

Abstract
Heparan sulfate (HS) regulates a wide range of biological events, including blood coagulation, cancer development, cell differentiation, and viral infections. It is generally recognized that structures of HS can critically impact its biological functions. However, with complex structures of naturally existing HS, systematic investigations into the structure-activity relationship (SAR) of HS and efforts to unlock their “sulfation code” have been largely limited due to the challenges in preparing diverse HS oligosaccharide sequences. Herein, we report an automated machine-aided solid-phase strategy that significantly expedited the assembly of HS disaccharides. The key strategically protected advanced disaccharide intermediates were immobilized onto Synphase lanterns. Divergent deprotections and sulfations of the disaccharides were achieved on the lanterns in high yields. In addition, the full synthetic process was automated, enabling the reproducible production of HS disaccharides. A library of 16 HS disaccharides with diverse sulfation patterns was prepared via this method. Compared to the traditional HS synthesis, this new strategy led to a reduction of 50% of the number of synthetic steps and over 80% of the number of column purification steps needed from the disaccharide intermediates, significantly improving the overall synthetic efficiency. The potential utility of the method was highlighted in a microarray study using the synthetic HS disaccharide library with fibroblast growth factor-2 (FGF-2), which yielded insights into the SAR of HS/FGF-2 interactions.
Keywords: Automated synthesis, fibroblast growth factor 2, heparan sulfate, library preparation, Synphase lanterns
Introduction:
Heparan sulfate (HS), a member of the glycosaminoglycan (GAG) family, is known to play important roles in a wide range of biological processes, including cancer development, inflammation, viral infections, and Alzheimer’s disease development.1–4 HS chains are composed of repeating disaccharide units of glucosamine (GlcN)-α−1→4 linked to a uronic acid (either d-glucuronic acid (GlcA) or l-iduronic acid (IdoA)). The sulfation may occur at O-2 of uronic acid and O-3, O-6 of GlcN, and the amine of GlcN can be sulfated, acetylated or unmodified. It is well known that the specific structure of HS can dictate their functions.5, 6 Sulfation patterns in naturally existing HS are highly heterogeneous, rendering it challenging to obtain sufficient amounts of pure HS from natural sources for detailed study of structure-activity relationship (SAR).7–9 The availability of HS oligosaccharides with diverse well-defined structures is critical to advance the knowledge on the SAR of HS.
During the last two decades, tremendous advances have been achieved in synthesis of HS oligosaccharides.1, 10–18 Chemical synthesis can enable access to well-defined HS molecules.19–21 However, the majority of the strategies were designed for target-oriented total synthesis with a relatively small number of HS structures generated in each study.1, 12, 13 One significant bottleneck for HS library preparation is the number of synthetic and purification steps required as it typically takes 6 – 8 synthetic steps to transform a protected oligosaccharide backbone to a fully deprotected sulfated HS glycan. Following each synthetic transformation, chromatography purifications need to be performed, which is tedious and challenging with the polar nature of sulfated glycan intermediates. Thus, the number of synthetic and purification steps needed for a library preparation presents a significant hurdle. With tremendous efforts, the Hung22, 23 and Wei24 groups have developed elegant synthesis to produce libraries of HS disaccharides. Boons and coworkers reported the synthesis of the largest HS tetrasaccharide library (47 compounds) to date based on a modular approach.25 Innovative new methods are needed to further accelerate HS synthesis.
Solid-phase strategies can offer an attractive approach to expedite synthesis. Although significant advances have been achieved in solid phase-supported glycosylation during the last two decades,26–31 a large excess (5 – 20 eq) of high value glycosyl donors is typically needed to obtain high yields, which presents significant roadblocks for library preparation. Furthermore, it has been challenging to synthesize sulfated glycans on solid phase. For example, although an automated synthesis of keratan sulfate tetrasaccharides was reported,28 extending the method to other classes of sulfated glycans has met challenges as suggested from the lack of fully deprotected chondroitin sulfate oligosaccharide products in the solid-phase synthesis.29 Recently, an on resin sulfation protocol was reported for several linear and branched glycans, which did not include HS oligosaccharides.32 While protected HS precursors have been assembled on solid phase,33, 34 to the best of our knowledge, no automated synthesis of sulfated HS oligosaccharides has been published.
To expedite HS assembly, herein, we report a new solid phase-based HS synthesis strategy to prepare a library of 16 HS disaccharides 1–16 (Figure 1). Our method integrates key disaccharide intermediates prepared from solution-phase synthesis with protective group removal and sulfation performed on solid phase assisted by microwave irradiation. Furthermore, the synthesis was automated on the commercially available Liberty Blue peptide synthesizer, which significantly enhanced the overall synthetic efficiency.
Figure 1:
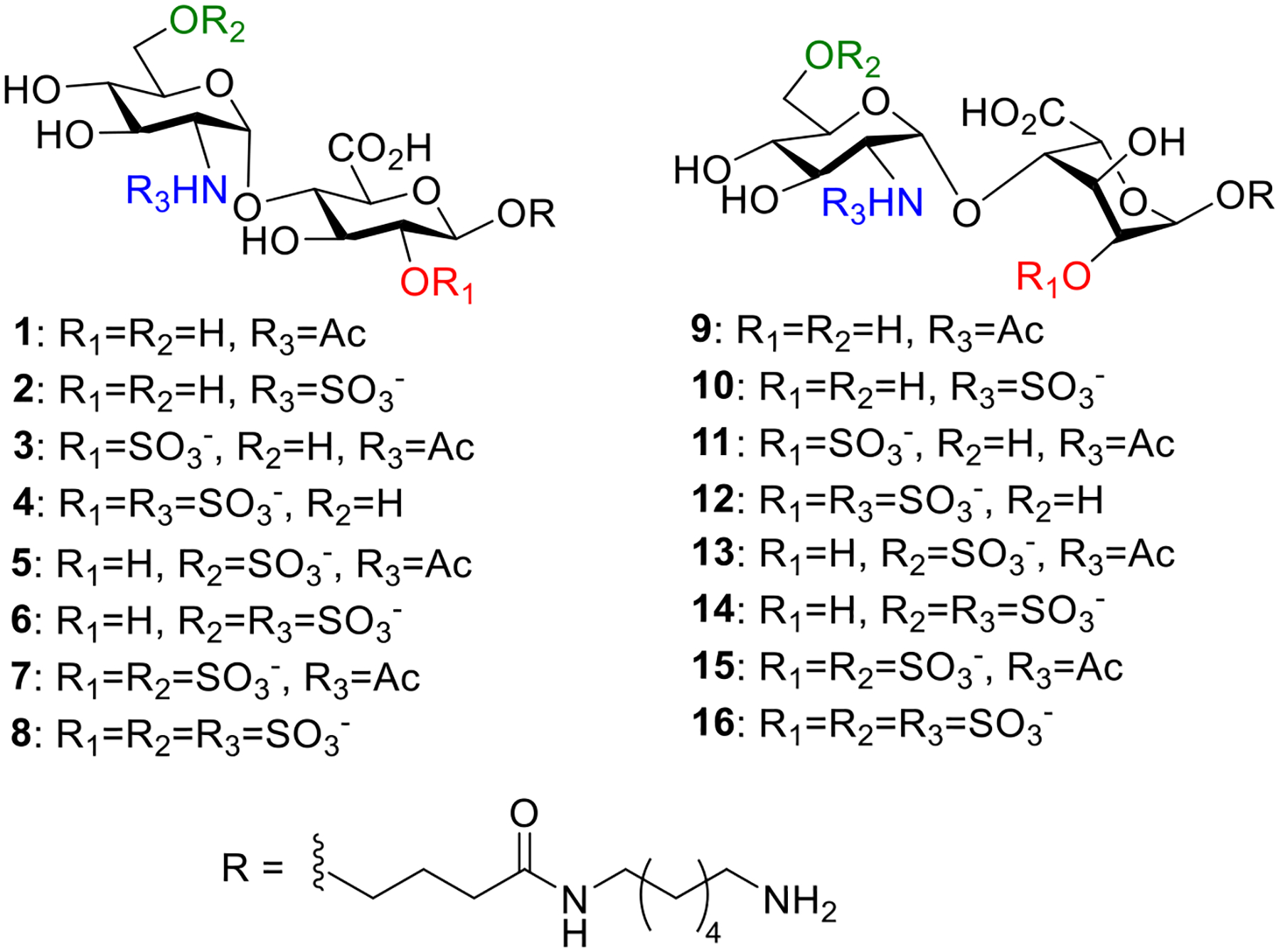
Structures of 16 HS disaccharides 1–16.
