

Abstract
Background and Objectives
Familial frontotemporal lobar degeneration (f-FTLD) is a phenotypically heterogeneous spectrum of neurodegenerative disorders most often caused by variants within chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), or granulin (GRN). The phenotypic association with each of these genes is incompletely understood. We hypothesized that the frequency of specific clinical features would correspond with different genes.
Methods
We screened the Advancing Research and Treatment in Frontotemporal Lobar Degeneration (ARTFL)/Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS)/ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration Consortium for symptomatic carriers of pathogenic variants in C9orf72, MAPT, or GRN. We assessed for clinical differences among these 3 groups based on data recorded as part of a detailed neurologic examination, the Progressive Supranuclear Palsy Rating Scale, Progressive Supranuclear Palsy–Quality of Life Rating Scale, Unified Parkinson's Disease Rating Scale Part III (motor items), and the Amyotrophic Lateral Sclerosis Functional Rating Scale, revised version. Data were analyzed using Kruskal-Wallis and Wilcoxon rank-sum tests and Fisher exact test.
Results
We identified 184 symptomatic participants who had a single pathogenic variant in C9orf72 (n = 88), MAPT (n = 53), or GRN (n = 43). Motor symptom age at onset was earliest in the MAPT participants followed by C9orf72, whereas the GRN pathogenic variant carriers developed symptoms later. C9orf72 participants more often had fasciculations, muscle atrophy, and weakness, whereas parkinsonism was less frequent. Vertical oculomotor abnormalities were more common in the MAPT cohort, whereas apraxia and focal limb dystonia occurred more often in participants with GRN variants.
Discussion
We present a large comparative study of motor features in C9orf72, MAPT, and GRN pathogenic variant carriers with symptomatic f-FTLD. Our findings demonstrate characteristic phenotypic differences corresponding with specific gene variants that increase our understanding of the genotype-phenotype relationship in this complex spectrum of neurodegenerative disorders.
Trial Registration Information
NCT02365922, NCT02372773, and NCT04363684.
Frontotemporal lobar degeneration (FTLD) is a group of phenotypically heterogeneous neurodegenerative disorders affecting cognitive, behavioral, and motor systems. Historically, 3 clinical syndromes were defined: behavioral variant frontotemporal dementia (bvFTD), progressive nonfluent aphasia, and semantic dementia.1 The latter syndromes are now classified as 2 of the 3 primary progressive aphasias (PPAs): nonfluent/agrammatic PPA (nfvPPA) and semantic PPA (svPPA).2 Motor involvement is common in FTLD and more often present with bvFTD than PPAs.3 Both typical parkinsonism and atypical parkinsonian syndromes, most commonly corticobasal syndrome (CBS) and progressive supranuclear palsy (PSP), comprise part of the FTLD phenotypic spectrum. Amyotrophic lateral sclerosis (ALS) is diagnosed in 5%–10% of patients with FTLD, and subclinical motor neuron degeneration approaches 50%.4-10
The complexity of FTLD genetics rivals that of the disease's phenotypic spectrum. Approximately 30% of FTLD is genetic, and those with bvFTD are 4 times more likely to have a strong family history compared with those with PPA.10-12 Pathogenic variants (hereafter referred to as variants) in chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and granulin (GRN) account for most familial FTLD.13,14 Variants in these genes have been associated with various motor phenotypes, but correlations between genotype and phenotype are imperfect making patient-level predictions unreliable. For instance, the MAPT N279K variant was described in 2 Japanese brothers with memory impairment, parkinsonism, and corticospinal disturbances with poor levodopa response, whereas others have observed Richardson syndrome (PSP-RS) with this variant.15,16 The unreliability of probabilistic phenotypic-genotype associations is likely due to clinical heterogeneity, small study sample sizes, and the limited use (and precision) of standardized clinical assessments. These findings highlight the need for detailed phenotypic assessments of large samples of genetic variant carriers to understand the frequency of phenotypic elements with respect to genetic alterations. This understanding may assist in the diagnostic pursuit and provide reliable clinical indicators for disease progression or response to therapy. Our study addresses the need for better understanding these phenotype-genotype associations by characterizing the motor phenotype of patients with FTLD and variants within C9orf72, MAPT, or GRN.
