

Abstract
Predicted highly expressed (PHX) genes are characterized for the completely sequenced genomes of the four fast-growing bacteria Escherichia coli, Haemophilus influenzae, Vibrio cholerae, and Bacillus subtilis. Our approach to ascertaining gene expression levels relates to codon usage differences among certain gene classes: the collection of all genes (average gene), the ensemble of ribosomal protein genes, major translation/transcription processing factors, and genes for polypeptides of chaperone/degradation complexes. A gene is predicted highly expressed (PHX) if its codon frequencies are close to those of the ribosomal proteins, major translation/transcription processing factor, and chaperone/degradation standards but strongly deviant from the average gene codon frequencies. PHX genes identified by their codon usage frequencies among prokaryotic genomes commonly include those for ribosomal proteins, major transcription/translation processing factors (several occurring in multiple copies), and major chaperone/degradation proteins. Also PHX genes generally include those encoding enzymes of essential energy metabolism pathways of glycolysis, pyruvate oxidation, and respiration (aerobic and anaerobic), genes of fatty acid biosynthesis, and the principal genes of amino acid and nucleotide biosyntheses. Gene classes generally not PHX include most repair protein genes, virtually all vitamin biosynthesis genes, genes of two-component sensor systems, most regulatory genes, and most genes expressed in stationary phase or during starvation. Members of the set of PHX aminoacyl-tRNA synthetase genes contrast sharply between genomes. There are also subtle differences among the PHX energy metabolism genes between E. coli and B. subtilis, particularly with respect to genes of the tricarboxylic acid cycle. The good agreement of PHX genes of E. coli and B. subtilis with high protein abundances, as assessed by two-dimensional gel determination, is verified. Relationships of PHX genes with stoichiometry, multifunctionality, and operon structures are also examined. The spatial distribution of PHX genes within each genome reveals clusters and significantly long regions without PHX genes.
Escherichia coli, Vibrio cholerae, and Haemophilus influenzae are gram-negative γ-proteobacteria that can grow in human tissue and produce or contribute to disease. The principal habitat of E. coli is the human gut, V. cholerae is mainly a freshwater microbe, and H. influenzae is found in the human lung. On the other hand, Bacillus subtilis is a gram-positive, nonpathogenic soil bacterium. The minimal doubling time for these four bacteria in cultures is significantly less than 1 h. Fast growth implies many ribosomes, and these four bacteria have large numbers of rRNA operons per genome.
Predicted highly expressed (PHX) genes are characterized for the rapidly dividing bacteria E. coli, H. influenzae, V. cholerae, and B. subtilis using a method based on codon usage differences among gene classes (21). For complete lists of PHX genes, consult the website ftp://gnomic.stanford.edu/pub (see also Table 2).
TABLE 2.
Top 20 PHX genes of the four fast-growing bacteria and their predicted E(g) valuea
| Proteins | Geneb |
E(g)
|
|||
|---|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | ||
| Ribosomal | rpsA | 2.12 | 2.07 | 1.66 | 1.20 |
| rpsB | 2.37 | 2.03 | 1.66 | 1.84 | |
| rpsC | 2.14 | 1.95 | 1.51 | 1.87 | |
| rpsD | 1.55 | 1.68 | 1.35 | 1.94 | |
| rpsE | 1.40 | 1.62 | 1.37 | 1.89 | |
| rpsI | 1.90 | 1.93 | 1.56 | 1.51 | |
| rpsM | 1.13 | 1.28 | 1.22 | 2.02 | |
| rplA | 2.08 | 2.07 | 1.63 | 1.91 | |
| rplC | 1.90 | 1.91 | 1.85 | 1.78 | |
| rplB | 2.44 | 2.12 | 1.71 | 2.02 | |
| rplD | 2.21 | 1.83 | 1.43 | 1.98 | |
| rplE | 1.81 | 1.46 | 1.41 | 1.92 | |
| rplK | 1.82 | 1.87 | 1.41 | 1.58 | |
| rplM | 1.79 | 1.68 | 1.75 | 1.91 | |
| rplN | 1.32 | 1.68 | 1.26 | 1.98 | |
| rplP | 1.41 | 1.66 | 1.56 | 1.90 | |
| rplQ | 1.43 | 1.66 | 1.33 | 1.87 | |
| rplT | 1.84 | 1.92 | 1.56 | 1.57 | |
| Chaperone-degradation | dnaK | 2.58 | 1.80 | 1.29 | 1.83 |
| groEL | 2.09 | 1.30, (0.45) | 1.47 | 1.87 | |
| pnp | 2.66 | 1.72 | 1.72 | (0.79) | |
| Translation-transcription | fusA | 2.24 | 2.02, (0.96) | 2.01 | 2.34 |
| tuf | 2.20, 2.14 | 1.72, 1.62 | 1.51, 1.51 | 1.97 | |
| tsf | 2.00 | 1.76 | 1.76 | 1.75 | |
| rpoB | 2.65 | 1.88 | 1.58 | 1.49 | |
| rpoC | 2.49 | 1.63 | 1.45 | 1.76 | |
| deaDc | 2.14 | 1.96 | (0.80) | 1.23, (0.45) | |
| Energy metabolism | fba | 2.38 | 1.88 | 1.46 | 1.99, (0.59) |
| pgk | 2.38 | 1.78 | 1.04 | 1.34 | |
| pykA/pykF | 2.58 (pykF), (0.95) (pykA) | 1.75, (0.53), (0.48) | 1.58 | 1.18 | |
| eno | 2.11 | 1.93 | 1.59 | 1.92 | |
| gapA | 2.07 | 1.82 | 1.72 | 1.80 | |
| pckA | 1.45 | (0.91) | 1.77 | (0.61) | |
| aceE | 2.43 | 1.81 | 1.51 | — | |
| pdhB | — | — | — | 1.91 | |
| aceF/pdhC | 1.92 | 1.34 | 1.23 | 2.05 | |
| lpdA/pdhD | 2.10 | 1.49 | 1.39 | 2.14 | |
| pfl | 2.41 (pflB), (0.67) (yhaS) | 2.25 | 1.94 | — | |
| adhE | 2.45 | 2.04 | — | — | |
| atpA | 2.37 | 1.78 | 1.20 | 1.30 | |
| atpD | 1.79 | 1.72 | 1.62 | 1.32 | |
| aspA | 2.32 | 1.23 | 1.29 | (0.66) | |
| Fatty acid and phospholipid metabolism | pta | 2.49 | 1.60 | 1.19 | 1.24 |
| Nucleotide or amino acid biosynthesis | purA | 2.20 | 2.02 | 1.08 | (0.78) |
| ilvC | 1.28 | (0.67) | 1.63 | 1.03 | |
| Outer membrane | ompA | 2.37 | 1.36 | 1.79 | — |
| ompC | 2.27 | — | — | — | |
| ompF | 2.23 | — | — | — | |
| ompU | — | 2.07 | — | — | |
| Other | pal | 1.84 | 1.80 | 1.66 | — |
| typA/bipA | 2.52 | 1.76 | 1.22 | 1.11 | |
| mglC | (0.59) | (0.59) | 1.67 | — | |
| ptsG | 1.74 | 1.91 | — | (0.80) | |
| yeiM/yeiJ | (0.43) (yeiM), (0.40) (yeiJ) | 2.00, (0.54), (0.37) | 1.07 | (0.38) | |
The genes listed include those that are found among the top 20 PHX genes in any of the four genomes and their homologs in the other genomes. Values in parentheses indicate that the gene is not PHX. For duplicated genes, all E(g) values are shown. —, gene does not exist in that genome.
Ribosomal protein genes: ribosomal protein S1 (rpsA), ribosomal protein S2 (rpsB), ribosomal protein S3 (rpsC), ribosomal protein S4 (rpsD), ribosomal protein S5 (rpsE), ribosomal protein S9 (rpsI), ribosomal protein S13 (rpsM), ribosomal protein L1 (rplA), ribosomal protein L3 (rplC), ribosomal protein L2 (rplB), ribosomal protein L4 (rplD), ribosomal protein L5 (rplE), ribosomal protein L11 (rplK), ribosomal protein L13 (rplM), ribosomal protein L14 (rplN), ribosomal protein L16 (rplP), ribosomal protein L16 (rplQ), and ribosomal protein L20 (rplT). Chaperone/degradation protein genes: heat shock protein 70 (dnaK), heat shock protein 60 (groEL), and polynucleotide phosphorylase (pnp) (mRNA degradation). Translation/transcription protein genes: elongation factor G (fusA), elongation factor Tu (tuf/tufA/tufB), elongation factor Ts (tsf), DNA-directed RNA polymerase β (rpoB), DNA-directed RNA polymerase β′ (rpoC), and ATP-dependent RNA helicase (deaD). Energy metabolism genes: fructose-1,6-bisphosphate aldolase (fba; glycolysis), phosphoglycerate kinase (pgk; glycolysis), pyruvate kinase (pykA/pykF; glycolysis), enolase (eno; multifunctional—glycolysis, mRNA degradation), glyceraldehyde-3-phosphate dehydrogenase (gapA; multifunctional), phosphoenolpyruvate carboxykinase (pckA), pyruvate dehydrogenase E1 (aceE), pyruvate dehydrogenase E1 β (pdhB), pyruvate dehydrogenase E2 (aceF/pdhC), dihydrolipoamide dehydrogenase (lpdA/pdhD), formate acetyltransferase (pfl; glucose metabolism), alcohol/acetaldehyde dehydrogenase (adhE), ATP synthase F1 α (atpA), ATP synthase F1 β (atpD), and aspartate-ammonia lyase (aspA). Fatty acid and phospholipid metabolism gene: phosphate acetyltransferase (pta). Nucleotide or amino acid biosynthesis genes: adenylosuccinate synthetase (purA; purine nucleotide biosynthesis) and ketol-acid reductoisomerase (ilvC). Outer membrane protein genes: outer membrane protein A (ompA), outer membrane protein C (ompC), outer membrane protein F (ompF), and outer membrane protein U (ompU). Other genes: peptidoglycan-associated lipoprotein (pal), GTP-binding protein (typA/bipA; similar to elongation factor), galactoside transport system permease (mglC), phosphotransferase system, glucose-specific IIBC component (ptsG; major carbohydrate transport system; also a chemoreceptor), and orf (E. coli yeiM and yeiJ, H. influenzae HI0519, and B. subtilis yutK)
The RNA helicase DeaD gene has multiple homologs in each genome, but only one is PHX. In B. subtilis, the gene annotated as deaD is not PHX, whereas deaD homolog ydbR is PHX.