Results and discussion
Design of the synthesis
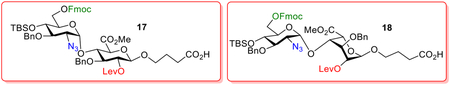
To access the 16 disaccharides, we designed two advanced common disaccharide building blocks, i.e., 17 and 18, for HS disaccharides containing IdoA and GlcA in the backbone respectively. Disaccharides 17 and 18 bear three strategically placed protective groups, i.e., fluorenylmethyloxycarbonyl (Fmoc), levulinoyl (Lev), and tert-butyl-dimethylsilyl (TBS) as well as the azide moiety to mask the amino group of GlcN. Each of these protective groups may be orthogonally removed without affecting others for divergent synthesis of the HS library.17
To further enhance the overall synthetic efficiency, we envision the key disaccharides can be immobilized on solid phase support followed by divergent deprotection and sulfation (Scheme 1a). With the ability to use an excess amount of reagent to drive the solid phase-supported reactions to completion without the need for intermediate purification, the overall production time may be significantly shortened.
Scheme 1.
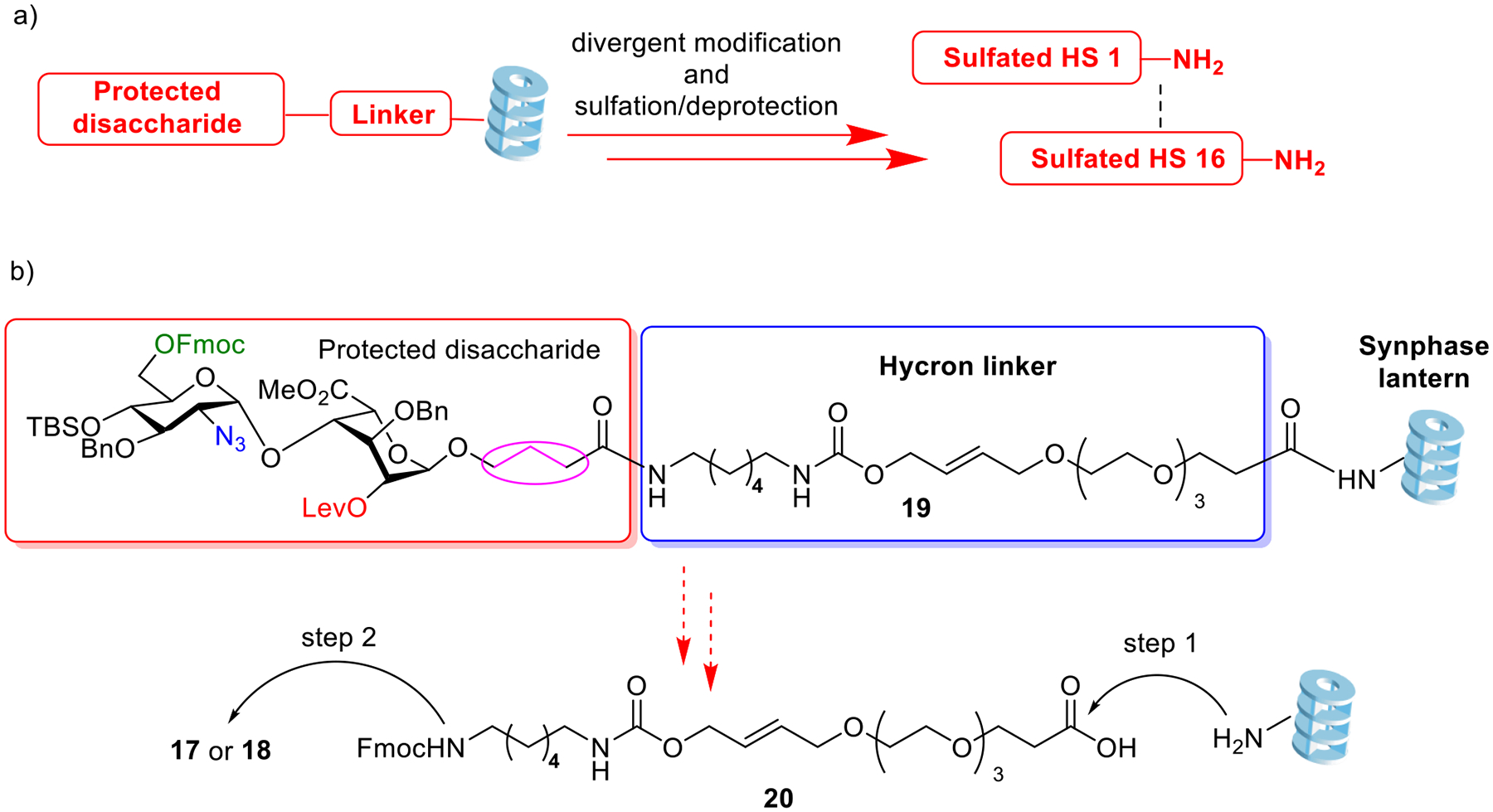
a) Schematic demonstration of the synthesis of sulfated HS disaccharides on Synphase lanterns; b) Retrosynthetic analysis of the HS synthesis on Synphase lanterns with the protected disaccharide linked through the Hycron linker.
We selected Synphase lanterns as the solid-phase support, which have a cylindrical shape (5 mm diameter and 5–17 mm length).35 Unlike the more common micron sized polymer beads, the lanterns can be easily transferred from flask to flask using a pair of tweezers without losing samples during the transfer. The lanterns have an unreactive rigid base polymer as the inner core, providing a robust and convenient framework, which is coated with a low cross-linked uniform polymer graft 50 μm thick as the outer layer where reactions take place. The unique “Lantern” shape provides high surface area, and enables free flow-through of reactants and rapid drainage of solutions. Furthermore, the lanterns can be coated with a hydrophilic polyamide surface36–38 suitable for HS modifications with high loading and reaction rates. Synphase lanterns can combine the advantages of solution-phase and solid-phase chemistry in one system, overcoming some of the limitations of either system alone.
In order to perform solid-phase synthesis, the linker used to immobilize the glycan is critical. This linker needs to be stable under the typical synthetic transformations encountered during HS synthesis including base treatment, oxidation, and sulfation. Yet, it should be readily cleavable under a mild condition without affecting the labile O- and N-sulfate moieties. After screening multiple structures, we selected the Hycron linker39 to attach the glycan. We originally chose an amino-oxy moiety to immobilize the glycan with the Hycron linker (Figure S1). However, the N-O bond of the oxime was cleaved during the final hydrogenolysis. We also investigated the usage of an ester linkage, which turned out to be too labile during the typical reactions employing base for HS functionalization (Figure S1). Finally, we designed the allyl carbamate linkage for attaching the glycan to the lantern through the Hycron linker (Scheme 1b). The three methylene units at the reducing end of the glycan (highlighted in cyan color oval in scheme 1b) turned out to be important as a shorter glycolic amide linker led to premature cleavage of the glycan by base (Figure S1). The multiple ethylene glycol units in linker 20 can increase the hydrophilicity of the lantern surface and facilitate the formation and reactions of highly polar sulfated glycans attached on the lantern. Furthermore, the glycan may be released from the lantern through the treatment of a mild Pd(0) catalyst39 to cleave the Hycron linker to afford the desired HS disaccharide.
Synthesis of GlcN-GlcA Building Block 17, GlcN-IdoA Building Block 18, and the Hycron linker 20.
Our synthesis started with preparation of the disaccharide 17 by the glycosylation reaction between donor 2140 (1.1 eq) and thioglycoside acceptor 2241 (1 eq) in the presence of a bulky base tri-tbutyl-pyrimidine (TTBP)42 (Scheme 2) following the pre-activation based glycosylation using the p-TolSCl/AgOTf promoter.43, 44 Disaccharide 23 was obtained in 70% yield as the desired α-anomer (1J(C1,H1) = 173.9 Hz suggesting α-linkage)45 without the need of resorting to 5–20 eq of the donor typically required for solid-phase glycosylation reactions. Disaccharide 23 underwent protective group manipulation to give tert-butyl-diphenylsilyl (TBDPS) protected disaccharide 24. Coupling between 24 and 4-pentene-1-ol gave disaccharide 25 in 82% yield as the β-isomer exclusively ((1J(C1,H1)= 160.5 Hz confirming β-linkage). It is important to switch the 6-O-PMB in 23 to 6-O-TBDPS, as direct usage of 23 as the thioglycoside donor gave the 1,6-anhydrosugar as a side product presumably due to participation of the 6-O-PMB moiety.41 The pentenyl glycoside 25 underwent silyl deprotection with HF/pyridine complex, and the resulting diol was subjected to selective oxidation,46 methyl esterification followed by TBS protection to give disaccharide 26 in 52.1% yield over 4 steps. Protective group manipulation on 26 installed a Fmoc group on 6-O of the GlcN and a Lev on 2-O of GlcA for future selective deprotection. Subsequent ozonolysis followed by Pinnick oxidation47, 48 of the resulting aldehyde afforded the desired disaccharide acid 17.