Methods
Participants
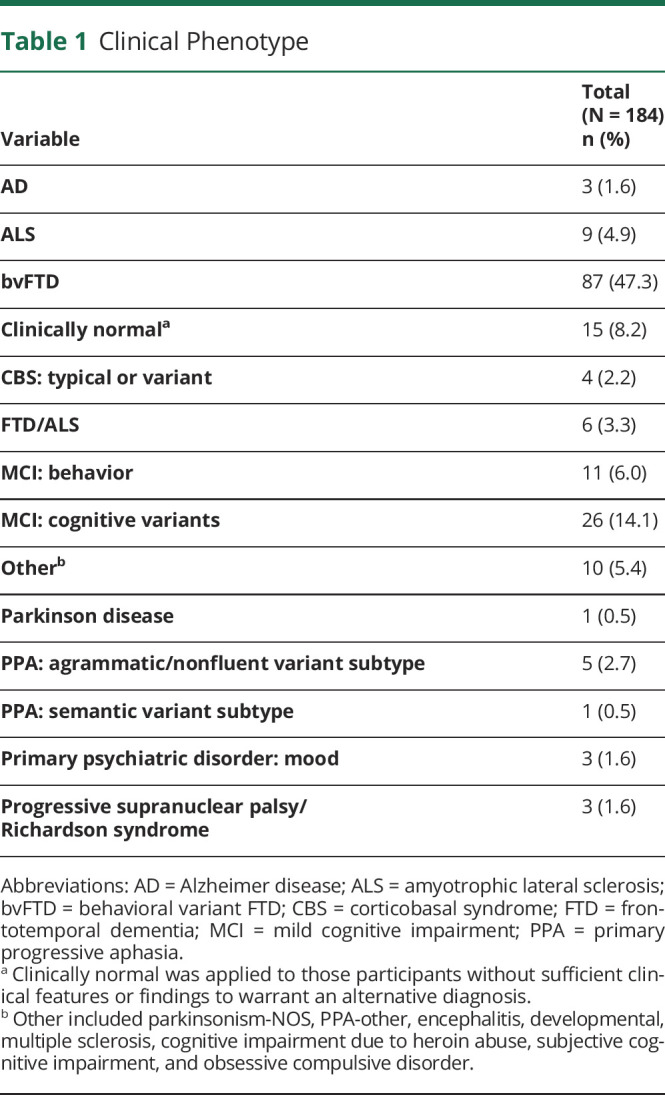
We screened participants in the Advancing Research and Treatment in Frontotemporal Lobar Degeneration (ARTFL)/Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS)/ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration Consortium (ALLFTD), from 14 study centers, for individuals with a single pathogenic variant in the C9orf72, MAPT, or GRN genes (Figure). Study participants ranged between the ages of 22 and 85 years at the time of evaluation and had no structural brain lesion or other known neurologic disorder. Inclusion criteria consisted only of pathogenic variant carriers who were symptomatic defined by CDR Dementia Staging Instrument PLUS National Alzheimer's Coordinating Center (NACC) Behavior and Language Domains (CDR plus NACC FTLD) >0, Amyotrophic Lateral Sclerosis Rating Scale (ALSFRS-R) sum score <48, Unified Parkinson's Disease Rating Scale (UPDRS) Part III >0, or Progressive Supranuclear Palsy Rating Scale (PSPRS) total >0. The ALSFRS-R, UPDRS Part III, and PSPRS allow for symptom quantification of ALS, Parkinson disease (PD), and PSP, which encompass anticipated FTLD motor phenotypes. The CDR plus NACC FTLD scale has 2 additional domains compared with the CDR, language and behavior, comportment and personality, making it more sensitive for detecting FTD.17,18 Participants were defined to have motor features if motor signs were documented on the detailed neurologic examination. Syndromic diagnoses were made using published criteria for bvFTD,19 svPPA, lvPPA, nfvPPA,2 CBS,20 PSP,21 Alzheimer disease,22 PD,23 and ALS/FTD-ALS (Table 1).24 Participants were classified as clinically normal in the absence of sufficient clinical features or findings to warrant an alternative diagnosis.
Figure. Participant Screening Flow Diagram.
