

Abstract
Background and aims:
Inflammatory bowel diseases (IBD) result in chronic inflammation of the gastrointestinal tract. Genetic studies have shown that the GPR65 gene, as well as its missense coding variant, GPR65*Ile231Leu, is associated with IBD. We aimed to define the signalling and biological pathways downstream of GPR65 activation and evaluate the impact of GPR65*231Leu on these.
Methods:
We used HEK 293 cells stably expressing GPR65 and deficient for either Gαs, Gαq/11 or Gα12/13, to define GPR65 signalling pathways, IBD patient biopsies and a panel of human tissues, primary immune cells and cell lines to determine biologic context, and genetic modulation of human THP-1-derived macrophages to examine the impact of GPR65 in bacterial phagocytosis and NLRP3 inflammasome activation.
Results:
We confirmed that GPR65 signals via the Gαs pathway, leading to cAMP accumulation. GPR65 can also signal via the Gα12/13 pathway leading to formation of stress fibers, actin remodelling and RhoA activation; all impaired by the IBD-associated GPR65*231Leu allele. Gene expression profiling revealed greater expression of GPR65 in biopsies from inflamed compared to non-inflamed tissues from IBD patients or control individuals, potentially explained by infiltration of inflammatory immune cells. Decreased GPR65 expression in THP-1-derived macrophages leads to impaired bacterial phagocytosis, increased NLRP3 inflammasome activation and IL-1β secretion in response to an inflammatory stimulus.
Conclusions:
We demonstrate that GPR65 exerts its effects through Gαs- and Gα12/13-mediated pathways, that the IBD-associated GPR65*231Leu allele has compromised interactions with Gα12/13 and that KD of GPR65 leads to impaired bacterial phagocytosis and increased inflammatory signaling via the NLRP3 inflammasome. This work identifies a target for development of small molecule therapies.
Keywords: GPR65, G proteins, actin remodeling, bacterial phagocytosis, NLRP3 inflammasome
Graphical Abstract
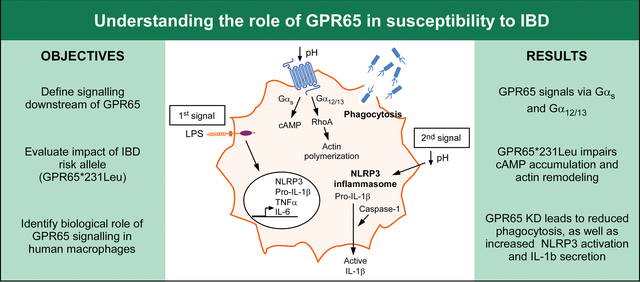
1. Introduction
Inflammatory bowel diseases (IBD) are characterized by chronic relapsing inflammation of the gastrointestinal tract. Crohn’s disease (CD; MIM 266600) and ulcerative colitis (UC; MIM 191390) are the two main subtypes of IBD. Although diagnosis and treatment options for IBD have improved over the years, patients with IBD still have a lower quality-of-life. It is therefore crucial to better understand the etiology of these debilitating diseases, and to identify putative therapeutic targets for drug development.
Genome-wide association studies (GWAS) have identified numerous loci implicated in complex diseases, and the success has been particularly notable for IBD where >200 genetic risk factors have been identified, with the majority being shared between CD and UC [1, 2]. High-density association mapping (or fine mapping) and targeted sequencing within these known GWAS regions have subsequently led to the identification of the most likely causal genes and variants at numerous additional loci, with many being confirmed by follow-up functional studies [3–6].
With the number of such confirmed susceptibility genes undoubtedly increasing in the coming years, there remains the significant challenge of identifying gene products with the potential of being “druggable”, prior to pursuing the arduous path of drug development. Importantly, with more than 800 different genes in humans, the family of G protein-coupled receptors (GPCRs) represent about 40% of protein targets for therapeutics currently on the market [7, 8]. GPCRs are comprised of seven transmembrane helices connected by alternating intracellular and extracellular loops with a N-terminal extracellular segment and a C-terminal cytosolic segment [7]. GPCRs translate signals from ligand binding via specific heterotrimeric G proteins that modulate different effector proteins. These are classically divided in four classes depending on the similarity in their sequence and the type of effector they activate; Gαs activates adenylyl cyclase (AC) to mediate production of cAMP, while Gαi inhibits AC; Gαq/11 activates phospholipase Cβ that leads, in part, to the release of intracellular Ca2+, which can also activate certain isoforms of AC, and Gα12/13 activates Rho-GTPases which regulate actin cytoskeletal remodeling [9].
In the current study, we focused on GPR65, located in an associated genomic region also containing the gene GALC and previously identified and validated as a genetic risk factor for IBD in a large international GWAS [1, 10]. GPR65 is part of the proton-sensing GPCR subfamily with GPR4, GPR68 and GPR132 [11, 12]. While all four of these proton-sensing GPCRs, when knocked out in mice, have been shown to either ameliorate (GPR4, GPR68) or aggravate (GPR65, GP132) DSS-induced colitis, only GPR65 is located within a known IBD locus [13–16]. Moreover, “fine mapping” of the association signal at this locus in individuals with IBD narrowed the associated region to 18 SNPs across ~65Kb having the greatest likelihood of being causal [6] and found only one non-synonymous coding variant (GPR65*Ile231Leu) among these 18 SNPs, further suggesting that this was the causal gene.
Given its role in susceptibility to developing IBD, it is important to determine its signalling cascade(s) and biological role. GPR65, when activated by acidification of the local pH, induces cAMP accumulation, partly via the Gαs pathway, and RhoA activation [11, 12, 17–20]. Specifically, overexpression of GPR65 in CHO cells, when exposed to low pH, led to the accumulation of cAMP, which was blocked by anti-Gαs antibody treatment in cell-free systems, or to the accumulation of stress fibers. GPR65 activation also leads to a reduction in the secretion of the pro-inflammatory cytokines TNFα and IL-6 in LPS-primed mouse peritoneal macrophages [17] and of IL-1β in LPS-primed mouse microglia [18] via a Gαs-PKA pathway. Its deficiency is also linked to an aggravated inflammation in mouse models of acute lung injury, chronic pain, rheumatoid arthritis and multiple sclerosis [21–24]. More importantly, loss of GPR65 was proven detrimental in mouse models of IBD and common GWAS associated variants in the GPR65 locus appear to be associated with a higher disease severity in IBD patients as well as aberrant lysosomal function, which affect important cellular functions like autophagy [19, 25–27].
Taking advantage of a panel of cell lines deleted for individual Gα subunits using CRISPR/Cas9 and by using a relevant human macrophage model, we were able to study the different pathways arising from GPR65 activation by extracellular acidic pH and pertinent cellular functions that result from these.
2. Materials and Methods
2.1. Materials
RNA from primary immune cells was obtained as described elsewhere [28]. RNA from a panel of different human tissues was purchased from Clontech Laboratories (Mountain View, CA). RNA from immortalized cell lines was obtained as described elsewhere [4]. The cDNA clones for human Gαs, Gαq and Gα12 were from the cDNA Resource Center (www.cdna.org). Anti-Flag M2 (F1804) was from Millipore Sigma (Oakville, CA), Anti-IL-1β (AF-201-NA) was from R&D Systems (Cedarlane, Burlington, CA), anti-capsase-1 (AG-20B-0048) and anti-NLRP3 (AG-20B-0014) were from Adipogen (Cedarlane), anti-β-actin (MA5–15739) was from Thermo Fisher Scientific (Montreal, CA). All secondary-HRP antibodies were from Abcam (Toronto, CA)
2.2. Gene expression in UC patients and healthy control biopsies
Samples were obtained as described previously [29]. Briefly, biopsies were collected from UC patients and healthy controls. Patients had to be older than 18 years of age and diagnosed with UC for at least 5 years. Patients undergoing treatment with biologic drugs (i.e., anti-TNFα) were excluded from the study. Biopsies were collected during routine exams (patients) and during cancer screening exams (controls). Written informed consent was obtained from all participants and ethics approval was granted at each of the participating institutions. Biopsies were taken from both inflamed (on the margin of ulcerations) and non-inflamed regions in the rectum (controls and most patients) and colon (some patients) and stored in RNAlater (Qiagen, Toronto, ON) until RNA extraction. RNA was extracted in the following 24 hours using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA was quantified using the RNA 6000 Nano Kit (Agilent) on a Bioanalyzer 2100 (Agilent). RNA Integrity Number (RIN) was above 8 for all samples and 1 μg of each RNA sample was used to evaluate the whole-genome expression using the Human Genome U133 Plus 2.0 Array (Affymetrix, Thermo Fisher Scientific).
2.3. Gene Expression in Tissue panel, Cell Lines and Primary Immune Cells
Gene expression in different human tissues, cell lines and primary immune cells was evaluated with a custom-made expression microarray (Agilent) as described elsewhere [4, 30].
2.4. Cell Culture
HEK 293T/17 cells, parental HEK 293 cells and HEK 293 cells deficient for different G proteins were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), and penicillin (100 units/mL)-streptomycin (100 μg/mL) (Thermo Fisher Scientific) at 37°C, in a 5% CO2 atmosphere. THP-1 cells were cultured in RPMI1640-Glutamax (Thermo Fisher Scientific) media supplemented with 10% FBS and 0.05 mM beta-mercaptoethanol (Thermo Fisher Scientific) at 37°C, in a 5% CO2 atmosphere.
2.5. CRISPR/Cas9 deletion of Gα subunits
Both members (Gαs and Gαolf subunits, encoded by the GNAS and the GNAL genes, respectively) of the Gαs family, two members (Gαq and Gα11 subunits, encoded by the GNAQ and the GNA11 genes, respectively) of the Gαq family and both members (Gα12 and Gα13 subunits, encoded by the GNA12 and the GNA13 genes, respectively) of the Gα12 family are expressed endogenously in HEK 293 cells and were deleted using the CRISPR/Cas9 system. Their generation and use has been described elsewhere [31–34].
2.6. Stable expression of GPR65 in HEK lines
A DNA fragment containing the coding region of GPR65 (MIM : 604620, ID : 8477), for the major allele or the minor allele (ATC→CTC, position 1249), were produced by GeneArt Gene Synthesis (Thermo Fisher Scientific). The different DNA fragments were cloned into the mammalian expression vector pcDNA3.1 containing a Zeocin™ resistance cassette (Thermo Fisher Scientific). The fragments were linearized by enzymatic digestion and HEK 293 parental cells, Gαs-KO, Gαq/11-KO or Gα12/13-KO cells were transfected using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific). Six hours later, media was changed with complete media for 24 hours then changed again for complete media containing 0.4 μg/mL zeocin (Thermo Fisher Scientific) for 7 days. Stable transfectants were maintained at 0.15 μg/mL of zeocin.
2.7. RNA Extraction, cDNA Synthesis and Quantitative RT-PCR
RNA was extracted and quantified as previously described [29]. 1μg of RNA from each sample was reverse transcribed into cDNA using the High Capacity cDNA RT kit (Thermo Fisher Scientific) with nuclease inhibitors (Invitrogen, Thermo Fisher Scientific). Real-time PCR was performed using the PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific) on a QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific) set to the Relative Standard Curve program. mRNA expression levels were normalized to HPRT (HEK-293 cells) or beta-actin (THP-1 cells) with QuantStudio Real-Time PCR Software. Primer sequences can be found in the supplementary methods.
2.8. Lentiviral particle production
All shRNA vectors were obtained from Sigma (MISSION®). GPR65 shRNA (TRCN0000357310 and TRCN0000011647, called KD1 and KD2 respectively) and shRNA empty control (SHC001) vectors were added to lentiviral packaging and envelope vectors in a ratio of 2: 2: 1. Vector mixtures were transfected in HEK 293T cells by the calcium phosphate precipitation method according to the Open BioSystems protocol. Forty-eight hours after transfection, lentivirus containing medium was collected and cell debris pelleted. Lentiviral particles were concentrated 10 times using the Lenti-X concentrator reagent (Clontech), resuspended in DMEM medium and frozen.