Scheme 2.
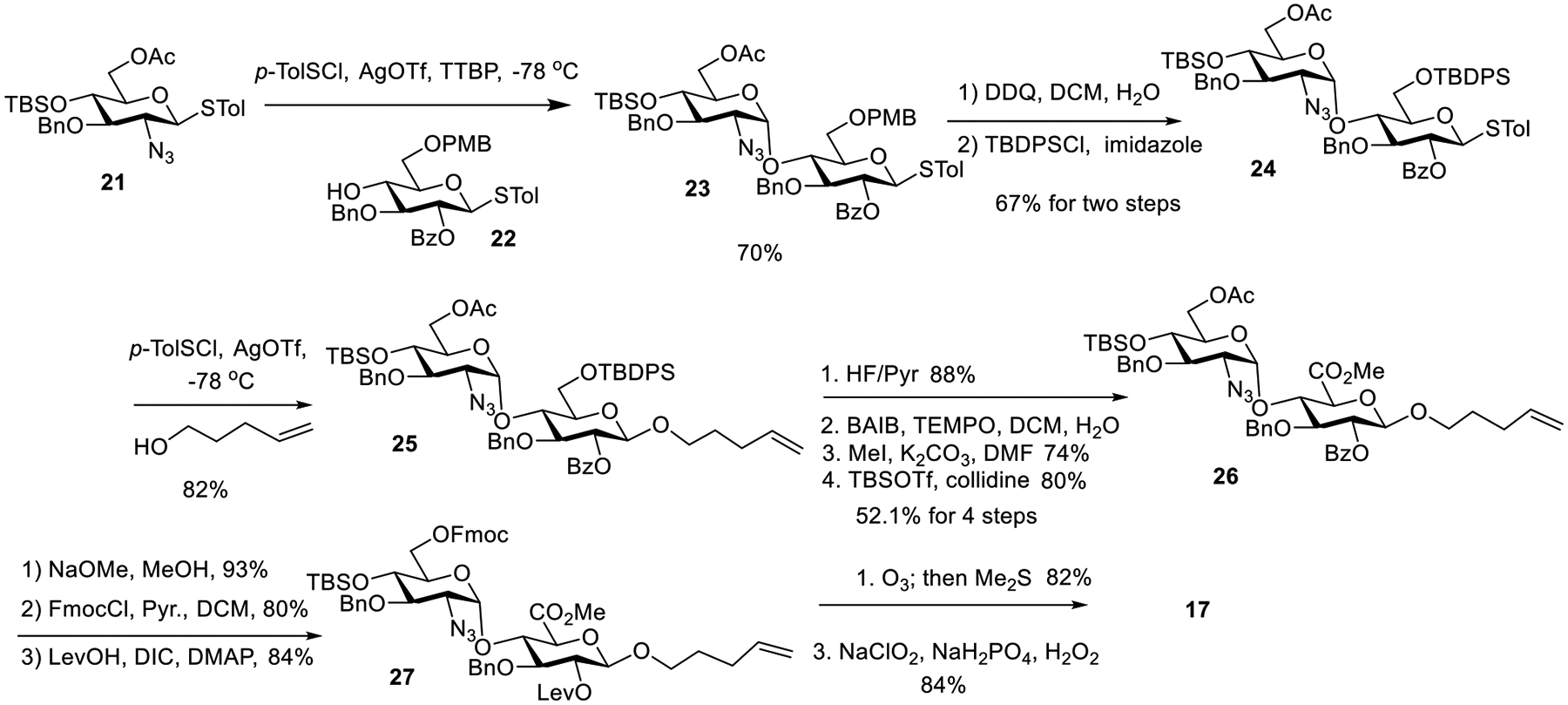
Synthesis of the GlcN-GlcA Building Block 17.
For GlcN-IdoA building block 18, the synthesis started with glycosylation between idose donor 2844 and 4-pentene-1-ol acceptor with the p-TolSCl/AgOTf promoter system, which gave the pentenyl glycoside 29 in 88% yield (Scheme 3). The newly formed α-linkage was confirmed by NMR analysis (1J(C1,H1)= 170.3 Hz). Subsequent treatment of 29 with p-toluene sulfonic acid (p-TSA) followed by oxidation and esterification afforded acceptor 30 in 77.4% yield over 3 steps. The coupling between donor 21 and acceptor 30 went smoothly and gave disaccharide 31 in 78% yield exclusively as the α-isomer ((1J(C1,H1)= 170.3 Hz).45 Similar sequence of protective group adjustments and oxidation of the pentenyl moiety of 31 as in the synthesis of disaccharide acid 17 led to the desired GlcN-IdoA disaccharide building block 18.
Scheme 3.
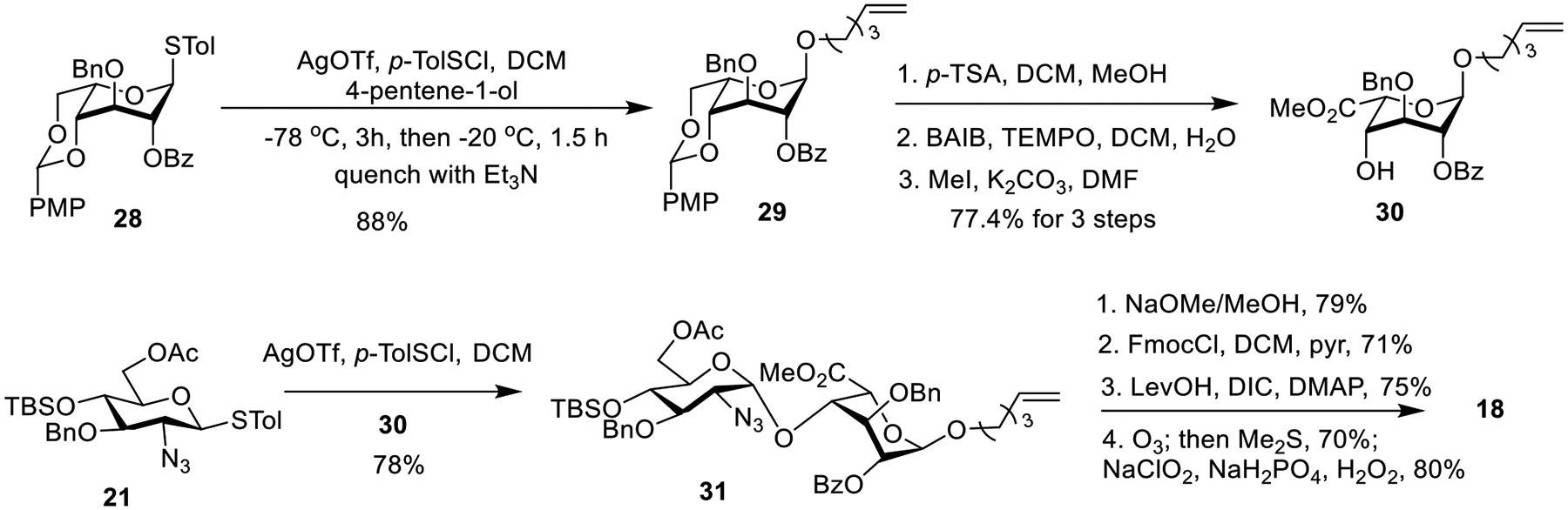
Synthesis of GlcN-IdoA Building Block 18.
After the synthesis of disaccharides 17 and 18, we turned to the preparation of the Hycron linker 20 (Scheme 4).39, 49–51 Coupling between 32 and the mono Fmoc-protected diamine 33, followed by acid treatment to deprotect the tBu ester gave the Hycron linker 20 in 77% yield.
