cells. Exposure of cells to NCS triggers more PLA events (white), which increase in time. Cells in S‐Phase show a greater fold increase in the number of PLA events in comparison with asynchronous cells. Phalloidin (green) stains actin filaments and DAPI (blue) stains the nucleus. Scale bar 10 μm. Antibodies as in (C).
-
FCo‐immunoprecipitation assays showing binding of BRCA2 and DPP9‐SWT. HEK293 DPP9 KO+DPP9WT cells, were treated with 1 μg/ml Dox (24 h) to induce the expression of DPP9‐FLAG. DNA damage was induced with 300 nM MMC treatment for 24 h. Control cells do not express DPP9 (− Dox). Bound proteins were eluted with a FLAG peptide and analyzed by western blotting (anti‐BRCA2: RRID:AB_2259370, anti‐DPP9: RRID:AB_731947, anti‐FLAG RRID:AB_262044).
Source data are available online for this figure.
BRCA2 is a 384 kDa protein that is crucial for the repair of DNA double‐strand breaks (DSBs) (Moynahan et al, 2001) by the high‐fidelity Homologous Recombination (HR) pathway (Chen et al, 2018; Scully et al, 2019). Briefly, DSB repair by HR starts with the generation of 3′ single‐strand DNA (ssDNA) overhangs in a process termed 5′ end resection. Next, protein filaments of RAD51 cover the ssDNA overhangs, and search for homologous sequences preferentially in the sister chromatid, that serve as a template for further repair by DNA polymerases (Jasin & Rothstein, 2013). BRCA2 is critical for HR by promoting the loading and assembly of RAD51 into protein filaments selectively on the ssDNA (Jensen et al, 2010; Liu et al, 2010; Thorslund et al, 2010). HR repair of DSBs occurs in S and G2 phases when sister chromatids are available for repair (Hustedt & Durocher, 2017).
Intriguingly, DNA damage triggers the proteasomal degradation of BRCA2 (Schoenfeld et al, 2004; Liu et al, 2017), a process that is antagonized by the ubiquitin‐specific protease USP21 (Liu et al, 2017). Notably, elevated expression of USP21, which leads to stabilization of BRCA2, is observed in hepatocellular carcinoma and inversely correlates with patient survival suggesting that regulated degradation of BRCA2 is critical for DNA repair (Liu et al, 2017). Given the presence of an evolutionary conserved N‐terminal sequence Met1‐Pro2‐Ile3/Val3 in BRCA2 and the rate‐limiting role of DPP9 in cleaving Pro‐containing peptide bonds (Geiss‐Friedlander et al, 2009), we asked whether DPP9 and the N‐terminal Met1‐Pro2‐Ile3 sequence determine the half‐life of BRCA2.
Here we show that BRCA2 stability is reduced by the removal of the N‐terminal residues Met1Pro2 and increased in cells depleted of DPP9 activity. Crystal structures combined with mass spectrometry analysis demonstrate that DPP9 hydrolyses the peptide bond between Pro2 and Ile3 in an N‐terminal peptide of BRCA2. In cells, the interaction between DPP9 and BRCA2 is observed mostly in the cytosol and is stimulated by DNA damage. Depletion of DPP9 leads to fewer RAD51 foci formation, a hypersensitivity of cells to genotoxic agents, and defects in HR.
Results
DNA damaging conditions trigger an interaction between BRCA2 and DPP9
As a starting point, we asked whether DPP9 and BRCA2 are in close proximity in cells, using proximity ligation assays (PLAs), which are especially well‐suited for the detection of dynamic and transient interactions, such as those between an enzyme and its substrate. Initially, only few DPP9‐BRCA2 PLA signals were detected in HeLa cells. However, exposure to Mitomycin C (MMC), a chemotherapeutic agent that forms inter‐strand DNA crosslinks that are converted into secondary DSBs, resulted in a clear increase in the number of DPP9‐BRCA2 PLA events (Figs 1B and C, and EV1A; Appendix Fig S1A and B). Although DPP9 and BRCA2 localize to both the nucleus and the cytoplasm, most of the MMC‐induced BRCA2‐DPP9 PLA signals were located in the cytosol (Fig 1B and C). Given its interactions with both DPP9 (Justa‐Schuch et al, 2016) and BRCA2 (Yuan & Shen, 2001; Mondal et al, 2012; Yue et al, 2012), cells were silenced for FLNA to test whether it is relevant for the observed DPP9‐BRCA2 proximity. Cell silenced for FLNA did not show an increase in the number of BRCA2‐DPP9 interactions in response to MMC, implying that the DNA damage triggered proximity of DPP9 and BRCA2 requires FLNA (Figs 1B and C, and EV1A; Appendix Fig S1A and B).
Figure EV1. Summaries of PLA experiments.
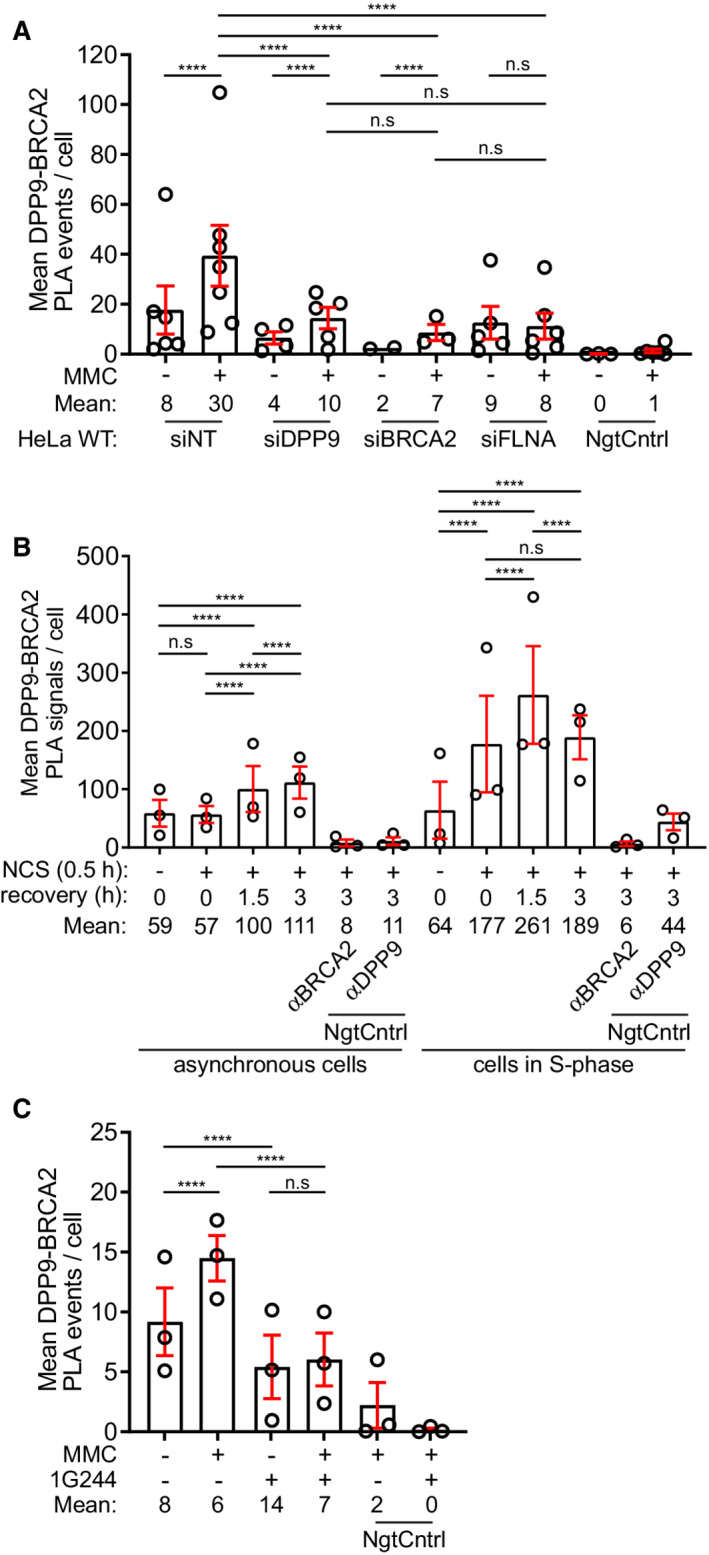
-
ASummary of PLAs between BRCA2 and DPP9 in control HeLa WT cells treated with nontargeting siRNA (siNT) or silenced with the indicated oligos (Fig 1B). Each dot represents the mean number of PLA events in a single repetition, from two to seven biological replicates. More than 100 cells were quantified per condition in each experiment. Data from the summary of all PLA events per cell were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (***P ≤ 0.001, ****P ≤ 0.0001).
-
BSummary of PLAs between BRCA2 and DPP9 in asynchronous HeLa WT cells and HeLa WT cells synchronized to S‐phase. DNA damage was induced by NCS (250 ng/ml for 30 min). Each dot represents the mean number of PLA events in a single repetition. More than 100 cells were quantified per condition in each experiment, from three biological replicates. Data from the summary of all PLA events per cell were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001).
-
CSummary of PLAs between BRCA2 and DPP9 in HeLa WT cells treated with 1G244 (Fig 2C). DNA damage was induced by MMC (300 ng/ml for 24 h). Each dot represents the mean number of PLA events in a single repetition, from three biological replicates. More than 100 cells were quantified per condition in each experiment. Data from the summary of all PLA events per cell were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001).
To extend these observations, cells were treated with another DSBs causing agent, the radiomimetic drug Neocarzinostatin (NCS). Similarly, NCS‐treated cells presented more DPP9‐BRCA2 PLA signals compared with mock‐treated cells, with most of these events in the cytosol (Figs 1D and E, and EV1B). The fold increase of DPP9‐BRCA2 PLA events in response to NCS was even higher in cells that were first arrested by double thymidine blocks and treated with NCS following release from the second block, validating a close proximity between DPP9 and BRCA2 in S‐phase (Figs 1D and E, and EV1B).
Additionally, we performed co‐immunoprecipitation against DPP9 and tested for the presence of BRCA2. Knockouts of DPP9 were established in HEK Flp‐In™ T‐REx™‐293 cells (HEK293 DPP9 KO) using CRISPR single guide RNAs. These were then used to establish a stable cell line with a Doxycycline (Dox) inducible expression of the cytoplasmic DPP9 isoform (FLAG‐ DPP9‐SWT), (cells characterized in Appendix Fig S2A–E). To enhance the interaction between DPP9 and BRCA2, DNA damage was induced by the addition of MMC. BRCA2 co‐purified with FLAG‐DPP9 (Fig 1F). Collectively, these data strongly suggest that DPP9 and BRCA2 interact in cells in response to DNA damage.
DPP9 removes the BRCA2 N‐terminal dipeptide Met1Pro2
To test whether DPP9 binds to the N‐terminus of BRCA2, a 40 amino acid peptide was selected. This peptide includes the binding site of PALB2, a protein that ensures the correct intra‐nuclear localization of BRCA2. A shorter BRCA221‐39 peptide was previously co‐crystallized in a complex with PALB2 (Xia et al, 2006; Oliver et al, 2009). Surface Plasmon Resonance (SPR) assays with the synthetic BRCA21‐40 peptide confirmed direct binding to immobilized recombinant DPP9 (Fig 2A). The SPR response curves obtained for DPP9 and BRCA21‐40 were not at perfect equilibrium, whereas the binding behavior of DPP9 to a shorter peptide lacking the Met‐Pro (BRCA23‐40) was less complex, suggesting that the curves for DPP9‐BRCA21‐40 reflect not only the binding of DPP9 and BRCA21‐40, but possibly also on‐chip processing of the BRCA21‐40 peptide by DPP9, and the release of the product (Fig 2A; Appendix Fig S3A–D). Additionally, we cloned and purified a recombinant protein containing the first 40 amino acids of BRCA2 followed by a C‐terminal HA tag, and examined whether DPP9 and BRCA2 show characteristics of an enzyme‐substrate interaction. Previously, we had shown that the binding of the competitive inhibitors 1G244 and SLRFLYEG (Wu et al, 2009; Pilla et al, 2013) leads to a rearrangement of the DPP8/9 active sites resulting in a closed conformation (Ross et al, 2018). Pull‐down assays with immobilized BRCA21‐40HA showed reduced binding to DPP9 in the presence of either 1G244 or SLRFLYEG (Fig 2B). Consistently, cells treated with 1G244 displayed fewer DPP9‐BRCA2 PLA events in response to MMC, compared with mock‐treated cells (Figs 2C and D, and EV1C; Appendix Fig S4A). Thus, both PLA in cells and pull‐down assays with purified recombinant components show the reduced association of BRCA2 and DPP9 in the presence of competitive inhibitors that occupy the active site of DPP9, suggesting that BRCA2 acts as a ligand that competes with these inhibitors for binding to DPP9. Direct processing of the BRCA21‐40 peptide by purified DPP9 was verified by mass spectrometry analysis, revealing the hydrolysis of the peptide bond between Pro2 and Ile3 (Met1Pro2↓Ile3) in the BRCA21‐40 peptide. The resulting BRCA23‐40 product presents a neo N‐terminus with isoleucine in the first position (Fig 2E). No cleavage was observed in control reactions that included the competitive inhibitor SLRFLYEG.
Figure 2. The BRCA2 N‐terminal dipeptide Met‐Pro is processed by DPP9.
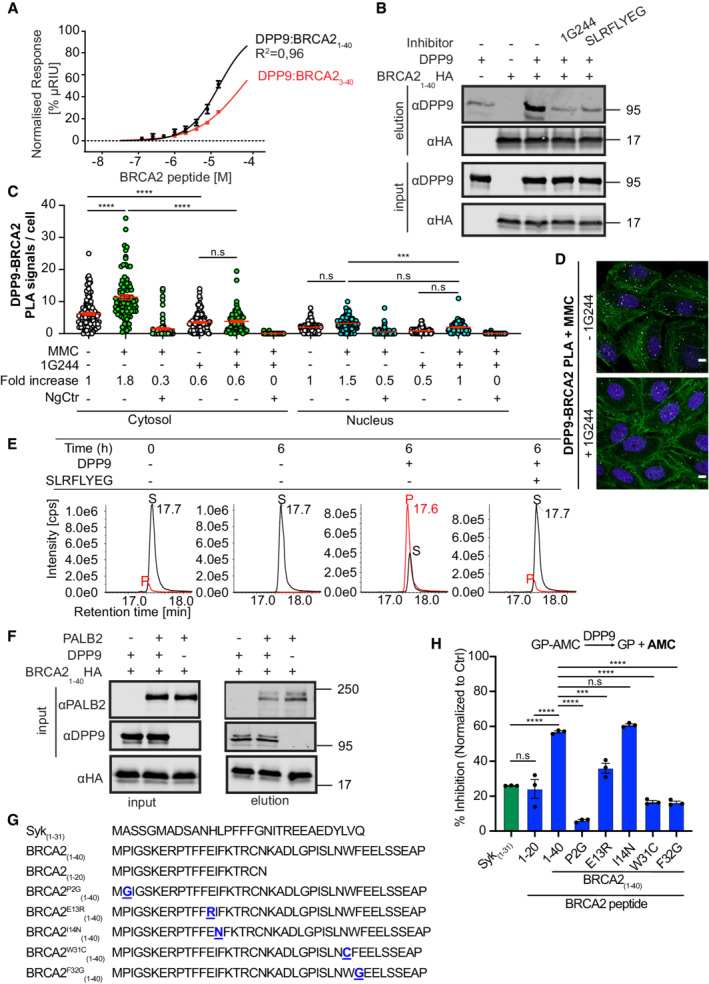
-
ASurface Plasmon Resonance (SPR) data showing a direct interaction of purified DPP9 with a BRCA21‐40 peptide and a truncated BRCA23‐40 peptide, which lacks the N‐terminal dipeptide Met‐Pro. A serial dilution of BRCA2‐derived peptides was injected over a surface covered with DPP9. Equilibrium binding isotherms obtained for interactions measured between DPP9 and BRCA21‐40 (black line) and BRCA23‐40 (red line). Data were fitted to a sigmoidal dose–response curve fit. Mean ± SEM of technical triplicates of a representative experiment out of 3.
-
BPull‐down assay showing direct binding of purified recombinant DPP9 to a BRCA2 N‐terminal1‐40 fragment immobilized on HA beads. The DPP9 inhibitors 1G244 or SLRFLYEG compete with BRCA21‐40HA for interaction with DPP9. Representative data of three technical replicates are shown. Anti‐HA: RRID:AB_2565334, anti‐DPP9: RRID:AB_2889071.
-
CQuantification of PLAs showing fewer MMC‐induced DPP9‐BRCA2 PLA events in HeLa cells treated with 10 μM 1G244. Each dot represents the number of PLA events in a single cell, from three biological replicates. The number of foci is shown based on their cellular localisation. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (***P ≤ 0.001, ****P ≤ 0.0001).