2.9. Transduction of THP-1 cells with lentiviral shRNA clones
THP-1 were transduced in RPMI containing 0.05 mM β-mercaptoethanol and 8ug/mL polybrene (minimal medium) in triplicate. Lentivirus-containing medium was added, and the cells were spinoculated in centrifuge for 30 minutes at 800 × g at room temperature. The infectious media was delicately removed, and the pellet resuspended in 100 μl of complete media and plated in a 96-wells plate. Forty-eight hours later, transduced THP-1 cells were selected in puromycin-containing (0.5 μg/ml) medium. This puromycin concentration was previously determined in-house, using a serial dilution of puromycin concentration, as the lowest concentration to efficiently killed uninfected THP-1 cells. Transduced cells were then cultured until all cells from a well of parental (non-transduced) THP-1 were killed (selection control). For each passage, cells were centrifuged to keep only viable cells and new medium was added. Cells were grown and expanded, until no dead cells could be detected in culture, prior to generating stock of stable frozen cells. Once the selection was completed, GPR65 expression was assessed by qPCR after cell differentiation.
2.10. cAMP measurements
Subconfluent HEK 293 cells were detached with non-enzymatic dissociation buffer. For each condition, cells were incubated for 30 minutes at room temperature in either DPBS, HEM-PSS (HEPES/EPPS/MES (8 mM each) with 130 mM NaCl, 0.9 mMNaH2PO4, 5.4 mM KCl, 0.8 mM MgSO4, 1.0 mM CaCl2, 25 mM glucose and 0.1% BSA) at different pH containing 0.5 mM IBMX (Sigma) or HEM-PSS pH 8.0 with IBMX and 50 μM forskolin (Millipore Sigma). The tubes were then centrifuged, the supernatant discarded and the cells lysed in ice cold 70% ethanol. The resulting pellets were decanted on ice and the extracts transferred to a clean tube. The pellets were washed with ice cold 70% ethanol and centrifuged. Ethanol extracts were then dried in a vacuum concentrator and stored frozen (−80°C) until assayed for cAMP. cAMP detection was made using the AlphaScreen cAMP assay kit according to the manufacturer’s instructions (PerkinElmer Life Sciences, Woodbridge, ON). Extracts and acceptor beads were diluted in HEM-PSS pH 7.5 and donor beads were diluted in 1X HEM pH 7.5, 0.1% BSA, 0.3% Tween-20 (Detection buffer). The plates (1/2 AreaPlate, PerkinElmer) were read in an Infinite M1000 (Tecan, Morissville, NC). The standard curve was fitted on the five-parameter logistic model with log transformed data using Prism 5 (GraphPad). Unknowns were then interpolated. Each Gα-KO HEK 293 experiment was done independently, together with parental HEK 293 cells that were repeated between batches. Batch effects were removed based on the repeated parental HEK 293, which were then combined. Direct comparison between the KO might still be slightly impacted by residual batch effects. Within batches analyses confirmed the results from the combined analysis (not shown).
2.11. Stress Fiber Detection
HEK 293 cells were seeded on laminin-treated (Life Technologies) coverslips and incubated for 24 hours. Cells were serum-starved (DMEM 1% BSA) for 24 hours before being incubated for 30 minutes in HEM-PSS pH 6.5 or pH 8.0 at 37°C. Cells were then fixed with 2% paraformaldehyde for 20 minutes at room temperature, washed with PBS and incubated for 30 minutes in permeabilization/blocking solution (0.2% Triton-X-100 and 2% BSA). The coverslips were incubated with 1 unit/mL of AlexaFluor555-Phalloidin (Invitrogen, Thermo Fisher Scientific), 0.1 μg/mL DAPI (Millipore Sigma), 1% BSA and 0.05% Triton-X-100 for 1 hour at room temperature. Images were obtained using a LSM510 laser-scanning confocal microscope (Carl Zeiss, North York, ON).
2.12. Protein isolation and western blots
All protein preparations were quantified using Pierce BCA Protein Assay kit according to the manufacturer’s instructions (Thermo Fisher Scientific) and the absorbance read on a Synergy 2 plate reader (Biotek, Winooski, VT). Western blots were performed as described [35]. Briefly, samples were loaded onto a denaturing SDS-PAGE and electro-transferred onto nitrocellulose membranes. After blocking the membrane with Tris-buffered saline (TBS) supplemented with 0.1% Tween-20 (TBST) and 5% skim milk for 1 hour, membranes were incubated in a diluted primary antibody O/N at 4°C or 1 hour at room temperature. The membranes were then washed three times with TBST and incubated in the diluted HRP-labeled secondary antibody for 1 hour at room temperature. After three additional washes with TBST, proteins were revealed by chemiluminescence using the Western Blot Lightning Plus-ECL according to the manufacturer’s instructions (PerkinElmer). Mandel Bioflex Blue Lite EC Film (Guelph, CA) was exposed to the membranes and revealed using an automated developer. Band densities were quantified using Image J software.
2.13. Measurement of F-actin and G-actin Levels
The amounts of filamentous (F) and globular (G) actin in cells were quantified using the F-actin/G-actin In Vivo Assay Biochem Kit according to the manufacturer’s instructions (Cytoskeleton, Cedarlane). HEK 293 cells were seeded and incubated for 24 hours at 37°C, in a 5% CO2 atmosphere. Cells were serum-starved for another 24 hours and were then incubated in HEM-PSS at pH 6.5 or pH 8.0 for 30 minutes before being lysed. Samples were run on a 12% SDS-PAGE gel and western blots performed as described above. Quantification of band density was performed using ImageJ software and then the ratio of F-actin on G-actin was calculated. Batch effects on ratios were removed by batches geometric mean before further analyses.
2.14. Measurement of RhoA activity
RhoA activation was measured using the G-LISA RhoA Activation Assay Biochem Kit according to the manufacturer’s instructions (Cytoskeleton). HEK 293 cells were seeded and incubated for 24 hours at 37°C, in a 5% CO2 atmosphere. In the G protein rescue experiments, cells were transiently transfected with supercoiled plasmid DNA using Lipofectamine 2000. The cells were serum-starved for another 24 hours and were then incubated in HEM-PSS at pH 6.5 for 2 minutes before being lysed. Batch effects on ratios were removed by batches geometric mean before further analyses.
2.15. Phagocytosis assay
THP-1 cells were seeded in the presence of 200 nM PMA for 3 days. The cells were returned to complete media for five days, changing the media every 2–3 days. On the day of the experiment, media was either replaced by RPMI 10% FBS at pH 8.0 or 6.5. After 3 hours, cytochalasin D (2.5 μg/mL, Millipore Sigma) or vehicle control (DMSO) was added for 1 hour. During that time, a vial of pHrodo Red E. coli BioParticle Conjugate (Thermo Fisher Scientific) was resuspended in 2 mL of HBSS, vortexed, sonicated for 10 minutes and finally left on ice for 10 min. Bacteria particles were added, the plate was briefly spun down and the cells were returned to the incubator for 45 minutes, after which cells were detached with non-enzymatic buffer. Cells were transferred in cold sort buffer (HBSS (Ca++/Mg++ free), 5mM EDTA, 25mM HEPES pH 7.5, 1% FBS). After two washes with cold sort buffer, cells were resuspended in cold sort buffer containing DAPI at pH 9.0 to quench the fluorescence from adherent particles and filtered on 70 μm filters. Phagocytosis was measured by flow cytometry with the FACSAria Fusion (BD) for the percentage of phagocytosis and the mean fluorescence. Data was analyzed using FlowJo®. Gating was made on non-phagocytic control for each cell line, and applied to the phagocytic or inhibitory cytochalasin D samples. Trend in time was evaluated from residuals of linear regression accounting for the conditions. Loess regression of these residuals on ranks was used to correct the trend.
2.16. Measuring active IL-1β and caspase-1 levels
THP-1 cells were seeded in the presence of 100 nM PMA for 3 days. The cells were then washed in PBS and incubated in RPMI-Glutamax with 1% FBS and 0.05 mM β-mercaptoethanol. After 6 hours, lipopolysaccharides from E. coli O26:B6 (Sigma) was added at a final concentration of 100 ng/mL for 18 hours. The cells were then washed again in PBS and incubated for 6 hours in custom RPMI media (Thermo Fisher) in which sodium bicarbonate concentrations were adjusted to pH 8.0, 7.5, 6.5 or 6.0 at 5% CO2. Media supernatants were added to cold ethanol 100% (1:3 ratio) and kept at −20°C O/N. The next day, tubes were centrifuged 15 minutes at 15000 xg at 4°C, the supernatant was carefully decanted and pellets were washed with ethanol 70%. Supernatant was aspirated and the pellets were let to dry. Finally, pellets were dissolved in RIPA buffer supplemented with 1X protease inhibitor cocktail (Roche Applied Science) and Laemmli buffer 4X (BioRad, Mississauga, CA) (1:1) and boiled for 10 minutes at 95°C. THP-1 cells were lysed in cold RIPA buffer. Proteins were quantified and western blot were performed as described above.
2.17. Statistical analyses
Unless otherwise specified, statistical analyses were performed (in R version 4.1.0) on the log scale, to account for plausible multiplicative effects and log-normal distribution. When technical replicates were available, these were combined by averaging before analyses. When batch effects were detected, these were corrected as described in each section. Repeated measure ANOVA and mixed-model analyses (nlme package R) were used for inference on effects. The results of these analyses are provided in supplementary tables for each figure. The mean and standard error of the mean (SEM) for each condition were computed and transformed back to the original scale as geometric mean (estimator of population median) with SEM, with all data points shown.
3. Results
3.1. GPR65 and Gαs are both required for pH-dependant cAMP accumulation in HEK 293
Taking advantage of cell lines deleted for individual G proteins, we were interested in mapping out the signalling pathways downstream of GPR65 activation. We first set out to confirm the GPR65-Gαs pathway and evaluate the implication of the other G proteins on cAMP accumulation. We also wanted to study the effect of the disease-associated GPR65*Ile231Leu variant on pH-driven activation of the cAMP signaling pathway in HEK 293 cell models, as this variant showed a significant reduction in the pH-dependant cAMP accumulation in HeLa cells overexpressing either allele [19] while there was no difference in human CD14+ cell from healthy controls and patients homozygous for either allele [26]. To do this, we expressed each allele of the GPR65*Ile231Leu variant or an empty vector in HEK 293 cells. We then assessed accumulation of cAMP in response to different pH levels in these lines. We observed an accumulation of cAMP in HEK 293 cells stably expressing either of the GPR65*Ile231Leu alleles at pH 8.0, while HEK 293 expressing the empty vector showed negligible cAMP accumulation (Fig. 1a). We detected significantly higher cAMP levels in samples cultured at a pH of 7.5 as compared to pH 8.0, although no further increases were detected at pH values of 7.0 or 6.5. When comparing the two alleles of the GPR65*Ile231Leu variant expressed in parental HEK 293 cells, we observed that cells expressing the GPR65*231Leu allele tended to show lower cAMP accumulation as compared to cells expressing GPR65*Ile231, although this was not statistically significant (Fig. 1a).
Figure 1 -. IBD-associated allele GPR65*231Leu impairs cAMP response following pH-mediated GPR65 activation.