Scheme 4:
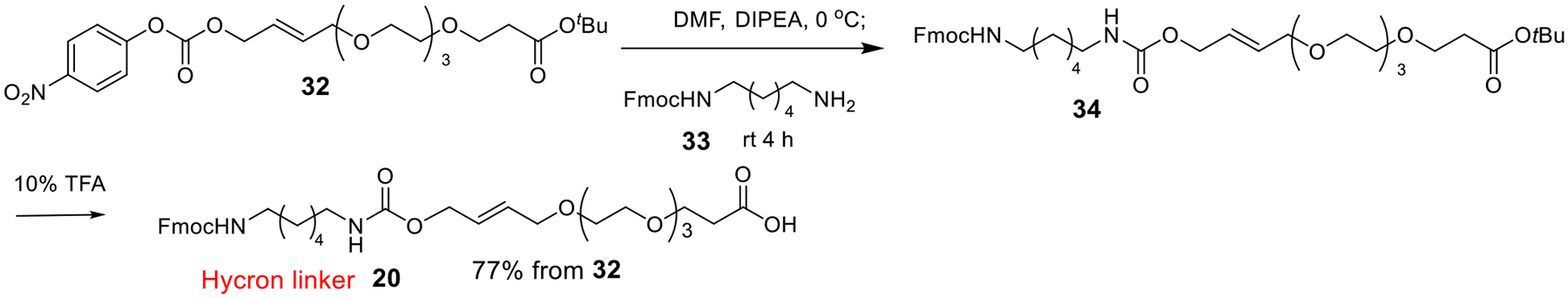
Synthesis of the Hycron Linker 20.
Lantern-supported automated synthesis of disaccharide 16
With all components in hands, we functionalized the amine bearing Synphase lanterns 35 with the Hycron linker 20 (Scheme 5). The coupling of Hycron linker 20 onto the lantern was promoted by 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU)/1-hydroxy-7-azabenzotriazole (HOAt) under microwave irradiation at 50 °C on a CEM Liberty Blue automated peptide synthesizer (Scheme 5). To quantify the loading of Hycron linker on the lanterns, the Fmoc moiety was deprotected by piperidine. Based on the UV-vis absorbance of the cleavage product dibenzofulvene, the loading level of Hycron linker was determined to be 6.4 μmol per lantern. Microwave (μW) irradiation significantly shortened the reaction time from 20 h with a traditional oil bath to 3 h, presumably due to better heating and agitation of the reaction mixture. Subsequently, disaccharides 17 and 18 (0.7 equiv. to amine on the lantern) were loaded respectively onto the lantern promoted by HOAt with microwave irradiation at 50 °C. 4.2 μmol of disaccharide 18 per lantern was immobilized corresponding to over 90% of the disaccharide added to the reaction, suggesting a high efficiency of the immobilization protocol. Following a similar procedure, 3.2 μmol of the disaccharide 17 was loaded per lantern.
Scheme 5.
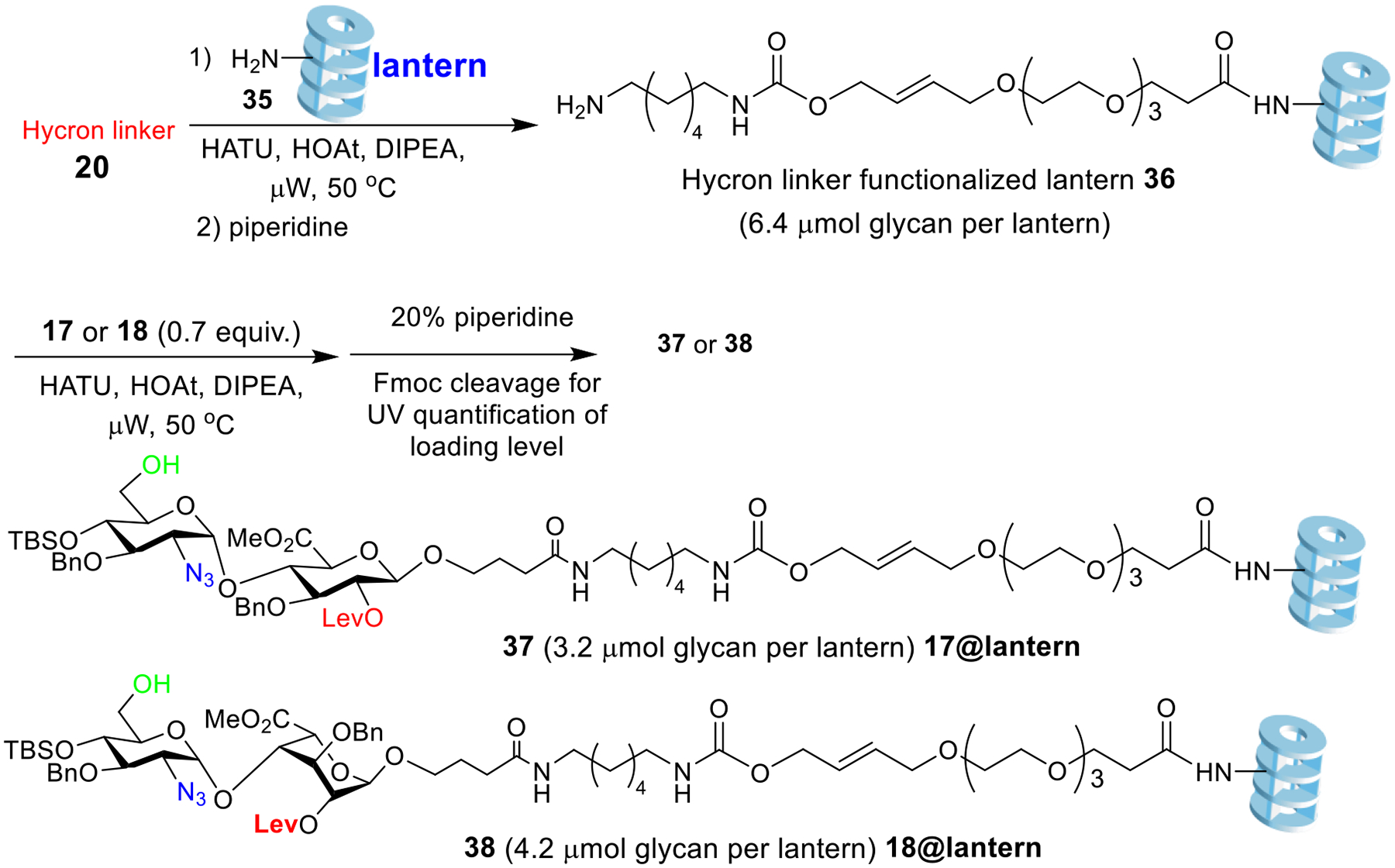
Disaccharides 17 and 18 can be efficiently loaded onto amine functionalized lantern 35 through the Hycron linker 20.
With the disaccharide successfully immobilized, synthetic transformations of the disaccharide were carried out on the lanterns. The sequence of reactions was optimized, and reagent compatibility with the Liberty Blue synthesizer was investigated. As each lantern is composed of 9 rings of polypropylene, half a ring of the lantern could be cut off easily with a razor blade. The attached glycans (~ 100 μg) could be released with a Pd(0) catalyst, which was sufficient for mass spectrometry (MS) analysis to enable monitoring reaction progress. For Lev deprotection, the reaction was performed using μW at 50 °C for 3h for 2 times, which was sufficient for complete Lev-cleavage as indicated by MS analysis. O-Sulfation was carried out using SO3.Et3N complex with Et3N in DMF using μW at 60 °C for 6h for 2 times. N-sulfation was performed in MeOH three times with the addition of NaOH (2M) to adjust pH to be around 8.5. Azide reduction was performed using the Staudinger reaction using μW at 60 °C for 30 minutes in 3 rounds to ensure its completion. Hydrolysis of the methyl ester was accomplished in two cycles using freshly prepared LiOH solutions. For TBS removal, while HF pyridine was efficient in promoting the reaction, it was corrosive to the tubing and the reaction vessel of the synthesizer. As a result, tetrabutylammonium fluoride (TBAF) was utilized to cleave TBS, the rate of which was expedited by microwave irradiation enabled by the synthesizer. As sulfates can be labile, we investigated the stability of sulfates during lantern supported deprotections. O-Sulfates were found to be stable to Lev, TBAF, and Staudinger reduction conditions on the lanterns. However, some N-sulfates were cleaved during the TBAF reaction. Thus, the reaction sequence was designed that the N-sulfate was to be installed at late stage of the synthesis. Throughout these transformations, no disaccharide products were observed to be prematurely cleaved suggesting the compatibility of the Hycron linker with protective group manipulation and sulfation reaction conditions typically encountered in HS synthesis.