-
DRepresentative images of DPP9‐BRCA2 PLA in the presence of 1G244—a competitive inhibitor of DPP9. Control cells were mock treated with DMSO. Phalloidin (green) stains actin filaments and DAPI (blue) stains the nucleus. Scale bar 10 μm. Anti‐BRCA2: RRID:AB_2259370, anti‐DPP9: RRID:AB_2889071 (these images, along with the corresponding controls can be found in Appendix Fig S1C).
-
EIn vitro processing of BRCA21‐40 synthetic peptide by purified recombinant DPP9. Samples were analyzed by high‐resolution liquid chromatography/tandem mass spectrometry, in quadruplicate. The panels show extracted MS1 ion chromatograms for both substrate BRCA21‐40 peptide (MPIGSKERPT…) (labeled S, [M+5H]5+ m/z 917.8637; retention time 17.7 min) and product BRCA23‐40 peptide (IGSKERPT…) (labeled P, [M+5H]5+ m/z 872.2451; retention time 17.6 min). The identity of the product peak was established both by accurate mass measurement to within 5 ppm and by product ion spectra.
-
FPull‐down assay showing a competition between DPP9 and PALB2 for binding to the BRCA2 N‐terminus. While each protein can bind to the BRCA2 N‐terminal peptide, in the presence of DPP9, the PALB2‐BRCA2 binding is negatively affected. Representative data of three technical replicates are shown. Anti‐PALB2: RRID:AB_890607, anti‐HA: RRID:AB_2565334, anti‐DPP9: RRID:AB_2889071.
-
GPeptide sequences used in the DPP9 competition assays shown in (H).
-
HDPP9 activity assays showing different competitive effects of BRCA2 peptides (G), hydrolysis of GP‐AMC is used as read out. Shown is % of inhibition in GP‐AMC processing normalized to the control reactions (DPP9 without competing peptides). Syk1‐31 was used as a positive control. n = 3 independent experiments. Data were analyzed by a one‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (***P ≤ 0.001, ****P ≤ 0.0001).
Source data are available online for this figure.
Since amino acids 1–40 in the N‐terminus of BRCA2 include the PALB2 interaction site, we asked whether DPP9 and PALB2 compete for binding to the N‐terminus of BRCA2, or whether they bind independently of each other. Pull‐down assays show reduced PALB2 binding in the presence of DPP9 implying that these two proteins sterically hinder mutual binding to BRCA21‐40HA (Fig 2F). To test for residues in BRCA2 that affect cleavage by DPP9, competition assays were performed in which we measured the hydrolysis of the artificial substrate H‐Gly‐Pro‐7‐amino‐4‐methylcoumarin (GP‐AMC) in the presence of the BRCA21‐40 peptide, or variants of this peptide (Fig 2G and H). For control, assays were carried out with the Syk1‐31 peptide (Justa‐Schuch et al, 2016) which as expected inhibited GP‐AMC cleavage. Stronger inhibition was observed with the BRCA21‐40 peptide, while a BRCA2 peptide in which the Pro in position 2 was substituted with a Gly (BRCA21‐40P2G) was a poor inhibitor. Interestingly, a BRCA21‐20 peptide was a less efficient competitor in comparison with BRCA21‐40 suggesting that residues between 20–40 contribute to the interaction with DPP9. We looked more closely into selected residues, based on natural mutations in BRCA2 that were depicted in cBioPortal databases. These assays show that substitution of the conserved Glu in position 13 with a Lys reduces the competitive capacity of the BRCA21‐40 peptide. In line with the pull‐down assays, which suggest a competition between DPP9 and PALB2 for interaction with BRCA2, a stronger effect was observed for the substitutions of Trp31 and Phe32, which are important for interaction with PALB2 (Xia et al, 2006; Oliver et al, 2009).
To better portray the cleavage event, DPP9 crystals were soaked with the BRCA21‐40 peptide. DPP9 crystals exhibited positive electron density at the active site corresponding to the N‐terminal Met1Pro2 dipeptide with the proline residue occupying the S1 subsite (Fig 3), in a dipeptide conformation that was identical to that seen earlier in the DPP9‐SLRFLYEG complex (Ross et al, 2018). No trace of electron density of a peptide continued after the proline residue was present (Fig 3; Appendix Table S1). Since DPP8 and DPP9 share very similar substrate‐binding mechanisms in vitro, and DPP9 crystals are notorious for being less well‐ordered (Ross et al, 2018), we also tested for a possible interaction between DPP8 and BRCA2. Similarly, in DPP8, all 3 copies of the asymmetric unit exhibit clearly interpretable electron density for the dipeptide. The well‐ordered R‐segment, a hallmark of ligand binding, was well‐defined and ordered and supports the full occupation of the active sites. These structures, with the dipeptide captured in the active site, clearly document intracrystalline enzymatic activity of DPP8 and DPP9 resulting in the hydrolysis of the BRCA21‐40 N‐terminal peptide, in accord with the mass spectrometry data (Fig 2E), and release of the BRCA23‐40 peptide.
Figure 3. The BRCA2 N‐terminal dipeptide Met‐Pro is captured in the active site of DPP9.
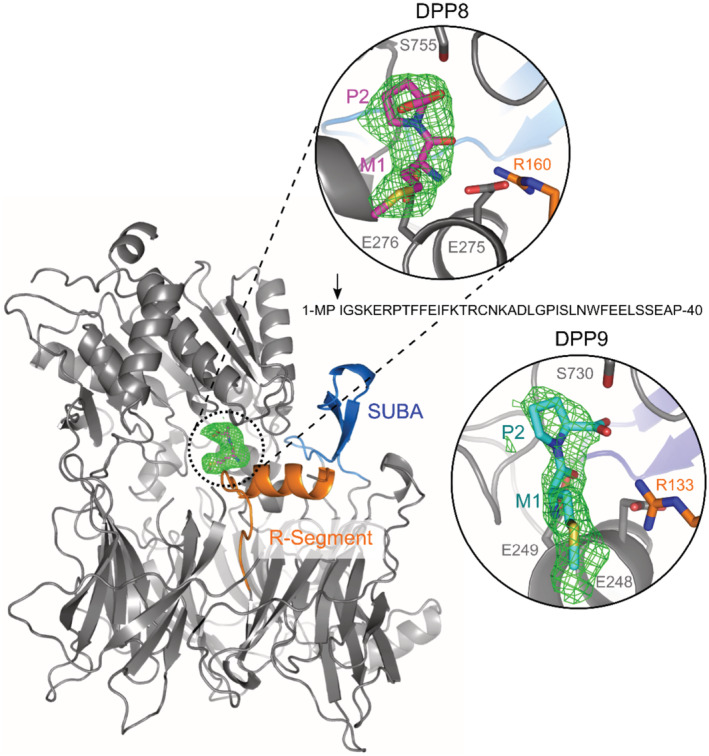
Shown is the N‐terminal dipeptide of BRCA2 bound to both DPP9 and DPP8. Omit map (Fo‐Fc; 3σ) of a BRCA21‐40 peptide soaked in DPP8 crystals (C2221) and in DPP9 crystals (P1211). The R‐Segment and SUMO‐binding arm (SUBA Pilla et al, 2012) are highlighted in orange and blue, respectively. The arrow marks the position where the BRCA2 peptide is cleaved. For simplification, shown is a monomer of DPP8. Both zoomed views are rotated 45° with respect to the monomer view to better display the ligand.
Source data are available online for this figure.
DPP9 targets BRCA2 for degradation following DNA damage
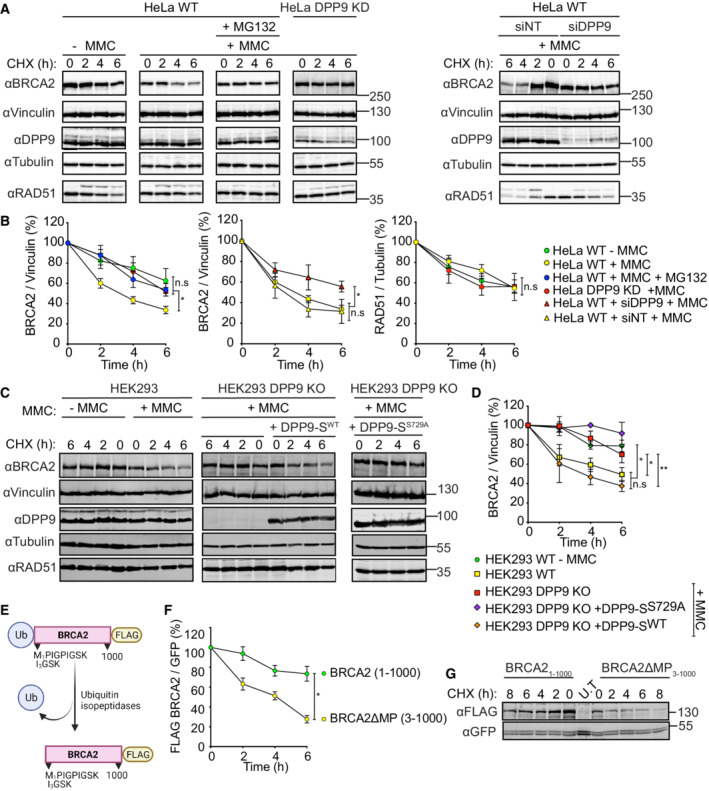
Based on our previous findings showing that DPP9 targets Syk and AK2 to the N‐degron pathway (Justa‐Schuch et al, 2016; Finger et al, 2020), we here asked whether DPP9 also influences the stability of BRCA2. Consistent with previous publications (Schoenfeld et al, 2004; Liu et al, 2017), exposure of cells to MMC induced an accelerated degradation of BRCA2, which was less pronounced in the presence of the proteasome inhibitor MG132 (Fig 4A and B). Importantly, cycloheximide (CHX) chase assays revealed a significantly lower turnover of BRCA2 in the HeLa DPP9 Knock‐Down cells (HeLa DPP9 KD; cells previously described in Justa‐Schuch et al (2016) and further characterized in Fig EV2A–C), compared with HeLa WT cells in response to MMC (Fig 4A and B). Similarly, we observed an increase in BRCA2 stability in HeLa WT cells transiently silenced for DPP9 (Fig 4A and B), and in the HEK 293 DPP9 KO cells (Fig 4C and D). In contrast to BRCA2, the stability of endogenous RAD51 was not affected by DPP9 (Fig 4A and B). To test whether the catalytic activity of DPP9 regulates BRCA2 stability, CHX chase assays were carried out in HEK 293 DPP9 KO cells in which we induced the expression of DPP9‐SWT. To test for the relevance of DPP9 activity, we generated a stable cell line HEK293 DPP9 KO+DPP9‐SS729A with an induced expression of the enzymatically inactive DPP9 (DPP9‐SS729A). Importantly, the re‐expression of DPP9‐SWT but not of DPP9‐SS729A restored the MMC‐dependent degradation of BRCA2 (Fig 4C and D; Appendix Fig S2A and B).
Figure 4. DPP9 triggers BRCA2 degradation in response to MMC.
-
A, BRepresentative western blots and accompanying graph from more than three biological replicates show that MMC (300 nM, 24 h) induces a rapid turnover of endogenous BRCA2 in HeLa WT cells, which is less pronounced in HeLa WT cells treated with the proteasome inhibitor MG132 (100 μM), in HeLa DPP9 KD cells and in cells transiently silenced for DPP9 (siDPP9). RAD51 stability is not altered in HeLa DPP9 KD cells. Vinculin is a loading control for BRCA2, Tubulin is a loading control for RAD51. Shown images originate from one representative cycloheximide (CHX) chase assay. The ratios of BRCA2 to Vinculin and RAD51 to Tubulin are defined as 100% at time 0 h. Mean ± SEM, data were analyzed by a paired two‐tailed t‐test. (*P ≤ 0.03). Anti‐BRCA2: RRID:AB_2259370, anti‐DPP9: RRID:AB_731947, anti‐Vinculin: RRID:AB_477629, anti‐Tubulin: RRID:AB_628412, anti‐RAD51: RRID:AB_1142428.
-
C, DRepresentative western blots and graph summarizing results of more than three biological replicates. CHX chase assays show that the MMC‐induced degradation of BRCA2 is less pronounced in HEK293 DPP9 KO cells and in HEK293 DPP9 KO+DPP9‐SS729A cells overexpressing an inactive DPP9‐S mutant. Expression of DPP9 was induced (+ Dox, 1 μg/ml) simultaneously with MMC (300 nM), 24 h. Cells that were overexpressing the active variant (DPP9‐SWT) show similar levels of MMC‐induced BRCA2 degradation. RAD51 stability is not altered by MMC and is similar in all cell lines. Vinculin is a loading control for BRCA2, Tubulin is a loading control for RAD51. Shown images originate from one representative cycloheximide (CHX) chase assay. The ratios of BRCA2 to Vinculin are defined as 100% at time 0 h. Mean ± SEM, data were analyzed by a paired two‐tailed t‐test (*P ≤ 0.03, **P ≤ 0.002). Antibodies as described in (A‐B).
-
EGraphical presentation of the plasmid used for the production of the N‐terminal‐truncated mutant BRCA2ΔMP3‐1000 that was transfected in H‐I. The BRCA21‐1000 insert was cloned in a similar manner. Both constructs were tagged with a C‐terminal FLAG. The ubiquitin moiety is removed in cells by endogenous ubiquitin isopeptidases.
-
F, GRepresentative western blots and graphs summarizing CHX assays from three biological replicates show that the BRCA2ΔMP3‐1000 truncation mutant is degraded at a higher rate compared with the untruncated BRCA21‐1000. Cells were co‐transfected with a GFP expressing plasmid, as transfection and loading control. Control cells were transfected with GFP only (U.T). Image originates from one representative experiment. BRCA2‐FLAG signals are related to the transfection and loading control GFP. BRCA2 levels in relation to GFP were defined as 100% at time 0 h. Mean ± SEM, data were analyzed by a paired two‐tailed t‐test (*P ≤ 0.03). Anti‐FLAG: RRID:AB_262044, anti‐GFP: RRID:AB_641123.
Source data are available online for this figure.
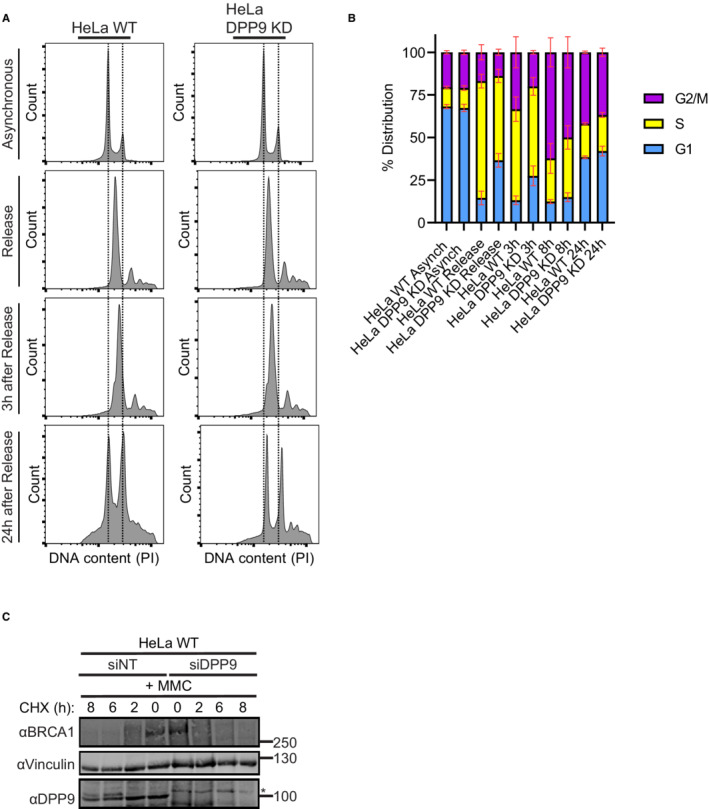
Figure EV2. Characterization of DPP9‐depleted HeLa cells.
-
ARepresentative histograms of HeLa WT and HeLa DPP9 KD cells investigating their relative DNA content via PI staining. Asynchronized cells and cells that were double thymidine‐treated were investigated. Shown are representatives of the asynchronized, double thymidine release, 3 h of recovery and 24 h of recovery samples.
-
BCell cycle distribution of the HeLa WT and HeLa DPP9 KD populations of asynchronized cells and synchronized cells at 0 h, 3, 8, and 24 after double thymidine release. Data from three to six biological replicates (asynchronized (n = 3), synchronized HeLa WT (n = 6), synchronized HeLa DPP9 KD (n = 4)) were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. No significant differences could be detected between the two cell lines. Shown are mean ± SEM.
-
CRepresentative western blots showing a CHX chase of HeLa WT cells transiently silenced for DPP9, and of control cells treated with nontargeting siRNA (siNT). Cells were treated with MMC (300 nM, 24 h) prior to the addition of CHX. Anti‐BRCA1: RRID:AB_626761, anti‐DPP9: RRID:AB_731947, anti‐Vinculin: RRID:AB_477629.
Source data are available online for this figure.