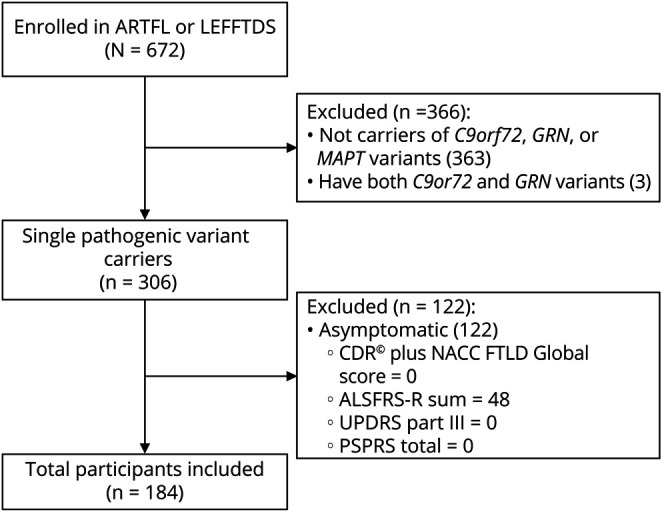
CDR Dementia Staging Instrument PLUS National Alzheimer's Coordinating Center (NACC) Behavior and Language Domains (CDR plus NACC FTLD), Amyotrophic Lateral Sclerosis Rating Scale (ALSFRS-R), Unified Parkinson's Disease Rating Scale (UPDRS), and Progressive Supranuclear Palsy Rating Scale (PSPRS).
Table 1.
Clinical Phenotype
Data Collection
Demographic and detailed clinical information was collected for each individual. A complete detailed semiquantitative neurologic examination was performed. Age at onset (AAO) was estimated by the evaluating clinician. Rating scales were administered including the PSPRS, Progressive Supranuclear Palsy–Quality of Life Rating Scale (PSP-QoL), UPDRS Part III (motor items), and ALSFRS-R. Here, we report data from the most recent study visits for each participant as of the latest data freeze on October 7, 2020 (n = 184) including 62 baseline and 122 follow-up evaluations. Written consent was obtained from all participants or their proxies before study enrollment. All procedures received ethics approval from a central review board at Johns Hopkins University, as well as local review at all sites.
Genetic Analysis
For each family member from MAPT and GRN kindreds, the exon harboring the known variant observed was sequenced as published previously.25,26 For individuals from C9orf72 kindreds, GGGGCC repeat lengths were determined using an established 2-step PCR assay; these participants had repeat lengths >30 repeats.27
Statistical Analyses
Continuous variables were summarized as medians and ranges. Categorical variables were summarized as counts and percentages. Only explicitly scored examination findings and rating scale items were included for analysis, and omitted items were not presumed to be normal. Comparisons of characteristics between the C9orf72, MAPT, and GRN groups were made using Kruskal-Wallis rank-sum tests (continuous and ordinal characteristics) or Fisher exact tests (categorical characteristics) in tests of overall difference between the 3 groups. For characteristics that differed among the 3 groups with a p value ≤0.05, subsequent pairwise comparisons between groups were made using Wilcoxon rank-sum tests (continuous/ordinal characteristics) or Fisher exact tests (categorical characteristics); p values ≤0.0167 were considered statistically significant after applying a Bonferroni correction for multiple testing. All statistical tests were 2 sided. Statistical analyses were performed using SAS (version 9.4; SAS Institute, Inc., Cary, NC).
Results
Demographic Characteristics
A total of 184 participants met the inclusion criteria for this study (Table 2). Patients with MAPT variants had the lowest age at visit compared with other variants (median: C9orf72: 61; GRN: 64; MAPT: 54 years, overall p < 0.001), and there was a statistically significant difference in disease duration (overall p = 0.019); however, pairwise comparison was only significant for C9orf72 vs GRN (p = 0.004). The overall AAO of cognitive and behavioral symptoms was recorded earlier in participants with MAPT variants (all overall p < 0.001). The AAO of motor signs was earliest for participants with MAPT variants and latest for those with C9orf72 repeat expansion (median, MAPT: 49 vs C9orf72: 59 years, overall p = 0.007). There were no significant differences regarding sex, race, years of education, or handedness across groups.
Table 2.
Comparison of Demographics Among C9orf72, GRN, and MAPT Groups

Genetic Data
Participants had pathogenic C9orf72 hexanucleotide repeat expansions (HREs) of GGGGCC (n = 88), GRN variants (n = 43), or MAPT variants (n = 53). Among GRN variants, 19 were unique, and 3 were novel (eTable 1, links.lww.com/WNL/C170). Among MAPT variants, 10 were unique (eTable 1).