Besides the possibility of using microwave irradiation to shorten the reaction time, , the reagent addition and washing steps can be programmed and automated on the CEM Liberty Blue system. As shown in Figure 3, the HS disaccharide 18 functionalized lantern 38 (18@lantern) was subjected to Fmoc cleavage (for quantification), O-acetylation, Lev-deprotection, O-sulfation, azide reduction, hydrolysis, TBS deprotection, and N-sulfation. The synthesizer was programmed to carry out the 7 synthetic steps from 38 in an automated fashion in 96 hours (Figure 2). Upon completion of the synthesis, the disaccharide product 39 was released from the lantern by Pd(PPh3)4 and borane, and purified by LH-20 size exclusion chromatography. It should be emphasized that from 38 to 39, only one chromatography purification is needed. The overall yield of 39 from 38 was an excellent 60% for the seven steps representing an average of 92% yield for each synthetic transformation. The high yield for the lantern supported synthesis could be due to 1) with the easy removal of unconsumed reagent by washing the lanterns, excess reagent was used to drive the reaction to completion; 2) the hydrophilic PA surface of the lantern enabling facile glycan sulfation on the solid phase; and 3) the avoidance of product loss during multiple purification steps as required in solution phase synthesis. Following hydrogenolysis to remove the benzyl protective groups from 39, the tri-sulfated HS disaccharide 16 was obtained in good purity as indicated by 1H-NMR analysis (Figure 3).
Figure 3.
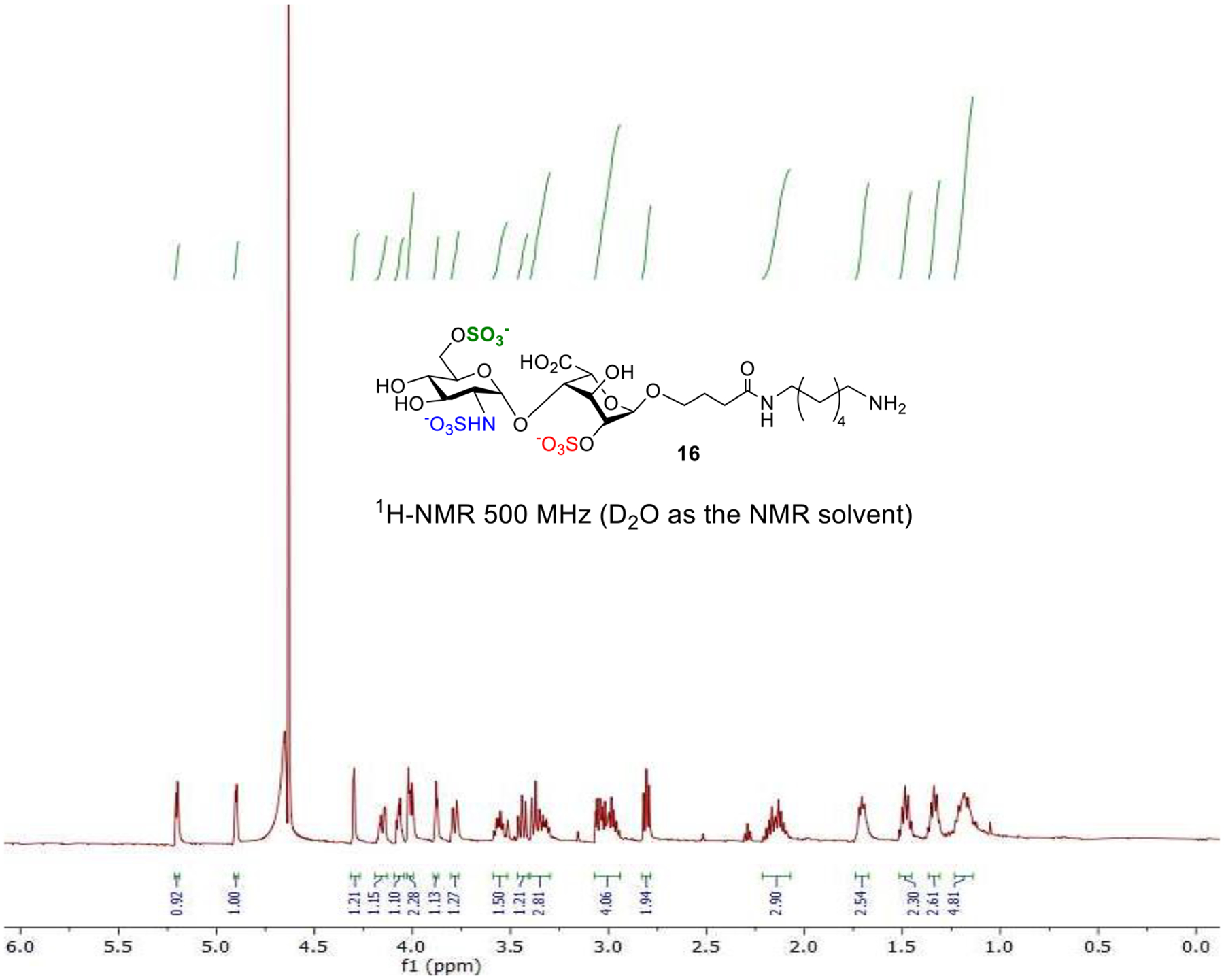
1H-NMR spectrum of compound 16.
Figure 2.
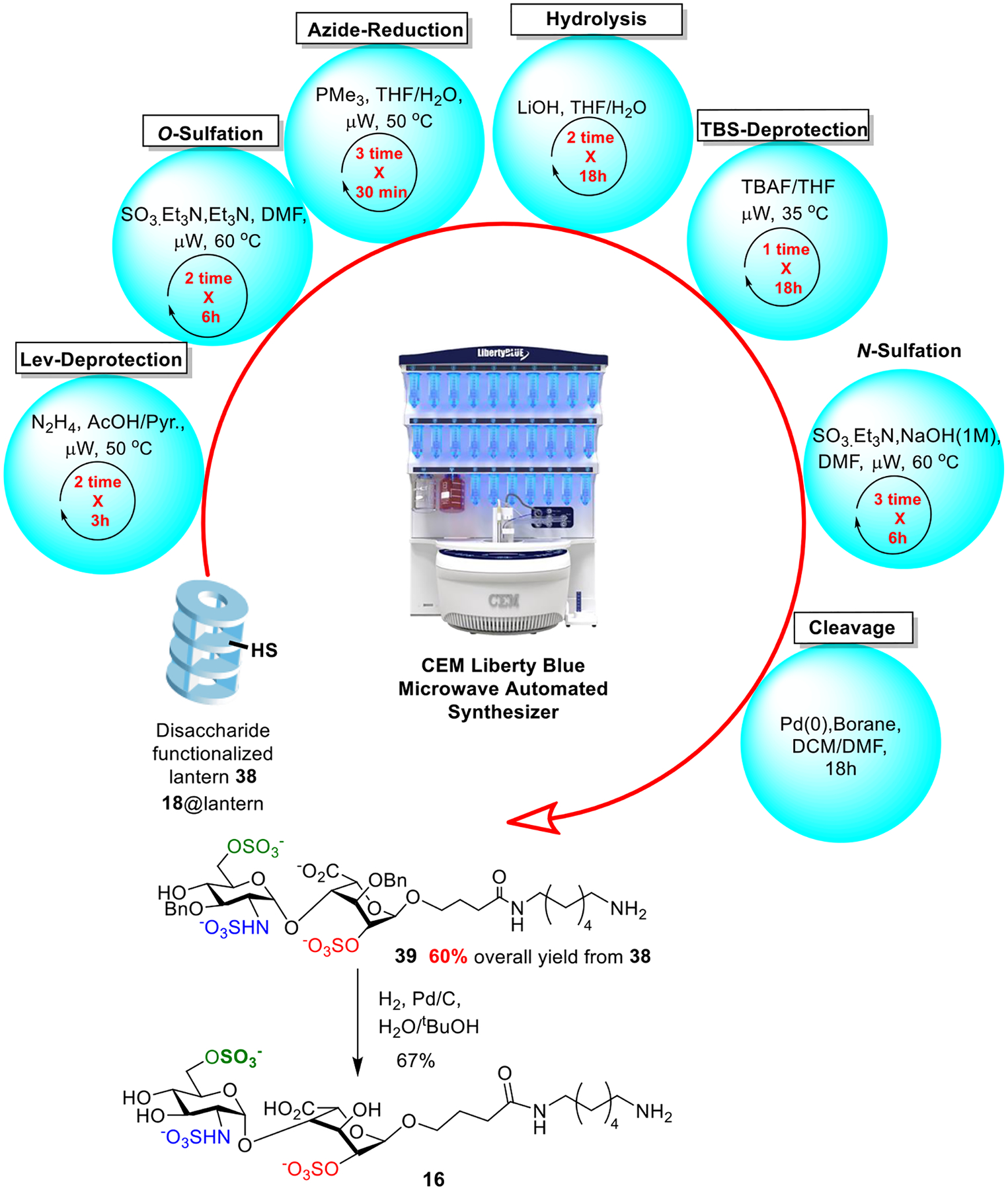
Automated synthesis of the trisulfated HS disaccharide 16 from disaccharide loaded lantern 38 (18@lantern) on the Liberty Blue system.