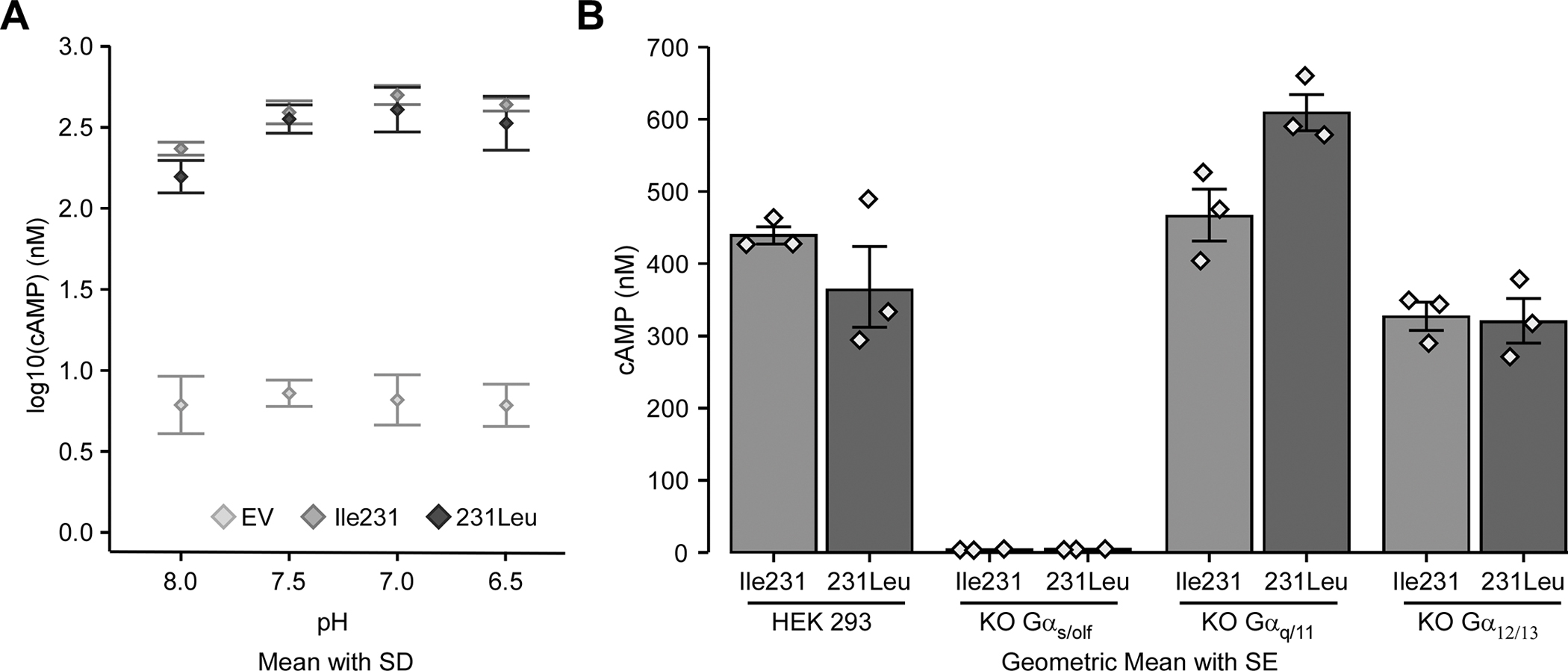
Parental HEK 293 cells and three different Gα-KO HEK 293 cell lines, stably transfected with the pcDNA3.1 expression vector containing the different GPR65*Ile231Leu alleles (Ile231 or 231Leu) or with empty vector (EV), were assessed for cAMP accumulation. cAMP accumulation was measured after a 30 minute incubation in HEM-PSS in the presence of IBMX at pH 8.0, 7.5, 7.0 and 6.5 in parental HEK 293 cells (A) and the G protein-KO cell lines (B). Detailed effect sizes analyses from ANOVA are presented in Table S1.
Next, to establish the role of different G proteins, GPR65 was expressed in HEK 293 cells depleted for different G proteins by CRISPR/Cas9 [31–34]. Specifically, we expressed the GPR65*Ile231 or GPR65*231Leu allele in parental HEK 293 cells, or in Gαs/olf (GNAS/GNAL)-KO, Gαq/11 (GNAQ/GNA11)-KO or Gα12/13 (GNA12/GNA13)-KO HEK 293 cells. We first confirmed that these different cell lines all expressed recombinant GPR65 at similar levels (Suppl. Fig. S1). We observed that when GPR65 was expressed in HEK 293 cell lines depleted for Gαs/olf, cAMP accumulation was completely abrogated, supporting a central role of Gαs/olf in GPR65 signaling (Fig. 1b). Interestingly, cAMP accumulation in Gα12/13-KO HEK 293 cells expressing the GPR65*231Leu allele was equivalent or slightly reduced as compared to the parental line expressing this allele. On the other hand, the GPR65*Ile231 allele, when expressed in the in Gα12/13-KO HEK 293 had levels of cAMP accumulation that were similar to the GPR65*231Leu allele (Fig. 1b). Although not significant, this may indicate that GPR65 can signal in part through Gα12/13 to activate the cAMP signaling pathway. In addition, given that both alleles appear to signal equivalently in the Gα12/13-KO lines, this suggests that the difference in signaling observed between these two alleles in the parental lines may be explained by reduced affinity of GPR65*231Leu allele for Gα12/13. In contrast, expression of the GPR65*231Leu allele in Gαq/11-KO HEK 293 cells shows increased cAMP accumulation as compared to GPR65*Ile231, which was similar to the parental HEK 293 expressing these alleles (Fig. 1b). This increase in cAMP accumulation could potentially be explained by an increase in GPR65 availability for signaling through Gαs/olf, possibly due to altered relative affinity of the GPR65*231Leu allele to Gα12/13 in cells lacking Gαq/11. Taken together these observations suggest a model where the affinity for different G proteins differs between GPR65 alleles, with the GPR65*231Leu allele showing either decreased affinity to bind or reduced capacity to activate Gα12/13.
3.2. IBD-associated allele GPR65*231Leu impairs stress fiber formation, actin remodeling and RhoA activation and Gα12/13 is necessary for those pathways
Given the observations described above and that activation of Rho-GTPases and subsequent regulation of actin cytoskeleton remodeling by GPCRs are known to occur through the activation of Gα12/13, we were interested to ascertain whether this was also the case for GPR65 using the same panel of G protein-deficient cells. We also wanted to study the effect of the IBD associated variant on those pathways. Thus, we analyzed pH-driven stress fiber formation using phalloidin staining in HEK 293 cells stably expressing GPR65*Ile231, GPR65*231Leu or an empty vector. Our results confirm that a reduction in pH increased stress fiber formation in HEK 293 cells expressing GPR65*Ile231, a process that was decreased in cells expressing the GPR65*231Leu variant (Fig. 2a). To quantify this decrease, we compared the ratio of filamentous actin (F-actin) versus free globular-actin (G-actin) content under the same conditions (Fig. 2b) and found that a reduction in pH resulted in a significant increase in the ratio of F-actin versus G-actin content in the cells expressing the GPR65*Ile231 variant, whereas this response was not observed with the GPR65*231Leu variant or in cells expressing the empty vector (Fig. 2c).
Figure 2 -. IBD-associated allele GPR65*231Leu impairs stress fiber formation.
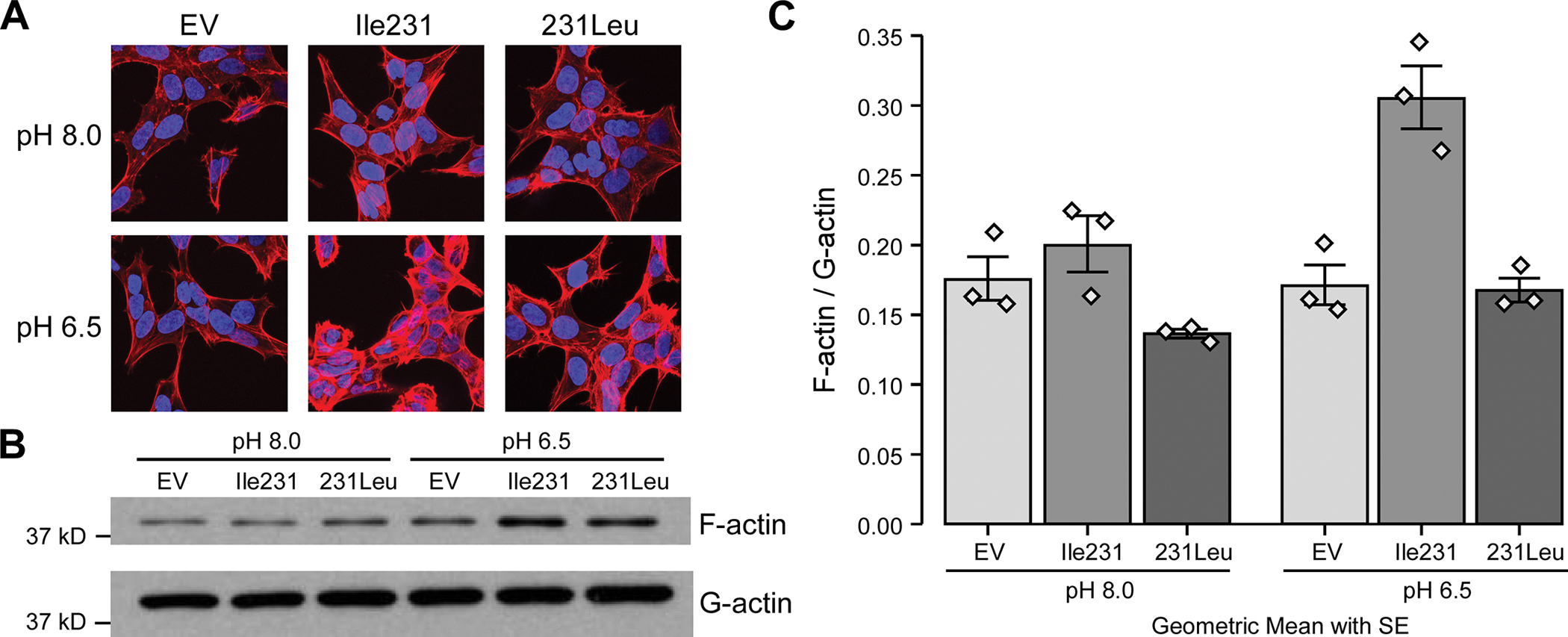
(A) HEK 293 cells stably expressing the different GPR65*Ile231Leu alleles (Ile231 or 231Leu) or empty vector (EV) were evaluated for stress fiber formation after 30 minutes incubation in HEM-PSS at pH 8.0 or 6.5 using AlexaFluor555-phalloidin detection by immunofluorescence on confocal microscopy. Representative images of 3 independent experiments are shown. (B) HEK 293 cells stably expressing the different GPR65*Ile231Leu alleles or empty vector were evaluated for F-actin accumulation through the ratio of cellular filamentous actin (F-actin) and globular actin (G-actin) levels detected via western blot. (C) Quantification of band density was performed. Barplots represent geometric mean with SE. Detailed effect sizes analyses from ANOVA are presented in Table S2.