Differential Motor Features Among Genetic Groups
All participants underwent a detailed neurologic examination (eTable 2, links.lww.com/WNL/C170), which revealed differences among the 3 groups (Table 3). Fasciculations were only observed in the C9orf72 cohort (overall p = 0.001). Muscle bulk was more often abnormal in C9orf72 patients (overall p < 0.001), and this is reflected by atrophy in cranial nerve distributions (overall p = 0.008), dominant lower extremity (overall p = 0.046), and dominant upper extremity (overall p = 0.001). Muscle strength was more often abnormal in C9orf72 (overall p = 0.001), and this reached statistical significance in the left and right lower extremities (overall p = 0.003 and p = 0.008, respectively). Apraxia was more frequent in GRN participants compared with C9orf72 (p = 0.015) and MAPT (p = 0.012). C9orf72 participants had less rest tremor of the face compared with GRN (p = 0.013). C9orf72 participants also had more rest tremor of the dominant lower extremity of MAPT patients (overall p = 0.030). Dominant-sided grasp reflexes were more frequent in the GRN group compared with C9orf72 (p = 0.012).
Table 3.
Clinical Differences Among C9orf72, GRN, and MAPT Groups Based on Neurologic Examination Informationa

PSPRS scores are summarized in Table 4. Neck rigidity/dystonia was less frequent in C9orf72 patients (overall p = 0.022). Consistent with findings from the neurologic examination, the GRN group had more apraxia of hand movements (overall p = 0.009) and limb dystonia (overall p = 0.013). Vertical oculomotor abnormalities were more common in participants with MAPT variants compared with those with GRN variants (p = 0.009). The PSP-QoL motor scores showed differences in mobility impairments, falling, and difficulties with eyelid opening, communication, and reading (Table 5). Results from analysis of the PSP-QoL, including nonmotor items, are reported in eTable 3 (links.lww.com/WNL/C170). The C9orf72 group had less difficulty moving compared with GRN patients (p = 0.002) and less difficulty communicating and reading than MAPT patients (p = 0.006 and p = 0.003, respectively).
Table 4.
Clinical Differences Among C9orf72, GRN, and MAPT Groups Based on PSPRS Informationa

Table 5.
Motor Differences Among C9orf72, GRN, and MAPT Groups Based on PSP-QoL Informationa

The UPDRS Part III scores are summarized in Table 6. There were no statistically significant differences between GRN and MAPT participants. Compared with C9orf72, the GRN group had more rest tremor of facial musculature (p = 0.013) and the right hand (p = 0.008). Compared with C9orf72, the MAPT group more often had abnormal finger taps (p = 0.011), abnormal posture (p = 0.003), and rigidity of the left lower extremity (p = 0.012) and neck (p = 0.012).
Table 6.
Clinical Differences Among C9orf72, GRN, and MAPT Groups Based on UPDRS Part III (Motor) Informationa

Discussion
Motor phenomena are common in patients with FTLD who have variants in C9orf72, MAPT, and GRN genes, but the relationships between genes and clinical motor manifestations have not been firmly established. We assessed the motor disturbances in this large cohort of familial FTLD and found several differences between carriers of variants in C9orf72, MAPT, and GRN genes.
Hexanucleotide repeat expansions within C9orf72 usually result in TDP-43 type B accumulation and are the most common genetic cause of FTD and ALS.27-29 Although the motor manifestations of C9orf72 variants are typically of motor neuron disease,30 movement disorders may occur.31-33 A recent retrospective study of 40 individuals with C9orf72 variants identified a movement disorder in >40% of patients.34 Among these, parkinsonism and tremor (resembling essential tremor) were the most common features, followed by myoclonus, dystonia, and chorea. An international study observing over 7,000 patients with PD identified C9orf72 variants in 0.06% of study participants using a hexanucleotide repeat cutoff of >60.35 Other studies have identified intermediate repeat expansions (usually defined as 20 to 30 repeats) as a risk factor for clinically diagnosed PD.33 Two studies of pathologically proven PD combined for over 800 cases and identified only a single patient with a C9orf72 HRE.31,36 Our cohort of C9orf72 repeat expansion carriers more often had features of motor neuron disease, for example, fasciculations, muscle atrophy, and weakness, and less often had parkinsonism compared with GRN and MAPT variant carriers.
GRN variants are primarily associated with the TDP-43 type A neuropathologic subtype most commonly leading to a clinical phenotype of bvFTD or nfvPPA.29 Although these phenotypes are most often sporadic, familial GRN variants may present with CBS.37-41 Despite not having pathologic tau deposition, these patients may appear phenotypically indistinguishable from corticobasal degeneration. Our GRN cohort was characterized by features of CBS, for example, parkinsonism and apraxia. Analysis of UPDRS Part III assessments showed that parkinsonian features such as neck rigidity, facial rest tremor, and right-hand rest tremor were more common in these participants than in carriers of C9orf72 variants. The frequency of these features was not significantly different from that observed for the MAPT cohort; however, apraxia was more common than was observed in the MAPT carriers. This is consistent with findings from a study that compared 13 GRN and 17 MAPT variant carriers with FTLD.42 There have been a few reports of GRN variant carriers with CBS and dystonia.37,39,43 Our GRN cohort also reported greater difficulty with movement, on the PSP-QoL, compared with the C9orf72 cohort.