To test whether the N‐terminal Pro determines BRCA2 stability, we determined the turnover of BRCA21‐1000 revealing a half‐life greater than 8 h, similar to the endogenous BRCA2 in the absence of MMC (Fig 4E–G). Additionally, we cloned an N‐terminal BRCA2 truncation mutant (BRCA2ΔMP3‐1000) with isoleucine in the first position that mimics the product generated in vitro upon DPP9 cleavage (MP↓IGSK → IGSK). The half‐life of BRCA2ΔMP3‐1000 was 2–3 h, similar to the turnover rate of endogenous BRCA2 in the presence of MMC, and significantly lower than BRCA2 1‐1000 (Fig 4F and G). To generate the desired N‐terminus both constructs were cloned in frame with an N‐terminal ubiquitin tag, which is removed in cells by endogenous ubiquitin isopeptidases based on the well‐established ubiquitin‐fusion technique (Varshavsky, 2005). We have also observed that the steady‐state levels of BRCA21‐1000 constructs with a P2H or P2G mutation are reduced (Appendix Fig S4B). In summary, Pro2 plays a stabilizing role for BRCA2, removal of which leads to accelerated turnover of BRCA2, phenocopying the MMC‐induced degradation of BRCA2.
DPP9‐deficient cells show defects in HR‐mediated repair and hypersensitivity to genotoxic agents
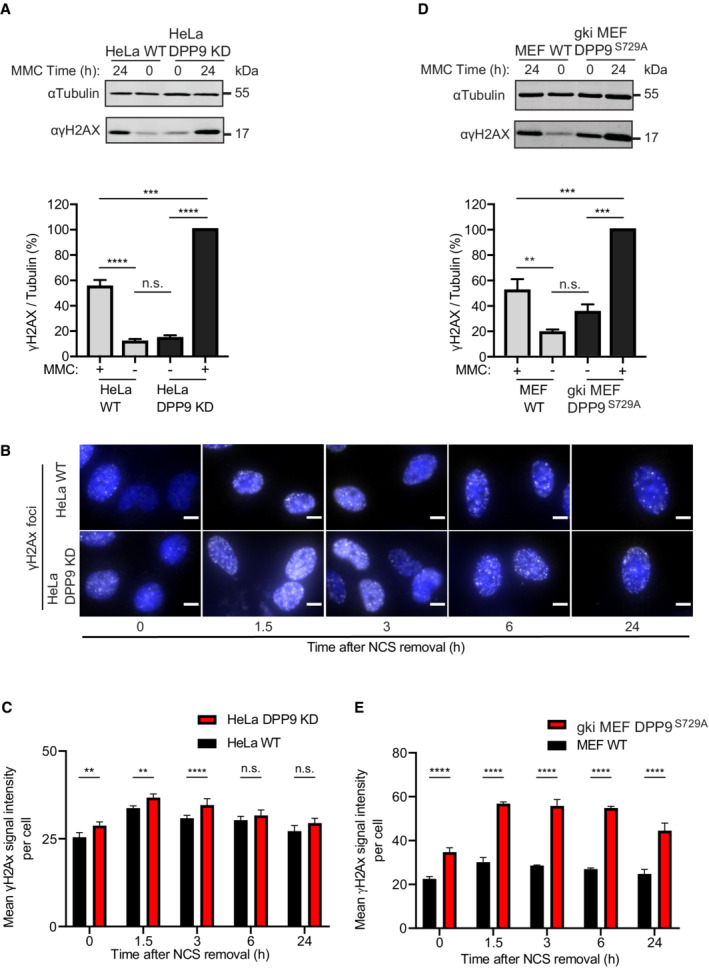
Previous studies have shown that high levels of wild‐type BRCA2 or the BRC4 repeat suppress DSB repair by homologous recombination and reduce RAD51 foci formation (Chen et al, 1999; Magwood et al, 2012; Abe & Branzei, 2014). Since DPP9 targets BRCA2 for degradation, we asked whether depletion of DPP9 and stabilization of BRCA2 alter the cellular response to DNA damage. First, we monitored the phosphorylation state of histone H2AX on serine 139 (γH2AX), which is an early and decisive step in the repair of DSBs, that is used as a marker for DSBs (Kuo & Yang, 2008). This analysis revealed significantly more γH2AX in HeLa DPP9 KD cells in response to MMC compared with the corresponding control cells (Fig EV3A). Likewise, γH2AX signals were higher in HeLa DPP9 KD cells following 30 min exposure to NCS (time 0 h) and remained higher also 3 h after removal of NCS (Fig EV3B and C), pointing to a delay in recovery of DSBs in HeLa DPP9 KD cells. At 6 h we observed that the γH2AX levels in the HeLa DPP9 KD cells were comparable to those of WT cells, suggesting that the DSBs have been resolved. To assess whether the enzymatic activity of DPP9 contributes to the elevated levels of γH2AX, we analyzed the response of the DPP9 gene knock‐in mouse embryonic fibroblasts (MEFs), which express enzymatically inactive DPP9 (gki MEF DPP9S729A cells were first described in Gall et al (2013)). Consistently, gki MEF DPP9S729A cells accumulated significantly more γH2AX in response to MMC and displayed a clear delay in the resolution of γH2AX that was formed in response to NCS (Figs 5A and B, and EV3D and E).
Figure EV3. DPP9‐deprived cells accumulate more γH2AX.
-
AIncreased γH2AX signals in HeLa DPP9 KD cells following exposure to 300 nM MMC for 24 h. Tubulin was a loading control. Quantification of the γH2AX / Tubulin signals in HeLa DPP9 KD cells and HeLa WT cells from three biological replicates. The γH2AX / Tubulin ratio in HeLa DPP9 KD cells at 24 h MMC was defined as 100%. Mean ± SEM. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test (****P ≤ 0.0001). Anti‐γH2AX: RRID:AB_2118009, anti‐Tubulin: RRID:AB_628412.
-
BRepresentative immunofluorescence images showing more γH2AX (white) in HeLa DPP9 KD cells following removal of Neocarzinostatin (NCS). Nuclei are shown in blue (DAPI). Scale bar 10 μm. HeLa DPP9 KD cells and HeLa WT cells were treated with 250 ng/ml Neocarzinostatin (NCS) for 30 min and allowed to recover for the indicated time points. γH2AX signals of each cell type at time 0, reflect 30 min of NCS, and no recovery time. Anti‐γH2AX: RRID:AB_309864.
-
CQuantification of mean γH2AX signals from HeLa WT and HeLa DPP9 KD cells. More than 1,300 cells were quantified per condition per experiment. Mean ± SEM from four biological replicates, each in technical duplicates. Data were analyzed by an unpaired two‐way ANOVA with the Sidak's multiple comparison test (**P ≤ 0.01, ****P ≤ 0.0001).
-
DHigher γH2AX signals in gki MEF DPP9S729A cells expressing enzymatically inactive DPP9 compared with MEF WT control cells following exposure to 300 nM MMC for 24 h. Tubulin was a loading control. Quantification of the γH2AX / Tubulin ratios in gki MEF DPP9S729A cells and MEF WT cells from three biological replicates. For normalization, the γH2AX / Tubulin ratio in gki MEF DPP9S729A cells at 24 h MMC was defined as 100%. Mean ± SEM. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test (****P ≤ 0.0001).
-
ESame as (C) for gki MEF DPP9S729A cells and MEF WT cells. Signals from more than 1,700 cells were quantified per condition per experiment. Mean ± SEM from six biological replicates. Data were analyzed by an unpaired two‐way ANOVA with the Sidak's multiple comparison test (****P ≤ 0.0001).
Source data are available online for this figure.
Figure 5. DPP9‐deficient cells accumulate more DNA Damage and are hypersensitive to genotoxic agents.
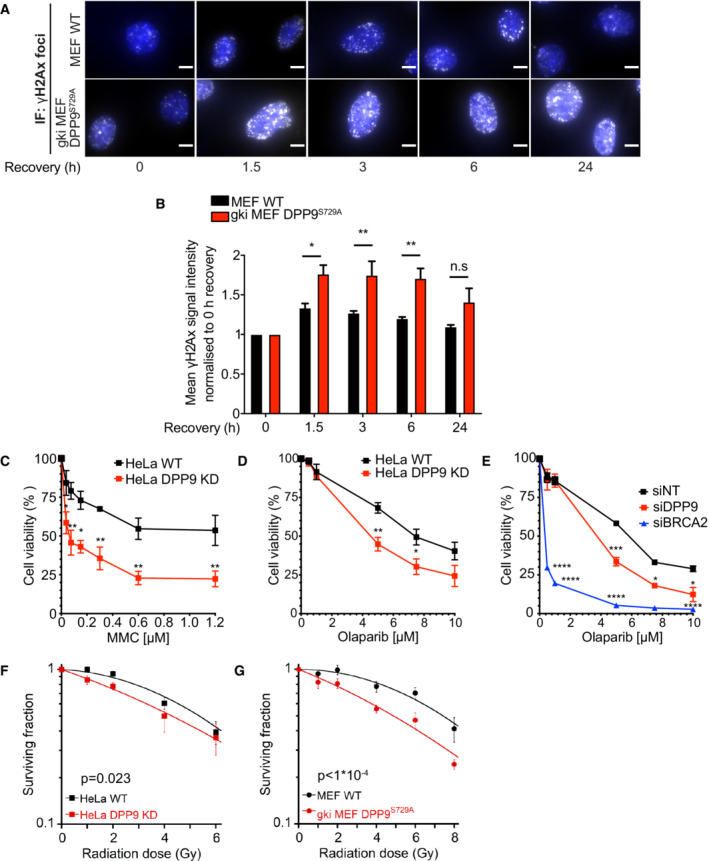
-
ARepresentative immunofluorescence images of γH2AX signals (white, RRID:AB_309864) in gki MEF DPP9S729A, showing more γH2AX in gki MEF DPP9S729A cells following removal of Neocarzinostatin (NCS), quantification in (B). MEF DPP9S729A and control MEF cells were treated with 250 ng/mL NCS for 30 min and allowed to recover for the indicated time points. γH2AX signals at time 0, reflect 30 min of NCS and no recovery time. Nuclei are shown in blue (DAPI). Scale bar 10 μm.
-
BQuantification of γH2AX in gki MEF DPP9S729A and control MEF cells as described in (A). Signals from more than 1,700 cells were quantified per condition per experiment. Mean ± SEM from six biological replicates, each in technical duplicates. Data were analyzed by an unpaired two‐way ANOVA with the Sidak's multiple comparison test (*P ≤ 0.05, **P ≤ 0.01).
-
C–EDose‐dependent viability assays show a higher sensitivity of HeLa DPP9 KD cells (C and D) and DPP9 silenced cells (siDPP9) (E) to MMC (C) and Olaparib (D, E). BRCA2 was silenced for control. Mean ± SEM of three biological replicates. Data were analyzed by an unpaired two‐way ANOVA with the Sidak's multiple comparison test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
-
F, GQuantification of colonies formed after γ‐radiation of HeLa WT and DPP9 KD (F), and MEF WT and gki MEF DPP9S729A cells (G), showing the mean ± SEM of the survival fraction (SF) from three biological replicates. Data were analyzed by an unpaired two‐way ANOVA.
We also tested the sensitivity of DPP9‐deprived cells to the genotoxic agents MMC and Olaparib, an inhibitor of poly(ADP‐ribose) polymerase (PARP). Since inhibition of PARP leads to the conversion of ssDNA breaks into DSBs, defects in HR‐mediated repair are associated with increased sensitivity to PARP inhibitors, and are applied for the treatment of germline BRCA1‐ and BRCA2‐mutated metastatic breast cancers (Menear et al, 2008; Rottenberg et al, 2008; Lord & Ashworth, 2016). We detected a hypersensitivity of HeLa DPP9 KD cells to MMC and Olaparib compared with the HeLa WT cells (Fig 5C and D). Similarly, DPP9‐silenced cells were more sensitive to Olaparib compared with control cells treated with nontargeting siRNA (Fig 5E), albeit not to the same extent as cells transiently silenced for BRCA2. Furthermore, HeLa DPP9 KD cells were slightly, but significantly more sensitive to ionizing radiation (γ‐radiation) compared with the corresponding control cells (Fig 5F). This effect was more pronounced in gki MEF DPP9S729A cells (Fig 5G), linking the hypersensitivity to ionizing radiation to the absence of DPP9 enzymatic activity. Thus, cells lacking DPP9 activity accumulate more unrepaired DNA damage (Fig 5A and B) and are hypersensitive to genotoxic stress caused by MMC, Olaparib, and IR (Fig 5C–G), phenocopying cells overexpressing BRCA2 or the BRC4 repeat (Chen et al, 1999; Magwood et al, 2012; Abe & Branzei, 2014).
DPP9 activity regulates the formation of RAD51 foci in response to DNA damage
To directly investigate whether DPP9 plays a role in HR, we applied the well‐established DR‐GFP reporter system (Pierce et al, 1999; Fig 6A and B) in the colon cancer cell line HCT116 stably expressing DR‐GFP (Kari et al, 2019). As expected, control cells silenced for BRCA2, RAD51, or FLNA were deficient in HR‐mediated repair of the GFP reporter constructs (Fig 6B; Moynahan et al, 2001; Stark et al, 2004; Yue et al, 2009). Importantly, depletion of DPP9 also resulted in a significant reduction in HR frequency, implying that DPP9 promotes the repair of DSBs by HR. Intriguingly, despite the in vitro structural similarities between DPP8 and DPP9 (Fig 3), depletion of DPP8 did not significantly reduce HR efficiency, suggesting that DPP8 and DPP9 are not biologically redundant for this pathway.
Figure 6. DPP9‐deficient cells show defects in HR repair DPP9 and form fewer RAD51 foci upon MMC treatment.
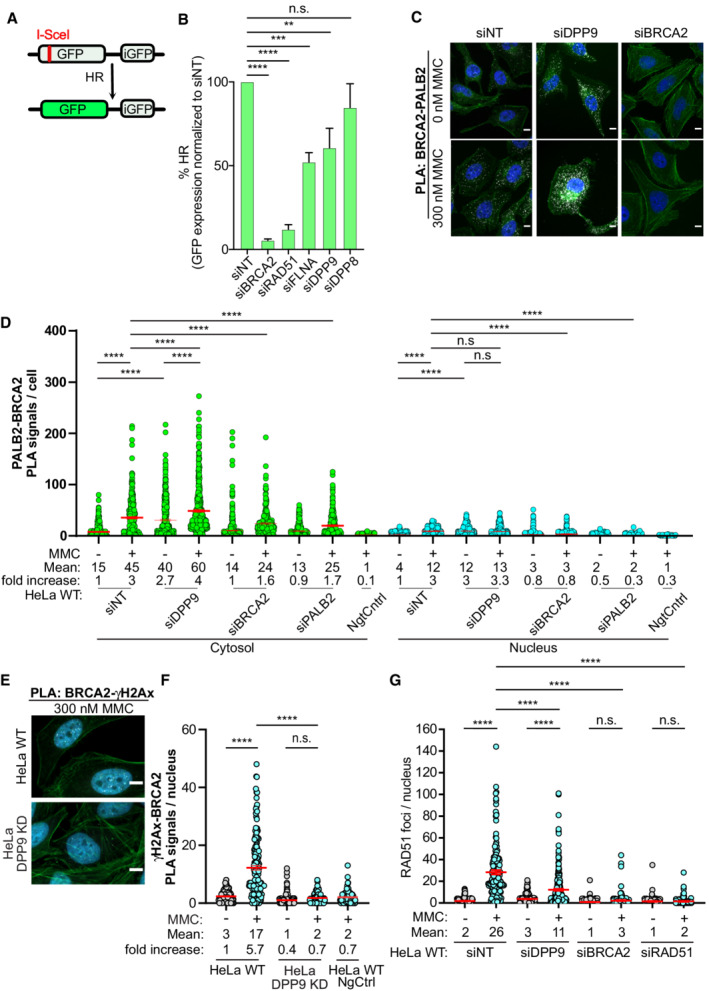
-
AGraphical presentation of the DR‐GFP reporter assay for HR. The construct includes two mutated GFP genes (SceGFP and iGFP) oriented as direct repeats. The SceGFP gene is mutated to contain the recognition site for the rare‐cutting endonuclease I‐SceI and a STOP codon, while the iGFP is a truncated form of GFP. Cells expressing the DR‐GFP are transfected with I‐SceI, which forms a DSB within the SceGFP. For HR‐mediated repair, the iGFP serves as a template for HR‐mediated repair of the DSB in GFP (Pierce et al, 1999).
-
BDPP9‐silenced cells are less efficient in the repair of DSBs by HR. HCT116 cells stably expressing the DR‐GFP reporter assay for HR efficiency were transiently transfected with the indicated siRNAs, and transfected with an I‐SceI‐expression vector. 48 h after transfection, the percentage of GFP‐positive cells was measured using flow cytometry analysis as an indication of HR efficiency. The graph shows the mean ± SEM from seven biological replicates. Data were analyzed by a paired two‐tailed t‐test (**P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
-
CRepresentative PLA images of BRCA2‐PALB2 PLA experiments showing more PLA events (white) in HeLa cells silenced for DPP9 (siDPP9) with respect to cells treated with nontargeting siRNA (siNT). Phalloidin (green) stains actin filaments and DAPI (blue) stains the nucleus. Scale bar 10 μm. Anti‐PALB2: RRID:AB_890607, anti‐BRCA2: RRID:AB_2259370.