MATERIALS AND METHODS
Assessments of gene or protein expression levels from codon usage were carried out as follows. High expression is predicted from codon usage as follows. Let G be a family of genes with average codon frequencies g(x, y, z) for the codon nucleotide triplet (x, y, z), normalized so that
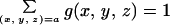 |
where the sum extends over all codons (x, y, z) translated to amino acid a. Let f(x, y, z) indicate the average codon frequencies for the gene family F, normalized to 1 in each amino acid codon family. The codon usage difference of the gene family F (or a single gene) relative to the gene family G, termed the codon bias of F with respect to G, is calculated with the following formula:
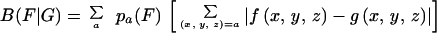 |
where pa(F) are the average amino acid frequencies of the genes of F (cf. references 19 and 20). Denoted by C is the collection of all genes, by RP the ribosomal protein genes, by CH chaperone/degradation protein genes, and by TF translation/transcription processing genes. Qualitatively, a gene g is deemed PHX if B(g|C) is appropriately high, whereas B(g|RP), B(g|CH), and B(g|TF) are suitably low. Predicted expression levels with respect to individual standards are based on the ratios
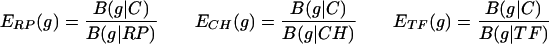 |
and the combined expression measure is
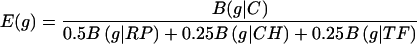 |
Other weighted combinations are also possible, but the results do not qualitatively change when different weights are used. We impose higher weight on the RP standard because the RP genes are generally the most PHX in all current completely sequenced genomes (21). The specification of the RP, CH, and TF gene classes as standards derives from the observation that these gene classes are consistently highly expressed in most genomes (21). Thus, these three gene classes (RP, CH, and TF) serve as representatives of highly expressed genes, and our method specifies genes with similar codon usages as PHX genes. These assignments are reasonable under fast growth conditions, where there is a need for many ribosomes, for proficient transcription and translation, and for many chaperone/degradation proteins needed to ensure correctly folded, modified, and translocated protein products.
A gene is predicted highly expressed (PHX) if the following two conditions are satisfied: at least two of the three expression values ERP(g), ECH(g), and ETF(g) exceed 1.05, and the general expression level E(g) is ≥1.00. We sometimes refer to genes that do not unequivocally satisfy this definition but that have an E(g) of approximately 1.00 as marginally PHX.
RESULTS
To expose the significance of the PHX gene classes, we plotted B(g|C) versus B(g|RP) traversing all individual genes g (≥100 codons in length). The plots are given in Fig. 1 for each of the four rapidly growing bacteria. The distribution of points reveals two horns. The left horn effectively corresponds to the PHX genes. The right horn we refer to as putative alien genes. It consists of genes that significantly differ in their codon usages from the four classes C, RP, CH, and TF and will be discussed in a separate publication. If we replace the horizontal axis B(g|RP) with the coordinates of B(g|TF) or B(g|CH), the plots in Fig. 1 remain largely unchanged (data not shown).
FIG. 1.

Genes of ≥100 codons in the four fast-growing bacteria. Each gene is represented by a single point. Its position is determined by its bias relative to all genes B(g|C) and by its bias relative to the RP genes B(g|RP). PHX genes are indicated by red circles. Partial overlaps among the PHX and normal gene clusters are due to minor differences among B(g|RP), B(g|CH), and B(g|TF), which all contribute to the PHX predictions (see Materials and Methods). The upper right horn corresponds to putative alien genes (22, 28).
Top 20 PHX genes.
The distribution of PHX genes among the four fast-growing bacteria is displayed in Table 1. The highest E(g) value exceeds 2 in all four genomes. Such high values are rare among the completely sequenced genomes (cf. reference 21). These four bacteria have a substantial number of PHX genes, ranging from 142 to 306.
TABLE 1.
Distribution of PHX genes in four fast-growing bacteria
| Genome | Length (kb) | No. of genes
|
Highest E(g) | |
|---|---|---|---|---|
| ≥100 codons | PHX (%) | |||
| E. coli | 4,639 | 3,898 | 306 (8) | 2.66 |
| H. influenzae | 1,830 | 1,529 | 142 (9) | 2.01 |
| B. subtilis | 4,215 | 3,612 | 148 (4) | 2.34 |
| V. choleraea | 4,036 | 3,253 | 172 (5) | 2.25 |
| Long chromosomeb | 2,963 | 2,393 | 158 (7) | 2.25 |
Both chromosome I (2.96 Mb) and chromosome II (1.07 Mb).
Chromosome I of V. cholerae.
Table 2 presents the 20 genes with the highest predicted expression levels in the genomes of E. coli, V. cholerae, H. influenzae, and B. subtilis. In those few instances when the homologous genes in the other genomes are not PHX, their E(g) values are shown in parentheses. The genes are segregated into functional categories. Almost all ribosomal proteins attain high expression levels in all rapidly growing bacteria (Tables 2 and 3). The S1 ribosomal protein gene (exceeding 500 codons in length in most bacteria) in B. subtilis is found at the diminished length of 327 codons but is still PHX, with an E(g) value of 1.20. Ribosomal protein genes are present in single copies, in contrast to rRNA genes, and are predominantly of a high expression level, presumably conforming with stoichiometric requirements for ribosome formation between proteins and RNA and among the proteins themselves.
TABLE 3.
Predicted expression levels for ribosomal protein genes among four fast-growing bacteriaa
| Ribosomal protein gene |
E(g)
|
|||
|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | |
| S1 | 2.12 | 2.07 | 1.66 | 1.20b |
| S2 | 2.37 | 2.03 | 1.66 | 1.84 |
| S3 | 2.14 | 1.95 | 1.51 | 1.87 |
| S4 | 1.55 | 1.68 | 1.35 | 1.94 |
| S5 | 1.40 | 1.62 | 1.37 | 1.89 |
| S6 | 1.52 | 1.71 | 1.15 | 1.67 |
| S7 | 1.45 | 1.80 | 1.56 | 1.73 |
| S8 | 1.40 | 1.70 | 1.54 | 1.51 |
| S9 | 1.90 | 1.93 | 1.56 | 1.51 |
| S10 | 1.22 | 1.65 | 1.41 | 1.25 |
| S11 | 1.57 | —c | 1.44 | 1.58 |
| S12 | 1.62 | 1.73 | 1.47 | 1.84 |
| S13 | 1.13 | 1.28 | 1.22 | 2.02 |
| S14 | 1.30 | 1.54 | 1.14 | 1.10 |
| L1 | 2.08 | 2.07 | 1.63 | 1.91 |
| L2 | 2.44 | 2.12 | 1.71 | 2.02 |
| L3 | 1.90 | 1.91 | 1.85 | 1.78 |
| L4 | 2.21 | 1.83 | 1.43 | 1.98 |
| L5 | 1.81 | 1.46 | 1.41 | 1.92 |
| L6 | 1.63 | 1.60 | 1.42 | 1.68 |
| L7/L12 | 1.61 | 1.69 | 1.41 | 1.62 |
| L9 | 1.88 | 1.76 | 1.56 | 0.78 |
| L10 | 1.63 | 1.71 | 1.46 | 1.46 |
| L11 | 1.82 | 1.87 | 1.41 | 1.58 |
| L13 | 1.79 | 1.68 | 1.75 | 1.91 |
| L14 | 1.32 | 1.68 | 1.26 | 1.98 |
| L15 | 1.73 | 1.66 | 1.43 | 1.83 |
| L16 | 1.41 | 1.66 | 1.56 | 1.90 |
| L17 | 1.43 | 1.66 | 1.33 | 1.87 |
| L18 | 1.59 | 1.36 | 1.27 | 1.47 |
| L19 | 1.59 | 1.85 | 1.46 | 1.84 |
| L20 | 1.84 | 1.92 | 1.56 | 1.57 |
| L21 | 1.71 | 1.75 | 1.34 | 1.44 |
| L22 | 1.23 | 1.54 | 1.25 | 1.23 |
| L24 | 1.69 | 1.57 | 1.22 | 1.69 |
Only genes of ≥100 codons are included. E. coli pairwise correlation coefficients were 0.75, 0.61, and 0.24 with V. cholerae, H. influenzae, and B. subtilis, respectively. V. cholerae pairwise correlation coefficients were 0.75, 0.68, and 0.15 with E. coli, H. influenzae, and B. subtilis, respectively. H. influenzae pairwise correlation coefficients were 0.61, 0.68, and 0.20 with E. coli, V. cholerae, and B. subtilis, respectively. B. subtilis pairwise correlation coefficients were 0.24, 0.15, and 0.20 with E. coli, V. cholerae, and H. influenzae, respectively.
B. subtilis has a weak homolog of S1 that is smaller than S1 in the other three genomes. Not included in the calculations of the correlation coefficients.
—, S11 is missing in V. cholerae. Not included in the calculation of the correlation coefficients.
The major (eubacterial) chaperone/degradation proteins HSP70 (DnaK) and HSP60 (GroEL) and the mRNA degradation protein polynucleotide phosphorylase (Pnp) are prominently PHX. Pnp in B. subtilis, however, is not PHX [E(g) = 0.79]. The corresponding genes in H. influenzae achieve E(g) values of 1.29, 1.47, and 1.72, respectively. The gene enolase (eno), listed under energy metabolism as part of the glycolysis pathway, is potently PHX. It is also a component of the mRNA degradosome in a multifunctional capacity (27) and so has reasons for being potently PHX.
Processing factors for protein synthesis are outstandingly PHX, especially the ATP-dependent DNA-directed RNA polymerase units RpoB and RpoC and the elongation factors EF-G (fus), EF-Tu (tuf), EF-Ts (tsf). The elongation factor EF-Tu often is present in two copies, both dramatically PHX. B. subtilis has but one copy, and it is PHX. EF-G (fusA) is present in two copies in V. cholerae, with E(g) values of 2.02 and 0.96. The DNA helicase DeaD is PHX in E. coli, V. cholerae, and B. subtilis but not in H. influenzae, nor is the second copy in B. subtilis PHX. It will be interesting to see if there are functional differences between the two copies of DeaD and the EF-G proteins. DeaD box proteins protect mRNA from endonucleases (27).
Many glycolysis genes are among the top PHX genes; they include genes for pyruvate kinase (pykA and pykF), fructose-1,6-bisphosphate aldolase (fba), phosphoglycerate kinase (pgk), enolase (eno), and glyceraldehyde-3-phosphate dehydrogenase (gap).