There were clinical features within our MAPT cohort that distinguished it from the other cohorts. MAPT variant carriers had more PSPRS-defined abnormalities with voluntary vertical eye movement abnormalities than carriers of other variant types. This likely contributed to their higher levels of reading difficulties, relative to C9orf72 carriers, identified in the PSP-QoL. These participants also had more parkinsonism than the C9orf72 carriers. These findings suggest a PSP phenotype, more specifically the Richardson syndrome (PSP-RS) given significant oculomotor abnormalities. These findings can also be understood as reflecting the propensity of various MAPT variants to result in an increased 4R/3R tau ratio. A case-control genome wide association study of PSP showed that the MAPT locus has a very strong effect.44,45 Notwithstanding phenotypic heterogeneity within mutation carrier class, considering the MAPT variants in aggregate facilitated differentiation of features from those of the C9orf72 variant carriers. This approach also facilitated distinction of features from those of GRN variant carriers, based on oculomotor abnormalities manifested by participants carrying MAPT variants, and apraxias in those carrying GRN variants. These characteristics suggest that MAPT is more often associated with a PSP-like phenotype, whereas GRN tends toward a CBS. The primary syndromic diagnoses in our sample reflect this as 75% (n = 3) of participants diagnosed with CBS had GRN variants (the other was MAPT) and 67% (n = 2) of participants with PSP-RS had MAPT variants (the other was C9orf72).
Although an FTLD syndrome can suggest mutation type, the syndrome is typically not fully developed at illness onset, and early diagnosis is challenging.46,47 Generally, the syndrome develops over a period of a few years, as abnormalities from various domains (of motor, cognitive, and behavioral function) accumulate. For example, a clinician cannot confidently predict the presence of a GRN variant on the basis of parkinsonism alone, but the clinical suspicion would substantially increase when apraxia develops. The temporal profile of clinical features is a key element for the clinician. We report a temporal relationship of overall AAO between variant groups (MAPT followed by C9orf72 and then GRN) that is in accordance with previous reports.48,49 We found that the mean AAO for cognitive symptoms was significantly different among cohorts beginning with MAPT (49 years) followed by C9orf72 (55 years) and then GRN (60 years). The same relationship was present for behavioral and motor features (Table 2). More importantly, GRN variant carriers most often presented with cognitive impairment, whereas the C9orf72 and MAPT usually presented with behavioral abnormalities. Motor features at onset were also more common in C9orf72 and MAPT compared with GRN patients. These characteristics can serve as valuable patterns when determining the genetic underpinnings of a patient's clinical presentation at the bedside.
Limitations of this study primarily relate to study size. This is a large study comparing motor features of familial FTLD (n = 184), but our subgroup sizes are relatively small precluding comparison of different types of variants within the same gene. Within the GRN cohort, there were 19 different variants (16 exonic) of which 3 are novel (see eTable 1, links.lww.com/WNL/C170). The MAPT cohort included 10 different variants (8 exonic). We previously reported these variants in our assessment of the entire ARTFL/LEFFTDS series, which also included patients without motor features.50 Some of our participants were from the same family, potentially skewing genotype-phenotype correlations. We do not have information on the temporal relationship between neurologic examinations and medication dosing. Finally, some patients may have received dopaminergic medication to address their parkinsonism. This may have diluted or even masked significant differences among the cohorts. It is unlikely that they benefitted from levodopa, although no conclusions on levodopa responsiveness in our subgroups can be made.
We present an analysis comparing the motor phenotypes of a large number of patients with symptomatic familial FTLD carrying a pathogenic variant in C9orf72, GRN, or MAPT. Our findings suggest that there are phenotypic elements that, while not specific, are more common with certain variant types. This study also highlights the importance of large prospective multicenter studies, which enable the collection of cohorts large enough to discern these types of phenotype-genotype relationships in complex neurodegenerative disorders in a standardized manner.
Acknowledgment
The authors extend their appreciation to their program officers from the National Institute on Aging and National Institute of Neurological Disorders and Stroke. The authors acknowledge the invaluable contributions of the study participants and families as well as the assistance of the support staffs at each of the participating sites. Daniel Kaufer is deceased.