-
DQuantification of PLAs experiments showing more BRCA2‐PALB2 PLA events in cells silenced for DPP9 (siDPP9) in comparison with nontargeting controls (siNT). Each dot represents the number of PLA events in a single cell, from four biological replicates. The technical control samples (NgtCntrl) omitted the BRCA2 antibody. The number of foci is shown based on their cellular localisation. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001).
-
ERepresentative PLA images of γH2Ax‐BRCA2 PLA experiment showing a reduction in the number of PLA events (white) between γH2Ax and BRCA2 in HeLa DPP9 KD cells. Phalloidin (green) stains actin filaments and DAPI (blue) stains the nucleus. Scale bar 10 μm. Anti‐γH2Ax: RRID:AB_2118009, anti‐BRCA2: RRID:AB_2259370 (these images, along with the corresponding controls can be found in Appendix Fig S1D).
-
FQuantification of PLAs showing fewer MMC‐induced γH2Ax‐BRCA2 PLA events in HeLa DPP9 KD cells, in comparison with HeLa WT cells. Each dot represents the number of PLA events in a single cell, from three biological replicates. The technical control samples (NgtCntrl) omitted the γH2Ax antibody. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001).
-
GGraph showing fewer RAD51 foci in DPP9‐silenced cells compared with control cells, following exposure to MMC. Each dot represents the number of RAD51 foci in a single cell, from two to six biological replicates: siNT‐MMC (n = 6), siNT+MMC (n = 6), siDPP9‐MMC (n = 5), siDPP9+MMC (n = 5), siBRCA2‐MMC (n = 3), siBRCA2+MMC (n = 3), siRAD51‐MMC (n = 2), siRAD51+MMC (n = 2). Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001).
To better understand the observed effect of DPP9 in the repair of DNA, we investigated BRCA2 in DPP9‐depleted cells, specifically its colocalization with γH2AX and with PALB2. In line with the higher stability of BRCA2 (Fig 4), and the observed partially overlapping binding site of both DPP9 and PALB2 to BRCA2 (Fig 2F and H), DPP9‐silenced cells contained significantly more BRCA2‐PALB2 PLA events both in the absence and presence of MMC (Fig 6C and D; Appendix Fig S4C). However, despite the increased colocalization with PALB2, and the higher γH2AX levels, fewer BRCA2‐γH2AX PLA events were formed in HeLa DPP9 KD cells in response to MMC (Fig 6E and F; Appendix Fig S4D and E). Similarly, fewer RAD51 foci were detected in HeLa DPP9 KD cells that were gated for EdU positive cells, HeLa DPP9‐silenced cells, and gki MEF DPP9S729A cells expressing inactive DPP9, in response to MMC compared with the number of RAD51 foci observed in the corresponding control cells (Figs 6G and EV4A–E; Appendix Fig S5A–C). Similarly, less RAD51 accumulated to chromatin fractions following 24 h of exposure to MMC in HeLa DPP9 KD cells (Appendix Fig S5D). To test whether the expression of DPP9 promotes the MMC‐induced appearance of RAD51, we monitored RAD51 in the HEK293 DPP9 KO, in HEK293 DPP9 KO+DPP9‐SWTand HEK293 DPP9 KO+DPP9‐SS729A. The re‐expression of DPP9‐SWT but not DPP9‐SS729A resulted in a significantly greater number of RAD51 foci in response to MMC (Figs 7A and B, and EV4F). Since DPP9‐S does not localize to the nucleus, we asked whether DPP9‐L which contains a nuclear localization signal (Justa‐Schuch et al, 2014) can rescue the formation of RAD51 foci. To address this question, we generated a stable cell line HEK293 DPP9 KO+DPP9‐L (Appendix Fig S2E). Comparing the RAD51 foci formed in the parental cell line and cells overexpressing the DPP9‐S or DPP9‐L isoform, we observed that the overexpression of the DPP9‐S results in more RAD51 foci formation compared with the DPP9‐L (Fig EV4G). Taken together, these results show that DPP9 activity promotes the formation of MMC‐induced RAD51 foci and a prominent role for the cytosolic form of DPP9 in the formation of RAD51 foci in response to MMC‐induced DNA damage.
Figure EV4. Summary of RAD51 experiments.
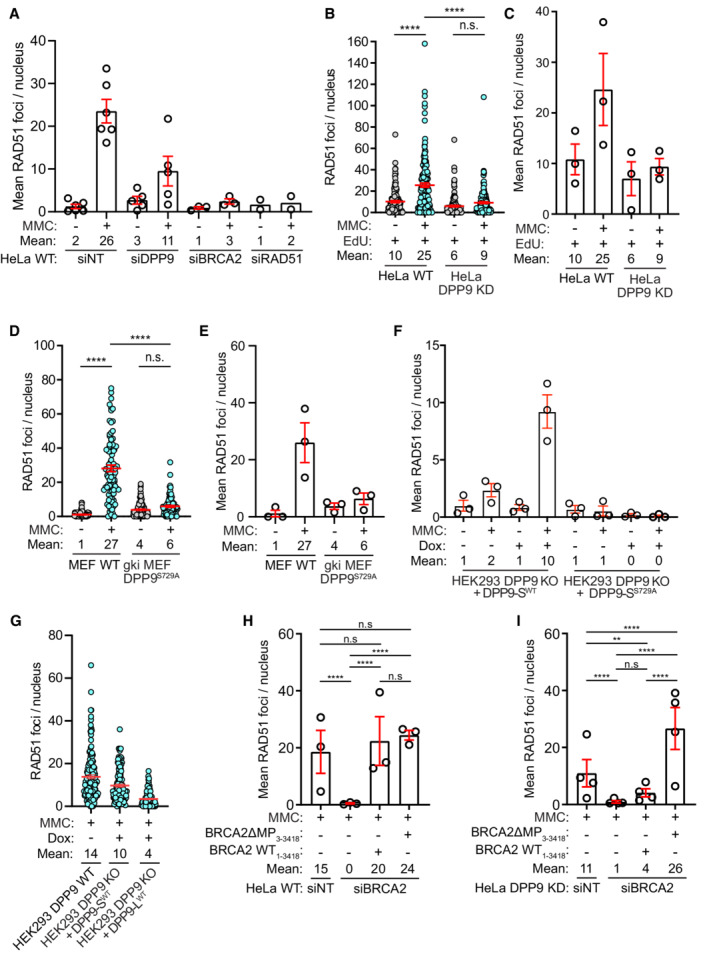
-
AA graph summarizing the mean number of RAD51 foci in HeLa WT cells treated with nontargeting siRNA (siNT) or silenced with the indicated oligos. Each dot represents the mean number of RAD51 foci in a single repetition, from two to six biological replicates. Statistical analysis on the data is shown in Fig 6G. Shown are mean ± SEM.
-
BA graph summarizing the number of RAD51 foci in HeLa WT and DPP9 KD cells treated with EdU and MMC. Shown is the quantification of RAD51 foci in EdU positive cells. HeLa DPP9 KD cells display fewer RAD51 foci in comparison with their WT counterparts. Each dot represents the number of RAD51 foci in a single cell, from three biological replicates. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM.
-
CA graph summarizing the mean number of RAD51 foci in EdU positive HeLa WT and DPP9 KD cells. Each dot represents the mean number of RAD51 foci in a single repetition, from three biological replicates. Statistical analysis on the data is shown in Fig EV4B. Shown are mean ± SEM.
-
DA graph summarizing the number of RAD51 foci in MEF WT and gki MEF DPP9S729A cells treated with MMC. Shown is the quantification of RAD51 foci. Each dot represents the number of RAD51 foci in a single cell, from three biological replicates. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM.
-
EA graph summarizing the mean number of RAD51 foci in MEF WT and gki MEF DPP9S729A cells upon MMC treatment. Each dot represents the mean number of RAD51 foci in a single repetition, from three biological replicates. Statistical analysis on the data is shown in Fig EV4D. Shown are mean ± SEM.
-
FA graph summarizing the mean number of RAD51 foci following induction of DPP9‐SWT expression, compared with uninduced HEK293 DPP9 KO+DPP9WT cells (‐Dox). Induction of HEK293 DPP9 KO+DPP9S729A for expression of DPP9‐SS729A did not result in more RAD51 foci. Each dot represents the mean number of RAD51 foci in a single repetition, from three biological replicates. Statistical analysis on the data is shown in Fig 7B. Shown are mean ± SEM.
-
GA graph summarizing the number of RAD51 foci following induction of DPP9‐SWT or DPP9‐LWT expression, compared with HEK293 DPP9WT cells. The number of RAD51 foci formed by the HEK293 DPP9 KO+DPP9‐SWT cells was similar to the HEK293 DPP9WT cells, compared with the HEK293 DPP9 KO+DPP9‐LWT cells. Each dot represents the mean number of RAD51 foci in a single repetition, from two biological replicates. Shown are mean ± SEM.
-
H, ISummary of RAD51 foci in HeLa WT and HeLa DPP9 KD cells as described in (Fig 7C and F). Each dot represents the mean number of RAD51 foci in a single repetition, from three or four biological replicates (HeLa WT (n = 3), HeLa DPP9 KD (n = 4)). Data from the summary of all RAD51 foci per nucleus upon MMC treatment, between biological replicates, were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001). Shown are mean ± SEM.
Figure 7. DPP9 activity and the BRCA2 N‐terminus promote RAD51 foci formation.
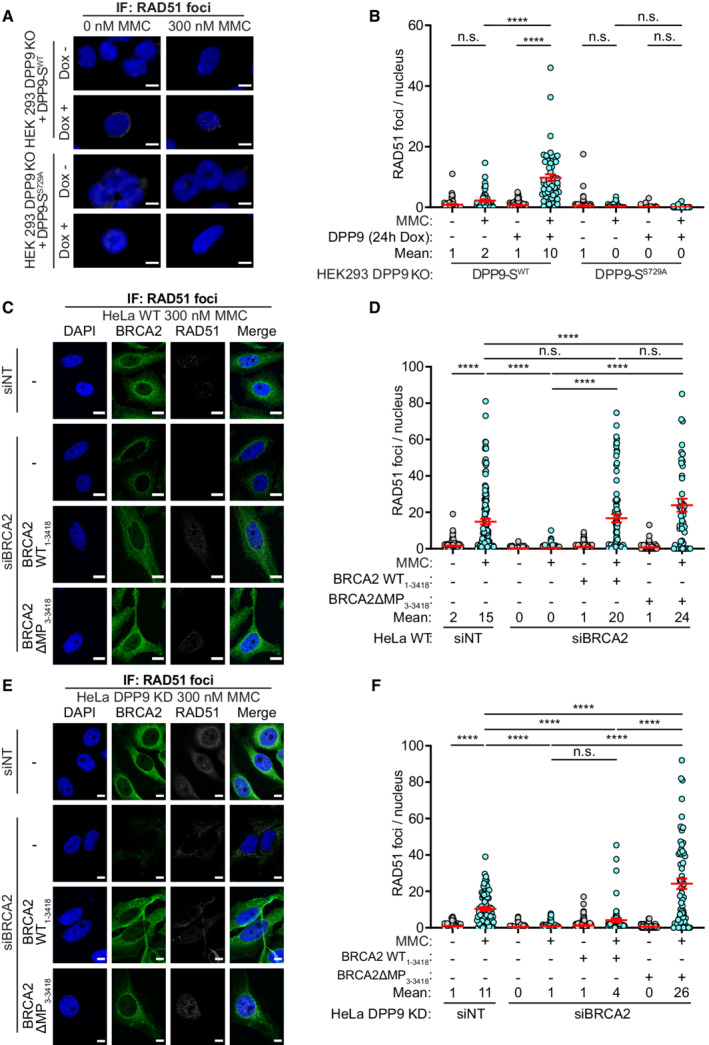
-
ARepresentative immunofluorescence images showing that re‐expression of DPP9‐SWT leads to an increase in the number of RAD51 foci formed following exposure to MMC in HEK293 DPP9 KO+DPP9‐SWT cells. Expression of DPP9 was induced (+ Dox, 1 μg/ml) simultaneously with MMC (300 nM), 24 h. RAD51 foci are shown in white, and nuclei (DAPI) are shown in blue. Scale bar 10 μm. Anti‐RAD51: RRID:AB_1142428.
-
BGraph showing the number of RAD51 foci following induction of DPP9‐SWT expression, compared with uninduced HEK293 DPP9 KO+DPP9WT cells (‐Dox). Induction of HEK293 DPP9 KO+DPP9S729A for expression of DPP9‐SS729A did not result in more RAD51 foci. Each dot represents the number of RAD51 foci in a single cell, from three biological replicates. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001).
-
C–FRepresentative images (C, E) and summarizing graph (D, F) showing the number of RAD51 foci per nucleus in HeLa WT cells (C, D) or HeLa DPP9 KD cells (E, F). Where stated, cells were treated with control siRNA (siNT) or silenced for BRCA2, and transiently transfected with the BRCA21‐3418 or BRCA23‐3418 constructs. Both BRCA2 constructs can rescue the RAD51 foci formation phenotype to the control levels in HeLa WT cells (C, D). In HeLa DPP9 KD cells significantly more RAD51 foci were present in cells transfected with BRCA2ΔMP3‐3418 compared with BRCA2 silenced cells and cells expressing the untruncated BRCA21‐3418 (E, F). Each dot represents the number of RAD51 foci in a single cell, from three (D) or four (F) biological replicates. Data were analyzed by a two‐way ANOVA, with the Tukey's multiple comparison test. Shown are mean ± SEM (****P ≤ 0.0001). RAD51 foci are shown in white, and nuclei (DAPI) are shown in blue. Scale bar 10 μm. Anti‐RAD51: RRID:AB_1142428; anti‐DPP9: RRID:AB_2889071; anti‐BRCA2: RRID:AB_2259370.
To directly test whether the N‐terminus of BRCA2 affects the appearance of the RAD51 foci, we constructed a BRCA2ΔMP truncation variant (BRCA2ΔMP3‐3418) lacking the N‐terminal dipeptide Met‐Pro. Both BRCA2ΔMP3‐3418 and full‐length BRCA21‐3418 were cloned into ubiquitin‐fusion vectors to express the two constructs with the desired N‐termini. To avoid possible toxicity by the overexpression of BRCA2, cells were first silenced for endogenous BRCA2 prior to transfection with either construct (Appendix Fig S6), and a silent mutation was introduced into both plasmids to acquire resistance to the BRCA2 siRNA. As expected, RAD51 foci were formed in HeLa WT cells following MMC treatment, a process that was down‐regulated by silencing of BRCA2 (Fig 7C and D). Expression of either BRCA2ΔMP3‐3418 or BRCA21‐3418 resulted in a significant increase in the number of RAD51 foci that appeared in response to MMC, indicating that both BRCA21‐3418 and BRCA2ΔMP3‐3418 can restore the silencing of BRCA2 (Figs 7C and D, and EV4H). However, since DPP9 is active in HeLa WT cells, BRCA21‐3418 may have been processed by DPP9 to BRCA2ΔMP3‐3418. Thus, the assay was also carried out in HeLa DPP9 KD cells to compare the capacities of both BRCA21‐3418 and BRCA2ΔMP3‐3418 constructs to promote RAD51 foci formation. Similar to HeLa WT cells, expression of BRCA2ΔMP3‐3418 into the HeLa DPP9 KD cells first silenced for BRCA2, was accompanied by a significant increase in the number of RAD51 foci in response to MMC. On the other hand, expression of WT BRCA21‐3418 did not lead to a significant increase in the number of MMC‐induced RAD51 foci in the BRCA2‐silenced HeLa DPP9 KD cells (Fig 7E and F, and EV4I). Taken together, these results strongly suggest that DPP9 promotes the formation of RAD51 foci by processing the N‐terminus of BRCA2.
Discussion
DPP9 regulates BRCA2 stability
This work identifies the rate‐limiting proline‐cleaving protease DPP9, as a regulator of BRCA2 stability and highlights the stabilizing role of the evolutionarily conserved Pro2 in the BRCA2 N‐terminus.
The imino acid proline is unique with its rigid ring‐like side chain. N‐terminal prolines confer high stability to proteins when found in the first or second N‐terminal position (Bachmair et al, 1986), since these are not identified by the classical N‐Recognins of the Arg/N‐degron and Ac/N‐degron pathways (Varshavsky, 2019). Therefore, the targeting and removal of such prolines can be utilized as major sites for regulating changes in protein stability. The best‐characterized example is the Pro/N‐degron pathway, which employs GID ubiquitin ligase complexes. These super‐complexes include substrate receptors that bind to prolines, with a preference for a four amino acid motif (Pro‐Gly‐Leu‐Trp; Dong et al, 2018; Sherpa et al, 2021). Regulated targeting of proline‐containing proteins by this pathway is accomplished by stimulation of complex assembly and expression of the receptor subunit (Menssen et al, 2018; Melnykov et al, 2019; Qiao et al, 2020). DPP9 presents an alternative pathway that cells apply to convert otherwise stable proteins with N‐terminal prolines such as AK2 and BRCA2 into substrates for proteasomal degradation. In contrast to the seemingly constitutive processing of AK2 by DPP9 (Finger et al, 2020), the interaction of BRCA2 with DPP9 is induced in response to genotoxic stress, which also promotes BRCA2 degradation. Another verified DPP9 substrate is Syk, with alanine in the second position. Similar to BRCA2, Syk degradation does not appear to be constitutive but instead is induced following stimulation of the B‐cell receptor, a process that relies on DPP9 activity (Justa‐Schuch et al, 2016).