PHX genes contributing to anaerobic fermentation include the alcohol/acetaldehyde dehydrogenase gene (adhE). Other PHX genes of energy metabolism include several, but significantly not all, genes of the tricarboxylic acid (TCA) cycle. Several subunits of the pyruvate dehydrogenase complex genes are among the top PHX genes. These include genes for multiple copies of the three enzymatic components, pyruvate dehydrogenase E1 (aceE), except in B. subtilis, dihydrolipoamide acetyltransferase E2 (aceF), and lipoamide dehydrogenase E3 (lpdA), all part of the pyruvate oxidation pathway. Genes contributing to proton gradient-driven ATP synthesis (namely, the genes for the two major subunits of the ATP synthase catalytic domain, atpA and atpD) are potently PHX. The PHX gene for adenylosuccinate synthetase (purA) stands out, except in B. subtilis. It participates in the de novo biosynthesis pathway of purine nucleotides and in the first step of AMP biosynthesis. However, the genes for the other enzymes of that pathway are not PHX.
Several porin genes of E. coli, H. influenzae, and V. cholerae are PHX. These are absent from B. subtilis, which is a gram-positive bacterium, lacking the distinctive gram-negative outer membrane. The peptidoglycan-associated lipoprotein (Pal) attached to the outer membrane by a lipid anchor is PHX in gram-negative bacteria. Several lipid biosynthesis PHX genes are among the top 20. The first enzyme of the glyoxylate shunt pathway, isocitrate lyase (AceA), is PHX in the moderately fast-growing Deinococcus radiodurans (90-min average doubling time) and the slow-growing Mycobacterium tuberculosis (24 to 36 h). It exists in E. coli and V. cholerae but is not PHX and has not been detected for most prokaryotic genomes. Isocitrate lyase is widespread in plant and fungal organisms. There is an open reading frame (ORF) (yeiM) of unknown function but possibly encoding a nucleoside transporter, with an E(g) value of 2.00, in V. cholerae, and there is a homolog with an E(g) value of 1.07 in H. influenzae but not PHX in E. coli and B. subtilis. Differences among genes in predicted expression levels present challenging questions for experimentation.
PHX genes in H. influenzae parallel PHX genes in E. coli. These include genes for mainstream glycolysis and TCA enzymes and genes for detoxification and DNA damage control, such as the sodA and catalase genes. The highest E(g) value is 2.01, attained by the elongation factor EF-G (fusA). The heat shock proteins GroEL and DnaK are among the most highly expressed. The ribosome release factor (Rrf) is the top PHX protein in H. influenzae. Rrf is responsible for the release of ribosomes from mRNA at the termination of protein synthesis (37). Rrf is present and generally highly expressed in all eubacterial organisms with completely sequenced genomes but is absent from archaea (35).
Comparison of predicted levels of expression in E. coli with 2D gel patterns.
For many E. coli proteins, two-dimensional (2D) gel electrophoresis data for their abundances during growth in minimal medium are available. We compared the molar abundances of 96 proteins (with lengths of ≥100 amino acids [aa] [45, 46]) with the set of PHX genes (Table 4). Among the 20 most abundant of the 96 proteins, 17 were identified as PHX by our method. Among the 20 least abundant proteins of the 96, only 7 qualified as PHX. Of the remaining 56 proteins, which have intermediate molar abundances on 2D gels, 28 were identified as PHX. This agreement between high 2D gel abundances and high E(g) values supports naming the genes “highly expressed.”
TABLE 4.
Comparison of 2D gel expression measurements (45) and predicted E(g) values
| Gene | RMBa | E(g) |
|---|---|---|
| tufA | 41.772 | 2.14 |
| metE | 22.281 | 0.69 |
| rplL | 18.270 | 1.61 |
| ompA | 17.291 | 2.37 |
| fabB | 14.005 | 1.86 |
| rpsA | 8.118 | 2.12 |
| rpsF | 7.727 | 1.52 |
| groEL | 7.377 | 2.09 |
| eno | 6.928 | 2.11 |
| fusA | 6.809 | 2.24 |
| hns | 6.739 | 1.45 |
| purC | 6.088 | 1.18 |
| glyA | 5.670 | 2.01 |
| ilvE | 5.323 | 0.88 |
| tsf | 4.965 | 2.00 |
| folA | 4.894 | 0.60 |
| dnaK | 4.380 | 2.58 |
| tig | 4.226 | 2.05 |
| atpA | 4.027 | 2.37 |
| glnA | 3.702 | 2.07 |
| atpD | 3.644 | 1.79 |
| livJ | 3.488 | 1.01 |
| metK | 3.429 | 2.21 |
| rpoA | 3.327 | 0.99 |
| lpdA | 2.905 | 2.10 |
| hisJ | 2.874 | 0.52 |
| aceE | 2.872 | 2.43 |
| pyrF | 2.683 | 0.62 |
| aspC | 2.645 | 0.86 |
| mdh | 2.492 | 1.42 |
| trpB | 2.111 | 0.66 |
| recA | 1.780 | 1.48 |
| grpE | 1.773 | 0.93 |
| ibpB | 1.676 | 0.77 |
| ptsI | 1.620 | 1.41 |
| aceF | 1.616 | 1.92 |
| carA | 1.567 | 0.51 |
| trpA | 1.561 | 0.53 |
| pflB | 1.537 | 2.41 |
| ppc | 1.459 | 0.73 |
| trxA | 1.406 | 1.06 |
| tyrS | 1.271 | 1.40 |
| carB | 1.257 | 1.24 |
| sucB | 1.256 | 1.41 |
| map | 1.245 | 0.91 |
| sucC | 1.234 | 1.74 |
| pnp | 1.224 | 2.66 |
| rpoB | 1.117 | 2.65 |
| argA | 1.081 | 0.38 |
| glpK | 1.074 | 0.79 |
| rimL | 1.000 | 0.65 |
| aroF | 0.924 | 0.59 |
| pheS | 0.915 | 1.30 |
| htpG | 0.864 | 1.85 |
| eda | 0.841 | 1.57 |
| trxB | 0.839 | 0.71 |
| ssb | 0.838 | 1.48 |
| lon | 0.803 | 0.91 |
| gltA | 0.771 | 1.29 |
| ileS | 0.767 | 1.17 |
| nusA | 0.756 | 1.15 |
| pheT | 0.754 | 1.16 |
| ackA | 0.748 | 2.11 |
| prfA | 0.748 | 0.77 |
| gltX | 0.699 | 1.35 |
| glyS | 0.696 | 1.32 |
| aspS | 0.660 | 1.95 |
| metH | 0.660 | 0.67 |
| hisS | 0.635 | 0.74 |
| pta | 0.629 | 2.49 |
| sucA | 0.610 | 1.30 |
| glnS | 0.595 | 1.26 |
| lysS | 0.593 | 1.84 |
| infB | 0.572 | 2.18 |
| sdhA | 0.560 | 1.08 |
| pfkA | 0.467 | 1.86 |
| trpD | 0.451 | 0.47 |
| xthA | 0.446 | 0.64 |
| pckA | 0.444 | 1.45 |
| nusB | 0.429 | 1.21 |
| argS | 0.415 | 1.04 |
| valS | 0.410 | 1.81 |
| ilvA | 0.408 | 0.76 |
| rplI | 0.400 | 1.88 |
| leuS | 0.383 | 1.57 |
| thrS | 0.373 | 1.04 |
| ppsA | 0.359 | 0.87 |
| trpE | 0.345 | 0.47 |
| gor | 0.333 | 0.63 |
| clpB | 0.280 | 0.62 |
| trmA | 0.245 | 0.53 |
| tyrA | 0.241 | 0.57 |
| polA | 0.194 | 0.48 |
| fmt | 0.190 | 0.76 |
| lysU | 0.119 | 0.68 |
| hisP | 0.116 | 0.54 |
RMB, Relative molecular abundance during growth on glucose minimal medium.
Three exceptions to the good agreement between high protein molar abundances and PHX status are MetE, FolA, and IlvE, which are involved in amino acid biosynthesis and methylation. These proteins are among the most abundant in 2D gel determinations but do not qualify as PHX. The enzymatic turnover rate for MetE, determined by kinetic studies, is low but is compensated for with a high molar abundance (12). In E. coli, the methionine biosynthesis pathway includes MetK, with a very high E(g) value, 2.21, whereas MetE has an E(g) value of 0.69 and MetH has an E(g) value of 0.60. MetE and MetH offer strict alternative pathways for l-methionine synthesis. MetK acts on homocysteine to produce S-adenosylmethionine, which serves as a methyl donor for a broad range of metabolites, lipids, and vitamins (41). It has been conjectured that the metE gene or the entire Met operon in E. coli, because of its codon usage, may be a newly laterally transferred gene analogous to the Cob operon of Salmonella enterica serovar Typhimurium (24). FolA (dihydrofolate reductase) registers high 2D gel assessments but has a low E(g) value, 0.60.
Hecker and colleagues (e.g., reference 3) have conducted extensive 2D gel assessments of B. subtilis proteins. Consulting their 2D database (http://microbio2.biologie.uni-greiswald.de:8880), we compared the brightest spots on their gels with the E(g) values for the corresponding proteins: RpS2, 1.84; SerA, 0.62; IlvC, 1.03; AroA, 0.93; Gap, 1.80; PdhC, 2.05; CitC, 1.33; TufA, 1.97; Fus, 2.34; YwjH, 1.01; RpL10, 1.46; ClpP, 1.05; SodA, 1.64; and CitH, 0.81. Most of these proteins are PHX, and several achieve an E(g) value of >1.8. Thus, there is a good correlation of PHX proteins with high 2D gel abundances in B. subtilis, as in E. coli.
Classes of PHX genes.
Tables 3 and 5 through 9 compare for the four fast-growing bacteria predicted levels of expression of all ribosomal protein genes, of the genes for the major transcription/translation processing factors, of the chaperone/degradation protein genes, and of the major energy metabolism genes. The extended repair gene repertoire of the four genomes and the vitamin biosynthesis genes of E. coli are evaluated in terms of E(g) levels (Tables 10 and 11). Each class is discussed in turn.
TABLE 5.