Glossary
- AAO
age at onset
- ALS
amyotrophic lateral sclerosis
- ARTFL
Advancing Research and Treatment in Frontotemporal Lobar Degeneration
- ALLFTD
ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration Consortium
- ALSFRS
Amyotrophic Lateral Sclerosis Rating Scale
- bvFTD
behavioral variant FTD
- C9orf72
chromosome 9 open reading frame 72
- CBS
corticobasal syndrome
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- GRN
granulin
- HRE
hexanucleotide repeat expansion
- LEFFTDS
Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects
- MAPT
microtubule-associated protein tau
- NACC
National Alzheimer's Coordinating Center
- nfvPPA
nonfluent/agrammatic variant PPA
- PD
Parkinson disease
- PPA
primary progressive aphasia
- PSP
progressive supranuclear palsy
- PSP-QoL
Progressive Supranuclear Palsy–Quality of Life Rating Scale
- PSPRS
Progressive Supranuclear Palsy Rating Scale
- svPPA
semantic PPA
- UPDRS
Unified Parkinson's Disease Rating Scale
Appendix. Authors
Study Funding
Data collection and dissemination of the data presented in this manuscript was supported by the ALLFTD Consortium (U19: AG063911, funded by the National Institute on Aging and the National Institute of Neurological Diseases and Stroke) and the former ARTFL and LEFFTDS Consortia (ARTFL: U54 NS092089, funded by the National Institute of Neurological Diseases and Stroke and National Center for Advancing Translational Sciences; LEFFTDS: U01 AG045390, funded by the National Institute on Aging and the National Institute of Neurological Diseases and Stroke), and the National Institute on Aging funded ADRCs: P30 AG62677 (PI Ronald Petersen, MD, PhD) and P50 AG023501 (PI Bruce Miller, MD).
Disclosure
A.B. Deutschlaender is supported by Allergan, Inc. (educational grant), by a gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, and by the Sol Goldman Charitable Trust. R. Savica is supported by the National Institute on Aging, the National Institute of Neurological Disorders and Stroke, the Parkinson's Disease Foundation, and Acadia Pharmaceuticals. N. Ghoshal has participated or is currently participating in clinical trials of anti-dementia drugs sponsored by the following companies: Bristol-Myers Squibb, Eli Lilly/Avid Radiopharmaceuticals, Janssen Immunotherapy, Novartis, Pfizer, and Wyeth. N.R. Graff-Radford receives royalties from UpToDate and has participated in multicenter therapy studies sponsored by Biogen, AbbVie, Novartis, and Lilly. M. Grossman is serving as a consultant to the Novartis Alzheimer's Prevention Advisory Board. G.R. Hsiung has served as an investigator for clinical trials sponsored by AstraZeneca, Eli Lilly, and Roche/Genentech. D. Knopman serves on the DSMB of the DIAN-TU study and is a site PI for clinical trials sponsored by Biogen, Lilly, and the University of Southern California. S. McGinnis has served as an investigator for clinical trials sponsored by AbbVie, Allon Therapeutics, Biogen, Bristol-Myers Squibb, C2N Diagnostics, Eisai Inc., Eli Lilly and Co., Genentech, Janssen Pharmaceuticals, Medivation, Merck, Navidea Biopharmaceuticals, Novartis, Pfizer, and TauRx Therapeutics. C. Onyike is a consultant for Alector and Acadia and has received research support from Alector and Biogen. K. Domoto-Reilly receives honoraria from MedBridge. E.D. Roberson has served as a consultant for AGTC, AVROBIO, Biogen, and Vida Ventures and has received research support from Alector. I. Litvan has served as consultant for Roche, AbbVie, Biogen, Centogene, EIP Pharma, Biohaven Pharmaceuticals, Novartis, Brain Neurotherapy Bio, and United Biopharma SRL–UCB, was a member of the Scientific Advisory Board of Lundbeck, is a scientific advisor for Amydis and Rossy Center for Progressive Supranuclear Palsy University of Toronto, and receives funding as Chief Editor of Frontiers in Neurology. B.S. Appleby received research funding from the NIH, CDC, Alector, and Ionis, has served as a consultant for Acadia, Ionis, & Sangamo, and has received royalties from Wolters Kluwer. D. Kaufer is deceased; disclosures are not included for this author. A.L. Boxer has served as a consultant for Aeton, AbbVie, Alector, Amgen, Arkuda, Ionis, Iperian, Janssen, Merck, Novartis, Samumed, Toyama, and UCB, and he received research support from Avid, Biogen, BMS, C2N, Cortice, Eli Lilly, Forum, Genentech, Janssen, Novartis, Pfizer, Roche, and TauRx. H.J. Rosen received research support from Biogen Pharmaceuticals and has consulting agreements with Wave Neuroscience and Ionis Pharmaceuticals. B.F. Boeve has served as an investigator for clinical trials sponsored by Alector, EIP Pharma, and Biogen and serves on the Scientific Advisory Board of the Tau Consortium. Z.K. Wszolek has served as an investigator for clinical trials sponsored by Biohaven Pharmaceuticals, Inc. (BHV4157-206 and BHV3241-301), Neuraly, Inc. (NLY01-PD-1), and Vigil Neuroscience, Inc. (VGL101-01.001) grants and serves as an external advisory board member for Vigil Neuroscience, Inc. The other authors report no relevant disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546-1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 2.