Using the HEK 293 cell lines gene-depleted for different G proteins as above, we next aimed to further characterise the signalling pathway mediating the pH-driven RhoA activation downstream of GPR65. As shown in Figure 3a, activation of GPR65*Ile231-expressing parental HEK 293 cells with low pH resulted in the activation of the RhoA pathway, while only very weak RhoA activation can be seen in cells expressing the GPR65*231Leu variant. In addition, the GPR65*Ile231-expressing Gα12/13 knockout HEK 293 cells revealed that the GPR65-driven RhoA activation is essentially abolished but there was no diminution of the signal in the GPR65*Ile231-expressing Gαs/olf or Gαq/11 knockout cells (Fig. 3a). Moreover, an analysis of pH-driven stress fiber formation staining in Gα12/13 knockout HEK 293 cells expressing the GPR65*Ile231 variant confirms that stress fiber formation was abolished supporting the critical role of Gα12/13 in this process (Suppl. Fig. S2). Taken together, these results suggest that the signalling pathway leading to pH-activation of RhoA by GPR65 primarily involves Gα12/13 and not Gαs/olf or Gαq/11 and that similar to what was suggested in the study of cAMP signalling above, the reduced impact of the GPR65*231Leu allele on this pathway likely occurs through reduced ability to bind or activate Gα12/13. It is noteworthy that GPR65*231Leu signaling was similar to GPR65*Ile231 in the absence of Gαs/olf or Gαq/11, suggesting that the IBD-associated GPR65*231Leu allele can still signal through Gα12/13-dependent pathways when competition for binding of GPR65*231Leu to Gαs/olf or Gαq/11 was removed. To test this, we examined GPR65-dependent RhoA activation at low pH in Gαs/olf-KO HEK 293 cells, Gα12/13 –KO HEK 293 cells and Gαq/11-KO HEK-293 cells where we reintroduced Gαs, Gα12 and Gαq respectively via expression. We tested different doses of plasmid for transient expression and every dose led to the same effect (Suppl. Fig. S3). As can be seen in Figure 3b and c, reintroduction of Gαs and Gαq resulted in reduced RhoA activation for both alleles of GPR65 illustrating that GPR65 may have higher affinity for these two G proteins when expressed at the expense of Gα12/13. With the Gα12/13-KO HEK 293 cells, the expression of Gα12 restored RhoA signaling, thus “rescuing” the effect of Gα12/13 deletion (Fig. 3d) for both GPR65 variants. There was no significant difference between both GPR65 alleles, which could mean that expressing any of those G proteins saturates the system and dampens any minor allele defect (Suppl. Fig. S3). We have previously shown that there is more Gαs and Gαq/11 than Gα12/13 in HEK 293 cells [36], which could also explain our results.
Figure 3 -. IBD-associated allele GPR65*231Leu impairs RhoA activation and is mediated via Gα12/13.
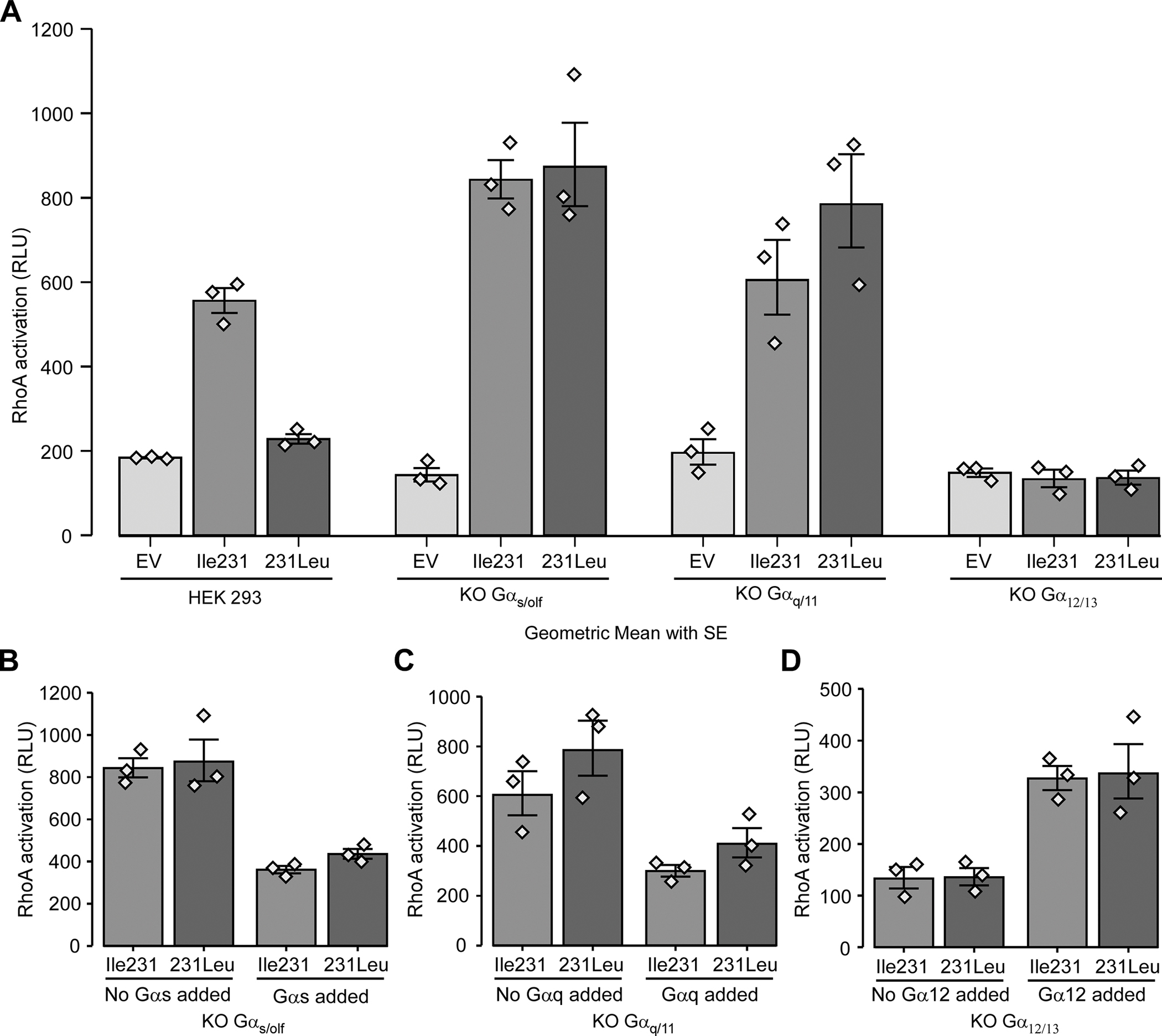
(A) Parental HEK 293 and three different Gα-KO HEK 293 cell lines stably expressing the different GPR65*Ile231Leu alleles (Ile231 or 231Leu) or empty vector (EV) were assessed for RhoA activation after two minutes incubation in HEM-PSS at pH 6.5. (B-D) Three different Gα-KO HEK 293 cell lines stably expressing the different GPR65*Ile231Leu alleles or empty vector were transiently transfected with 200ng of Gα expressing plasmid in order to rescue the expression their respective knocked-out Gα subunit and then assessed for RhoA activation after two minutes incubation in HEM-PSS at pH 6.5. A dose-response experiment was performed before-hand (Suppl. Fig. S3) and 200ng of plasmid was chosen as it was the lowest dose of plasmid that led to an effect. Barplots represent geometric mean with SE. Detailed effect sizes analyses from ANOVA are presented in Table S3.
3.3. Expression profiling of GPR65 suggests that its role in IBD is primarily mediated by immune cells
In order to validate clinical implications of GPR65 in IBD, we assessed its expression in a panel of human intestinal biopsies. GPR65 expression was significantly increased in biopsies from inflamed tissues compared to biopsies from non-inflamed tissues or tissues originating from healthy controls (Fig. 4a). This could be either due to an increased expression of GPR65 in epithelial cells, in immune cells or both, or to an increased migration of immune cells to sites of inflammation. We next examined the expression of GPR65 in human tissues, cell lines and primary immune cells. Our results indicate that GPR65 was expressed in lymphoid and mucosal tissues, as well as in immortalized and primary immune cells (Fig. 4b–c). Its expression increased in the THP-1 monocytic cell line upon differentiation with TNFα/IFNγ and in the Jurkat T lymphocyte cell line with PMA/ionomycin treatment. Strong expression of GPR65 was also seen in primary immune cells, especially in NK cells and neutrophils. In contrast, GALC, which is also in the IBD-associated region on chromosome 14q31, is expressed in all tissues tested and in certain epithelial and immune cell lines (Suppl. Fig. S4b–c). Its expression was not altered by stimulation indicated above in monocyte or T lymphocyte cell lines and there was no significant difference in expression levels between the biopsies from UC patients and healthy controls (Suppl. Fig. S4a).
Fig. 4 -. Expression profiling of GPR65 suggests that its role in IBD is primarily mediated by immune cells.
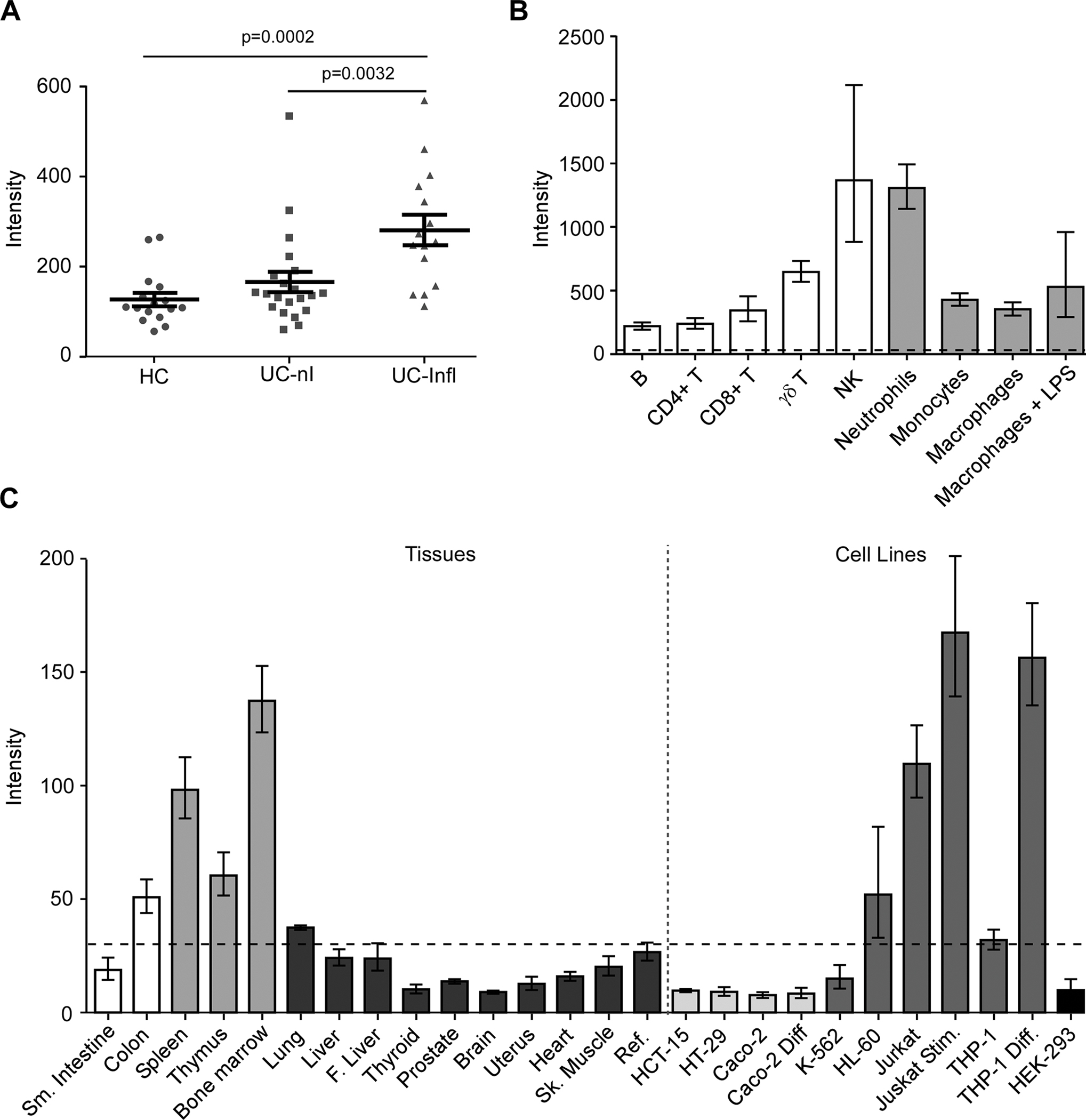
(A) GPR65 expression in biopsies from healthy controls (HC, ●), UC patients in non-inflamed tissues (UC-nI, ■) or inflamed tissues (UC-Infl, ▲) measured using the Human Genome U133 Plus 2.0 Array (Affymetrix). The lines in panel A represent the mean and SEM of each group. P values (p) were calculated using a Student’s t test. For panel B and C, GPR65 expression was evaluated with a custom-made expression microarray (Agilent). (B) Gene expression in primary immune cells, cells were isolated from peripheral blood of healthy donors (n=6). Macrophages were generated in vitro following a seven-day treatment of monocytes with M-CSF (Millipore, Cedarlane, 50 ng/mL). Macrophage activation was achieved by a one-day treatment with 1 μg/mL LPS (macrophages + LPS) for a total incubation period of eight days. Barplots represent geometric mean with SE. (C) Expression levels of GPR65 in a panel of human tissues (bone marrow, heart, skeletal muscle, uterus, liver, fetal liver, spleen, thymus, thyroid, prostate, brain, lung, small intestine, and colon). Cell lines represent models of human T lymphocytes (Jurkat), monocytes (THP-1), erythroleukemia cells (K562), promyelocytic cells (HL-60), colonic epithelial cells (HCT-15, HT-29, Caco-2), and cells from embryonic kidney (HEK 293). For gene expression in immortalized cell lines, a differentiation model of colonic epithelium was obtained by culture of Caco-2 cells after 21 days of confluence. Jurkat cells were stimulated using 40 ng/mL PMA and 1 μg/mL ionomycin (Millipore Sigma) for 6 hours (Jurkat Stim) and THP-1 cells were differentiated using 10 ng/mL TNFα and 400 U/mL IFNγ (Biolegend, Cedarlane) for 24 hours (THP-1 Diff). Intensity values for each tissue/cell line represent the geometric mean with geometric standard deviation of 3 independent (technical) measurements; each measurement represents the geometric mean of all probes (one per exon) for each gene followed by a median normalization across all genes on the array. Dotted line indicates the threshold level for detection of basal expression. The reference sample is composed of a mixture of RNAs derived from 10 different human tissues.