Predicted expression levels for translation/transcription processing genes among four fast-growing bacteriaa
| Geneb |
E(g)
|
|||
|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | |
| rpoB | 2.65 | 1.88 | 1.58 | 1.49 |
| rpoC | 2.49 | 1.63 | 1.45 | 1.76 |
| fus | 2.24 | 2.02 | 2.01 | 2.34 |
| tuf | 2.20 | 1.72 | 1.51 | 1.97 |
| infB | 2.18 | 1.38 | 1.28 | 0.79 |
| tsf | 2.00 | 1.76 | 1.76 | 1.75 |
| efp | 1.93 | 1.23 | 1.23 | 1.27 |
| rho | 1.64 | 1.18 | 0.67 | 0.77 |
| gyrA | 1.39 | 0.93 | 0.94 | 0.95 |
| nusG | 1.38 | 0.87 | 1.00 | 0.71 |
| gyrB | 1.27 | 0.89 | 0.67 | 0.39 |
| frr | 1.21 | 1.11 | 1.31 | 1.19 |
| nusB | 1.21 | 0.95 | 0.69 | 0.62 |
| rbfA | 1.20 | 0.91 | 0.87 | 0.76 |
| nusA | 1.15 | 1.16 | 0.66 | 0.59 |
| rpoA | 0.99 | 1.56 | 0.95 | 1.38 |
| infC | 0.70 | 1.02 | 1.13 | 1.01 |
Only genes that qualify as PHX genes in at least one genome are included. For duplicated genes, the highest E(g) value was used. E. coli pairwise correlation coefficients were 0.75, 0.70, and 0.63 with V. cholerae, H. influenzae, and B. subtilis, respectively. V. cholerae pairwise correlation coefficients were 0.75, 0.83, and 0.88 with E. coli, H. influenzae, and B. subtilis, respectively. H. influenzae pairwise correlation coefficients were 0.70, 0.83, and 0.90 with E. coli, V. cholerae, and B. subtilis, respectively. B. subtilis pairwise correlation coefficients were 0.63, 0.88, and 0.90 with E. coli, V. cholerae, and H. influenzae, respectively.
DNA-directed RNA polymerase β (rpoB), DNA-directed RNA polymerase β′ (rpoC), elongation factor G (fus), elongation factor Tu (tuf), translation initiation factor IF-2 (infB), elongation factor Ts (tsf), elongation factor P (efp), transcription termination factor rho (rho), DNA gyrase subunit A (gyrA), transcription antitermination protein (nusG), DNA gyrase subunit B (gyrB), ribosome recycling factor (frr), N utilization substance protein B (nusB), ribosome-binding factor A (rbfA), N utilization substance protein A (nusA), DNA-directed RNA polymerase α (rpoA), and translation initiation factor IF-3 (infC).
TABLE 9.
Major energy metabolism genes of the four fast-growing bacteria and their predicted E(g) valuesa
| Pathway | Geneb |
E(g)
|
|||
|---|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | ||
| Glycolysis | pgi | 1.41 | (0.66) | (0.60) | (0.92) |
| pfkA | 1.86 | 1.06 | 1.09 | (0.89) | |
| fba | 2.38 | 1.88 | 1.46 | 1.99, (0.59) | |
| tpi | 2.08 | 1.32 | 1.30 | 1.48 | |
| gap | 2.07 | 1.82 | 1.72 | 1.80 | |
| pgk | 2.38 | 1.78 | 1.04 | 1.34 | |
| gpm | 1.24 (gpmA), 1.69 (yibO) | 1.46 | 1.23 | 1.08 | |
| eno | 2.11 | 1.93 | 1.59 | 1.92 | |
| pykA/pykF | 2.58 (pykF), (0.95) (pykA) | 1.75, (0.53), (0.48) | 1.58 | 1.18 | |
| Pyruvate oxidation of acetyl-CoA | aceE | 2.43 | 1.81 | 1.51 | — |
| pdhA | — | — | — | 1.57 | |
| pdhB | — | — | — | 1.91 | |
| aceF/pdhC | 1.92 | 1.34 | 1.23 | 2.05 | |
| lpdA/pdhD | 2.10 | 1.49 | 1.39 | 2.14 | |
| TCA cycle | gltA | 1.29 | (0.96) | — | (0.57), (0.63), (0.46) |
| acnA/acnB | (0.29), 1.40 | (0.42), (0.69) | — | 1.08 | |
| icdA/citC | 1.65 | — | — | 1.33 | |
| sucA | 1.30 | (0.58) | (0.44) | (0.39) | |
| sucD | 1.73 | 1.00 | (0.57) | (0.67) | |
| sucC | 1.74 | 1.12 | (0.49) | (0.63) | |
| sdhA | 1.08 | 1.12 | — | (0.57) | |
| sdhB | (0.55) | (0.51) | — | (0.63) | |
| sdhC | (0.81) | (0.67) | — | (0.64) | |
| sdhD | (0.88) | (0.90) | — | — | |
| fumABC/citG | (0.56), 1.10, (0.32) | (0.39) | (0.92) | (0.54) | |
| mdh/citH | 1.42 | (0.73) | 1.18 | (0.81) | |
| Pentose phosphate pathway | zwf | (0.60) | (0.66) | (0.81) | (0.95) |
| devB | — | (0.79) | (0.75) | — | |
| gnd | 1.33 | 1.66 | 1.16 | 1.12, (0.65) | |
| rpiA | (0.80) | (0.89) | (0.78) | — | |
| tkt | 2.00, (0.45) | 1.29, 1.21 | 1.26 | 1.37 | |
| tal | 1.58, (0.46) | 1.52 | 1.17 | 1.01 | |
Values in parentheses indicate that the gene is not PHX. For duplicated genes, all E(g) values are shown. —, gene does not exist in that genome. E. coli pairwise correlation coefficients were 0.92, 0.68, and 0.70 with V. cholerae, H. influenzae, and B. subtilis, respectively. V. cholerae pairwise correlation coefficients were 0.92, 0.76, and 0.78 with E. coli, H. influenzae, and B. subtilis, respectively. H. influenzae pairwise correlation coefficients were 0.68, 0.76, and 0.78 with E. coli, V. cholerae, and B. subtilis, respectively. B. subtilis pairwise correlation coefficients were 0.70, 0.78, and 0.78 with E. coli, V. cholerae, and H. influenzae, respectively.
Glucose-6-phosphate isomerase (pgi), 6-phosphofructokinase (pfkA), fructose-1,6-bisphosphate aldolase (fba), triosephosphate isomerase (tpi), glyceraldehyde-3-phosphate dehydrogenase (gap), phosphoglycerate kinase (pgk), phosphoglycerate mutase (gpm), enolase (eno), pyruvate kinase (pykA/pykF), pyruvate dehydrogenase E1 (aceE), pyruvate dehydrogenase E1 α (pdhA), pyruvate dehydrogenase E1 β (pdhB), pyruvate dehydrogenase E2 (aceF/pdhC), dihydrolipoamide dehydrogenase (lpdA/pdhD), citrate synthase (gltA), aconitate hydratase (acnA/acnB), isocitrate dehydrogenase (icdA/citC), 2-oxoglutarate dehydrogenase (sucA), succinyl-CoA synthetase alpha chain (sucD), succinyl-coA synthetase beta chain (sucC), succinate dehydrogenase flavoprotein subunit (sdhA), succinate dehydrogenase iron-sulfur protein (sdhB), succinate dehydrogenase cytochrome b556 subunit (sdhC), succinate dehydrogenase 13-kDa hydrophobic protein (sdhD), fumarate hydratase (fumA, fumB, fumC, or citG), malate dehydrogenase (mdh/citH), glucose-6-phosphate dehydrogenase (zwf), phosphogluconolactonase (devB), 6-phosphogluconate dehydrogenase (gnd), ribose-5-phosphate isomerase (rpiA), transketolase (tkt), and transaldolase (tal).
TABLE 10.
Repair proteinsa
| Group |
E(g)
|
Gene | |||
|---|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | ||
| Direct repair | 0.40 | 0.38 | — | — | Deoxyribodipyrimidine photolyase |
| 0.51 | — | — | 0.79 | O6—Methylguanine DNA alkyltransferase | |
| — | 0.62 | 0.68 | 0.59 | Methylated DNA-protein-cysteine methyltransferase | |
| 0.53 | — | — | 0.76 | Regulatory protein, methyltransferase, alkyltransferase Ada | |
| Base excision repair | 0.47 | 0.47 | 0.69 | 0.53 | Uracil DNA glycosylase |
| 0.53 | 0.46 | 0.55 | 0.49 | A/G-specific adenine glycosylase | |
| 0.57 | 0.57 | 0.62 | 0.50 | Endonuclease III | |
| 0.53 | 0.51 | 0.58 | 0.54 | Formamidopyrimidine DNA glycosylase | |
| 0.61 | 0.54 | 0.68 | — | 3-Methyladenine DNA glycosylase I | |
| 0.56 | — | — | 0.69 | DNA 3-methyladenine glycosidase II | |
| AP endonucleases | 0.64 | 0.53 | 0.70 | — | Exonuclease III, apurinic-apyrimidinic endonuclease |
| 0.53 | 0.47 | — | 0.57 | Endonuclease IV | |
| Mismatch excision repair | 0.46 | 0.35 | 0.48 | 0.35 | DNA mismatch repair protein MutS |
| 0.40 | 0.37 | 0.47 | 0.42 | DNA mismatch repair protein MutL | |
| 0.44 | 0.53 | 0.62 | — | GATC endonuclease | |
| 0.43 | 0.44 | 0.63 | — | GATC methylase | |
| 0.46 | 0.48 | 0.56 | 0.39 | Exodeoxyribonuclease large subunit | |
| 0.85 | 0.78 | 0.88 | 1.09 | Exodeoxyribonuclease small subunit | |
| Nucleotide excision repair | 0.66 | 0.35 | 0.44 | 0.40 | Excision nuclease UvrA |
| 0.57 | —b | 0.56 | 0.50 | Excision nuclease UvrB | |
| 0.40 | 0.38 | 0.53 | 0.61 | Excision nuclease UvrC | |
| 0.68 | 0.50 | 0.51 | 0.51 | DNA helicase II | |
| 0.38 | 0.41 | 0.45 | 0.37 | Transcription repair coupling factor | |
| Recombinational repair | 1.48 | 0.88 | 0.69 | 0.80 | RecA protein |
| 0.28 | 0.41 | 0.39 | — | ExoV helicase RecB | |
| 0.32 | 0.46 | 0.37 | — | ExoV nuclease RecC | |
| 0.39 | 0.41 | 0.42 | — | ExoV helicase RecD | |
| 0.51 | 0.47 | 0.64 | 0.43 | Single-stranded DNA binding protein RecF | |
| 0.39 | 0.44 | 0.48 | 0.37 | 5′ → 3′ single-stranded DNA exonuclease RecJ | |
| 0.59 | 0.43 | 0.63 | — | RecO protein | |
| 0.71 | 0.55 | 0.75 | 0.72 | RecR protein | |
| 0.56 | 0.49 | 0.53 | 0.39 | ATP binding protein RecN | |
| 0.45 | 0.53 | 0.53 | 0.48 | DNA helicase RecQ | |
| 0.40 | 0.43 | 0.53 | — | Exodeoxyribonuclease I | |
| 0.35 | 0.37 | — | 0.39 | Double-stranded DNA exonuclease SbcC | |
| 0.45 | 0.47 | — | 0.52 | Double-stranded DNA exonuclease SbcD | |
| 0.50 | 0.61 | 0.63 | 0.51 | DNA repair protein RadC | |
| Branch migration resolution | 0.59 | 0.55 | 0.65 | 0.55 | RuvA protein |
| 0.56 | 0.47 | 0.65 | 0.64 | RuvB protein | |
| 0.48 | 0.40 | 0.52 | — | RecG protein | |
| 0.61 | 0.50 | 0.58 | — | Holliday junction endonuclease RuvC | |
| DNA ligases | 0.56 | 0.39 | 0.44 | 0.51 | NAD-dependent DNA ligase |
| Nucleotide pools | 0.72 | 0.63 | 0.74 | 0.54 | GTPase triphosphatase MutT |
| 0.97 | — | 0.71 | 0.89 | Deoxyuridine 5′-triphosphate nucleotide hydrolase | |
| Replication | 0.48 | 0.32 | 0.42 | 0.40 | DNA polymerase I |
| 0.43 | 0.36 | — | — | DNA polymerase II | |
| 0.64 | 0.34 | 0.41 | 0.30 | DNA polymerase III alpha chain | |
| Other repair proteins | 0.78 | 0.56 | 0.78 | 0.53 | SOS regulon transcription repressor LexA |
| 0.50 | 0.53 | 0.56 | 0.56 | ATP-dependent protease Sms/RadA | |
| 0.40 | 0.37 | 0.48 | 0.36 | Primosomal protein replication factor PriA | |
| 1.48 | 0.66 | 0.94 | 0.80 | Single-stranded DNA binding protein Ssb | |
| 0.99 | 0.52 | 0.49 | — | ATP-dependent helicase HepA | |
The classification of repair proteins was adopted in part from data in reference 9. Only genes that occur in at least two species are listed. E(g) values of PHX genes are shown in bold. —, gene does not exist in that genome.