Synthesis of the 16-membered HS disaccharide library
As disaccharide 18 bears orthogonally removable protective groups at the strategic positions, the deprotection and sulfation reaction sequence can be varied to divergently create multiple HS disaccharide. As an example, starting from 38 (18@lantern), skipping the Lev deprotection step while keeping the rest of the reactions in the synthesis of 16 produced disaccharide 14 in 41.5% overall yield.
The lantern supported automated synthesis process is robust. Without the need for further optimization, simply by adjusting the sequence of the reactions, a panel of 8 disaccharides 9–- 16 were produced in good yields with each synthesis taking about 90 hours on the synthesizer starting from 18@lantern (Table 1). Similarly, the GlcN-GlcA disaccharide bearing 37 (17@lantern) was converted to 8 disaccharides 1 – 8 with the GlcN-GlcA backbone in good overall yields (Table 1).
Table 1.
Overall yields for the multi-step synthesis of 1–16 from 37 (for compounds 1– 8) and 38 (for compounds 9–- 16) respectively.
| Compound number | Overall yields from 37 (17@lantern) % | Compound number | Overall yields from 38 (18@lantern) % |
|---|---|---|---|
| 1 | 57.7 | 9 | 38.5 |
| 2 | 57 | 10 | 39 |
| 3 | 38.9 | 11 | 49 |
| 4 | 33.6 | 12 | 24 |
| 5 | 33 | 13 | 37.5 |
| 6 | 41 | 14 | 41.5 |
| 7 | 60 | 15 | 47.6 |
| 8 | 42 | 16 | 40.2 |
Each of the 16 disaccharides (1–16) was prepared on 1– 3 mg scales with >95% purity. Thus, the overall operation for library preparation was significantly expedited by Synphase lantern aided synthesis using the CEM microwave synthesizer, which reduced the number of purification steps by over 80% for each member of the library with the synthesis automated.
The disaccharide library yielded insights on structural features important for fibroblast growth factor 2 (FGF-2) binding
To demonstrate the utility of the disaccharides, we investigated the binding of the disaccharides with a representative member of the fibroblast growth factor (FGF) family, the FGF-2. FGFs play crucial roles in cellular development, which can bind with endogenous HS facilitating the complex formation with FGF receptors to induce signal transduction and promote cell proliferation and growth. A better understanding of the structural requirements of HS for FGF-2 interactions can enhance our knowledge and facilitate the development of therapeutics in areas including wound healing, angiogenesis, and cancer therapy.52, 53
To investigate HS disaccharide binding to FGF-2, the disaccharides 1–16 were immobilized onto an N-hydroxysuccinimide (NHS) ester functionalized glass slide through amide linkages at three different concentrations (0.25 mM, 0.5 mM, and 1 mM) to fabricate the disaccharide microarray. Fluorescently labeled FGF-2 was incubated with the microarray. Upon washing off unbound protein, the microarray was scanned by a fluorescence slide reader to semi-quantify the amounts of FGF-2 remaining bound.
HS disaccharides exhibited structure dependent binding with FGF-2. As shown in Figure 4, compound 12 (GlcNSIdoA2S) bound the strongest with FGF-2, with the corresponding GlcA disaccharide 4 bearing the same sulfation patterns also binding well when printed at 1 mM suggesting the backbone structure in this context does not drastically impact FGF-2 binding. Interestingly, addition of the 6-O sulfate to GlcN (compound 16 GlcNS6SIdoA2S) decreased the binding by about 50% indicating higher sulfation density on the disaccharide could negatively influence the FGF-2 interaction. Compounds 12 (GlcNSIdoA2S) and 13 (GlcNS6SIdoA) both have two sulfates in their structures. Yet, 12 exhibited significantly stronger binding than 13 suggesting the positions of the sulfates can be important for binding. The N-sulfation was important as GlcNAcIdoA2S 11 and GlcNAc6SIdoA2S 15 are much weaker binders than the corresponding N-sulfated disaccharides GlcNSIdoA2S 12 and GlcNS6SIdoA2S 16. The importance of 2-O sulfation and N-sulfation is consistent with literature reports on HS-FGF-2 interactions obtained using HS oligosaccharides including HS disaccharides,54–58 suggesting the HS disaccharide library prepared via the current automated synthesis can be used to probe HS interactions.
Figure 4.
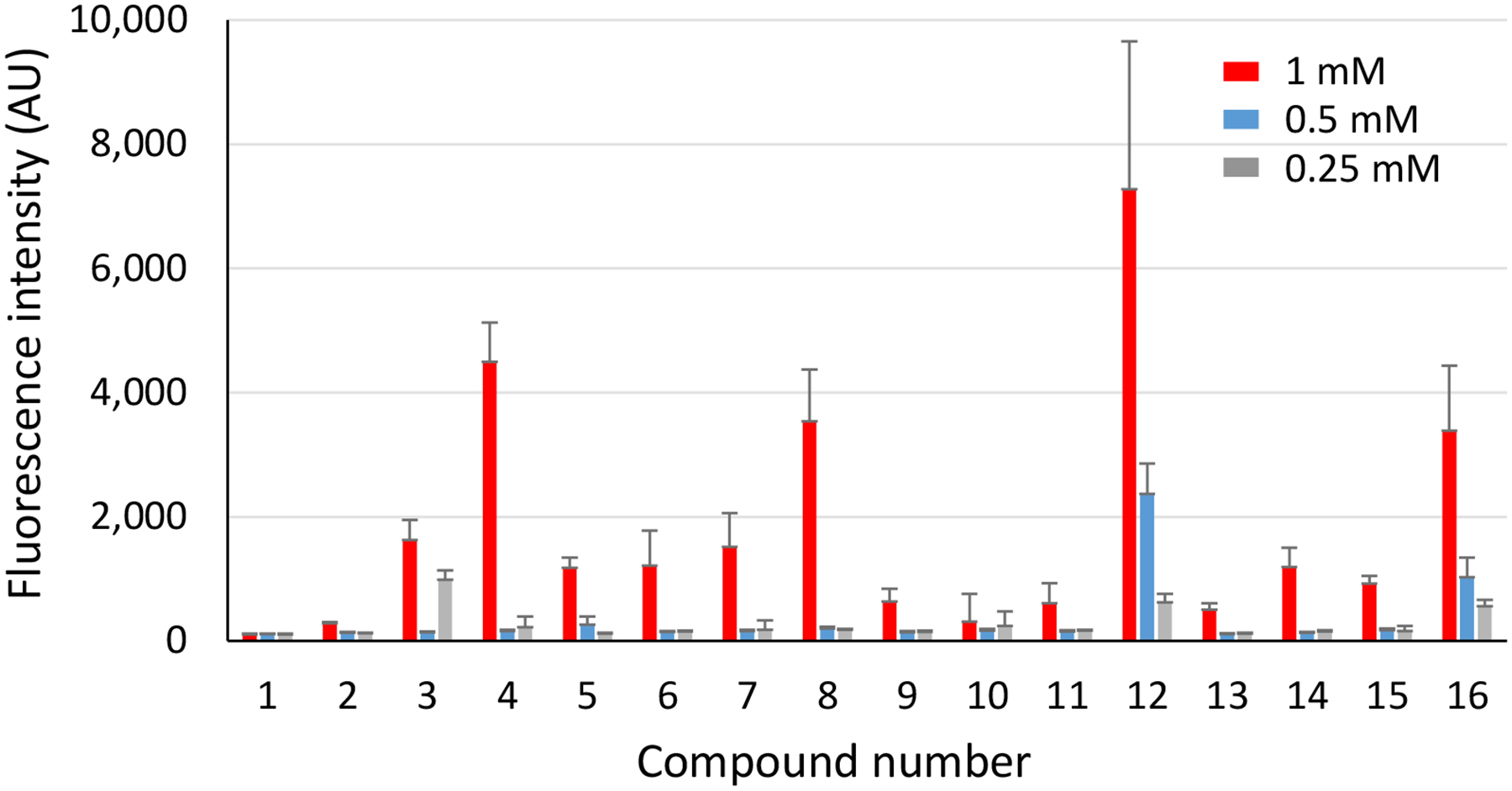
Fluorescence signal intensities observed for each arrayed HS disaccharide binding to FGF-2. Three different concentrations of each disaccharide, ranging from 1 mM to 0.25 mM from left to right were printed on the microarray. All samples were printed in replicates of sixteen.