3.4. GPR65 knockdown in macrophages alters phagocytosis
Previous studies showed that macrophages have decreased bacteria killing in IBD, suggested to be caused by an aberrant monocyte to macrophage differentiation [37]. It has also previously been shown that GPR65-null HeLa cells and macrophages from Gpr65−/− mice have reduced intracellular bacterial clearance caused by an aberrant lysosomal function [19]. Given these studies and our results described above, we decided to study the role of GPR65 on bacterial phagocytosis, a key macrophage function relevant to IBD pathogenesis. For this, we opted to use PMA-differentiated THP-1 cells, as a surrogate for a human macrophage model, to assess the role of GRP65 on macrophage phagocytic activity. More specifically, we studied the effect of extracellular pH on phagocytosis in cells where GPR65 expression was reduced via shRNA knockdown. We used PMA-differentiated THP-1 cells stably transduced with either one of two different GPR65 shRNA-expressing vector generating ~80% reduction in GPR65 expression (named KD1 and KD2) and compared them with THP1 cells transduced with similar vector but containing no shRNA (empty vector) (Suppl. Fig. S5a). In order to confirm that both shRNAs only affected GPR65 expression since it was shown that the GPR132 response to protons is weak compared to the three other acid-sensing GPCRs [20], we tested the impact of the two shRNAs on GPR4 and GPR68 expression by qPCR. GPR4 was not detected in those cells (data not shown) and GPR68 expression was not affected under any knockdown conditions (Suppl. Fig. S5b). Upon differentiation of THP-1 cells into macrophages with PMA, morphological changes were observable in THP1 KD1 and KD2 compared with empty vector, with cells of the KD1 and KD2 conditions showing a less elongated morphology, suggesting an altered pattern of macrophage differentiation (Suppl. Fig. S5c). To study phagocytosis, PMA-differentiated THP1 cells were incubated in pH 8.0 or 6.5 for four hours with the addition of pH-Rodo E. coli during the last hour. Given that these bacteria present a bright red fluorescence when in an acidic environment, fluorescence should be observed when bacteria-containing phagocytic vesicles fuse with lysosomes. As this process is dependent on actin remodelling, we used cytochalasin D to inhibit phagocytosis as a negative control. Using this approach, we were able assess both the percentage of phagocytic cells for each cell line and also the mean fluorescence intensity (MFI) of positive cells by FACS analysis. Our analyses showed a significant decrease in the mean fluorescence intensity (MFI) of positive cells for GPR65 KD1 (decrease of 26.5%) and KD2 (decrease of 47.7%) (Fig. 5a) when compared to controls. Those results indicate a strong reduction in the number of phagocytosed bacteria per cell and/or the acidity of the lysosomes containing bacteria in GPR65 KD cells. We also saw that the fraction of phagocytic cells in GPR65 KD2 decreased by a factor of 31.2% compared to control (0.688 fold change Fig. 5b). There was also an observable decrease in KD1 compared to control cells which did not reach significance. Next, we evaluated the pH sensitivity of this process by exposing our different THP1-KD cells to pH 8.0 or 6.5; we chose to use pH 8.0 instead of 7.4 as our baseline, as it is known that GPR65 is activated at physiological pH but not at pH 8.0 [11, 17]. Although we can detect a trend where pH 6.5 increased MFI and fraction of positive cells for all cell lines, it did not reach significance. These results tend to implicate GPR65 in the phagocytosis process as its deficiency impacts on both the number of phagocytic cells and the acidification of the phagosome.
Figure 5 -. GPR65 knockdown in macrophages alters phagocytosis.
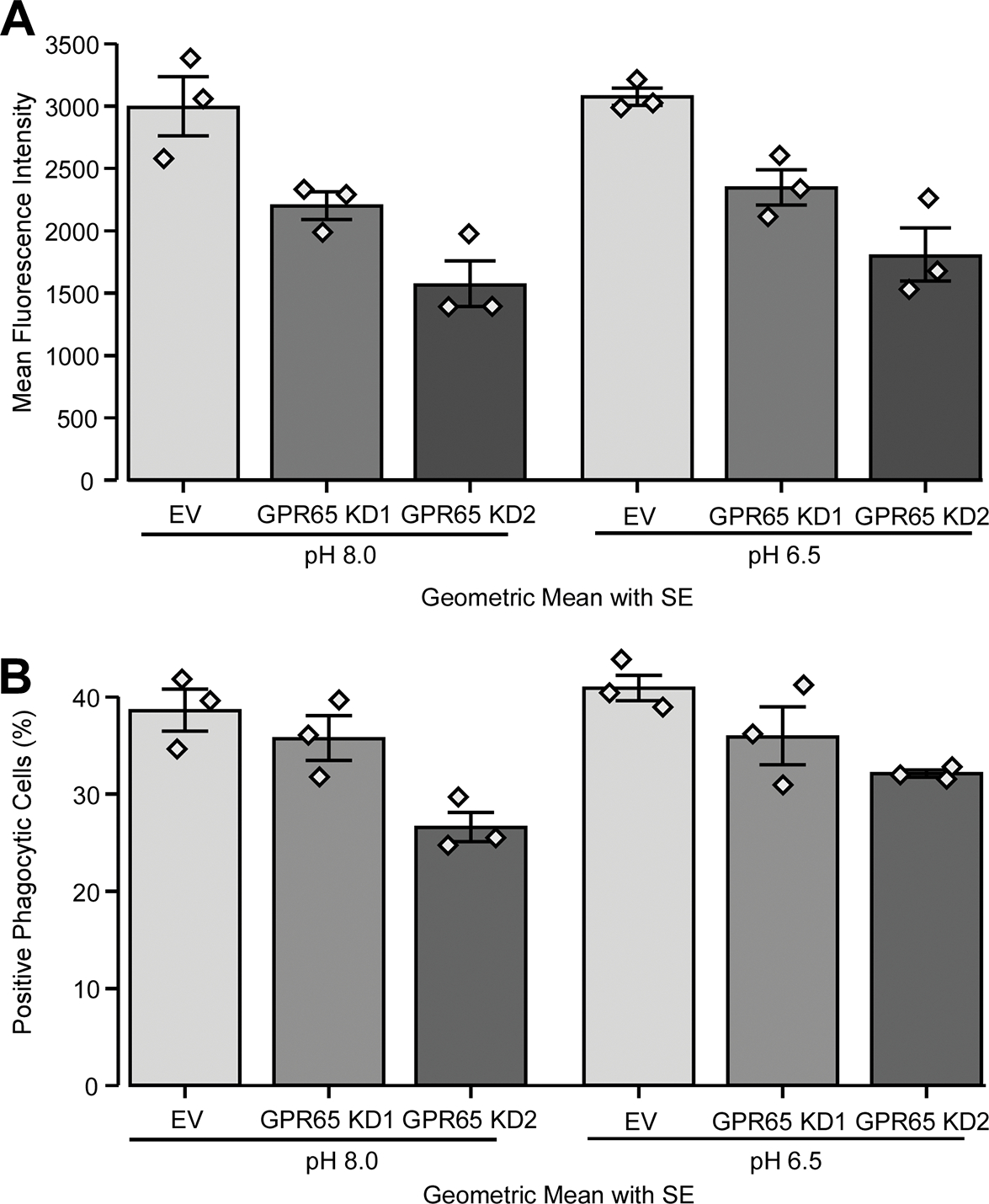
THP-1 cells were transduced with two different GPR65 shRNAs or empty vector (EV) control. PMA-differentiated THP-1 cells were placed in two different pHs (6.5 and 8.0) for four hours, with cytochalasin D or DMSO for the last hour. Cells were then incubated for one hour with pH sensitive pHRodo E. coli (A) GPR65 KD and pH effect on the mean fluorescence intensity (MFI) of positive cells. (B) GPR65 KD and pH effect on the percentage of positive phagocytic cells. Barplots represent geometric mean with SE. Detailed effect sizes analyses from ANOVA are presented in Table S4.
3.5. GPR65 modulates caspase-1 activation and IL-1β secretion in response to acidic pH
cAMP accumulation and RhoA-driven cytoskeletal remodeling, both downstream of the pH-driven activation of GPR65, have been shown to impact the expression of proinflammatory cytokines and the activation of the NLRP3 inflammasome [38–41]. Moreover, NLRP3 inflammasome activation is a two-step process, requiring priming and activation steps, the latter being generated by a danger signal like PAMPs and DAMPs. During the priming step, genes necessary for the NLRP3 inflammasome, pro-IL1β and NLRP3, are expressed. The activation step is necessary for formation of the inflammasome complex containing active caspase-1 cleaved from pro-caspase-1, which in turn cleaves pro-IL1β into active IL-1β. In this context it has been shown that extracellular acidosis can also be described as a danger signal, with LPS-primed human macrophages and PMA-differentiated THP-1 cells responding to low pH by activation of the NLRP3 inflammasome with the subsequent caspase-1 activation and IL-1β secretion [42].
To study the pH-driven impact of GPR65 on the inflammasome pathway, we used LPS as the priming ligand to induce the inflammasome in PMA-differentiated THP-1 cells, either native or depleted for GPR65, followed by a 6 hr activation step at pH 8.0, 7.5, 6.5 or 6.0. Compared to the empty vector control, knockdown of GPR65 caused a pH-dependent increase in caspase-1 activation (p20 Capsase-1) and secretion of active IL-1β (p17 IL-1beta) (Fig. 6a–b). The increase in active IL-1β in the supernatant seems to correlate to an overall increase in pro-IL-1β in the cell lysates, indicating that the elevated active IL-1β could be caused by an increase in overall expression on the inflammasome components in macrophages (Fig. 6a).
Figure 6 -. GPR65-knockdown macrophages secrete more IL-1β in response to acidic pH.