UvrB is present in V. cholerae with a frameshift.
TABLE 11.
Major vitamin biosynthesis genes of E. coli
| E(g) | Length | Positiona | Geneb |
|---|---|---|---|
| 0.50 | 428 | 808480 − | bioA |
| 0.62 | 345 | 808567 + | bioB |
| 0.43 | 383 | 809604 + | bioF |
| 0.53 | 250 | 810745 + | bioC |
| 0.61 | 224 | 811493 + | bioD |
| 0.51 | 255 | 3542480 − | bioH |
| 0.66 | 197 | 442871 − | thiJ |
| 0.45 | 376 | 4189446 − | thiH |
| 0.47 | 280 | 4190288 − | thiG |
| 0.51 | 244 | 4191136 − | thiF |
| 0.62 | 210 | 4191782 − | thiE |
| 0.49 | 630 | 4193677 − | thiC |
| 0.46 | 366 | 432679 + | ribD |
| 1.24c | 155 | 433871 + | ribH |
| 0.65 | 195 | 1337184 − | ribA |
| 0.54 | 212 | 1741266 − | ribE |
| 0.95 | 216 | 3182482 − | ribB |
| 0.71 | 320 | 659439 − | lipA |
| 0.69 | 190 | 661435 − | lipB |
| 0.41 | 328 | 53416 − | pdxA |
| 0.66 | 217 | 1716031 − | pdxH |
| 0.41 | 377 | 2435871 − | pdxB |
| 0.68 | 242 | 2699749 − | pdxJ |
| 0.59 | 358 | 2062489 − | cobT |
| 0.50 | 246 | 2063244 − | cobS |
| 0.64 | 180 | 2063786 − | cobU |
Position in the genome is shown as the translation initiation site and orientation of the gene (+, direct strand; −, complementary strand).
Adenosylmethionine-8-amino-7-oxononanoate aminotransferase (bioA), biotin synthetase (bioB), 8-amino-7-oxononanoate synthase (bioF), biotin synthesis protein (bioC), dethiobiotin synthetase (bioD), biotin synthesis block prior to pimeloyl-CoA (bioH), 4-methyl-5(β-hydroxyethyl)-thiazole monophosphate biosynthesis (thiJ), thiamine biosynthesis (thiH), thiamine biosynthesis protein (thiG), thiamine biosynthesis protein (thiF), thiamine biosynthesis protein (thiE), thiamine biosynthesis protein (thiC), riboflavin biosynthesis protein (ribD), riboflavin synthase beta chain (ribH), GTP cyclohydrolase II (ribA), riboflavin synthase alpha chain (ribE), 3,4-dihydroxy-2-butanone-4-phosphate synthase (ribB), lipoic acid synthetase (lipA), lipoate biosynthesis protein B (lipB), pyridoxal phosphate biosynthetic protein (pdxA), pyridoxamine-5′-phosphate oxidase (pdxH), erythronate-4-phosphate dehydrogenase (pdxB), pyridoxal phosphate biosynthetic protein (pdxJ), nicotinate-nucleotide-dimethylbenzimidazole phosphoribosyltransferase (cobT), cobalamin (5′-phosphate) synthase (cobS), and cobinamide kinase and guanylyltransferase (cobU).
PHX gene.
(i) Ribosomal protein genes (Table 3).
Ribosomes of the four fast-growing bacteria have practically the same numbers of small- and large-subunit proteins. However, among all prokaryotic genomes, that number ranges from 50 to 65, while in eukaryotes, the number is constant at 79 (except in yeast, 78) (48, 50). This information suggests a greater range of variation in the patterns of protein synthesis among prokaryotes, consistent with the constrained phylogenetic origin of eukaryotic cells compared with the less constrained origin of prokaryotic species.
Thirty-five RP genes are shown in Table 3 (only those ≥100 codons long). Unlike those of yeast and Drosophila, many of the bacterial RP genes are concatenated to form a large operon encompassing 20 to 40% of all RP genes. Genes for some of the major translation/transcription processing factors, including tuf, fus, rpoA, rpoB, and rpoC, are within or near the large RP operon. Other RP operons typically consist of two to five genes. In E. coli, the cluster of L7/L12, L10, L1, L11, rpoB, and rpoC is noteworthy. B. subtilis possesses an RP cluster that effectively combines the two largest clusters of E. coli. In these fast-growing bacteria, most of the eubacterial RP genes are positioned near the origin of replication, oriC. It is evident from Table 3 that virtually all RP genes are PHX. The EF-Tu gene is often duplicated, with both copies being PHX and incorporated near or in an RP cluster. groEL, rpoB, and rpoC also tend to localize to the vicinity of the main RP cluster. Many eukaryotic and eubacterial ribosomal proteins are multifunctional (50).
The “giant” RP (labeled S1 or RpsA, generally exceeding 500 amino acids in length) has a remarkable phylogeny. It is recognized in most eubacteria but is not part of an RP operon, and it generally reaches among the highest expression levels. In B. subtilis, there is an S1 homolog, but it is only 327 codons long, and the S1 gene is entirely missing from the three current completely sequenced mycoplasma genomes. The S1 gene is essential in E. coli, where it is thought to contribute to the initiation of polypeptide synthesis. The absence of an S1 protein in B. subtilis can possibly be compensated for by a strong ribosome binding site (34). The evolutionarily deep branching bacterium Aquifex aeolicus has a giant S1 gene. Thermotoga maritima, allowing for a frameshift, also has an S1 homolog. None of the archaeal genomes has an S1 homolog, and eukaryotic genomes also lack an S1 homolog.
The origin of replication (oriC) for E. coli is identified within the 232-bp interval from 3923372 to 3923603. The major RP cluster is proximal to oriC at 3436600 to 3476134 and contains, in addition to RP genes, genes for the elongation factors EF-Tu and EF-G and two flanking chaperones of the peptidyl-prolyl cis-trans isomerase (PPIase) family. Proximity to oriC implies a higher-than-average gene copy number per rapidly growing cell. A second RP cluster occurs proximally on the other side of oriC and includes genes for a duplicate copy of EF-Tu (tufB) and the DNA-directed RNA polymerase units rpoB and rpoC. The E(g) values for RP genes (≥100 codons long) in E. coli range from 2.44 to 1.13. All but one of the RP genes are PHX; the single exception is L9 in B. subtilis. The majority have E(g) values exceeding 1.50. The correlations of E(g) values among the RP genes of E. coli, V. cholerae, and H. influenzae are high (Table 3).
Does stoichiometry matter? For example, among the RP genes, why aren't all 50S units PHX at the same expression level? A partial answer may be that not all ribosomal proteins play an exclusive role in determining ribosome structure. Some may have a regulatory role (e.g., S1 is proposed to function in translation initiation) (M. Nomura, personal communication) (34). The acidic ribosomal protein component P0 is PHX in archaea but is absent from eubacteria. L7/L12 is also acidic and is thought to act in adapting mRNA chains to the ribosome. Actually, L7/L12 forms dimers with an elongated shape. Two dimers associate with a copy of L10 to form a very strong complex (4). Very relevant is that several ribosomal proteins are multifunctional (50). For example, S9 provides ancillary utility in certain repair activities (49); S16, in part, acts as an endonuclease (31).
(ii) Genes for transcription/translation processing factors (Table 5).
The majority of protein synthesis factors are PHX over all prokaryotic genomes. Expression levels correlate highly across species (Table 5, footnote a). As with the ribosomal proteins, the E(g) values cover a wide range. Elongation factor EF-G (fus) is distinctive, with an E(g) value exceeding 2 for each genome. The highest expression levels in E. coli occur for the RpoB and RpoC subunits of the core RNA polymerase. RpoA is PHX in B. subtilis but not in E. coli, V. cholerae, and H. influenzae. Why are the predicted expression levels for the RpoB and RpoC subunits higher than that for RpoA? Based on the RNA polymerase stoichiometry (one copy of RpoB, one copy of RpoC, but two RpoA units), should one expect elevated expression levels for RpoA compared to RpoB and RpoC? A possible explanation relates to the differences in protein sizes, RpoB and RpoC being larger proteins than RpoA. It has been observed for E. coli that codon choices in long genes tend to be more biased than those in short genes (10). Interestingly, Mycoplasma genitalium, its relative Ureaplasma urealyticum, and the spirochete Treponema pallidum feature PHX RpoA but not RpoB and RpoC.
(iii) Chaperone/degradation protein genes (Table 6).
TABLE 6.