How DNA damage signals for increased interaction between DPP9 and BRCA2 remains to be shown. However, we note that the N‐terminus of BRCA2 is intrinsically disordered (Le et al, 2020; Sidhu et al, 2020; Paul et al, 2021) and is involved in the formation of BRCA2 dimers and multimers through self‐interactions (Shahid et al, 2014; Reuter et al, 2015; Sánchez et al, 2017; Le et al, 2020; Sidhu et al, 2020). Thus, it is tempting to speculate that BRCA2 is shielded from DPP9 in the multimeric form, and becomes available as a DPP9 substrate by a transition of BRCA2 multimers to monomers, a process that is favored by ssDNA, RAD51, increased temperature, and the BRCA2 chaperone DSS1 (Le et al, 2020; Sidhu et al, 2020). Since FLNA supports the MMC‐induced proximity between DPP9 and BRCA2 it is tempting to speculate that FLNA binds to both proteins thereby increasing their local concentration to support cleavage.
Finally, N‐terminally tagged constructs of BRCA2 are applied in several studies and have allowed important discoveries in the field of HR. We can speculate that these complementation assays with N‐terminally tagged BRCA2 constructs were possible due to differences in the expression levels of the tagged BRCA2 protein compared with its endogenous levels, which thus compensate for the lack in DPP9‐mediated turnover. We raise awareness to the role of the BRCA2 N‐terminus in regulating BRCA2 stability.
DPP9 promotes HR‐mediated DNA repair
Here, we show that DPP9‐depleted cells are hypersensitive to genotoxic agents, are less efficient in HR repair, and accumulate more γH2AX in response to DNA damage. The involvement of proteases in DNA maintenance has been shown for example for the DNA‐binding metalloprotease SPRTN and the trypsin‐like protease FAM111A that remove DNA‐protein crosslinks (Lopez‐Mosqueda et al, 2016; Stingele et al, 2016; Vaz et al, 2016; Kojima et al, 2020). The ubiquitin protease system facilitates HR by ubiquitinating Rad51 and the replication protein A (RPA) on DSBs, leading to the removal of both proteins from these sites (Elia et al, 2015; Feeney et al, 2017; Gong & Chen, 2011; Inano et al, 2017; Liu et al, 2011). While we observed a similar fold increase in the number of BRCA2‐DPP9 PLA events in response to MMC‐ and NCS‐induced DNA damage in both, the nucleus and the cytosol, the majority of the interaction events are in the cytosol, suggesting that the processing of BRCA2 by DPP9 occurs predominantly in this compartment, and not necessarily on the chromatin. Previous work has shown that the ubiquitin‐specific protease USP21, which is also found in cytosol, counteracts the degradation of BRCA2 (Liu et al, 2017). Thus, N‐terminal processing and deubiquitination of BRCA2 appear to occur preferentially in the cytoplasm, presenting a sensitive system of opposing forces that fine‐tune the cellular concentrations of BRCA2. The importance of regulating BRCA2 steady‐state levels can be observed in patients with sporadic breast cancer where BRCA2 is overexpressed (Bièche et al, 1999; Egawa et al, 2002; Wang et al, 2018). Similarly, the expression levels of USP21, which deubiquitinates and stabilizes BRCA2 negatively correlate with the survival of patients with hepatocellular carcinoma (Liu et al, 2017). Similarly, low DPP9 expression, which should allow higher BRCA2 levels, correlates with a poorer prognosis for patients with breast cancer, an effect that is not seen for DPP8 (Fig EV5A and B). That lower BRCA2 levels impair HR is well‐established. A possible scenario is that the cellular levels of BRCA2 are tightly regulated and fine‐tuned to allow productive molecular interactions of BRCA2 with PALB2 and RAD51. Consistent with the higher levels of BRCA2 in DPP9‐deficient cells, more PALB2‐BRCA2 interactions are observed in these cells, suggesting that DPP9 limits this interaction by lowering the cellular concentration of BRCA2. DPP9 may additionally limit this interaction by competing with PALB2 for interaction with BRCA2. Nonetheless, despite the increase in the BRCA2‐PALB2 association, DPP9‐depleted cells show a decrease in the colocalization of BRCA2 with γH2AX. Consistently, DPP9‐depleted cells accumulate fewer RAD51 foci, a phenotype that can be partially restored by the overexpression of active DPP9‐SWT, less efficiently by DPP9‐LWT, but not by the enzymatically inactive DPP9‐SS720A mutant. Additionally, the expression of the BRCA2ΔMP truncation mutant results in a better recovery of RAD51 foci in DPP9 KD cells silenced for BRCA2, compared with the expression of the WT BRCA2 construct. Since both constructs compensate for BRCA2 silencing in cells expressing endogenous levels of DPP9, these results imply that DPP9 promotes the formation of RAD51 foci by modifying the N‐terminus of BRCA2. Similarly, fewer RAD51 foci were reported for cells expressing an excess of BRCA2 or the BRC4 repeat (Chen et al, 1999; Magwood et al, 2012; Abe & Branzei, 2014).
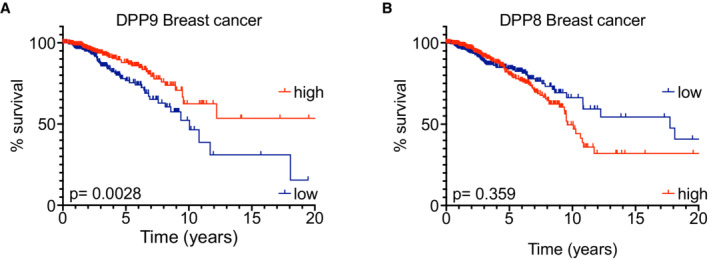
Figure EV5. Low DPP9 mRNA expression correlates with poor overall survival for patients with breast cancer.
-
A, BKaplan–Meier survival curves of Breast cancer patients from The Human Protein Atlas into “high DPP9 or DPP8” (n = 282, 445) and “low DPP9 or DPP8” (n = 408, 630) mRNA expression (greater than or less than 9.678 or 4.36 reads per kilobase per million, respectively). P‐values calculated by log‐rank (Mantel–Cox) indicated greater overall survival in patients with high levels of DPP9 expression (A), while differences in the DPP8 levels were not statistically significant (B).
The interaction between BRCA2 and RAD51 involves eight BRC repeats in the centre of BRCA2 and an additional domain in the C‐terminus (CTD) allowing BRCA2 to bind multiple copies of RAD51 (Davies et al, 2001; Pellegrini et al, 2002; Esashi et al, 2005, 2007; Galkin et al, 2005; Davies & Pellegrini, 2007). Different studies estimate a ratio of one BRCA2 monomer binding simultaneously at least 5–7 molecules of RAD51 (Yang et al, 2005; Jensen et al, 2010; Liu et al, 2010; Shahid et al, 2014; Sidhu et al, 2020). In vitro studies suggest that by binding to multiple monomers of RAD51, BRCA2 provides a rapid mechanism for nucleating the RAD51 filaments on the ssDNA and promoting their growth (Jensen et al, 2010; Liu et al, 2010; Thorslund et al, 2010; Carreira & Kowalczykowski, 2011; Shahid et al, 2014; Sánchez et al, 2017). Thus, the DNA damage‐induced degradation of BRCA2 that we and others observe (Schoenfeld et al, 2004; Liu et al, 2017) may serve to establish an optimal stoichiometric ratio between BRCA2 and RAD51 for repair. In this scenario, DPP9 facilitates efficient repair by fine‐tuning the cellular concentration of BRCA2.
The interaction between RAD51 and BRCA2 is cell‐cycle‐dependent (Ayoub et al, 2009), through the CDK‐mediated phosphorylation of the BRCA2 C‐terminus at S3291 (Esashi et al, 2005) and ubiquitination of RAD51, which interfere with the interaction (Luo et al, 2016). Fine‐tuning the protein levels of BRCA2 by DPP9 presents an additional layer of regulation cells applied to ensure efficient repair by HR.
Finally, our results set the ground for future analysis of DPP9 activity in breast cancer and suggest that DPP9 inhibition in combination with Olaparib or radiation presents future potential therapies for patients with breast cancer.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| HeLa wt | Justa‐Schuch et al (2016) | |
| HeLa DPP9 KD | Justa‐Schuch et al (2016) | |
| MEF wt | Gall et al (2013) | |
| gki MEF DPP9S729A | Gall et al (2013) | |
| Sf9 | Gibco™ | Cat#11496015 |
| HEK293 Flp‐In T‐REx‐293 | Thermo Fisher Scientific | Cat #R78007, RRID:CVCL_U427 |
| HEK293 Flp‐In T‐REx‐293 DPP9 KO | This study | |
| HEK293 Flp‐In T‐REx‐293 DPP9 KO‐DPP9‐S WT | This study | |
| HEK293 Flp‐In T‐REx‐293 DPP9 KO‐DPP9‐SS729A | This study | |
| HEK293 Flp‐In T‐REx‐293 DPP9 KO‐DPP9‐L WT | This study | |
| HCT116 cells, stably transfected with pDRGFP | Kari et al (2019) | |
| Recombinant DNA | ||
| pcDNA3 236HSC WT (hBRCA2) | Addgene, (Wang et al, 2002) | Cat #16246 |
| 3xHA‐DHFR‐Ubiquitin‐BRCA2‐eGFP | This paper | |
| 3xHA‐DHFR‐Ubiquitin‐BRCA2ΔMP‐FLAG‐eGFP | This paper | |
| 3xHA‐DHFR‐Ubiquitin‐BRCA21‐1000‐FLAG‐eGFP | This paper | |
| pSpCas9(BB)‐2A‐GFP | Addgene, Ran et al (2013) | Cat #48138 |
| 3xHA‐DHFR‐Ubiquitin‐BRCA21‐1000 ΔMP 1‐1000‐FLAG‐eGFP | This paper | |
| Flag tagged DPP9‐SWT in pUC57‐BsaI‐Free | BioCat | Custom made |
| Flag tagged DPP9‐SS729A in pUC57‐BsaI‐Free | BioCat | Custom made |
| Flag tagged DPP9‐SWT in pcDNA5/FRT/TO | This paper | |
| Flag tagged DPP9‐SS729A in pcDNA5/FRT/TO | This paper | |
| Flag tagged DPP9‐LWT in pcDNA5/FRT/TO | This paper | |
| SUMO1‐BRCA2,1‐39_pet11a | This paper | |
| SUMO1‐BRCA2ΔMP,3‐39_pet11a | This paper | |
| BRCA21‐1000 in pcDNA3.1(+) P2A eGFP | Genscript | Custom made |
| BRCA21‐1000 P2G in pcDNA3.1(+) P2A eGFP | Genscript | Custom made |
| BRCA21‐1000 P2H in pcDNA3.1(+) P2A eGFP | Genscript | Custom made |
| Antibodies | ||
| Mouse anti‐BRCA2 (monoclonal), IF/PLA (1:100), WB (1:1,000) | R&D | Cat #MAB2476; RRID:AB_2259370 |
| Rabbit anti‐DPP9 (polyclonal), WB (1:500) | Abcam | Cat #AB42080; RRID:AB_731947 |
| Goat anti‐DPP9 (polyclonal), IF and PLA (1:20) | Justa‐Schuch et al (2016) | RRID:AB_2889071 |
| Rabbit anti‐Filamin A (polyclonal), WB (1:500) | Novus | Cat #nb‐100‐58812; RRID:AB_877728 |
| Mouse anti‐FLAG (monoclonal), WB (1:500) | Sigma‐Aldrich | Cat #F1804; RRID:AB_262044 |
| Rabbit anti‐GFP (polyclonal), WB (1:200) | Santa Cruz Biotechnology | Cat #sc‐8334; RRID:AB_641123 |
| Rabbit anti‐H2AX‐P (polyclonal), IF (1:500), WB (1:1,000) | Cell Signaling | Cat #9718; RRID:AB_2118009 |
| Mouse anti‐H2AX‐P (monoclonal), IF and PLA (1:500) | Millipore | Cat #05‐636; RRID:AB_309864 |
| Mouse anti‐HA, WB (1:1,000) | Biolegend | Cat #901515, RRID:AB_2565334 |
| Rabbit anti‐H3 (polyclonal), WB (1:100) | Cell Signaling | Cat #9715; RRID:AB_331563 |
| Rabbit anti‐PALB2 (polyclonal), PLA (1:100), WB (1:500) | Bethyl | Cat #A301‐246A; RRID:AB_890607 |
| Rabbit anti‐RAD51 (polyclonal), IF (1:500) WB (1:1,000) | Abcam | Cat #ab63801; RRID:AB_1142428 |
| Mouse anti‐Tubulin (monoclonal), WB (1:5,000) | Santa Cruz | Cat #sc‐32293; RRID:AB_628412 |
| Mouse anti‐Vinculin (monoclonal), WB (1:6,000) | Sigma‐Aldrich | Cat #V9131; RRID:AB_477629 |
| Anti‐FLAG® M2 Magnetic Beads antibody | Sigma‐Aldrich | Cat #M8823, RRID:AB_2637089 |
| Pierce Anti‐HA magnetic beads | Thermo Scientific | Cat #88836; RRID:AB_2749815 |
| Ni‐NTA Agarose | Qiagen | Cat #1018244 |
| Chemicals, enzymes and other reagents | ||
| 1G244 (in DMSO) | AK Scientific, Inc. | Cat #Y0432 |
| Cycloheximide | Sigma‐Aldrich | Cat #C7698 |
| Doxycycline | Ratiopharm | Cat #PZN4314646 |
| Lipofectamine P3000 | Thermo Fisher Scientific | Cat #L3000015 |
| Lipofectamine RNAimax | Thermo Fisher Scientific | Cat #13778150 |
| Mayer's Hemalum | Merck | Cat #1.09249.0500 |
| Mitomycin C | Sigma‐Aldrich | Cat #M4287 |
| Neocarcinostatin | Sigma‐Aldrich | Cat #N9162 |
| Propidium Iodide (PI) solution | Sigma‐Aldrich | Cat #P4864 |
| MG132 | Sigma‐Aldrich | Cat #C2211 |
| DAPI | Thermo Fisher Scientific | Cat #D1306 |
| Phalloidin‐iFluor 488 | Abcam | Cat #ab176753 |
| Olaparib | Selleck Chemicals | Cat #AZD2281 |
| Puromycin | Sigma‐Aldrich | Cat #P8833 |
| Aprotinin | Carl ROTH | Cat #162.3 |
| Leupeptin | Carl ROTH | Cat #N33.4 |
| Pepstatin | Carl ROTH | Cat #2936.3 |
| Phusion DNA polymerase | Thermo Fisher Scientific | Cat #F530S |
| BRCA21‐40 peptide | Genscript | MPIGSKERPTFFEIFKTRCNKADLGPISLNWFEELSSEAP, > 85% pure |
| BRCA23‐40 peptide | Genscript | IGSKERPTFFEIFKTRCNKADLGPISLNWFEELSSEAP, > 85% pure |
| BRCA21‐20 peptide | Genscript | MPIGSKERPTFFEIFKTRCN, > 85% pure |
| BRCA21‐40 P2G peptide | Genscript | MGIGSKERPTFFEIFKTRCNKADLGPISLNWFEELSSEAP, > 85% pure |
| BRCA21‐40 E13R peptide | Genscript | MPIGSKERPTFFRIFKTRCNKADLGPISLNWFEELSSEAP, > 85% pure |
| BRCA21‐40 I14N peptide | Genscript | MPIGSKERPTFFENFKTRCNKADLGPISLNWFEELSSEAP, > 85% pure |
| BRCA21‐40 W31C peptide | Genscript | MPIGSKERPTFFEIFKTRCNKADLGPISLNCFEELSSEAP, > 85% pure |
| BRCA21‐40 F32G peptide | Genscript | MPIGSKERPTFFEIFKTRCNKADLGPISLNWGEELSSEAP, > 85% pure |
| SLRFLYEG peptide | Genscript | SLRFLYEG, > 85% pure |
| Software | ||
| DuoLink software | Sigma‐Aldrich | RRID:SCR_015574 |
| ImageStudio Lite v4.0.21 | LI‐COR | RRID:SCR_013715 |
| LSM Image Examiner | Zeiss | RRID:SCR_014344 |
| LSM 510 Release Version 4.0 SP2 | Zeiss | |
| Celigo Software v2.0 | Nexlecom | |
| GraphPad Prism 8 | GraphPad | RRID:SCR_002798 |
| XDS Program Package | RRID:SCR_015652 | |
| Refmac | RRID:SCR_014225 | |
| FlowJo v10.8.1 | BD Biosciences | RRID:SCR_008520 |
| Microsoft Excel | Microsoft | RRID:SCR_016137 |
| Empiria Studio v2.2.0.141 | LI‐COR | |
| Biorender | https://biorender.com/ | RRID:SCR_018361 |
| Fiji |
https://imagej.net/software/fiji/ Schindelin et al (2012) doi:10.1038/nmeth.2019 |
|
| The Human Protein Atlas | https://www.proteinatlas.org/ | |
| Other | ||
| CellTiter‐Glo® Luminescent Cell Viability Assay | Promega | Cat # G7571 |
| DuoLink in Situ PLA probe Mouse plus | Sigma‐Aldrich | Cat #DUO92001; RRID:AB_2810939 |
| DuoLink in Situ PLA probe Rabbit plus | Sigma‐Aldrich | Cat #DUO92002; RRID:AB_2810940 |
| DuoLink in Situ PLA probe Mouse minus | Sigma‐Aldrich | Cat #DUO92004; RRID:AB_2713942 |
| DuoLink in Situ PLA probe Goat minus | Sigma‐Aldrich | Cat #DUO92006 |
| DuoLink in Situ Detection Reagents Red | Sigma‐Aldrich | Cat #DUO92008 |
| CytoFLEX S | Beckman Coulter | RRID:SCR_019627 |
| Enspire Multimode Plate Reader | PerkinElmer | |
| Celigo | Nexlecom | RRID:SCR_018808 |
| LI‐COR Odyssey CLX | LI‐COR | |
Methods and Protocols
Method details
See Reagents Tools table and Table EV1 for Primers and siRNA.