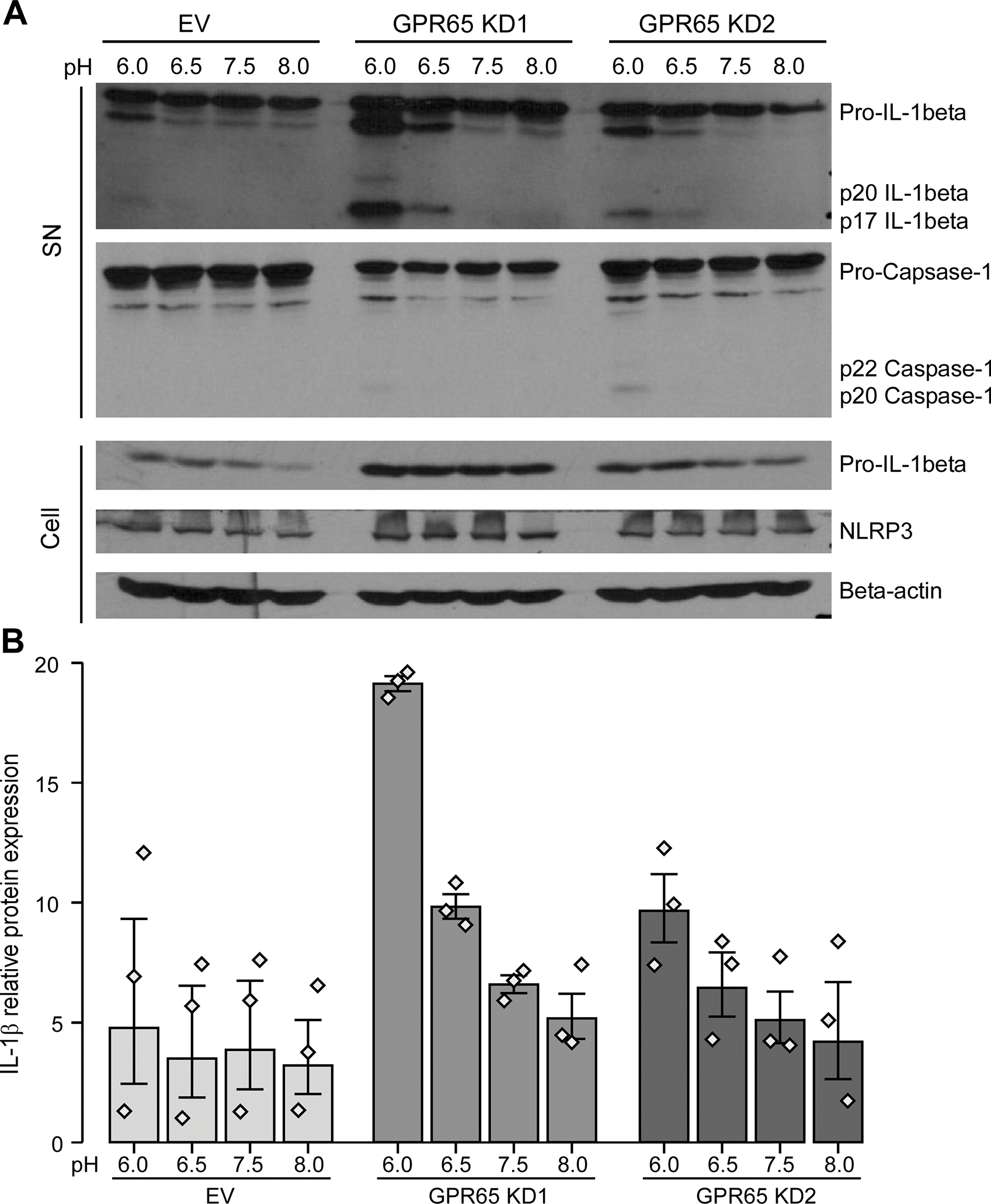
THP-1 cells were transduced with two different GPR65 shRNAs or empty vector (EV) control. PMA-differentiated THP-1 cells were incubated with LPS and then in custom RPMI media at different pHs for 6 hours. (A) Supernatants (SN) and cell lysates (Cell) were assessed by western blot for active IL-1β and caspase-1 and pro-IL-1β, NLRP3 and β-actin respectively. Representative blots of three experiments are shown. (B) Band density for active p17 IL-1β was obtained with ImageJ. Barplots represent geometric mean with SE. Detailed effect sizes analyses from ANOVA are presented in Table S5.
As GPR65 KD affected active IL-1β secretion as well as pro-IL1β expression at the protein level, we wanted to evaluate whether GPR65 was also implicated in the transcriptional (or priming) step of inflammasome complex formation, as well as in the regulation of pro-inflammatory cytokines. We took LPS-primed PMA-differentiated THP-1 cells that were washed and then incubated for 6 hours at pH 7.5 or 6.0 without LPS and extracted mRNA at each step of the protocol. In terms of mRNA expression, LPS priming of PMA-differentiated THP-1 cells induced expression of inflammasome components NLRP3 (Fig. 7a) and pro-IL1β (Fig. 7b). We were also interested in examining genes regulated by NF-κB, but not linked to the inflammasome pathway. We noted that the levels of the pro-inflammatory cytokines IL6 (Fig. 7c) and TNFα (Fig. 7d) were also induced by LPS treatment. Interestingly, the induction of these four genes was increased in the GPR65 knockdown lines, especially KD1. Given that cAMP accumulation has previously been shown to impact expression of proinflammatory cytokines, we evaluated cAMP accumulation in GPR65 knockdown THP-1 lines. We noted that GPR65 knockdown led to a significant reduction in cAMP accumulation compared to control cell lines, supporting its role in the observed increase in proinflammatory cytokine expression (Fig. 8).
Figure 7 -. GPR65 affects the inflammasome pathway, at least partly, at the transcriptional level.
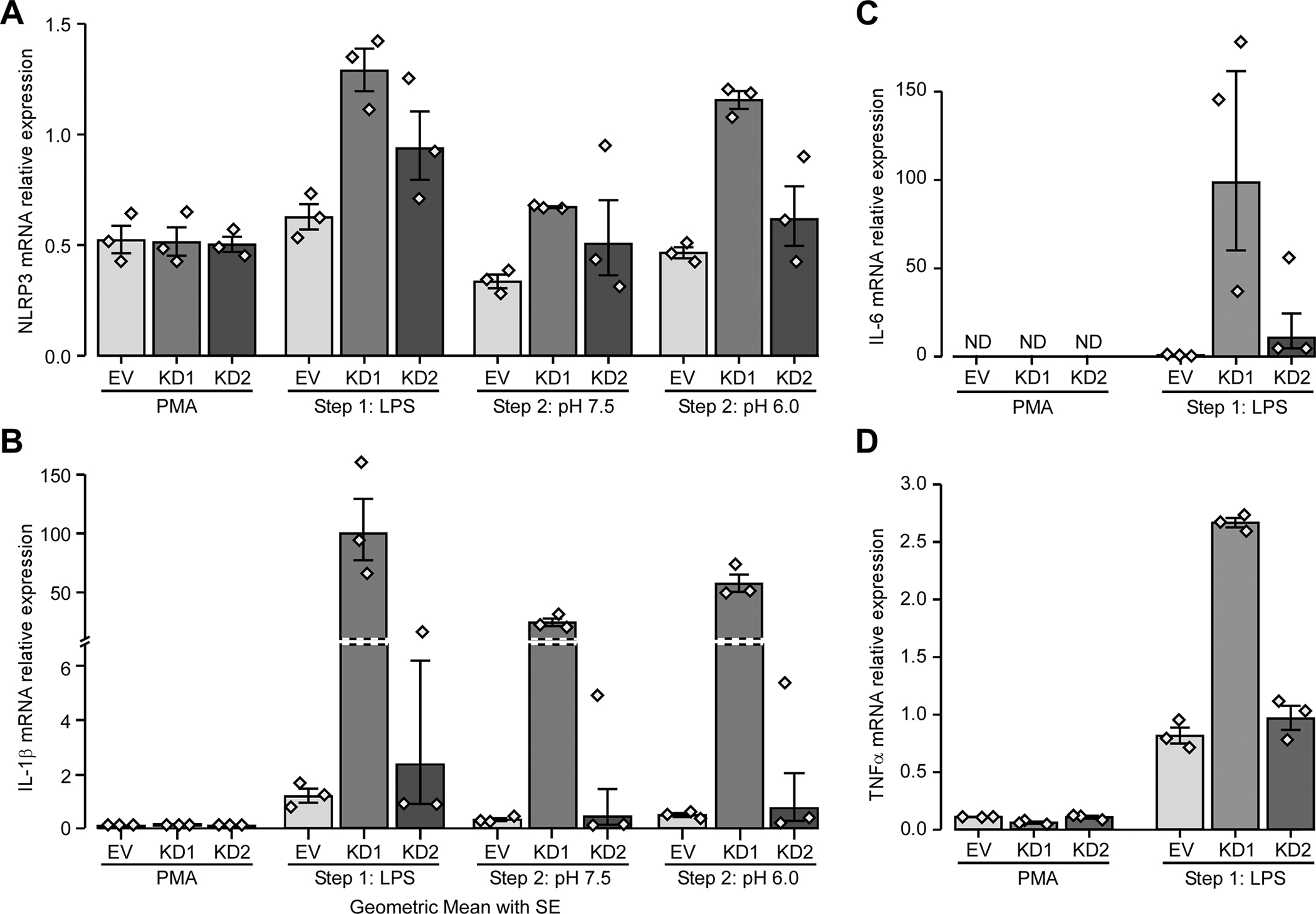
THP-1 cells were transduced with two different GPR65 shRNAs or empty vector (EV) control. RNA samples were taken after each of the three steps (PMA, LPS and incubation in different pH) and (A) NLRP3, (B) pro-IL1β, (C) IL-6 and (D) TNFα and expression were evaluated by RT-qPCR. Barplots represent geometric mean with SE. Detailed effect sizes analyses from ANOVA are presented in Table S6.
Figure 8 – GPR65 is at least partly responsible for cAMP accumulation in THP-1-derived macrophages.
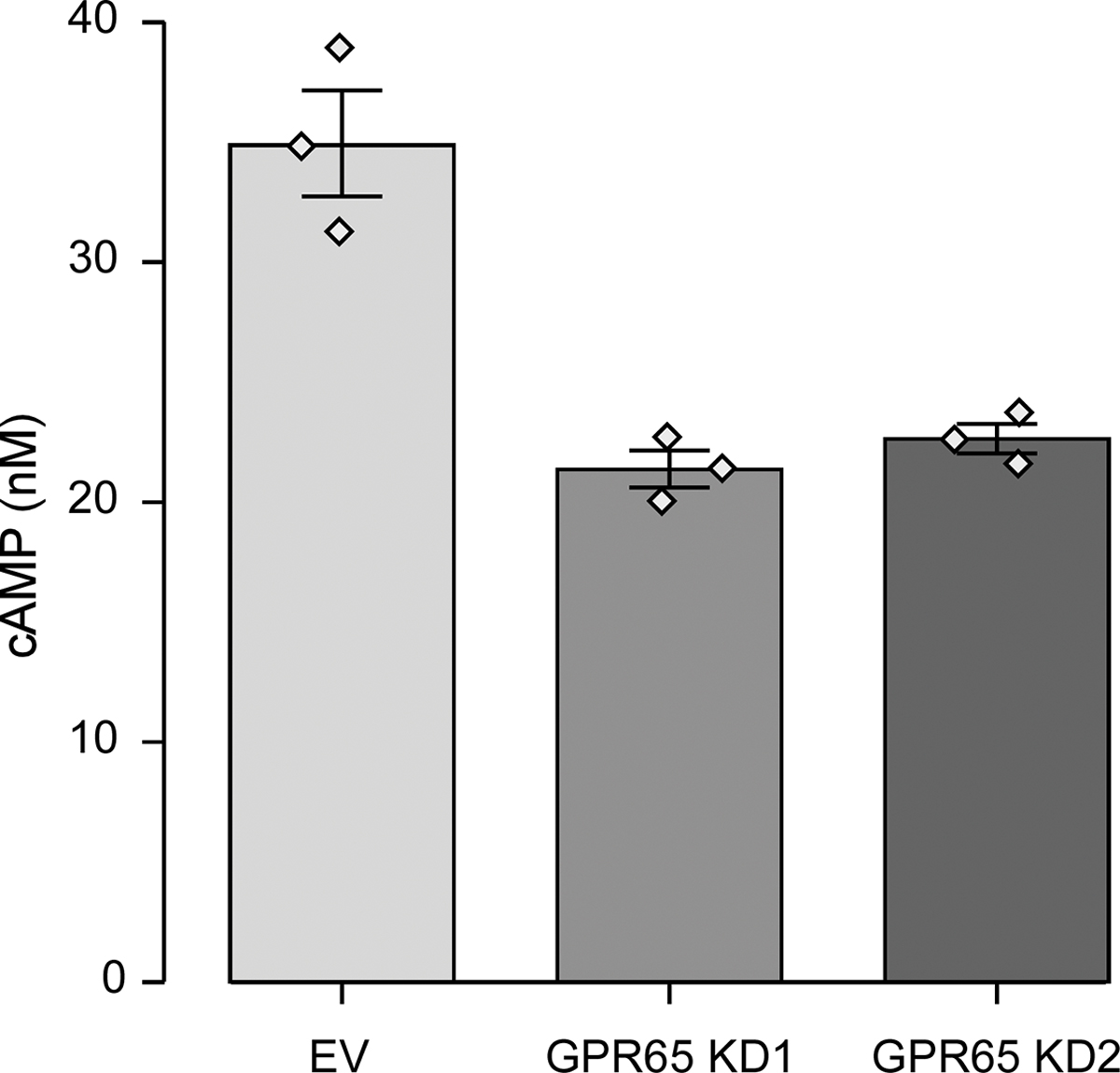
THP-1 cells were transduced with two different GPR65 shRNAs or empty vector (EV) control. PMA-differentiated THP-1 cells were assessed for cAMP accumulation after an incubation at pH 6.0 with IBMX for 30 minutes. Barplots represent geometric mean with SE. Detailed effect sizes analyses from ANOVA are presented in Table S7.