Predicted expression levels for chaperone/degradation genes among four fast-growing bacteriaa
| Geneb |
E(g)
|
|||
|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | |
| pnp | 2.66 | 1.72 | 1.72 | 0.79 |
| dnaK | 2.58 | 1.80 | 1.29 | 1.83 |
| groEL | 2.09 | 1.30 | 1.47 | 1.87 |
| tig | 2.05 | 1.71 | 1.21 | 1.85 |
| htpG | 1.85 | 0.50 | 0.62 | 0.67 |
| hslU | 1.67 | 0.50 | 0.63 | 0.55 |
| ppiB | 1.53 | 0.80 | 0.86 | 1.12 |
| ftsH | 1.52 | 0.88 | 0.59 | 0.78 |
| secA | 1.49 | 0.83 | 0.73 | 0.75 |
| ftsZ | 1.16 | 0.95 | 0.67 | 1.01 |
| secG | 1.14 | 0.97 | 0.95 | 0.81 |
| trxA | 1.06 | 1.21 | 1.11 | 1.35 |
| clpX | 1.04 | 0.78 | 0.49 | 1.08 |
| grpE | 0.93 | 0.79 | 0.64 | 1.05 |
| clpP | 0.58 | 0.83 | 0.88 | 1.05 |
| slyD | 2.08 | 1.11 | 1.28 | |
| htrA | 1.26 | 0.46 | 0.64 | |
| secB | 1.23 | 1.25 | 0.92 | |
| surA | 1.10 | 0.46 | 0.78 | |
Only genes that qualify as PHX genes in at least one genome are included. For duplicated genes, the highest E(g) value was used. E. coli pairwise correlation coefficients were 0.64, 0.66, and 0.31 with V. cholerae, H. influenzae, and B. subtilis, respectively. V. cholerae pairwise correlation coefficients were 0.64, 0.85, and 0.70 with E. coli, H. influenzae, and B. subtilis, respectively. H. influenzae pairwise correlation coefficients were 0.66, 0.85, and 0.54 with E. coli, V. cholerae, and B. subtilis, respectively. B. subtilis pairwise correlation coefficients were 0.31, 0.70, and 0.54 with E. coli, V. cholerae, and H. influenzae, respectively. Genes missing from B. subtilis (slyD, htrA, secB, and surA) were not included in calculations of the correlation coefficients.
Polynucleotide phosphorylase (pnp), heat shock protein 70 (dnaK), heat shock protein 60 (groEL), trigger factor (tig), heat shock protein 90 (htpG), heat shock protein (hslU), PPIase B (ppiB), cell division metallopeptidase (ftsH), preprotein translocase subunit (secA), cell division protein (ftsZ), protein export membrane protein (secG), thioredoxin (trxA), Clp protease ATP-binding subunit (clpX), HSP70 cofactor (grpE), Clp protease proteolytic subunit (clpP), PPIase (slyD), protease DO and heat shock protein (htrA), protein translocase (secB), and PPIase survival protein (surA).
Among the top PHX genes in most eubacterial genomes are those for the major chaperone protein archetypes, DnaK and GroEL. These reach E(g) values exceeding 1.3 (>2 in E. coli). The gene for the multifunctional enzyme Pnp, fundamental in RNA processing and mRNA degradation, attains the highest predicted E(g) value, 2.66, among all E. coli genes. Pnp is PHX in many eubacterial genomes but not in B. subtilis.
Thioredoxin (trxA) implements protein folding by catalyzing the formation or disruption of disulfide bonds. The eukaryotic thioredoxin homolog is protein disulfide isomerase, operating in the endoplasmic reticulum. It has been verified experimentally that protein disulfide isomerase augments protein folding needs (7, 15, 47). The highest E(g) values for thioredoxin occur in B. subtilis (1.35) and then in other fast-growing bacteria in the order D. radiodurans (1.23) (data not shown), V. cholerae (1.21), H. influenzae (1.11), and E. coli (1.06).
Peptidyl-prolyl cis-trans isomerases (PPIases) accelerate the proper folding of proteins by promoting the cis-trans isomerization of imide bonds in proline within oligopeptides. E. coli has at least nine PPIases defined by sequence similarity. One of these, the survival protein SurA, enhances the folding of periplasmic and outer membrane proteins. As expected, SurA does not exist in gram-positive B. subtilis, which has neither compartment. Trigger factor (Tig) is a ribosome-associated chaperone that can complement DnaK (8). Tig and DnaK cooperate in the folding of newly synthesized proteins. Simultaneous deletion of Tig and DnaK is lethal under usual growth conditions (43). Tig is broadly PHX for eubacterial genomes but is not found for archaeal genomes. Expression levels of Tig in fast-growing bacteria are quite similar (Table 6).
DegP is a chaperone folding factor that is significantly PHX, with an E(g) value of 1.26; it acts primarily in degrading misfolded proteins in the periplasm. Also associated with periplasmic and cytoplasmic chaperones are several PPIases, including PpiC [E(g) = 1.02], PpiB (1.53), FkpA (1.40), SlyD (2.08), PpiA (0.95), PpiD (1.11), SurA (1.10), FhlB (0.85), and YaaD (0.77); four are active in the periplasm, and five are active in the cytoplasm. Another relevant chaperone protein is disulfide oxidase (DsbA), which is marginally PHX, with an E(g) value of ≈1.02; it senses misfolded proteins in the periplasm.
Correlations among the fast-growing bacteria for levels of expression of major chaperone genes are generally significantly high (Table 6, footnote a). However, E. coli and B. subtilis are marginally correlated (0.3). In E. coli, degradation proteins are mostly PHX, but this is not consistently the case for the other fast-growing bacteria. Why are the major chaperone genes so often PHX? Chaperone/degradation proteins are vitally needed both during rapid growth and in stationary phase. In normal cell physiology, these proteins have multiple functions: they contribute decisively in ensuring correct protein folding, in remedying misfolded structures, in directing protein trafficking, and in coordinating protein secretion. Chaperone proteins also contribute to conformational changes and to minimizing protein damage during stress.
(iv) Levels of expression of aminoacyl-tRNA synthetases (Table 7).
TABLE 7.
Predicted expression levels for aminoacyl-tRNA synthetase genes among four fast-growing bacteriaa
| Gene |
E(g)
|
|||
|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | |
| aspS | 1.95 | 0.98 | 1.01 | 0.73 |
| lysS | 1.84 | 1.35 | 1.05 | 1.09 |
| valS | 1.81 | 1.17 | 1.02 | 0.65 |
| serS | 1.64 | 1.16 | 0.98 | 0.70 |
| proS | 1.64 | 0.89 | 0.96 | 0.58 |
| leuS | 1.57 | 1.08 | 0.78 | 0.66 |
| tyrS | 1.40 | 0.42 | 0.97 | 1.15 |
| gltX | 1.35 | 0.90 | 0.72 | 0.60 |
| glyS | 1.32 | 0.87 | 1.07 | 0.59 |
| pheS | 1.30 | 1.08 | 0.90 | 0.70 |
| glnS | 1.26 | 0.74 | 0.91 | |
| glyQ | 1.18 | 1.25 | 1.14 | 0.60 |
| asnS | 1.17 | 1.25 | 1.10 | 0.83 |
| metG | 1.17 | 0.76 | 0.73 | 0.69 |
| ileS | 1.17 | 0.84 | 0.78 | |
| pheT | 1.16 | 1.02 | 0.77 | 0.52 |
| alaS | 1.15 | 0.83 | 0.63 | 0.73 |
| thrS | 1.04 | 0.87 | 1.40 | 1.12 |
| argS | 1.04 | 0.82 | 0.61 | 0.89 |
Only genes that qualify as PHX in at least one genome are included. For duplicated genes, the highest E(g) value was used. E. coli pairwise correlation coefficients were 0.32, 0.16, and −0.04 with V. cholerae, H. influenzae, and B. subtilis, respectively. V. cholerae pairwise correlation coefficients were 0.32, 0.29, and −0.24 with E. coli, H. influenzae, and B. subtilis, respectively. H. influenzae pairwise correlation coefficients were 0.16, 0.29, and 0.35 with E. coli, V. cholerae, and B. subtilis, respectively. B. subtilis pairwise correlation coefficients were −0.04, −0.24, and 0.35 with E. coli, V. cholerae, and H. influenzae, respectively.
There are 19 PHX tRNA synthetase polypeptides in E. coli, including two subunits of phenylalanyl-tRNA synthetase (PheS-α and PheT-β) and two subunits of glycyl-tRNA synthetase (GlyQ-α and GlyS-β). However, there are only eight in V. cholerae, seven in H. influenzae, and three in B. subtilis. IleS is missing from H. influenzae, and GlnS is missing from B. subtilis, which uses amidotransferase modifications to produce Gln-tRNAGln from Glu-tRNAGlu synthetase. Actually, the GlnS gene is absent from most prokaryotic genomes (14).
Expression level correlations for the tRNA synthetase genes among the three rapidly dividing gram-negative genomes are generally positive but low. On the other hand, the corresponding relationship of B. subtilis with E. coli is uncorrelated (−0.04) and that of B. subtilis with V. cholerae is modestly negatively correlated (−0.24). LysS is the only PHX tRNA synthetase for all four genomes.
There are three aminoacyl-tRNA synthetases in E. coli which occur at only moderate predicted expression levels: CysS, with an E(g) of 0.89; TrpS, with an E(g) of 0.91; and HisS, with an E(g) of 0.74. The average amino acid usage frequencies for E. coli genes correlate positively with the predicted expression levels for tRNA synthetases. Interestingly, the three lowest amino acid usage frequencies in E. coli are for Cys (1.2%), Trp (1.5%), and His (2.3%) (Table 8).
TABLE 8.
Relationship between aminoacyl-tRNA synthetase expression levels and amino acid frequencies in E. coli proteinsa
| Gene | % Amino acid frequencyb | E(g) |
|---|---|---|
| aspS | 5.15 | 1.95 |
| lysS | 4.42 | 1.84 |
| valS | 7.08 | 1.81 |
| serS | 5.84 | 1.64 |
| proS | 4.44 | 1.64 |
| leuS | 10.68 | 1.57 |
| tyrS | 2.86 | 1.40 |
| gltX | 5.76 | 1.35 |
| glyS | 7.39 | 1.32 |
| pheS | 3.91 | 1.30 |
| glnS | 4.44 | 1.26 |
| glyQ | 7.39 | 1.18 |
| ileS | 6.02 | 1.17 |
| asnS | 3.96 | 1.17 |
| metG | 2.54 | 1.17 |
| pheT | 3.91 | 1.16 |
| alaS | 9.51 | 1.15 |
| thrS | 5.42 | 1.04 |
| argS | 5.56 | 1.04 |
| trpS | 1.53 | 0.91 |
| cysS | 1.17 | 0.89 |
| hisS | 2.28 | 0.74 |
The correlation between amino acid frequencies and tRNA synthetase expression levels is 0.41. The two-subunit aminoacyl-tRNA synthetases Gly and Phe were excluded.
Average frequency of the corresponding amino acid in the annotated E. coli proteins.
(v) Levels of expression of major energy metabolism genes (Table 9).