Cell lines and cell culture
HeLa DPP9 stable Knock‐Down cells (DPP9 KD) and the corresponding HeLa WT cells (Genscript) (Justa‐Schuch et al, 2016), MEF WT and gki MEF DPP9S729A (Gall et al, 2013), HEK293 DPP9 KO+DPP9‐SWT or DPP9‐SS729A, HEK293 DPP9 KO+DPP9‐LWT (this study), and the corresponding HEK 293 DPP9 WT cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM L‐glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. To maintain the selection pressure on the DPP9 KD cells, 1.5 μg/ml puromycin (Sigma‐Aldrich, Germany) was added to the growth medium. HCT 116 cells, stably expressing pDRGFP were a kind gift from Holger Bastians (Kari et al, 2019). Cells were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum. All cells were grown at 37°C and 5% CO2 and routinely tested for mycoplasma contamination.
Generation of DPP9 knockout (HEK293 DPP9 KO) cells and complementation cell lines (HEK293 DPP9 KO+DPP9‐SWT , HEK293 DPP9 KO+DPP9‐SS729A , and HEK293 DPP9 KO+DPP9‐LWT )
For the generation of the HEK Flp‐In™ T‐REx™‐293 DPP9 knockout clone (HEK293 DPP9 KO), guide RNA sequences (CRISPR guide 3 on exon 9, see Table EV1) targeting human DPP9 were cloned into the pSpCas9(BB)‐2A‐GFP (PX458) vector, which was a gift from Feng Zhang (Addgene plasmid # 48138) (Ran et al, 2013). HEK Flp‐In™ T‐REx™‐293 (ThermoFisher Scientific) cells were transfected using PEI. After 24 h, FACS sorting was used to collect GFP‐positive cells. Single cells were seeded into 96‐well plates. Clones were screened using western blot.
For complementation, the Flp‐In™T‐REx™ system (Invitrogen) was used to create stable, inducible cell lines expressing DPP9‐SWT (HEK293 DPP9 KO+DPP9‐SWT), DPP9‐SS729A (HEK293 DPP9 KO+DPP9‐SS729A) or DPP9‐LWT (HEK293 DPP9 KO+DPP9‐LWT) in pcDNA5/FRT/TO. For the selection of positive clones, DMEM complete containing 100 μg/ml hygromycin and 10 μg/ml blasticidin was used. Unless otherwise stated, DPP9 expression was induced by the addition of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM L glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin containing 1 μg/ml doxycycline for 24 h.
Plasmids
The following plasmids were kind gifts, obtained from Addgene: the pCBASce I endonuclease expression vector (Addgene plasmid # 26477) from Maria Jasin (Pierce et al, 1999), the template pcDNA3 236HSC WT BRCA2 (Addgene plasmid # 16246) from Mien‐Chie Hung (Wang et al, 2002). BRCA21‐3418, BRCA21‐1000, and the corresponding mutants BRCA23‐3418, and BRCA23‐1000 that lack the N‐terminal dipeptide Met1Pro2, were cloned into pcDNA3.1+ vector with a Ubiquitin tag fused at the N‐terminus to provide a defined N‐terminus, as first described by Bachmair and Varshavsky (Bachmair et al, 1986). Silent mutations were introduced at the binding sites of the silencing oligos targeting BRCA2 (1721GAAGAATGCAGGTTTAATA1740) at the sites 1731A‐>T and 1734T‐>A. SUMO1‐BRCA21‐40‐HA‐His constructs were cloned by adding SUMO1 to the N‐terminus of BRCA21‐39‐3HA‐His pET11a (custom‐made from Genscript), using the Gibson Chew Back and Anneal Assembly (CBA) as described in (Torella et al, 2014) Primers are listed in extended view Table EV1. Flag‐tagged DPP9‐SWT, Flag‐tagged DPP9‐SS729A, and DPP9‐LWT were cloned into pcDNA5/FRT/TO (ThermoFisher Scientific #V601020). All plasmids have been verified by sequencing. HeLa cDNA was used as a template for PCR amplification of human PALB2. The PALB2 PCR product was cloned into pENTR 3C using the BamH1 and NotI sites. PALB2 was further cloned from pENTR3C into BaculoDirect, using the LR Clonase (ThermoFisher Scientific). BRCA21‐1000 WT, BRCA21‐1000 P2G, and BRCA21‐1000 P2H were custom synthesized by GenScript.
Co‐immunoprecipitation
HEK293 DPP9 KO+DPP9‐SWT cells were treated for 24 h with 300 nM MMC. For induced expression of FLAG‐tagged DPP9‐SWT, 1 μg/ml of Doxycyline was added together with the MMC for 24 h. Control cells were not treated with Doxycycline. Cells were trypsinized and lysed in Lysis buffer (50 mM HEPES pH 7.4, 100 mM NaCl, 0.5% NP40, 5 mM EDTA, 1 mM DTT, PMSF and 10 mM MG132). Following a 30‐min incubation at 4°C, cell lysates were centrifuged at 20,000 g at 4°C for 20 min. Cleared cell lysates were diluted in 50 mM HEPES pH 7.4, 100 mM NaCl, 5 mM EDTA to a final NP‐40 concentration 0.1%. Diluted lysates were incubated with magnetic FLAG‐beads for 3 h at 4°C. Bound proteins were eluted with 0.5 mg/ml FLAG peptide and analyzed by western blotting.
Chromatin fractionation
HeLa WT and HeLa DPP9 KD were treated with MMC (300 nM) for 0, 8, or 24 h. Chromatin fractionation was performed essentially as described in (Kari et al, 2016). Briefly, cells were resuspended in lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton‐X‐100, 1 mM DTT, and protease inhibitors) and centrifuged at 1,500 g for 5 min. The nuclear pellet was lysed in nuclear lysis buffer (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, and protease inhibitors) for 30 min on ice. Soluble chromatin fractions were separated by centrifuging at 1,700 g for 5 min. Chromatin fractions were sonicated in a water sonicator (BioRad) for 15 min before loading on SDS–PAGE electrophoresis.
Immunofluorescence
HeLa, MEF, HEK293 DPP9 WT, HEK293 DPP9 KO+DPP9‐SWT/+DPP9‐SS729A, and HEK293 DPP9 KO+DPP9‐LWT cells were grown on coverslips in 24‐well plates, fixed with 4% formaldehyde in PBS for 10 min and permeabilized with 0.2% Triton‐X‐100 in PBS for 5 min. Cells were washed with PBS and blocked with 2% BSA in PBS for 10 min. Cells were incubated with primary antibodies for 90 min at 37°C. Following a PBS wash, cells were incubated for 45 min at room temperature with the respective secondary antibodies. Cells were washed with PBS and water and mounted in fluorescent mounting medium (DAKO) with DAPI. EdU stained cells were treated with EdU (abcam) 30 min prior to DNA damage induction according to the manufacturer's specifications. Cells were analyzed and images were taken using either an LSM 510‐Meta confocal microscope, oil immersion objective 63x/1.3 (Carl Zeiss MicroImaging, Inc), or a Nikon Eclipse Ti2‐E Inverted microscope, Plan Apoλ oil immersion objective 60x NA1.40 WD = 0.13 (Nikon Instruments Inc). Images were processed using LSM Image Browser (Carl Zeiss MicroImaging, Inc) or NIS‐Elements AR 5.02.00 (Nikon Instruments Inc), based on the microscope used, and FIJI for the preparation of figures. RAD51 foci were quantified using the Duolink ImageTool (Sigma‐Aldrich).
Proximity ligation assay
Proximity Ligation Assay (PLA) was performed using the DUOLINK In Situ PLA Kit (Sigma‐Aldrich) according to the manufacturer's protocol. Briefly, HeLa cells were grown on coverslips and fixed as described above for immunofluorescence assays. In the case of DPP9 inhibition studies, 10 μM 1G244 was added to the cells for 30 min before fixation. Control cells were mock treated with DMSO. HeLa cells were incubated with primary antibodies for 90 min at 37°C and actin filaments were simultaneously counterstained with CytoPainter Phalloidin‐iFluor 488 Reagent (Abcam ‐ #ab176753). Coverslips were washed with PBS and treated with PLA reagents. Control coverslips (NgtCtrl) were treated with one primary antibody to estimate background staining in each experiment. Cells were mounted in DAKO with DAPI fluorescent mounting medium and analyzed using an LSM 510‐Meta confocal microscope, oil immersion objective 63x/1.3 (Carl Zeiss MicroImaging, Inc) or a Nikon Eclipse Ti2‐E Inverted microscope, Plan Apoλ oil immersion objective 60x NA1.40 WD = 0.13 (Nikon Instruments Inc). Images were processed using LSM Image Browser (Carl Zeiss MicroImaging, Inc) or NIS‐Elements AR 5.02.00 (Nikon Instruments Inc), based on the microscope used and subsequently analyzed using the Duolink ImageTool (Sigma).
Viability assay
Cells were seeded in 96‐well White Microplates (Perkin Elmer cat# 6005680) and treated with different concentrations of Olaparib for 96 h. For the MMC assays, cells were first incubated for 18–24 h to allow cell attachment. Next, different MMC concentrations were added and cells were analyzed 72 h later. Control cells were treated with DMSO. Cell viability was measured using the CellTiter‐Glo®Luminescence Cell Viability Assay (Promega, cat# Cat G7571). CellTiter‐Glo®Reagent was added in a 1:1 ratio to the cell culture medium. The microplate was shaken on an orbital shaker for 10 min for induction of cell lysis. Subsequently, the luciferase signal was measured on a LuminometerDLReady™ Centro LB 960 reader. Each experiment was performed three times, in triplicates or quadruplicates.
Colony formation assay
To calculate the respective surviving fractions (SF) after γ radiation (0, 1, 2, 4, 6, and 8G), a standard colony‐forming assay was performed, as previously described (Rave‐Fränk et al, 2007). Briefly, cells were exposed to γ‐ radiation and incubated for 7 days, fixed with 70% ethanol, and stained with Mayer's hemalum (Cat#1.09249.0500, Merck). Nonirradiated cultures were used for normalization. Colonies with > 50 cells were scored as survivors. Three biological replicates were performed, each containing three technical triplicates, and medians were calculated. To validate statistical differences between control and treatment groups, analysis of variance (ANOVA: two‐factor with replication) was performed using Microsoft Excel software (version 2016 MSO). P‐values < 0.05 were considered significant.
Analysis of DSB repair by GFP‐based reporter assay
To measure HR frequency, HCT116 cells expressing stably integrated pHPRT‐pDR‐GFP reporter plasmid were transfected with the indicated siRNAs. Following 24 h of siRNA transfection, cells were either mock‐transfected or transfected with 2 μg pCBASceI (Addgene) to induce DSBs. 48 h after transfection, the expression of GFP was quantified by flow cytometry using a CytoFLEX S (Beckman Coulter). Cell debris and dead cells were identified using propidium iodide staining (Sigma‐Aldrich P4864). The data were analyzed using the FlowJo software (BS Bioscience). The HR frequency was determined as the percentage of GFP‐positive cells in the total number of alive cells.
Protein purification
Human recombinant DPP9 was expressed from Sf9 cells and purified essentially as described previously (Pilla et al, 2012). Human recombinant His‐tagged PALB2 was expressed from Sf9 cells. Cells were resuspended in PALB2 lysis buffer (25 mM Hepes pH7.5, 200 mM NaCl, 5 mM imidazole, 10% glycerol, 0.2% Triton, 5 mM ß‐mercaptoethanol, 2 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin), and lysed by homogenization and sonication. 15 U/ml Benzonase and 1 mM MgCl2 were added to the cell lysate. Following a 1 h incubation at 4°C, cell lysate was centrifuged at 50,000 g for 90 min at 4°C. The supernatant was incubated was bound to Ni‐NTA Agarose (Qiagen) for 2 h. Following extensive washing, PALB2 was eluted with Elution Buffer (25 mM Hepes pH7.5, 50 mM NaCl, 300 mM imidazole, 5 mM ß‐mercaptoethanol) and further purified on a Superdex 200 (GE Healthcare). For purification of N‐terminally His‐HA‐tagged fragments corresponding to the N‐terminus of BRCA2 (BRCA2 1‐40HA‐His), SUMO1BRCA21‐40HA‐His was transformed into BL21 (DE3 codon+) cells and induced by 1 mM IPTG for 2 h at 30°C. Cells were lysed in 25 ml Lysis Buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM Imidazole pH 8.0, 1 mM ß‐Mercaptoethanol and protease inhibitors: 1 μg/ml Aprotinin, 1 μg/ml Leupeptin and 1 μg/ml Pepstatin), by sonification. The 100,000 g supernatant was bound to Ni‐NTA Agarose (Qiagen) for 2 h. Beads were thoroughly washed with Lysis buffer lacking protease inhibitors and incubated with 20 μg SENP overnight to release SUMO1. Following extensive washing, BRCA21‐40HA‐His was eluted with Elution buffer (50 mM Na‐Phosphate pH 8.0, 300 mM NaCl, 250 mM Imidazole, 1 mM ß‐Mercaptoethanol).
Pull‐down assay of purified proteins
Purified BRCA21‐40HA‐His (2 μg) was immobilized on magnetic anti‐HA beads (Pierce Cat# 88836, Thermo Scientific) in a binding buffer containing (25 mM Tris–pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.01% (v/v) IGEPAL, 1 mM DTT), and incubated overnight at 4°C with recombinant DPP9 (2 μg), and/or recombinant PALB2 (9 μg). Where stated, DPP9 was preincubated for 1 h with the following inhibitors: 100 μM 1G244 or 110 μM SLRFYEG. Bound proteins were eluted with the HA peptides and analyzed by western blot.
Surface plasmon resonance (SPR)
Direct interactions between recombinant DPP9 and BRCA2‐derived N‐terminal peptides (1–40 and 3–40 amino acids) were analyzed employing surface plasmon resonance (SPR). The surface of a NiHC1000m sensorchip (Xantec Bioanalytics, Duesseldorf, Germany) was rinsed with 0.5 M EDTA (pH 8.0) solution and afterwards conditioned in SPR running buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 50 μM EDTA, 0.005% TWEEN‐20). The chip surface was loaded with Ni2+‐ions, injecting 300 mM NiSO4 solution. A 200 nM solution of His‐tagged DPP9 (ligand) was injected over the chip surface and immobilized to a surface density of 800–1,000 μRIU (Refractive index units; response observed), at a flow rate of 15 μl/min. A serial dilution of the analyte (BRCA2 peptides) diluted in SPR running buffer was injected, and the association was followed for 4.5 min. Dissociation of the analyte was monitored for 7 min. For each analyte concentration, the surface was regenerated, injecting 0.5 M EDTA pH 8.0 and fresh ligand was immobilized as described above (one immobilization/regeneration cycle for each analyte concentration). All binding experiments were performed on a Reichert SR 7500 DC biosensor at 20°C and a flow rate of 40 μl/min. Obtained data were analyzed with Scrubber2.0c. Raw data were double referenced (reference channel, specificity check, and buffer blanks/injections). Equilibrium‐binding response data were exported and plotted for further analysis with GraphPad Prism 6.0.