Next, we examined the activation step of inflammasome activation, to evaluate the effect of an acidic extracellular pH on expression of inflammasome components NLRP3 and pro-IL1β. The expression of both NLRP3 (Fig. 7a, S5a) and pro-IL1β (Fig. 7b, S5b) were higher following an incubation at pH 6.0 compared to pH 7.5.
Taken together, our results suggest that GPR65 KD macrophages showed a greater inflammatory response to LPS which could be caused by increased expression of NLRP3 inflammasome components and pro-inflammatory cytokines.
4. Discussion
Large-scale genetic studies have previously shown that the chromosomal region containing the GPR65 gene is associated with IBD at genome-wide significance (Pval = 2.35×10−14) [1, 10]. “Fine mapping” of this association signal in over 34,000 individuals with IBD narrowed the associated region to 18 SNPs across ~65Kb having the greatest likelihood of being causal [6]. This region included the entire GPR65 gene and the first 10 exons of GALC (of a total of 17). This study also found that there was only one non-synonymous coding variant (GPR65*Ile231Leu) among these 18 SNPs, suggesting that this was the causal gene. In the current study, we performed RNA expression analyses of intestinal biopsies from IBD patients and controls where we demonstrated that GPR65 was most highly expressed in intestinal biopsies from inflamed tissues from IBD patients as compared with non-inflamed samples from these patients or with biopsies from healthy controls (Fig. 4a). In contrast, GALC was ubiquitously expressed in the same samples, with no evidence of increased expression in inflamed colon biopsies (Suppl. Fig. S4). This analysis of clinical samples provided further evidence that GPR65 is a likely causal gene within this associated region. Moreover, our analyses of a panel of human tissues, cell lines and primary immune cells suggested that this increase in expression in inflamed tissues was due to the immune cell component of these biopsies rather than the epithelial cell component (Fig. 4). Finally, our observation that GPR65 expression is increased in the human monocytic cell line THP-1 following differentiation with pro-inflammatory cytokines TNFα/IFNγ, provided the scientific foundation for our functional studies using this cell line.
GPR65 is part of the proton-sensing GPCR subfamily. It was known that activation of GPR65 via a reduction of extracellular pH leads to the accumulation of cAMP in different murine and human cells lines and primary immune cells, and to the activation of RhoA with subsequent stress fiber formation in CHO-S cells [11, 12, 17–20]. It was shown that Gαs is in part responsible for accumulation of cAMP following GPR65 activation by acidic pH. However, other G proteins and the specific signalling cascades downstream of GRP65 for Rho activation had not yet been studied. To address this, we first evaluated GPR65 signaling in HEK 293 cells in which Gαs, Gα12/13 and Gαq/11 were knocked-out using CRISPR/Cas9. We were able to demonstrate that cAMP accumulation and RhoA activation in response to acidic pH are mediated primarily via Gαs (Fig. 1b) and Gα12/13 (Fig. 3a) signalling, respectively. Next, in order to assess the impact of the IBD-associated GPR65*Ile231Leu variant on these signalling pathways, we expressed each allele in the parental HEK 293 cell line. For cAMP accumulation, we saw that although the minor allele result in reduced cAMP accumulation, this effect did not reach statistical significance. However, when we tested a panel of HEK 293 cell lines where individual G proteins had been deleted, we observed that Gα12/13 deletion eliminated that trend, in fact bringing levels of cAMP accumulation from the major allele similar to the minor allele in both the parental and the Gα12/13 –KO HEK 293 cells (Fig. 1b), suggesting a potential role for Gα12/13, in addition to Gαs, in the activation of cAMP formation. While the synthesis of cAMP by membrane-bound AC is primarily regulated by Gαs, it has been shown that the Gα12/13 could also regulate cAMP synthesis via the AC7 isoform [43], although little is know about the mechanistic aspects of this. In addition, as Gα12/13 are primarily known to link cell surface GPCRs with guanine nucleotide exchange factors for small Rho GTPases that regulate the actin cytoskeleton, the impact of GPR65*Ile231Leu on this function was also examined. Specifically, we found that the GPR65 minor allele also abrogated the pH driven effect of GPR65 on Gα12/13-dependent RhoA activation and the subsequent actin remodelling in parental cells (Fig. 3a). These results lead us to hypothesize that the GPR65 minor allele may have altered affinity for the different G proteins, and more specifically a reduced affinity to Gα12/13. To explore this possibility, we reintroduced each G protein in their respective KO cell lines to determine if transient expression of the G protein would impact RhoA activation. As expected, expressing Gα12 in the Gα12/13-KO lines essentially rescued the phenotype. Somewhat surprisingly, transient expression of Gαq or Gαs reduced the Gα12/13-dependent RhoA activation, suggesting that they compete GPR65 signalling away from Gα12/13 (Fig. 3b–d). This could indicate that the variant reduces, but does not abolish, binding of and/or activation by GPR65 to Gα12/13 and that modulating Gα12/13 expression could overcome the effects of the IBD-associated GPR65 variant.
Interestingly, Lassen et al. noted a reduction of about 50% in cAMP accumulation in HeLa cells expressing the minor allele of GPR65 when compared to the major allele [19], while Tcymbarevich et al. [26], saw no difference in the pH-dependent cAMP accumulation between CD14+ cells from healthy controls and patients homozygous for either alleles. Our own results show only a modest impact of the GPR65 minor allele on cAMP accumulation compared to the major allele following expression in HEK 293 cells. Given that GPR65 may show variable affinity for different G proteins and that the minor allele may have reduced affinity for Gα12/13, it is possible that relative levels of the different G proteins across the different cellular models used will likely impact on the ability to detect an effect of the GPR65 minor allele.
Given our observed effects of the GPR65*231Leu allele on Gα12/13 signalling, we were interested in determining the impact that GPR65 might have on monocyte/macrophage-related functions of relevance to IBD pathogenesis. Although it is clear that multiple immune cells are implicated in IBD, macrophages play an important role in the resolution of inflammation by maintaining tolerance toward commensal microbiota as well as clearing bacteria and apoptotic cells, such as neutrophils, at sites of inflammation [37]. Importantly, intestinal macrophages are highly bactericidal without engaging in an exaggerated pro-inflammatory response. In IBD patients, however, there appears to be an increased accumulation of monocytes and immatures macrophages at sites of inflammation, an enhanced bacterial survival following macrophage engulfment, and increased production of pro-inflammatory cytokines in response to commensal bacteria [37, 44]. Lassen et al [19] showed that cells where GPR65 was either knocked down (HeLa) or knocked out (bone marrow-derived macrophages from Gpr65−/− mice) have dysfunctional autophagy that affected how cells responded to an intracellular bacterial infection.
It is known that Rho GTPase-dependent actin remodelling plays an important role in phagocytosis [45]. It was also suggested that cAMP has a major role in key macrophage function in inflammation resolution such as efferocytosis, which is in fact the phagocytosis of apoptotic cells by macrophages without engaging in a pro-inflammatory response [38]. Given that the GPR65*231Leu allele was showed reduced Gα12/13 mediated signalling central to these processes, we chose to knock down endogenous levels of GPR65 in THP-1-derived macrophages to model the effect of this IBD-associated allele. Specifically, we measured the ability of these cells to phagocytose E. coli. In these experiments we detected a significant decrease in MFI in bacteria-positive cells when GPR65 was knocked down by ~80% as compared to controls (Fig. 5c). This suggests that either fewer bacteria are engulfed per cell and/or a less effective fusion of phagosomes and lysosomes, as the intensity of fluorescent dye increases as the pH is decreased upon fusion of lysosomes. This is consistent with a previous study of the impact of reducing GPR65 expression in HeLa cells on bacterial autophagy where it was observed that GPR65 deficiency caused an aberrant lysosomal function which led to an increased survival of intracellular pathogens [19]. While phagocytosis and autophagy are distinct cellular processes, both are important for clearing bacteria, both involve fusion with lysosomes and have been associated with IBD [5, 46].
cAMP is a well-known regulator of inflammation. It functions in immune cells by reducing secretion of pro-inflammatory cytokines like TNFα and drives macrophage polarization towards anti-inflammatory functions [47]. In particular, multiple GPCRs that stimulate cAMP production have been shown to enhance anti-inflammatory M2 marker expression in different environments [39]. In fact, GPR65-dependent cAMP accumulation has been shown to inhibit LPS-induced IL-1β and TNFα production in murine microglia and peritoneal macrophages [17, 18]. Furthermore, the GPR65 minor allele was also associated with susceptibility to atopic dermatitis (AD) and GPR65 deficiency in an AD mice was shown to lead to an increase in infiltration at the inflammation site of neutrophils and T cells and in TNFα secretion by T cells [48].
RhoA signaling and its subsequent actin remodelling can affect transcription through localization and phosphorylation of transcription factors. Thus RhoA activation has been proposed to play a role in both innate and adaptive immunity [41, 49, 50]. More specifically, it can activate NF-κB, which results in increased expression of genes encoding proteins involved in pro-inflammatory pathways, such as TNF-α and IL-6, but also priming of the NLRP3 inflammasome, via increased production of NLRP3 and pro-IL-1β. It has been shown that NLRP3 inflammasome activation is a two-step process with a “priming” signal coming from PAMPS like LPS, followed by a “danger” signal from numerous PAMPs and DAMPs [41]. The priming signal results in NF-κB-mediated expression of NLRP3 and pro-IL-1β, whereas the danger signal results in assembly of NLRP3 inflammasomes, which contain active caspase-1, that converts pro-IL-1β into biologically active IL-1β. As it has been previously proposed that extracellular acidosis can constitute a danger signal [42], and that increased protein expression of NLRP3 inflammasome components, NLRP3 and IL-1β has been seen in mucosal biopsies from patients with Crohn disease [51, 52], we have used THP-1-derived macrophages to examine the potential role of GPR65 deficiency on pro-inflammatory cytokines secretion as well as activation of the NLRP3 inflammasome. Here, we observed that LPS-activated macrophages derived from THP-1 cells, where we had knocked down GPR65 and placed in an acidic environment, showed an increase in the transcription of genes that respond to the priming step of the inflammasome, such as NLRP3 and the proinflammatory cytokines IL-6 and TNFα (Fig. 7). We also saw an increase in both pro-IL-1β mRNA and protein levels, as well as increases in caspase-1 activity and conversion of pro-IL-1β to IL-1β (Fig. 6a–b, Fig. 7b). Although GPR65*231L variant showed a larger effect via the Gα12/13 pathway in our model, with RhoA signaling that could lead to impaired NF-κB transcription, it is possible that the observed exacerbation of pro-inflammatory cytokine expression may also be due to reduced cAMP production and its regulatory effect on inflammation. The current model of the impact of the IBD-risk allele GPR65*231Leu is consistent with the increased levels of circulating pro-inflammatory observed in patients [53, 54].