Enzymes of major catabolic pathways can be divided into four groups: glycolysis, pyruvate metabolism, the pentose phosphate pathway, and the TCA cycle. The glycolysis genes are predominantly PHX in all four fast-growing bacteria, with very high E(g) values, >2.00, for several of these genes in E. coli. Hexokinase and glucokinase are prominent glycolysis proteins in most eukaryotes, but the former is not found in most prokaryotes, including the four fast-growing bacteria under analysis in this study. Why? In glycolysis, hexokinase converts glucose to glucose-6-phosphate. However, glucose-6-phosphate arises from other hexoses and from glucose transported into the cell via the phosphotransferase system. Perhaps the multiplicity of sources means that glucokinase need not be PHX. Glucokinase occurs in many (but not all) eubacteria, normally at low to moderate E(g) values, 0.3 to 0.8.
The genes for pyruvate dehydrogenase are commonly PHX in the four genomes. The TCA genes are generally PHX in E. coli but generally not PHX in H. influenzae and B. subtilis. In B. subtilis, two TCA genes are PHX and the others cover the range 0.4 to 1.0. Many prominent TCA genes appear to be absent from H. influenzae. Why are TCA genes in B. subtilis mostly not PHX? The TCA cycle, apart from energy (ATP) production, can contribute in myriad ways to cellular needs, especially in making precursors and intermediates to macromolecules, e.g., in amino acid, vitamin, and heme biosyntheses (see Discussion). The order of actions in the TCA cycle is as follows: citrate synthase (GltA; in B. subtilis, there are two versions, designated CitZ and CitA), aconitate hydratase (AcnA/AcnB), isocitrate dehydrogenase (Icd), 2-oxoglutarate dehydrogenase (SucA), succinyl coenzyme A (succinyl-CoA) synthetase (SucD and SucC), succinate dehydrogenase (SdhB, SdhC, and SdhD), fumarate hydratase (FumA, FumB, FumC, or CitG), and malate dehydrogenase (Mdh/CitH). The initial enzymes of the TCA pathway in E. coli are all PHX, with E(g) values ≥1.29, whereas those beyond succinyl-CoA synthetase (except for Mdh) all have E(g) values ≤1.10, and most are not PHX. Apart from the differences in the expression levels among the TCA cycle genes, correlations among genomes for energy metabolism gene expression levels across all four fast-growing bacteria are high, suggesting similar uses for this set of enzymes (Table 9, footnote a).
Certain gene groups generally not PHX.
Specific regulatory proteins or proteins responding to special demands and used few times, as in the highly specialized DNA repair processes, are not expected to be PHX. Also, specific transcription proteins and DNA replication proteins, because the cell assembles few replication machines, tend not to be PHX.
(i) Genomic repair proteins.
Table 10 reports predicted expression levels for the main collection of repair proteins for the four genomes. Only two repair proteins of E. coli reach PHX levels: RecA and Ssb (single-stranded DNA binding protein) [E(g) for both, 1.48]. Two other repair proteins are borderline PHX: Dut (deoxyuridine 5′-triphosphate nucleotide hydrolase) and HepA [E(g) = 0.97 and 0.99, respectively]. Other repair proteins have low to moderate predicted expression levels, the E(g) values almost always in the range from 0.35 to 0.80. These evaluations parallel those for D. radiodurans, in which RecA [E(g), 2.04] has a dramatically high predicted expression level and MutT (gene no. DR2358) reaches an E(g) of 1.29, these being the only two proteins qualifying as PHX (22). The other repair proteins of D. radiodurans have E(g) values in the range 0.40 to 0.80.
(ii) Vitamin biosynthesis proteins (Table 11).
Pathways to the synthesis of vitamins, of which only small amounts are needed to provide adequate cofactor function, have largely low predicted expression levels, with E(g) values of about 0.40 to 0.75. In E. coli, the genes acting in the synthesis of six vitamin cofactors, biotin, thiamine, riboflavin, lipoate, pyridoxal, and cobalamin, were examined. Only RibH, which participates in riboflavin biosynthesis, is PHX in E. coli. Although the enzymes of the biosynthetic pathways are poorly expressed, some of the enzymes that utilize the vitamins as cofactors are highly expressed, for example, biotin carboxylase (a subunit of E. coli acetyl-CoA carboxylase). In B. subtilis, RibE, which is not PHX, in the same pathway forms an oligomer complex with RibH in which the structural union (RibE-RibH) combines 3 units of RibE with 60 units of RibH (23). This anomalous stoichiometry makes it likely that RibH furnishes structural support and, for this reason, is PHX; in this guise, RibH may be used in other capacities. Paradoxically, RibH is not PHX in B. subtilis.
Interestingly, M. tuberculosis features nine PHX proteins among the vitamin biosynthesis pathways. Synechocystis and A. aeolicus each have three PHX vitamin biosynthesis genes, Borrelia burgdorferi has one, Archaeoglobus fulgidus has two, T. pallidum has one, and D. radiodurans has one. The biotin carboxylase protein is PHX in the E. coli, H. influenzae, V. cholerae, Helicobacter pylori, Synechocystis, Chlamydia trachomatis, and A. fulgidus genomes.
(iii) Genes of signal transduction pathways.
In Table 8 of reference 21, the predicted expression levels for several two-component sensor genes (histidine kinases) of E. coli and B. subtilis are reported. In all of those examples, the predicted expression levels were low, the E(g) values ranging from 0.30 to 0.70.
One particular example is the Cpx regulon of the sensor kinase/phosphatase periplasmic family, which encompasses the genes encoding CpxA and CpxR (components of a histidine kinase), CpxP (down regulates the Cpx pathway), and NlpE (membrane lipoprotein), believed to eliminate abnormal proteins in the periplasm and to recover amino acids during nitrogen starvation (32). These proteins regulate a hierarchy of ς factors, including ς32 and ςE, active in autoregulation and repression. The predicted expression levels are low [for CpxA, E(g) = 0.70; for CpxR, E(g) = 0.57; for CpxP, E(g) = 0.62; and for NlpE, E(g) = 0.61], as is common with specific regulatory proteins. Cpx is a sensor kinase acting in the periplasm. The Cpx pathway apparently also monitors pilus assembly during infection of tissues by uropathogenic E. coli (17).
(iv) Principal starvation genes of E. coli and their predicted levels of expression (Table 12).
TABLE 12.
Genes induced under starvation conditions in E. coli
| E(g) | Length | Positiona | Geneb |
|---|---|---|---|
| 1.13c | 166 | 848134 − | dps |
| 0.56 | 265 | 1980411 − | otsB |
| 0.43 | 473 | 1979636 − | otsA |
| 0.62 | 329 | 2865574 − | rpoS |
| 1.46c | 283 | 3598414 − | rpoH |
| 0.52 | 476 | 3342358 + | rpoN |
| 0.60 | 190 | 2708032 − | rpoE |
| 0.69 | 248 | 582904 + | appY |
| 0.71 | 115 | 453663 + | bolA |
| 1.10c | 427 | 54702 − | surA |
| 0.42 | 752 | 1811891 + | katE |
Position in the genome is shown as the translation initiation site and orientation of the gene (+, direct strand; −, complementary strand).
DNA protection during starvation protein (dps; induced by oxidative damage), trehalose phosphatase (otsB), alpha trehalose phosphate synthase (otsA; osmoprotection), RNA polymerase ς-38 subunit (rpoS; principal ς factor in stationary phase), RNA polymerase ς-32 subunit (rpoH; main ς factor of major chaperones), RNA polymerase ς-54 factor (rpoN; effective in nitrogen regulation), RNA polymerase ς-E (ς-24) factor (rpoE; contributes to stress response), M5 polypeptide (appY; induced by phosphate starvation), BolA protein (bolA; rod-shaped protein; ς-S regulon), survival protein (surA; PPIase), and catalase HPII (katE; ς-S dependent).
PHX gene.
The genes shown in Table 12 are associated with starvation states, as discussed in the review (26). Three genes in this category are PHX: dps, also labeled pexB [E(g), 1.13], which provides protection from oxidative radicals; rpoH, which encodes ς32 [E(g),1.46]; and the survival protein, SurA [E(g),1.10], a chaperone which is a member of the PPIase family. We expect these proteins, by virtue of their codon usage patterns, to be capable of high levels of expression, especially when induced by starvation. Other starvation proteins (Table 12) have low to moderate E(g) values. The ςE factor, which regulates the activity of other periplasmic proteins, is not PHX, and the same is true for ς54 and ς38, which respond to nitrogen and/or carbon starvation, respectively. However, ς32 (rpoH), the principal chaperone sigma factor, pervasively registers as PHX, presumably to establish high levels of chaperone production.
Homologous PHX genes among the fast-growing bacteria.
Table 13 compares the numbers of homologous PHX gene families among the four rapidly dividing bacteria. There are 60 gene families common to the four fast-growing bacteria, with each member PHX. Thirty-two of these are families of RP genes, eight are families of TF genes, and nine are families of genes essential for energy metabolism. Twenty-three gene families distinguish E. coli with PHX representatives, but these are not PHX in the other three fast growers, including five CH genes and five TF genes.
TABLE 13.
Families of homologous genes among the four fast-growing bacteria with at least one PHX gene
| Family typea
|
No. of gene familiesb
|
|||||||
|---|---|---|---|---|---|---|---|---|
| E. coli | V. cholerae | H. influenzae | B. subtilis | All | RP | TF | CH | EN |
| + | + | + | + | 60 | 32 | 8 | 4 | 9 |
| + | + | + | − | 12 | 1 | 0 | 1 | 1 |
| + | + | − | + | 4 | 0 | 0 | 0 | 0 |
| + | − | + | + | 2 | 0 | 0 | 0 | 0 |
| − | + | + | + | 0 | 0 | 0 | 0 | 0 |
| + | + | − | − | 12 | 0 | 2 | 0 | 2 |
| + | − | + | − | 7 | 0 | 0 | 0 | 1 |
| − | + | + | − | 1 | 0 | 0 | 0 | 0 |
| + | − | − | + | 7 | 0 | 0 | 3 | 0 |
| − | + | − | + | 1 | 0 | 1 | 0 | 0 |
| − | − | + | + | 2 | 0 | 1 | 0 | 0 |
| + | − | − | − | 23 | 0 | 5 | 5 | 3 |
| − | + | − | − | 2 | 0 | 0 | 0 | 0 |
| − | − | + | − | 3 | 0 | 0 | 0 | 0 |
| − | − | − | + | 2 | 0 | 0 | 2 | 0 |
+, the gene is PHX in the genome; −, the gene is not PHX. For example, the family type +++− applies to all families of genes that have a homolog in all four genomes, and these homologous genes are PHX in E. coli, V. cholerae, and H. influenzae but not in B. subtilis. Genes that are missing from one or more of the four genomes are not counted.
All, the number of homologous gene families of a specified type. EN, energy metabolism.