Peptidase activity assay by liquid chromatography–tandem mass spectrometry (LC/MS/MS)
The assay was performed as described in (Justa‐Schuch et al, 2016) by incubating 50 μM of the BRCA21‐40 peptide with 125 nM DPP9. To test for inhibition, a 10 μM peptide inhibitor (SLRFLYEG) was added. Reactions were stopped after 6 h by dilution and acidification in aqueous 0.1% formic acid and 2% acetonitrile (1/20,000, v:v). In addition, bovine insulin was added to the dilution buffer at a concentration of 1 pmol/μl to prevent analyte loss due to adsorption. The resulting samples were analyzed on a nanoLC425 nanoflow chromatography system coupled to a TripleToF 5600+ Plus mass spectrometer of QqToF geometry (both AB SCIEX). 5 μl of the sample was preconcentrated on a self‐packed Reversed Phase‐C18 precolumn (Reprosil C18‐AQ, Pore Size 120 Å, Particle Size 5 μm, 4 cm length, 0.15 cm I.D., Dr. Maisch) and separated on a self‐packed Reverse Phase‐C18 microcolumn (Reprosil C18‐AQ, 120 Å, 3 μm, 15 cm, 0.075 cm) using a 15 min linear gradient (5 to 50% acetonitrile, 0.1% formic acid modifier, flow rate 300 nl/min, column temperature 50°C) followed by a 5 min high organic cleaning step and a 15 min column re‐equilibration. The eluent was introduced to the mass spectrometer using a Nanospray III ion source with Desolvation Chamber Interface (AB SCIEX) via a commercial Fused Silica tip (FS360‐20‐10‐N‐C15, New Objective) at a spray voltage of 2.2 kV, a sheath gas setting of 15 and an interface heater temperature of 150°C. The MS acquisition cycle consisted of a 500 ms TOF MS survey scan that was used for profiling substrate and product concentrations followed by the data‐dependent triggering of up to 5,100 ms TOF product ion spectra to confirm the identity of detected analytes. Data analysis was performed using the Analyst TF 1.7 and the PeakView 2.1 softwares (AB SCIEX). Analyses were performed in quadruplicate.
Peptide competition assay
75 ng of purified DPP9 was incubated with 200 μM of a test peptide (Syk1‐31, BRCA21‐20, BRCA21‐40, BRCA2P2G 1‐40, BRCA2E13R 1‐40, BRCA2I14N 1‐40, BRCA2W31C 1‐40, BRCA2F32G 1‐40) for 5 min at 24°C in 20 mM HEPES/KOH pH 7.3, 110 mM potassium acetate, 2 mM magnesium acetate, 1 mM EGTA, 0.02% Tween‐20 supplemented with 1 mM DTT. The peptide‐enzyme mixture was added to 100 μM H‐Gly‐Pro‐7‐amino‐4‐methylcoumarin (GP‐AMC) substrate in a final reaction volume of 20 μl. Fluorescence was analyzed using an EnSpire microplate fluorimeter (Perkin Elmer) with 360‐nm (excitation) and 460‐nm (emission) filters. Percentage inhibition was normalized to the corresponding mock‐treated DPP9 controls. All peptides (> 85% purity) were purchased from GenScript.
Protein crystallography
Purification, crystallization, and structure solution of DPP8 and DPP9 were performed as published in our previous report (Ross et al, 2018). Briefly, the DPP8 isoform 1 (Uni‐ProtKB Q6V1X1) and DPP9 isoform 2 (Uni‐ProtKB Q86TI2–2) were expressed in Spodoptera frugiperda cells and purified. The crystallization was performed in both cases using the hanging drop method with 0.46 M Na‐citrate pH 6.75 as precipitant solution and 10 mg/ml of DPP8 at 4°C. DPP9 crystallization occurred at 20°C, using 20 mg/ml of protein and 10% PEG 8000, 25% glycerol, 0.16 M Calcium acetate, and 0.08 M cacodylate pH 6.25 as precipitant solution. Crystals of space group C2221 (DPP8) and P1211 (DPP9) were soaked with BRCA21‐40 peptide by performing dropping using a pico‐dropper (Ross et al, 2018). Data were collected at SLS‐X10SA and solved to 3.2 Å and 3.0 Å, respectively, by molecular replacement using a DPP8 structure (PDB: 6EOO). Structure solution and refinement were performed as described previously (Ross et al, 2018). Data processing was performed with XDS (Kabsch, 2010); Molecular Replacement with Phaser (McCoy et al, 2007); data refinement with Refmac5 (Murshudov et al, 1997). Data processing, statistics, and refinement values are summarized in Appendix Table S1.
Cycloheximide (CHX) chase assays
HeLa WT, DPP9 KD, HEK293 DPP9 WT, HEK293 DPP9 KO+DPP9‐SWT / + DPP9‐SS729A cells were treated with 300 nM MMC for 24 h. The CHX chase was started by the addition of 100 μg/ml CHX. Where stated, 100 μM of MG132 was added to the cells 30 min before the addition of the CHX. Samples were harvested at the indicated time points directly in Laemmli Sample Buffer. Immunoblotting and incubation with primary antibodies were performed according to standard protocols. Secondary fluorophore‐coupled antibodies were applied. Signals were developed in the Odyssey Sa Infrared Imaging System (LI‐COR) and analyzed with the ImageStudio (version 4.0.21, LI‐COR) software.
Cell synchronization
To obtain a majority population of cells in S‐phase, cells were synchronized using a double thymidine block. Briefly, HeLa WT & HeLa DPP9 KD cells were treated with 2 mM of thymidine. After the 18 h, the cells were washed, and replenished with fresh DMEM for 9 h, prior to the second thymidine block (2 mM) for another 15 h. For detection of the cell cycle stages, cells were trypsinized and collected at 0, 2, 3, 4, 5, 6, 8, 24, and 26 h after release. The cell pellet was washed once in PBS and fixed in 70% Ethanol. Cells pellets were resuspended in PBS supplemented with 0.25% Triton‐X‐100 and incubated on ice for 15 min. The residual Triton was washed off with PBS and the cells were incubated in PBS supplemented with 100 μg/ml RNAse A and 50 μg/ml propidium iodide, in the dark, overnight at 4°C. The cells were sorted in a Cytoflex S cell sorter and analyzed using the FlowJo Software.
Accumulation and recovery of γH2AX signals
Cells were seeded on 96‐well, black/clear, tissue culture‐treated plates (Corning Falcon cat# 353219). Following 18 h incubation at 37°C, 5% CO2, cells were treated with 250 ng/ml NCS for 30 min. NCS was then removed and replaced with a fresh medium to allow cells to recover. Cells were fixed at the indicated time points in 4% formaldehyde in PBS for 20 min and permeabilized with 0.2% Triton‐X‐100 in PBS for 20 min. Cells were washed with PBS, blocked with 10% FBS in PBS for 30 min, and incubated with primary antibodies for 2 h. Following three PBS wash steps, cells were incubated for 60 min with secondary antibodies and 1:5,000 DAPI. γH2AX signal intensities were measured on a Celigo 4 Channel Imaging Cytometer (Nexlecom Bioscience) and quantified using the Celigo Software. Images were taken using a Nikon Eclipse Ti2‐E Inverted microscope, Plan Apoλ oil immersion objective 100x NA1.45 WD = 0.13 (Nikon Instruments Inc), and processed using NIS‐Elements AR 5.02.00 (Nikon Instruments Inc), and FIJI (ImageJ) for the preparation of figures. At least three biological replicates were analyzed, each with technical duplicates, and the means calculated.
Conservation and logo plot analysis
Amino acid conservation was assessed by aligning the human BRCA2 orthologs from Ensembl. The sequences of BRCA2 orthologs from 108 placental mammals were aligned with the ClustalW algorithm (Thompson et al, 1994). The conservation was illustrated as a logo plot with web logo (Schneider & Stephens, 1990; Crooks et al, 2004). This plot illustrates the alignment as letters, where the height of each letter is in proportion to the observed frequency of the corresponding amino acid, which is measured in bits.
Statistical analysis
Graphs were generated using the PRISM8 software, statistical analysis was carried out by unpaired or paired two‐tailed t‐test, one‐way or two‐way ANOVA. ImageStudio software was used for the quantification of western blots, which were visualized with LI‐COR. Bands were quantified relative to the respective loading control. Mean and SEM values were calculated by the PRISM software and represented together with SEM error bars. For Colony Formation and Cell Viability assays, measurements from three biological replicates each with technical triplicates per data point are represented as Mean ± SEM. For microscopy images, dots from PLAs or dots from foci formation assays were quantified using DUO‐LINK software.
Author contributions
Oguz Bolgi: Data curation; validation; investigation; visualization; formal analysis, writing – review and editing. Maria Silva‐Garcia: Data curation; investigation; visualization; writing – review and editing. Breyan Ross: Data curation; investigation; visualization; writing – original draft; writing – review and editing. Esther Pilla: Investigation; writing – review and editing. Vijayalakshmi Kari: Investigation; writing – review and editing. Markus Killisch: Investigation; formal analysis, visualization; writing – review and editing. Melanie Spitzner: Formal analysis, investigation; visualization; writing – review and editing. Nadine Stark: Investigation; writing – review and editing. Christof Lenz: Formal analysis, visualization, investigation; writing – review and editing. Konstantin Weiss: Investigation; Resources; writing – review and editing. Laura Donzelli: Investigation; Resources; writing – review and editing. Mark D Gorrell: Resources; supervision; writing – review and editing. Marian Grade: Resources; supervision; writing – review and editing. Jan Riemer: Resources; supervision; methodology; writing – original draft; writing – review and editing. Henning Urlaub: Supervision; methodology; writing – original draft; writing – review and editing. Matthias Dobbelstein: Conceptualization; supervision; investigation; methodology; writing – review and editing. Robert Huber: Conceptualization; data curation; supervision; methodology; writing – original draft; project administration; writing – review and editing. Ruth Geiss‐Friedlander: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; visualization; methodology; writing – original draft; project administration; writing – review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization: R.G‐F; laboratory experiments: OB, MS‐G, BR, EP, VK, MK, MS, NS and CL. Writing original draft preparation: R.G‐F, R.H; writing, review, and editing: all authors. Visualization: OB, MK, BR, MS, CL. supervision: MG, JR, HU, MD, RH and RG‐F.
Disclosure and competing interest statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
The authors thank Bettina Mayer and Ulrike Möller for excellent technical assistance. R.G‐F is thankful to Frauke Melchior for critical reading of this manuscript, and Thomas Reinheckel for inspiring discussions. This work was supported by the Deutsche Forschungsgemeinschaft Grant 2234/1‐3 and by the Deutsche Forschungsgemeinschaft–423813989/GRK2606 ProtPath to R.G‐F; and Deutsche Forschungsgemeinschaft–411422114/RTG2550 RELOC to J.R. We thank the Lighthouse Core Facility of the University of Freiburg for their support with confocal microscopy. The LCF is funded in part by the Medical Faculty, University of Freiburg (Project Numbers 2021/A2‐Fol; 2021/B3‐Fol) and the DFG (Project Number 450392965). Open Access funding enabled and organized by Projekt DEAL.
EMBO reports (2022) 23: e54136
Data availability
The structure of DPP8 bound to a dipeptide (MP) from the N‐terminus of BRCA2 is available in RCSB under the accession code (PDB ID): PDB: 6QZW (https://www.rcsb.org/structure/6QZW). The structure of DPP9 bound to a dipeptide (MP) from the N‐terminus of BRCA2 is available in RCSB under the accession code (PDB ID): PDB: 6QZV (https://www.rcsb.org/structure/6QZV).
References
- Abe T, Branzei D (2014) High levels of BRC4 induced by a Tet‐on 3G system suppress DNA repair and impair cell proliferation in vertebrate cells. DNA Repair 22: 153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami K, Abbott CA, McCaughan GW, Gorrell MD (2004) Dipeptidyl peptidase 9 has two forms, a broad tissue distribution, cytoplasmic localization and DPIV‐like peptidase activity. Biochim Biophys Acta 1679: 18–28 [DOI] [PubMed] [Google Scholar]
- Arnesen T, Damme PV, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F et al (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N‐terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A 106: 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub N, Rajendra E, Su X, Jeyasekharan AD, Mahen R, Venkitaraman AR (2009) The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol 19: 1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmair A, Finley D, Varshavsky A (1986) In vivo half‐life of a protein is a function of its amino‐terminal residue. Science 234: 179–186 [DOI] [PubMed] [Google Scholar]
- Bièche I, Noguès C, Lidereau R (1999) Overexpression of BRCA2 gene in sporadic breast tumours. Oncogene 18: 5232–5238 [DOI] [PubMed] [Google Scholar]
- Brower CS, Piatkov KI, Varshavsky A (2013) Neurodegeneration‐associated protein fragments as short‐lived substrates of the N‐end rule pathway. Mol Cell 50: 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira A, Kowalczykowski SC (2011) Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci U S A 108: 10448–10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha‐Molstad H, Yu JE, Feng Z, Lee SH, Kim JG, Yang P, Han B, Sung KW, Yoo YD, Hwang J et al (2017) p62/SQSTM1/Sequestosome‐1 is an N‐recognin of the N‐end rule pathway which modulates autophagosome biogenesis. Nat Commun 8: 102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C‐F, Chen P‐L, Zhong Q, Sharp ZD, Lee W‐H (1999) Expression of BRC repeats in breast cancer cells disrupts the BRCA2‐Rad51 complex and leads to radiation hypersensitivity and loss of G2/M checkpoint control*. J Biol Chem 274: 32931–32935 [DOI] [PubMed] [Google Scholar]
- Chen S‐J, Wu X, Wadas B, Oh J‐H, Varshavsky A (2017) An N‐end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 355: eaal3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C‐C, Feng W, Lim PX, Kass EM, Jasin M (2018) Homology‐directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol 2: 313–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA et al (2019) N‐terminal degradation activates the NLRP1B inflammasome. Science 364: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J‐M, Brenner SE (2004) WebLogo: A Sequence Logo Generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies OR, Pellegrini L (2007) Interaction with the BRCA2 C terminus protects RAD51–DNA filaments from disassembly by BRC repeats. Nat Struct Mol Biol 14: 475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AA, Masson J‐Y, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC (2001) Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 7: 273–282 [DOI] [PubMed] [Google Scholar]
- Ditzel M, Wilson R, Tenev T, Zachariou A, Paul A, Deas E, Meier P (2003) Degradation of DIAP1 by the N‐end rule pathway is essential for regulating apoptosis. Nat Cell Biol 5: 467–473 [DOI] [PubMed] [Google Scholar]
- Dong C, Zhang H, Li L, Tempel W, Loppnau P, Min J (2018) Molecular basis of GID4‐mediated recognition of degrons for the pro/N‐end rule pathway. Nat Chem Biol 14: 466–473 [DOI] [PubMed] [Google Scholar]
- Egawa C, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S (2002) High BRCA2 mRNA expression predicts poor prognosis in breast cancer patients. Int J Cancer 98: 879–882 [DOI] [PubMed] [Google Scholar]
- Eldeeb MA, Fahlman RP (2014) The anti‐apoptotic form of tyrosine kinase Lyn that is generated by proteolysis is degraded by the N‐end rule pathway. Oncotarget 5: 2714–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia AEH, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, Lowry E, Gygi SP, Scully R, Elledge SJ (2015) RFWD3-dependent ubiquitination of RPA regulates repair at stalled replication forks. Mol Cell 60: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC (2005) CDK‐dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434: 598–604 [DOI] [PubMed] [Google Scholar]
- Esashi F, Galkin VE, Yu X, Egelman EH, West SC (2007) Stabilization of RAD51 nucleoprotein filaments by the C‐terminal region of BRCA2. Nat Struct Mol Biol 14: 468–474 [DOI] [PubMed] [Google Scholar]
- Feeney L, Muñoz IM, Lachaud C, Toth R, Appleton PL, Schindler D, Rouse J (2017) RPA-mediated recruitment of the E3 ligase RFWD3 is vital for interstrand crosslink repair and human health. Molecular Cell 66: 610–621.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger Y, Habich M, Gerlich S, Urbanczyk S, Logt E, Koch J, Schu L, Lapacz KJ, Ali M, Petrungaro C et al (2020) Proteasomal degradation induced by DPP9‐mediated processing competes with mitochondrial protein import. EMBO J 39: e103889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, Esashi F, Yu X, Yang S, West SC, Egelman EH (2005) BRCA2 BRC motifs bind RAD51‐DNA filaments. Proc Natl Acad Sci U S A 102: 8537–8542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall MG, Chen Y, de Ribeiro AJV, Zhang H, Bailey CG, Spielman DS, Yu DMT, Gorrell MD (2013) Targeted inactivation of dipeptidyl peptidase 9 enzymatic activity causes mouse neonate lethality. Plos One 8: e78378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss‐Friedlander R, Parmentier N, Möller U, Urlaub H, de Eynde BJV, Melchior F (2009) The cytoplasmic peptidase DPP9 is rate‐limiting for degradation of proline‐containing peptides. J Biol Chem 284: 27211–27219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Chen J (2011) E3 ligase RFWD3 participates in replication checkpoint control. Journal of Biological Chemistry 286: 22308–22313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig AO, Gauci S, Raijmakers R, van Breukelen B, Slijper M, Mohammed S, Heck AJR (2010) Profiling of N‐acetylated protein termini provides in‐depth insights into the N‐terminal nature of the proteome. Mol Cell Proteomics 9: 928–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, Durocher D (2017) The control of DNA repair by the cell cycle. Nat Cell Biol 19: 1–9 [DOI] [PubMed] [Google Scholar]