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006-1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siuda J, Fujioka S, Wszolek ZK. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat Disord. 2014;20(9):957-964. doi: 10.1016/j.parkreldis.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipton AM, White CL, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108(5):379-385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 5.Seelaar H, Jurgen Schelhaas H, Azmani A, et al. TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain. 2007;130(5):1375-1385. doi: 10.1093/brain/awm024. [DOI] [PubMed] [Google Scholar]
- 6.Johnson JK, Diehl J, Mendez MF, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62(6):925-930. doi: 10.1001/archneur.62.6.925. [DOI] [PubMed] [Google Scholar]
- 7.Rosso SM, Kaat LD, Baks T, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(9):2016-2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie IRA, Feldman HH. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol. 2005;64(8):730-739. doi: 10.1097/01.jnen.0000174335.27708.0a. [DOI] [PubMed] [Google Scholar]
- 9.Ferrari R, Kapogiannis D, Huey ED, Momeni P. FTD and ALS: a tale of two diseases. Crr Alzhemier Res. 2011;8(3):273-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 2005;65(11):1817-1819. doi: 10.1212/01.wnl.0000187068.92184.63. [DOI] [PubMed] [Google Scholar]
- 11.Wood EM, Falcone D, Suh ER, et al. Development and validation of pedigree classification criteria for frontotemporal lobar degeneration. JAMA Neurol. 2013;70(11):1411-1417. doi: 10.1001/jamaneurol.2013.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73(18):1451-1456. doi: 10.1212/WNL.0b013e3181bf997a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075-2086. doi: 10.1007/s00415-019-09363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672-1682. doi: 10.1016/S0140-6736(15)00461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arima K, Kowalska A, Hasegawa M, et al. Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology. 2000;54(9):1787-1795. doi: 10.1212/WNL.54.9.1787. [DOI] [PubMed] [Google Scholar]
- 16.Ogaki K, Motoi Y, Li Y, et al. Visual grasping in frontotemporal dementia and parkinsonism linked to chromosome 17 (microtubule-associated with protein tau): a comparison of N-Isopropyl-p-[123I]-iodoamphetamine brain perfusion single photon emission computed tomography analysis with pr. Mov Disord. 2011;26(3):562-563. doi: 10.1002/mds.23461. [DOI] [PubMed] [Google Scholar]
- 17.Knopman DS, Kramer JH, Boeve BF, et al. Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain. 2008;131(11):2957-2968. doi: 10.1093/brain/awn234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyagawa T, Brushaber D, Syrjanen J, et al. Utility of the global CDR plus NACC FTLD rating and development of scoring rules: data from the ARTFL/LEFFTDS Consortium. Alzheimers Dement. 2020;16(1):106-117. doi: 10.1002/alz.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456-2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54(suppl 5):S15-S19. doi: 10.1002/ana.10570. [DOI] [PubMed] [Google Scholar]
- 21.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP International Workshop. Neurology. 1996;47(1):1-9. doi: 10.1212/WNL.47.1.1. [DOI] [PubMed] [Google Scholar]
- 22.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group⋆ under the auspices of department of health and human services task force on Alzheimer's disease. Neurology. 1984;34(7):939-944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 23.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56(1):33-39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 24.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 25.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916-919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 26.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702-704. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 27.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245-256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257-268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panza F, Lozupone M, Seripa D, et al. Development of disease-modifying drugs for frontotemporal dementia spectrum disorders. Nat Rev Neurol. 2020;16(4):213-228. doi: 10.1038/s41582-020-0330-x. [DOI] [PubMed] [Google Scholar]