Conclusion:
We report the establishment of a new method to rapidly access a library of HS disaccharides. The two key strategically protected intermediates were efficiently assembled in solution without the need for 5–20 equiv. of high-valued glycosyl donors typical for solid-phase synthesis. These advanced disaccharides could be divergently deprotected to enable production of a library of HS disaccharides. To expedite the library preparation, the two advanced disaccharides were immobilized onto Synphase lanterns through an allyl carbamate bearing Hycron linker, which was found to be compatible with a wide range of typical reactions encountered in HS synthesis, including acid, base, Staudinger reduction, and silyl removal. Multiple sulfates could be installed on the disaccharides immobilized on the lanterns in high yields. In addition, the synthetic process was automated on a microwave-assisted peptide synthesizer, enabling the expedient production of the 16-membered HS disaccharide library. Compared to the traditional HS synthesis, this new strategy led to a reduction of 50% of the number of synthetic steps and over 80% of the number of column purification steps needed from the disaccharide intermediates, thus significantly enhancing the overall synthetic efficiency. The disaccharide library obtained was transformed into a glycan microarray and screened against FGF-2, which yielded insights into the SAR of HS/FGF-2 interactions. This is the first time that Synphase lanterns and the microwave-assisted peptide synthesizer have been utilized for HS synthesis, presenting an attractive platform for automated glycan synthesis.
Supplementary Material
Acknowledgments
We are grateful for financial supports from the National Institute of General Medical Sciences, NIH (R01GM072667, U01GM116262, and R44GM134738) and Michigan State University for financial support of our work.
Footnotes
Conflicts of Interests
JL is a founder for Glycan Therapeutics. GS is an employee of Glycan Therapeutics. The authors declare no other conflicts of interests.
References:
- 1.Petitou M; van Boeckel CAA A Synthetic Antithrombin III Binding Pentasaccharide is Now a Drug! What Comes Next? Angew. Chem. Int. Ed 2004, 43, 3118–3133 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 2.Liu J; Thorp SC Cell Surface Heparan Sulfate and Its Roles in Assisting Viral Infections. Med. Res. Rev 2002, 22, 1–25. [DOI] [PubMed] [Google Scholar]
- 3.Sasisekharan R; Shriver Z; Venkataraman G; Narayanasami U Roles of Heparan-Sulphate Glycosaminoglycans in Cancer. Nat. Rev. Cancer 2002, 2, 521–528. [DOI] [PubMed] [Google Scholar]
- 4.Gama CI; Hsieh-Wilson LC Chemical Approaches to Deciphering the Glycosaminoglycan Code. Curr. Opin. Chem. Biol 2005, 9, 609–619. [DOI] [PubMed] [Google Scholar]
- 5.Xu D; Esko JD Demystifying Heparan Sulfate-Protein Interactions. Annu. Rev. Biochem 2014, 83, 129–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capila I; Linhardt RJ Heparin-Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 390–412. [DOI] [PubMed] [Google Scholar]
- 7.Shi X; Zaia J Organ-Specific Heparan Sulfate Structural Phenotypes. J. Biol. Chem 2009, 284, 11806–11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feyzi E; Saldeen T; Larsson E; Lindahl U; Salmivirta M Age-Dependent Modulation of Heparan Sulfate Structure and Function. J. Biol. Chem 1998, 273, 13395–13398. [DOI] [PubMed] [Google Scholar]
- 9.Brickman YG; Ford MD; Gallagher JT; Nurcombe V; Bartlett PF; Turnbull JE Structural Modification of Fibroblast Growth Factor-binding Heparan Sulfate at a Determinative Stage of Neural Development. J. Biol. Chem. 1998, 273, 4350–4359. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X; Lin L; Huang H; Linhardt RJ Chemoenzymatic Synthesis of Glycosaminoglycans. Acc. Chem. Res 2020, 53, 335–346. [DOI] [PubMed] [Google Scholar]
- 11.Tsai CT; Zulueta MML; Hung S-C Synthetic Heparin and Heparan Sulfate: Probes in Defining Biological Functions. Curr. Opin. Chem. Biol 2017, 40, 152–159. [DOI] [PubMed] [Google Scholar]
- 12.Mende M; Bednarek C; Wawryszyn M; Sauter P; Biskup MB; Schepers U; Bräse S Chemical Synthesis of Glycosaminoglycans. Chem. Rev 2016, 116, 8193–8255. [DOI] [PubMed] [Google Scholar]
- 13.Dulaney SB; Huang X Strategies in Synthesis of Heparin Heparan Sulfate Oligosaccharides: 2000 - Present. Adv. Carbohydr. Chem. Biochem 2012, 67, 95–136 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karst NA; Linhardt RJ Recent Chemical and Enzymatic Approaches to the Synthesis of Glycosaminoglycan Oligosaccharides. Curr. Med. Chem 2003, 10, 1993–2031. [DOI] [PubMed] [Google Scholar]
- 15.Codée JDC; Overkleeft HS; van der Marel GA; van Boeckel CAA The Synthesis of Well-defined Heparin and Heparan Sulfate Fragments. Drug Discovery Today: Technol. 2004, 1, 317–326. [DOI] [PubMed] [Google Scholar]
- 16.Noti C; Seeberger PH Synthetic Approach to Define Structure-Activity Relationship of Heparin and Heparan Sulfate. In: Garg HG, Linhardt RJ, Hales CA, editors. Chemistry and Biology of Heparin and Heparan Sulfate Oxford: Elsevier; 2005. p. 79–142. [Google Scholar]
- 17.Pawar NJ; Wang L; Higo T; Bhattacharya C; Kancharla PK; Zhang F; Baryal K; Huo C-X; Liu J; Linhardt RJ; Huang X; Hsieh-Wilson LC Expedient Synthesis of Core Disaccharide Building Blocks from Natural Polysaccharides for Heparan Sulfate Oligosaccharide Assembly. Angew. Chem. Int. Ed 2019, 58, 18577–18583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pongener I; O’Shea C; Wootton H; Watkinson M; Miller GJ Developments in the Chemical Synthesis of Heparin and Heparan Sulfate. Chem. Rec 2021, 21, 3238–3255. [DOI] [PubMed] [Google Scholar]
- 19.Hansen SU; Miller GJ; Cliff MJ; Jayson GC; Gardiner JM Making the longest sugars: a chemical synthesis of heparin-related [4]n oligosaccharides from 16-mer to 40-mer. Chem. Sci 2015, 6, 6158–6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baleux F; Loureiro-Morais L; Hersant Y; Clayette P; Arenzana-Seisdedos F; Bonnaffé D; Lortat-Jacob H A Synthetic CD4-Heparan Sulfate Glycoconjugate Inhibits CCR5 and CXCR4 HIV-1 Attachment and Entry. Nat. Chem. Biol 2009, 5, 743–748 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 21.Hansen SU; Miller GJ; Cole C; Rushton G; Avizienyte E; Jayson GC; Gardiner JM Tetrasaccharide Iteration Synthesis of a Heparin-like Dodecasaccharide and Radiolabelling for in vivo Tissue Distribution Studies. Nature Commun 2013, 4, DOI: 10.1038/ncomms3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu Y-P; Zhong Y-Q; Chen Z-G; Chen C-Y; Shi Z; Zulueta MML; Ku C-C; Lee P-Y; Wang C-C; Hung S-C Divergent Synthesis of 48 Heparan Sulfate-Based Disaccharides and Probing the Specific Sugar–Fibroblast Growth Factor-1 Interaction. J. Am. Chem. Soc 2012, 134, 20722–20727. [DOI] [PubMed] [Google Scholar]
- 23.Lu L-D; Shie C-R; Kulkarni SS; Pan G-R; Lu X-A; Hung S-C Synthesis of 48 Disaccharide Building Blocks for the Assembly of a Heparin and Heparan Sulfate Oligosaccharide Library. Org. Lett 2006, 8, 5995–5998. [DOI] [PubMed] [Google Scholar]
- 24.Fan R-H; Achkar J; Hernandez-Torres JM; Wei A Orthogonal Sulfation Strategy for Synthetic Heparan Sulfate Ligands. Org. Lett 2005, 7, 5095–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zong C; Venot A; Li X; Lu W; Xiao W; Wilkes J-SL; Salanga CL; Handel TM; Wang L; Wolfert MA; Boons G-J Heparan Sulfate Microarray Reveals That Heparan Sulfate–Protein Binding Exhibits Different Ligand Requirements. J. Am. Chem. Soc 2017, 139, 9534–9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panza M; Pistorio SG; Stine KJ; Demchenko AV Automated Chemical Oligosaccharide Synthesis: Novel Approach to Traditional Challenges. Chem. Rev 2018, 118, 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panza M; Stine KJ; Demchenko AV HPLC-assisted automated oligosaccharide synthesis: the implementation of the two-way split valve as a mode of complete automation. Chem. Commun 2020, 56, 1333–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kandasamy J; Schuhmacher F; Hahm HS; Kleina JC; Seeberger PH Modular Automated Solid Phase Synthesis of Dermatan Sulfate Oligosaccharides. Chem. Commun 2014, 50, 1875–1877. [DOI] [PubMed] [Google Scholar]
- 29.Eller S; Collot M; Yin J; Hahm HS; Seeberger PH Automated Solid-Phase Synthesis of Chondroitin Sulfate Glycosaminoglycans. Angew. Chem. Int. Ed 2013, 52, 5858–5861. [DOI] [PubMed] [Google Scholar]
- 30.Plante OJ; Palmacci ER; Seeberger PH Automated Solid-phase Synthesis of Oligosaccharides. Science 2001, 291, 1523–1527. [DOI] [PubMed] [Google Scholar]
- 31.Hahm HS; Broecker F; Kawasaki F; Mietzsch M; Heilbronn R; Fukuda M; Seeberger PH Automated Glycan Assembly of Oligo-N-Acetyllactosamine and Keratan Sulfate Probes to Study Virus-Glycan Interactions. Chem 2017, 2, 114–124. [Google Scholar]
- 32.Tyrikos-Ergas T; Sletten ET; Huang J-Y; Seeberger PH; Delbianco M On resin synthesis of sulfated oligosaccharides. Chem. Sci 2022, 13, 2115–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guedes N; Kopitzki S; Echeverria B; Pazos R; Elosegui E; Calvo J; Reichardt N-C Solid-phase assembly of glycosaminoglycan oligosaccharide precursors. RSC Adv 2015, 5, 9325–9327. [Google Scholar]
- 34.Budhadev D; Saxby K; Walton J; Davies G; Tyler PC; Schwörer R; Fascione MA Using automated glycan assembly (AGA) for the practical synthesis of heparan sulfate oligosaccharide precursors. Org. Biomol. Chem 2019, 17, 1817–1821. [DOI] [PubMed] [Google Scholar]
- 35. http://www.mimotopes.com/knowledgeBaseDocument.asp?did=722014.