- Hwang C‐S, Shemorry A, Varshavsky A (2010) N‐terminal acetylation of cellular proteins creates specific degradation signals. Science 327: 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, Nakada S, Miyoshi H, Knies K, Takaori-Kondo A et al (2017) RFWD3-mediated ubiquitination promotes timely removal of both RPA and RAD51 from DNA damage sites to facilitate homologous recombination. Mol Cell 66: 622–634.e8 [DOI] [PubMed] [Google Scholar]
- Jasin M, Rothstein R (2013) Repair of Strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5: a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, Kowalczykowski SC (2010) Purified human BRCA2 stimulates RAD51‐mediated recombination. Nature 467: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A et al (2018) DPP8/DPP9 inhibitor‐induced pyroptosis for treatment of acute myeloid leukemia. Nat Med 24: 1151–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justa‐Schuch D, Möller U, Geiss‐Friedlander R (2014) The amino terminus extension in the long dipeptidyl peptidase 9 isoform contains a nuclear localization signal targeting the active peptidase to the nucleus. Cell Mol Life Sci 71: 3611–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justa‐Schuch D, Silva‐Garcia M, Pilla E, Engelke M, Kilisch M, Lenz C, Möller U, Nakamura F, Urlaub H, Geiss‐Friedlander R (2016) DPP9 is a novel component of the N‐end rule pathway targeting the tyrosine kinase Syk. Elife 5: e16370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, Sirma H, Simon R, Will H, Dobbelstein M et al (2016) Loss of CHD1causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep 17: 1609–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari V, Raul SK, Henck JM, Kitz J, Kramer F, Kosinsky RL, Übelmesser N, Mansour WY, Eggert J, Spitzner M et al (2019) The histone methyltransferase DOT1L is required for proper DNA damage response, DNA repair, and modulates chemotherapy responsiveness. Clin Epigenetics 11: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H‐K, Kim R‐R, Oh J‐H, Cho H, Varshavsky A, Hwang C‐S (2014) The N‐terminal methionine of cellular proteins as a degradation signal. Cell 156: 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Machida Y, Palani S, Caulfield TR, Radisky ES, Kaufmann SH, Machida YJ (2020) FAM111A protects replication forks from protein obstacles via its trypsin‐like domain. Nat Commun 11: 1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo LJ, Yang L‐X (2008) Gamma‐H2AX ‐ a novel biomarker for DNA double‐strand breaks. In Vivo 22: 305–309 [PubMed] [Google Scholar]
- Le HP, Ma X, Vaquero J, Brinkmeyer M, Guo F, Heyer W‐D, Liu J (2020) DSS1 and ssDNA regulate oligomerization of BRCA2. Nucleic Acids Res 48: 7818–7833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Tasaki T, Moroi K, An JY, Kimura S, Davydov IV, Kwon YT (2005) RGS4 and RGS5 are in vivo substrates of the N‐end rule pathway. Proc Natl Acad Sci U S A 102: 15030–15035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chu J, Yucer N, Leng M, Wang S-Y, Chen BPC, Hittelman WN, Wang Y (2011) RING finger and WD repeat domain 3 (RFWD3) associates with replication protein A (RPA) and facilitates RPA-mediated DNA damage response. J Biol Chem 286: 22314–22322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Doty T, Gibson B, Heyer W‐D (2010) Human BRCA2 protein promotes RAD51 filament formation on RPA‐covered single‐stranded DNA. Nat Struct Mol Biol 17: 1260–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kruswick A, Dang H, Tran AD, Kwon SM, Wang XW, Oberdoerffer P (2017) Ubiquitin‐specific protease 21 stabilizes BRCA2 to control DNA repair and tumor growth. Nat Commun 8: 137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Mosqueda J, Maddi K, Prgomet S, Kalayil S, Marinovic‐Terzic I, Terzic J, Dikic I, Harper W (2016) SPRTN is a mammalian DNA‐binding metalloprotease that resolves DNA‐protein crosslinks. Elife 5: e21491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16: 110–120 [DOI] [PubMed] [Google Scholar]
- Luo K, Li L, Li Y, Wu C, Yin Y, Chen Y, Deng M, Nowsheen S, Yuan J, Lou Z (2016) A phosphorylation–deubiquitination cascade regulates the BRCA2–RAD51 axis in homologous recombination. Gene Dev 30: 2581–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwood AC, Mundia MM, Baker MD (2012) High levels of wild‐type BRCA2 suppress homologous recombination. J Mol Biol 421: 38–53 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnykov A, Chen S‐J, Varshavsky A (2019) Gid10 as an alternative N‐recognin of the pro/N‐degron pathway. Proc Natl Acad Sci U S A 116: 15914–15923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menear KA, Adcock C, Boulter R, Cockcroft X, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S et al (2008) 4‐[3‐(4‐cyclopropanecarbonylpiperazine‐1‐carbonyl)‐4‐fluorobenzyl]‐2H‐phthalazin‐1‐one: A novel bioavailable inhibitor of poly(ADP‐ribose) polymerase‐1. J Med Chem 51: 6581–6591 [DOI] [PubMed] [Google Scholar]
- Menssen R, Bui K, Wolf DH (2018) Regulation of the Gid ubiquitin ligase recognition subunit Gid4. FEBS Lett 592: 3286–3294 [DOI] [PubMed] [Google Scholar]
- Mondal G, Rowley M, Guidugli L, Wu J, Pankratz VS, Couch FJ (2012) BRCA2 localization to the midbody by filamin a regulates cep55 signaling and completion of cytokinesis. Dev Cell 23: 137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, Jasin M (2001) BRCA2 is required for homology‐directed repair of chromosomal breaks. Mol Cell 7: 263–272 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nguyen KT, Kim J‐M, Park S‐E, Hwang C‐S (2019) N‐terminal methionine excision of proteins creates tertiary destabilizing N‐degrons of the Arg/N‐end rule pathway. J Biol Chem 294: 4464–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH et al (2017) DPP8 and DPP9 inhibition induces pro‐caspase‐1‐dependent monocyte and macrophage pyroptosis. Nat Chem Biol 13: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, Johnson DC, Bachovchin DA (2018) Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem Biol 25: 262–267.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH (2009) Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep 10: 990–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S‐E, Kim J‐M, Seok O‐H, Cho H, Wadas B, Kim S‐Y, Varshavsky A, Hwang C‐S (2015) Control of mammalian G protein signaling by N‐terminal acetylation and the N‐end rule pathway. Science 347: 1249–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Lee J‐Y, Nguyen YTK, Kang N‐W, Oh EK, Jang DM, Kim H‐J, Kim D‐D, Han BW (2020) Structural analyses on the deamidation of N‐terminal Asn in the human N‐degron pathway. Biomolecules 10: 163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul MW, Sidhu A, Liang Y, van Rossum‐Fikkert SE, Odijk H, Zelensky AN, Kanaar R, Wyman C (2021) Role of BRCA2 DNA‐binding and C‐terminal domain in its mobility and conformation in DNA repair. Elife 10: e67926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR (2002) Insights into DNA recombination from the structure of a RAD51‐BRCA2 complex. Nature 420: 287–293 [DOI] [PubMed] [Google Scholar]
- Piatkov KI, Brower CS, Varshavsky A (2012) The N‐end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc Natl Acad Sci U S A 109: E1839–E1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkov KI, Oh J‐H, Liu Y, Varshavsky A (2014) Calpain‐generated natural protein fragments as short‐lived substrates of the N‐end rule pathway. Proc Natl Acad Sci U S A 111: E817–E826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M (1999) XRCC3 promotes homology‐directed repair of DNA damage in mammalian cells. Genes Dev 13: 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla E, Möller U, Sauer G, Mattiroli F, Melchior F, Geiss‐Friedlander R (2012) A novel SUMO1‐specific interacting motif in dipeptidyl peptidase 9 (DPP9) that is important for enzymatic regulation. J Biol Chem 287: 44320–44329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla E, Kilisch M, Lenz C, Urlaub H, Geiss‐Friedlander R (2013) The SUMO1‐E67 interacting loop peptide is an allosteric inhibitor of the dipeptidyl peptidases 8 and 9. J Biol Chem 288: 32787–32796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S, Langlois CR, Chrustowicz J, Sherpa D, Karayel O, Hansen FM, Beier V, von Gronau S, Bollschweiler D, Schäfer T et al (2020) Interconversion between anticipatory and active GID E3 ubiquitin ligase conformations via metabolically driven substrate receptor assembly. Mol Cell 77: 150–163.e9 [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao H, Uhlmann F, Nasmyth K, Varshavsky A (2001) Degradation of a cohesin subunit by the N‐end rule pathway is essential for chromosome stability. Nature 410: 955–959 [DOI] [PubMed] [Google Scholar]
- Rave‐Fränk M, Schmidberger H, Christiansen H, Boll C, Lehmann J, Weiss E (2007) Comparison of the combined action of oxaliplatin or cisplatin and radiation in cervical and lung cancer cells. Int J Radiat Biol 83: 41–47 [DOI] [PubMed] [Google Scholar]
- Reuter M, Zelensky A, Smal I, Meijering E, van Cappellen WA, de Gruiter HM, van Belle GJ, van Royen ME, Houtsmuller AB, Essers J et al (2015) BRCA2 diffuses as oligomeric clusters with RAD51 and changes mobility after DNA damage in live cells. J Cell Biol 208: 857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross B, Krapp S, Augustin M, Kierfersauer R, Arciniega M, Geiss‐Friedlander R, Huber R (2018) Structures and mechanism of dipeptidyl peptidases 8 and 9, important players in cellular homeostasis and cancer. Proc Natl Acad Sci U S A 115: E1437–E1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AOH, Zander SAL, Derksen PWB, de Bruin M, Zevenhoven J, Lau A et al (2008) High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A 105: 17079–17084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez H, Paul MW, Grosbart M, van Rossum‐Fikkert SE, Lebbink JHG, Kanaar R, Houtsmuller AB, Wyman C (2017) Architectural plasticity of human BRCA2‐RAD51 complexes in DNA break repair. Nucleic Acids Res 45: 4507–4518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santt O, Pfirrmann T, Braun B, Juretschke J, Kimmig P, Scheel H, Hofmann K, Thumm M, Wolf DH (2008) The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol Biol Cell 19: 3323–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saso K, Miyoshi N, Fujino S, Sasaki M, Yasui M, Ohue M, Ogino T, Takahashi H, Uemura M, Matsuda C et al (2020) Dipeptidyl peptidase 9 increases chemoresistance and is an indicator of poor prognosis in colorectal cancer. Ann Surg Oncol 27: 4337–4347 [DOI] [PubMed] [Google Scholar]
- Savoy RM, Ghosh PM (2013) The dual role of filamin a in cancer: can't live with (too much of) it, can't live without it. Endocr Relat Cancer 20: R341–R356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM (1990) Sequence logos: A new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld AR, Apgar S, Dolios G, Wang R, Aaronson SA (2004) BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits Prosurvival function in the cellular response to DNA damage. Mol Cell Biol 24: 7444–7455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, Willis NA (2019) DNA double‐strand break repair‐pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 40: 179–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahid T, Soroka J, Kong EH, Malivert L, McIlwraith MJ, Pape T, West SC, Zhang X (2014) Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat Struct Mol Biol 21: 962–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemorry A, Hwang C‐S, Varshavsky A (2013) Control of protein quality and stoichiometries by N‐terminal acetylation and the N‐end rule pathway. Mol Cell 50: 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa D, Chrustowicz J, Qiao S, Langlois CR, Hehl LA, Gottemukkala KV, Hansen FM, Karayel O, von Gronau S, Prabu JR et al (2021) GID E3 ligase supramolecular chelate assembly configures multipronged ubiquitin targeting of an oligomeric metabolic enzyme. Mol Cell 81: 2445–2459.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A, Grosbart M, Sánchez H, Verhagen B, van der Zon NLL, Ristić D, van Rossum‐Fikkert SE, Wyman C (2020) Conformational flexibility and oligomerization of BRCA2 regions induced by RAD51 interaction. Nucleic Acids Res 48: 9649–9659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smebye ML, Agostini A, Johannessen B, Thorsen J, Davidson B, Tropé CG, Heim S, Skotheim RI, Micci F (2017) Involvement of DPP9 in gene fusions in serous ovarian carcinoma. BMC Cancer 17: 642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnuolo PA, Hurren R, Gronda M, MacLean N, Datti A, Basheer A, Lin F‐H, Wang X, Wrana J, Schimmer AD (2013) Inhibition of intracellular dipeptidyl peptidases 8 and 9 enhances parthenolide's anti‐leukemic activity. Leukemia 27: 1236–1244 [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M (2004) Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24: 9305–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjær S, Tainer JA et al (2016) Mechanism and regulation of DNA‐protein crosslink repair by the DNA‐dependent metalloprotease SPRTN. Mol Cell 64: 688–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li J, Shen Q, Feng J, Liu H, Wang W, Xu L, Shi G, Ye X, Ge M et al (2017) Contribution of upregulated dipeptidyl peptidase 9 (DPP9) in promoting tumoregenicity, metastasis and the prediction of poor prognosis in non‐small cell lung cancer (NSCLC). Int J Cancer 140: 1620–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC (2010) The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single‐stranded DNA. Nat Struct Mol Biol 17: 1263–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torella JP, Lienert F, Boehm CR, Chen J‐H, Way JC, Silver PA (2014) Unique nucleotide sequence‐guided assembly of repetitive DNA parts for synthetic biology applications. Nat Protoc 9: 2075–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A (2005) Ubiquitin fusion technique and related methods. Meth Enzymol 399: 777–799 [DOI] [PubMed] [Google Scholar]
- Varshavsky A (2019) N‐degron and C‐degron pathways of protein degradation. Proc Natl Acad Sci U S A 116: 358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vasconcelos NM, Vliegen G, Gonçalves A, Hert ED, Martín‐Pérez R, Opdenbosch NV, Jallapally A, Geiss‐Friedlander R, Lambeir A‐M, Augustyns K et al (2019) DPP8/DPP9 inhibition elicits canonical Nlrp1b inflammasome hallmarks in murine macrophages. Life Sci Alliance 2: e201900313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I et al (2016) Metalloprotease SPRTN/DVC1 orchestrates replication‐coupled DNA‐protein crosslink repair. Mol Cell 64: 704–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S‐C, Shao R, Pao AY, Zhang S, Hung M‐C, Su L‐K (2002) Inhibition of cancer cell growth by BRCA2. Cancer Res 62: 1311–1314 [PubMed] [Google Scholar]
- Wang H, Piatkov KI, Brower CS, Varshavsky A (2009) Glutamine‐specific N‐terminal amidase, a component of the N‐end rule pathway. Mol Cell 34: 686–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xia X, Yang X, Zhang X, Liu Y, Wu D, Fang Y, Liu Y, Xu J, Qiu Y et al (2017) A picorna‐like virus suppresses the N‐end rule pathway to inhibit apoptosis. Elife 6: e30590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang J, Zhang Y, Deng Q, Liang H (2018) Expression and mutations of BRCA in breast cancer and ovarian cancer: Evidence from bioinformatics analyses. Int J Mol Med 42: 3542–3550 [DOI] [PubMed] [Google Scholar]
- Weaver BP, Weaver YM, Mitani S, Han M (2017) Coupled caspase and N‐end rule ligase activities allow recognition and degradation of pluripotency factor LIN‐28 during non‐apoptotic development. Dev Cell 41: 665–673.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J‐J, Tang H‐K, Yeh T‐K, Chen C‐M, Shy H‐S, Chu Y‐R, Chien C‐H, Tsai T‐Y, Huang Y‐C, Huang Y‐L et al (2009) Biochemistry, pharmacokinetics, and toxicology of a potent and selective DPP8/9 inhibitor. Biochem Pharmacol 78: 203–210 [DOI] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22: 719–729 [DOI] [PubMed] [Google Scholar]
- Xu H, Shi J, Gao H, Liu Y, Yang Z, Shao F, Dong N (2019) The N‐end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J 38: e101996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Li Q, Fan J, Holloman WK, Pavletich NP (2005) The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA–ssDNA junction. Nature 433: 653–657 [DOI] [PubMed] [Google Scholar]
- Yuan Y, Shen ZY (2001) Interaction with BRCA2 suggests a role for filamin‐1 (hsFLNa) in DNA damage response. J Biol Chem 276: 48318–48324 [DOI] [PubMed] [Google Scholar]
- Yue J, Wang Q, Lu H, Brenneman M, Fan F, Shen Z (2009) The cytoskeleton protein filamin‐a is required for an efficient recombinational DNA double strand break repair. Cancer Res 69: 7978–7985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Lu H, Liu J, Berwick M, Shen Z (2012) Filamin‐a as a marker and target for DNA damage based cancer therapy. DNA Repair 11: 192–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Maqsudi S, Rainczuk A, Duffield N, Lawrence J, Keane FM, Justa‐Schuch D, Geiss‐Friedlander R, Gorrell MD, Stephens AN (2015) Identification of novel dipeptidyl peptidase 9 substrates by two‐dimensional differential in‐gel electrophoresis. FEBS J 282: 3737–3757 [DOI] [PubMed] [Google Scholar]