- 30.Hsiung GYR, Dejesus-Hernandez M, Feldman HH, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135(3):709-722. doi: 10.1093/brain/awr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooper-Knock J, Frolov A, Highley JR, et al. C9ORF72 expansions, parkinsonism, and Parkinson disease A clinicopathologic study. Neurology. 2013;81(9):808-811. doi: 10.1212/WNL.0b013e3182a2cc38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck J, Poulter M, Hensman D, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet. 2013;92(3):345-353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bourinaris T, Houlden H. C9orf72 and its relevance in parkinsonism and movement disorders: a comprehensive review of the literature. Mov Disord Clin Pract. 2018;5(6):575-585. doi: 10.1002/mdc3.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Estevez-Fraga C, Magrinelli F, Hensman Moss D, et al. Expanding the spectrum of movement disorders associated with C9orf72 hexanucleotide expansions. Neurol Genet. 2021;7(2):e575. doi: 10.1212/nxg.0000000000000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theuns J, Verstraeten A, Sleegers K, et al. Global investigation and meta-analysis of the C9orf72 (G4C2)n repeat in Parkinson disease. Neurology. 2014;83(21):1906-1913. doi: 10.1212/WNL.0000000000001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nuytemans K, Inchausti V, Beecham GW, et al. Absence of C9ORF72 expanded or intermediate repeats in autopsy-confirmed Parkinson's disease. Mov Disord. 2014;29(6):827-830. doi: 10.1002/MDS.25838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masellis M, Momeni P, Meschino W, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129(11):3115-3123. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- 38.Tartaglia MC, Sidhu M, Laluz V, et al. Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol. 2010;119(3):365-374. doi: 10.1007/s00401-009-0605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taghdiri F, Sato C, Ghani M, Moreno D, Rogaeva E, Tartaglia MC. Novel GRN mutations in patients with corticobasal syndrome. Sci Rep. 2016;6(1):22913. doi: 10.1038/srep22913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohrer JD, Beck J, Warren JD, et al. Corticobasal syndrome associated with a novel 1048_1049insG progranulin mutation. J Neurol Neurosurg Psychiatry. 2009;80(11):1297-1298. doi: 10.1136/jnnp.2008.169383. [DOI] [PubMed] [Google Scholar]
- 41.Mackenzie IRA, Baker M, Pickering-Brown S, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129(11):3081-3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 42.Pickering-Brown SM, Rollinson S, du Plessis D, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain. 2008;131(3):721-731. doi: 10.1093/BRAIN/AWM331. [DOI] [PubMed] [Google Scholar]
- 43.Spina S, Murrell JR, Huey ED, et al. Corticobasal syndrome associated with the A9D Progranulin mutation. J Neuropathol Exp Neurol. 2007;66(10):892-900. doi: 10.1097/nen.0b013e3181567873. [DOI] [PubMed] [Google Scholar]
- 44.Höglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43(7):699-705. doi: 10.1038/NG.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen Y, Zhou Y, Jiao B, Shen L. Genetics of progressive supranuclear palsy: a review. J Parkinsons Dis. 2021;11(1):93. doi: 10.3233/JPD-202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barc K, Kuźma-Kozakiewicz M. Positron emission tomography neuroimaging in neurodegenerative diseases: Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. Neurol Neurochir Pol. 2019;53(2):99-112. doi: 10.5603/PJNNS.a2019.0013. [DOI] [PubMed] [Google Scholar]
- 47.Nojszewska M, Potulska-Chromik A, Jamrozik Z, Janik P, Zakrzewska-Pniewska B. Electrophysiological and clinical assessment of dysautonomia in multiple system atrophy (MSA) and progressive supranuclear palsy (PSP): a comparative study. Neurol Neurochir Pol. 2018;53(1):26-33. doi: 10.5603/PJNNS.a2019.0005. [DOI] [PubMed] [Google Scholar]
- 48.Moore KM, Nicholas J, Grossman M, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020;19(2):145-156. doi: 10.1016/S1474-4422(19)30394-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caswell C, McMillan CT, Xie SX, et al. Genetic predictors of survival in behavioral variant frontotemporal degeneration. Neurology. 2019;93(18):e1707. doi: 10.1212/WNL.0000000000008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramos EM, Dokuru DR, van Berlo V, et al. Genetic screening of a large series of North American sporadic and familial frontotemporal dementia cases. Alzheimer Dement. 2020;16(1):118-130. doi: 10.1002/ALZ.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]