- 36.Dandapani S; Germain AR; Jewett I; le Quement S; Marie J-C; Muncipinto G; Duvall JR; Carmody LC; Perez JR; Engel JC; Gut J; Kellar D; Siqueira-Neto JL; McKerrow JH; Kaiser M; Rodriguez A; Palmer MA; Foley M; Schreiber S; Munoz B Diversity-Oriented Synthesis Yields a New Drug Lead for Treatment of Chagas Disease. ACS Med. Chem. Lett 2014, 5, 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerard B; Duvall JR; Lowe JT; Murillo T; Wei J; Akella LB; Marcaurelle LA Synthesis of a Stereochemically Diverse Library of Medium-Sized Lactams and Sultams via SNAr Cycloetherification. ACS Comb. Sci 2011, 11, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duvall JR; Vrcic A; Marcaurelle LA Small-Molecule Library Synthesis on SiliconFunctionalized SynPhase Lanterns. Curr. Protoc. Chem. Biol 2010, 2, 135–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seitz O; Kunz H HYCRON, an Allylic Anchor for High-Efficiency Solid Phase Synthesis of Protected Peptides and Glycopeptides. J. Org. Chem 1997, 62, 813–826. [Google Scholar]
- 40.Yang W; Yoshida K; Yang B; Huang X Obstacles and solutions for chemical synthesis of syndecan-3 (53–62) glycopeptides with two heparan sulfate chains. Carbohydr. Res 2016, 435, 180–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramadan S; Yang W; Zhang Z; Huang X Synthesis of Chondroitin Sulfate A Bearing Syndecan-1 Glycopeptide. Org. Lett 2017, 19, 4838–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crich D; Smith M; Yao Q; Picione J 2,4,6-Tri-tert-butylpyrimidine (TTBP): A Cost Effective, Readily Available Alternative to the Hindered Base 2,6-Di-tert-butylpyridine and Its 4-Substituted Derivatives in Glycosylation and Other Reactions. Synthesis 2001, 323–326. [Google Scholar]
- 43.Huang X; Huang L; Wang H; Ye X-S Iterative One-pot Oligosaccharide Synthesis. Angew. Chem. Int. Ed 2004, 43, 5221–5224. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z; Xu Y; Yang B; Tiruchinapally G; Sun B; Liu R; Dulaney S; Liu J; Huang X Preactivation-based One-pot Combinatorial Synthesis of Heparin-like Hexasaccharides for the Analysis of Heparin-protein Interactions. Chem. Eur. J 2010, 16, 8365–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bock K; Pedersen C A Study of 13CH Coupling Constants in Hexopyranoses. J. Chem. Soc., Perkin Trans 2. 1974, 293–297. [Google Scholar]
- 46.van den Bos LJ; Codee JDC; van der Toorn JC; Boltje TJ; van Boom JH; Overkleeft HS; van der Marel GA Thio-glycuronides: synthesis and application in the assembly of acidic oligosaccharides. Org. Lett 2004, 6, 2165–2168. [DOI] [PubMed] [Google Scholar]
- 47.Bal BS; Childers WE; Pinnick HW Oxidation of α,β-un saturated aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar]
- 48.Dalcanale E; Montanari F Selective oxidation of aldehydes to carboxylic acids with sodium chlorite-hydrogen peroxide. J. Org. Chem 1986, 51, 567–569. [Google Scholar]
- 49.Seitz O Solid-Phase Synthesis of Doubly Labeled Peptide Nucleic Acids as Probes for the Real-Time Detection of Hybridization. Angew. Chem. Int. Ed 2006, 39, 3249–3252. [DOI] [PubMed] [Google Scholar]
- 50.Seitz O; Wong C-H Chemoenzymatic Solution- and Solid-Phase Synthesis of O-Glycopeptides of the Mucin Domain of MAdCAM-1. A General Route to O-LacNAc, O-Sialyl-LacNAc, and O-Sialyl-Lewis-X Peptides. J. Am. Chem. Soc 1997, 119, 8766–8776. [Google Scholar]
- 51.Seitz O; Kunz H A Novel Allylic Anchor for Solid-Phase Synthesis—Synthesis of Protected and Unprotected O-Glycosylated Mucin-Type Glycopeptides. Angew. Chem. Int. Ed 1995, 34, 803–805. [Google Scholar]
- 52.Yingkai L; Kristi LK Heparin-functionalized polymeric biomaterials in tissue engineering and drug delivery applications. Acta Biomater 2014, 10, 1588–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hasan J; Shnyder SD; Clamp AR; McGown AT; Bicknell R; Presta M; Bibby M; Double J; Craig S; Leeming D; Stevenson K; Gallagher JT; Jayson GC Heparin Octasaccharides Inhibit Angiogenesis In vivo. Clin. Cancer Res 2005, 11, 8172–8179. [DOI] [PubMed] [Google Scholar]
- 54.Maccarana M; Casufl B; Lindahl U Minimal Sequence in Heparin/Heparan Sulfate Required for Binding of Basic Fibroblast Growth Factor. J. Biol. Chem 1993, 268, 23898–23905. [PubMed] [Google Scholar]
- 55.Noti C; de Paz JL; Polito L; Seeberger PH Preparation and use of microarrays containing synthetic heparin oligosaccharides for the rapid analysis of heparin-protein interactions. Chem. Eur. J 2006, 12, 8664–8686. [DOI] [PubMed] [Google Scholar]
- 56.Ashikari-Hada S; Habuchi H; Kariya Y; Itoh N; Reddi AH; Kimata K Characterization of Growth Factor-binding Structures in Heparin/Heparan Sulfate Using an Octasaccharide Library. J. Biol. Chem 2004, 279, 12346–12354. [DOI] [PubMed] [Google Scholar]
- 57.Li Y-C; Ho IH; Ku C-C; Zhong Y-Q; Hu Y-P; Chen Z-G; Chen C-Y; Lin WC; Zulueta MML; Hung S-C; Lin M-G; Wang C-C; Hsiao C-D Interactions That Influence the Binding of Synthetic Heparan Sulfate Based Disaccharides to Fibroblast Growth Factor-2. ACS Chem. Biol 2014, 9, 1712–1717. [DOI] [PubMed] [Google Scholar]
- 58.Zulueta MML; Chyan CL; Hung SC Structural analysis of synthetic heparan sulfate oligosaccharides with fibroblast growth factors and heparin-binding hemagglutinin. Curr. Opin. Struct. Biol 2018, 50, 126–133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.