Taken together the results reported herein, generated by comparing the function of the IBD-associated GPR65*231Leu allele to its wildtype GPR65*Ile231 allele in HEK 293 cells or by examining the impact of decreasing the endogenous GPR65 levels to mimic the impact of the GPR65*231Leu allele in THP-1-derived macrophages, provide strong evidence of causality. Finally, work has already been done in order to find an agonist for GPR65 in terms of cAMP accumulation [55]. Our data suggests that pharmacologic intervention to increase GPR65 signalling via Gα12/13 could also potentially improve many processes that are affected in some IBD patients, including phagocytosis and inflammasome.
Supplementary Material
7. ACKNOWLEDGEMENTS
The authors wish to acknowledge the contribution of Dr. Alain Bitton (Royal Victoria Hospital) and Drs. Guy Aumais and Gilles Jobin (Maisonneuve-Rosemont Hospital) for the collection of intestinal biopsies. The authors would like to thank Dr. Asuka Inoue for the generous gift of the Gα subunit CRISPR/Cas9 knockout lines. We would like to acknowledge the contribution of the staff of the McGill University and Génome Québec Innovation Centre, for the whole-genome expression analysis service. Terence E. Hébert holds the Canadian Pacific Chair in Biotechnology.
Funding
J.D.R. holds a Canada Research Chair and this work was supported by grants from the U.S. National Institute of Diabetes and Digestive and Kidney Diseases (DK064869; DK062432) to J.D.R. as well as a Canadian Institutes of Health Research grant (PJT-156298) to T.E.H. This project also benefited from infrastructure supported by the Canada Foundation for Innovation (JDR: 202695, 218944, and 20415). T.E.H. holds the Canadian Pacific Chair in Biotechnology.
Abbreviations
- AC
Adenylyl cyclase
- cAMP
cyclic adenosine monophosphate
- CD
Crohn’s disease
- DAMPs
damage-associated molecular patterns
- EV
empty vector
- F-ACTIN
filamentous actin
- IBD
Inflammatory bowel disease
- G-ACTIN
Monomeric (or globular) actin
- GPCR
G protein-coupled receptors
- GWAS
Genome-wide association studies
- KD
knockdown
- KO
knockout
- LPS
lipopolysaccharide
- NLRP3
NLR family pyrin domain containing 3
- PAMPs
pathogen-associated molecular patterns
- PMA
phorbol 12-myristate 13-acetate
- UC
Ulcerative colitis
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest related to the contents of this article.
8. REFERENCES
- 1.Jostins L, et al. , Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 2012. 491(7422): p. 119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu JZ, et al. , Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet, 2015. 47(9): p. 979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rivas MA, et al. , Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet, 2011. 43(11): p. 1066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohanan V, et al. , C1orf106 is a colitis risk gene that regulates stability of epithelial adherens junctions. Science, 2018. 359(6380): p. 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rioux JD, et al. , Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet, 2007. 39(5): p. 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang H, et al. , Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature, 2017. 547(7662): p. 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang D, et al. , G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct Target Ther, 2021. 6(1): p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevens RC, et al. , The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discov, 2013. 12(1): p. 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozasa T, et al. , Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J Biochem, 2011. 150(4): p. 357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke A, et al. , Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 42(12): p. 1118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang JQ, et al. , TDAG8 is a proton-sensing and psychosine-sensitive G-protein-coupled receptor. J Biol Chem, 2004. 279(44): p. 45626–33. [DOI] [PubMed] [Google Scholar]
- 12.Ishii S, Kihara Y, and Shimizu T, Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J Biol Chem, 2005. 280(10): p. 9083–7. [DOI] [PubMed] [Google Scholar]
- 13.Zeng Z, et al. , Roles of G protein-coupled receptors in inflammatory bowel disease. World J Gastroenterol, 2020. 26(12): p. 1242–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, et al. , The Proton-activated Receptor GPR4 Modulates Intestinal Inflammation. J Crohns Colitis, 2018. 12(3): p. 355–368. [DOI] [PubMed] [Google Scholar]
- 15.de Valliere C, et al. , G Protein-coupled pH-sensing Receptor OGR1 Is a Regulator of Intestinal Inflammation. Inflamm Bowel Dis, 2015. 21(6): p. 1269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frasch SC, et al. , G2A Signaling Dampens Colitic Inflammation via Production of IFN-gamma. J Immunol, 2016. 197(4): p. 1425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mogi C, et al. , Involvement of proton-sensing TDAG8 in extracellular acidification-induced inhibition of proinflammatory cytokine production in peritoneal macrophages. J Immunol, 2009. 182(5): p. 3243–51. [DOI] [PubMed] [Google Scholar]
- 18.Jin Y, et al. , Inhibition of interleukin-1beta production by extracellular acidification through the TDAG8/cAMP pathway in mouse microglia. J Neurochem, 2014. 129(4): p. 683–95. [DOI] [PubMed] [Google Scholar]
- 19.Lassen KG, et al. , Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity, 2016. 44(6): p. 1392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radu CG, et al. , Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc Natl Acad Sci U S A, 2005. 102(5): p. 1632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsurumaki H, et al. , Protective Role of Proton-Sensing TDAG8 in Lipopolysaccharide-Induced Acute Lung Injury. Int J Mol Sci, 2015. 16(12): p. 28931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai SP, et al. , TDAG8 involved in initiating inflammatory hyperalgesia and establishing hyperalgesic priming in mice. Sci Rep, 2017. 7: p. 41415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh WS, et al. , TDAG8, TRPV1, and ASIC3 involved in establishing hyperalgesic priming in experimental rheumatoid arthritis. Sci Rep, 2017. 7(1): p. 8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wirasinha RC, et al. , GPR65 inhibits experimental autoimmune encephalomyelitis through CD4(+) T cell independent mechanisms that include effects on iNKT cells. Immunol Cell Biol, 2018. 96(2): p. 128–136. [DOI] [PubMed] [Google Scholar]
- 25.Tcymbarevich I, et al. , Lack of the pH-sensing Receptor TDAG8 [GPR65] in Macrophages Plays a Detrimental Role in Murine Models of Inflammatory Bowel Disease. J Crohns Colitis, 2019. 13(2): p. 245–258. [DOI] [PubMed] [Google Scholar]
- 26.Tcymbarevich IV, et al. , The impact of the rs8005161 polymorphism on G protein-coupled receptor GPR65 (TDAG8) pH-associated activation in intestinal inflammation. BMC Gastroenterol, 2019. 19(1): p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marie MA, et al. , GPR65 (TDAG8) inhibits intestinal inflammation and colitis-associated colorectal cancer development in experimental mouse models. Biochim Biophys Acta Mol Basis Dis, 2021. 1868(1): p. 166288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mola S, et al. , A transcriptome-based approach to identify functional modules within and across primary human immune cells. PLoS One, 2020. 15(5): p. e0233543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labbe C, et al. , Genome-wide expression profiling implicates a MAST3-regulated gene set in colonic mucosal inflammation of ulcerative colitis patients. Inflamm Bowel Dis, 2012. 18(6): p. 1072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shutinoski B, et al. , Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci Transl Med, 2019. 11(511). [DOI] [PubMed] [Google Scholar]
- 31.Milligan G and Inoue A, Genome Editing Provides New Insights into Receptor-Controlled Signalling Pathways. Trends Pharmacol Sci, 2018. 39(5): p. 481–493. [DOI] [PubMed] [Google Scholar]
- 32.O’Hayre M, et al. , Genetic evidence that beta-arrestins are dispensable for the initiation of beta2-adrenergic receptor signaling to ERK. Sci Signal, 2017. 10(484). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Hayre M, et al. , Inactivating mutations in GNA13 and RHOA in Burkitt’s lymphoma and diffuse large B-cell lymphoma: a tumor suppressor function for the Galpha13/RhoA axis in B cells. Oncogene, 2016. 35(29): p. 3771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sergeev E, et al. , A single extracellular amino acid in Free Fatty Acid Receptor 2 defines antagonist species selectivity and G protein selection bias. Sci Rep, 2017. 7(1): p. 13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ausubel FM, Current Protocols in Molecular Biology, ed. Ausubel FM, et al. 2003: John Wiley & Sons, Inc.. [Google Scholar]
- 36.Lukasheva V, et al. , Signal profiling of the beta1AR reveals coupling to novel signalling pathways and distinct phenotypic responses mediated by beta1AR and beta2AR. Sci Rep, 2020. 10(1): p. 8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Na YR, et al. , Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol, 2019. 16(9): p. 531–543. [DOI] [PubMed] [Google Scholar]
- 38.Negreiros-Lima GL, et al. , Cyclic AMP Regulates Key Features of Macrophages via PKA: Recruitment, Reprogramming and Efferocytosis. Cells, 2020. 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polumuri S, Perkins DJ, and Vogel SN, cAMP levels regulate macrophage alternative activation marker expression. Innate Immun, 2021. 27(2): p. 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerlo S, et al. , Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol Life Sci, 2011. 68(23): p. 3823–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swanson KV, Deng M, and Ting JP, The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol, 2019. 19(8): p. 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajamaki K, et al. , Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem, 2013. 288(19): p. 13410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang LI, et al. , Regulation of cAMP responses by the G12/13 pathway converges on adenylyl cyclase VII. J Biol Chem, 2008. 283(34): p. 23429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caer C and Wick MJ, Human Intestinal Mononuclear Phagocytes in Health and Inflammatory Bowel Disease. Front Immunol, 2020. 11: p. 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao Y and Finnemann SC, Regulation of phagocytosis by Rho GTPases. Small GTPases, 2015. 6(2): p. 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolfkamp SC, et al. , ATG16L1 and NOD2 polymorphisms enhance phagocytosis in monocytes of Crohn’s disease patients. World J Gastroenterol, 2014. 20(10): p. 2664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raker VK, Becker C, and Steinbrink K, The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front Immunol, 2016. 7: p. 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie L, et al. , pH and Proton Sensor GPR65 Determine Susceptibility to Atopic Dermatitis. J Immunol, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mostowy S and Shenoy AR, The cytoskeleton in cell-autonomous immunity: structural determinants of host defence. Nat Rev Immunol, 2015. 15(9): p. 559–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bros M, et al. , RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells, 2019. 8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, et al. , The Pathogenic Role of NLRP3 Inflammasome Activation in Inflammatory Bowel Diseases of Both Mice and Humans. J Crohns Colitis, 2017. 11(6): p. 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhen Y and Zhang H, NLRP3 Inflammasome and Inflammatory Bowel Disease. Front Immunol, 2019. 10: p. 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee SH, Kwon JE, and Cho ML, Immunological pathogenesis of inflammatory bowel disease. Intest Res, 2018. 16(1): p. 26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neurath MF, Cytokines in inflammatory bowel disease. Nat Rev Immunol, 2014. 14(5): p. 329–42. [DOI] [PubMed] [Google Scholar]
- 55.Huang XP, et al. , Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature, 2015. 527(7579): p. 477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.