E. coli and V. cholerae share 124 homologous genes that are both PHX and in total 236 homologous genes with one or both genes being PHX; the respective values for E. coli and H. influenzae are 105 and 226, and the values for V. cholerae and H. influenzae are 94 and 156. Paired PHX genes between fast-growing bacteria and non-fast-growing bacteria are fewer in numbers (Table 14). Of homologous genes among genomes with at least one PHX gene, the expression levels for E. coli versus archaeal genomes and E. coli versus H. pylori and M. genitalium genomes are uncorrelated or negatively correlated (Table 14). Similarly, V. cholerae, H. influenzae, and B. subtilis expression levels correlate negatively with homologous genes of archaeal genomes, possibly reflecting differences in lifestyles, habitats, and energy sources.
TABLE 14.
Numbers of pairs of homologousa genes with one or both genes PHX and correlations between their E(g) valuesb
| Organism | eco | hin | bsu | vch | dra | rpr | ctr | cpn | mge | mpn | bbu |
|---|---|---|---|---|---|---|---|---|---|---|---|
| eco | 0.42 (226) | 0.23 (152) | 0.54 (236) | 0.14 (135) | 0.11 (92) | 0.26 (66) | 0.23 (71) | −0.28 (39) | 0.35 (42) | −0.05 (70) | |
| hin | 0.42 (226) | 0.52 (90) | 0.40 (156) | 0.26 (87) | 0.35 (51) | 0.27 (50) | 0.25 (56) | 0.16 (33) | 0.30 (38) | 0.03 (59) | |
| bsu | 0.23 (152) | 0.52 (90) | 0.46 (103) | 0.30 (130) | 0.27 (61) | 0.36 (58) | 0.19 (59) | 0.23 (42) | 0.32 (48) | 0.00 (68) | |
| vch | 0.54 (236) | 0.40 (156) | 0.46 (103) | 0.18 (102) | 0.31 (64) | 0.33 (52) | 0.32 (58) | 0.18 (32) | 0.22 (38) | 0.20 (56) |
| tpa | hpy | mtu | scy | aae | tma | mja | mth | afu | pho | pab |
|---|---|---|---|---|---|---|---|---|---|---|
| 0.14 (63) | −0.05 (102) | 0.08 (150) | 0.29 (140) | 0.02 (137) | 0.09 (105) | −0.53 (15) | 0.14 (19) | −0.60 (37) | −0.15 (18) | −0.20 (24) |
| 0.22 (46) | 0.30 (62) | −0.01 (95) | 0.12 (90) | 0.00 (94) | 0.15 (76) | — (5) | — (7) | −0.61 (18) | — (8) | −0.59 (11) |
| 0.09 (69) | 0.16 (68) | −0.09 (139) | 0.12 (129) | −0.10 (115) | 0.21 (101) | −0.17 (12) | 0.30 (15) | −0.57 (29) | −0.44 (24) | −0.47 (26) |
| 0.11 (48) | 0.09 (73) | −0.05 (119) | 0.18 (108) | −0.11 (106) | 0.10 (84) | — (8) | 0.13 (12) | −0.61 (31) | −0.35 (18) | −0.41 (18) |
Genes are deemed homologous if they are at least 40% similar (based on SSPA scores [5]).
Correlation coefficient of E(g) values for pairs of homologous genes between the two genomes (number of homologous pairs of genes with at least one PHX between the two species). Correlation coefficients were calculated only if ≥10 pairs were available. Species: E. coli (eco), H. influenzae (hin), B. subtilis (bsu), V. cholerae (vch), D. radiodurans (dra), Rickettsia prowazekii (rpr), C. trachomatis (ctr), C. pneumoniae (cpn), M. genitalium (mge), Mycoplasma pneumoniae (mpn), B. burgdorferi (bbu), T. pallidum (tpa), H . pylori (hpy), M. tuberculosis (mtu), Synechocystis (scy), A. aeolicus (aae), T. maritima (tma), M. jannaschii (mja), Methanobac terium themoautotrophicum (mth), A. fulgidus (afu), P. horikoshii (pho), and P. abyssi (pab).
Codon usages along the gene and expression levels.
For relatively long genes (≥600 codons long), we determined expression levels with the gene length divided into three equal parts (5′, middle, and 3′ parts). The pairwise correlations among the three parts of the E. coli genes are high, 0.86, 0.85, and 0.88, respectively, indicating that expression levels calculated from codon biases are effectively the same for the three parts of genes.
Independent of gene size, we observed (20) that the middle and 3′ end of the genes show quite similar codon frequencies, whereas the 5′ third-codon ensemble possesses somewhat different codon frequencies. This finding may reflect differences in translation initiation versus later stages of translation elongation. A prominent example concerns encoding of arginine with major codons (CGN) versus minor codons (AGR). The AGR codons are scarce in E. coli genes and are restricted mostly to the 5′ end of the genes (especially to the initial 30 bp), whereas CGN codons are preferred elsewhere in the genes (6).
PHX ORFs shared by the four fast-growing genomes.
Genes are considered homologous if their SSPA (significant segment pair alignment) score (percent similarity; see reference 5) is ≥40%. Examples include three ORFs (yaaH, yajC, and yeeX) common to E. coli and V. cholerae, three similar ORFs (yfiD, yjjK, and yebC) present in the genomes of E. coli, V. cholerae, and H. influenzae, respectively, and one ORF (ybaB) common to E. coli and B. subtilis. These PHX genes of unknown function offer attractive candidates for mutagenesis and knockout studies to determine their functions.
Distributions of PHX genes over the chromosomes.
Clusters of PHX genes are displayed in Table 15. Statistical significance was assessed using the r-scan analysis protocol described elsewhere (18).
TABLE 15.
Clusters of PHX genes
| Organism | Positiona (kb) | Gene(s) |
|---|---|---|
| E. coli (oriC at kb 3,923) | 190 → 194 | rps2, tsf, pyrH, frr |
| 447 ← 450 | cyoB, cyoC, cyoD; aerobic respiration | |
| 763 ← 775 | TCA cycle genes: gltA, sdhA, sucA, sucB, sucC, sucD | |
| 1279 → 1285 | Respiratory nitrate reductase α, β, and γ chains; use nitrate as electron acceptor during anaerobic growth | |
| 1793 ← 1801 | tRNA synthetase genes: pheT, pheS, rpL20, thrS | |
| 2385 ← 2402 | NADH dehydrogenase I chains C, F, G, I, L, and N | |
| 3306 ← 3315 | nusA, infB, ribosome binding factor A, pnp, deaD | |
| 3437 ← 3451 | Elongation factors tufA and fusA, 2 PPIase genes, fkpA, slyD, 2 RP genes | |
| 3468 ← 3476 | Long RP operon | |
| 3905 ← 3918 | ATP synthase subunits b, α, and β, almS, pstS, pstB | |
| 4174 → 4183 | tufB, nusG, rpL11, rpL1, rpL10, rpL7/L12, rpoB, rpoC | |
| 4376 ← 4380 | Fumarate reductase operon | |
| H. influenzae (oriC near kb 603) | 180 → 184 | nif, ORF, nqrE, ORF (on lagging strand) |
| 530 ← 542 | ORF, deoD, rpL11, rpL1, rpoB, rpoC | |
| 596 ← 600 | rpS12, rpS7, fusA, tufB | |
| 838 → 853 | 20 RP operons | |
| 883 ← 885 | Fumarate reductase (lagging strand) | |
| 1300 ← 1306 | Proteins of pyruvate dehydrogenase complex | |
| B. subtilis (oriC is near kb 0) | 118 → 154 | 26 RP genes and genes rpoB, rpoC, fus, tufA, and rpoA |
| 1528 → 1531 | Pyruvate dehydrogenases E1, α, β, and E3, pdhC | |
| 3475 ← 3482 | five major glycolysis genes: eno, pgm, tpi, pgk, and gap | |
| 3798 ← 3811 | Ctp synthetase ctrA, fba, transaldolase, malate oxidoreductase | |
| V. cholerae long chromosome I (2.96 Mb, oriC located at about kb 10) | 358 → 379 | 4 RP genes, 1 copy of tuf; secE, rpoB, rpoC |
| 401 → 409 | 4 RP genes, second copy of tuf | |
| 817 ↔ 825 | 5 Genes of mixed transcription orientation | |
| 2266 ← 2268 | Succinyl-CoA synthetases α and β | |
| 2468 ← 2472 | Na+-translocating NADH: ubiquinone oxidoreductase units B, C, D, E, and F | |
| 2772 ← 2788 | 19 RP genes surrounded by 2 PPIase genes with rpoA enclosed | |
| 2848 ↔ 2861 | 7 genes of mixed orientations, including 2 units of fumarate reductase, plus groEL, tpi, efp, and secB |
Arrows indicate transcription orientation.
The PHX genes in each cluster generally possess the same transcription orientation, mostly that of the leading strand. However, E. coli features the PHX fumarate reductase operon genes (kb 4380 → 4376) frdD, frdB, and frdA untypically located in the lagging strand (the direction of transcription is indicated by the arrow). The genes encoding the principal units of NADH dehydrogenase I, N, L, I, G, F, and C cover positions 2402 → 2387 (about a 5-kb extent) on the leading strand.
The PHX gene clusters of E. coli, apart from the segments at kb 450 → 447 and kb 4380 → 4376 of the cytochrome o ubiquinol oxidase operon and the fumarate reductase operon, respectively, are all located in the leading strand. Note that the two RP clusters near oriC (kb 3476 → 3437 and kb 4174 → 4183) include a number of TF genes and some PPIase genes. There are no extended intervals devoid of PHX genes in the E. coli genome.
The V. cholerae large chromosome contains two significantly long segments, at kb 43 to 327 and kb 1657 to 1985, each devoid of PHX genes and positioned antipodal in the chromosome. The main PHX clusters correspond to long RP operons located in the leading strand. These descriptions indicate that PHX genes are irregularly distributed in the V. cholerae chromosomes. The V. cholerae genome has two chromosomes (chromosome I, 2.96 Mb, and chromosome II, 1.07 Mb) containing 138 PHX genes and 14 PHX genes, respectively. The PHX genes in the large chromosome comprise 7% of its genes. V. cholerae has a single PHX RP gene on chromosome II.
In H. influenzae, the PHX clusters are of RP genes and protein synthesis genes.
B. subtilis contains a PHX cluster which features a conglomerate of 27 RP genes (kb 118 → 154) intermeshed with the protein synthesis genes rpoB, rpoC, fus, tuf, and rpoA. A compact operon of PHX genes distinguishes five glycolysis genes (kb 3482 → 3475), enolase (eno), phosphoglycerate mutase (pgm), triosephosphate isomerase (tpi), phosphoglycerate kinase (pgk), and glyceraldehyde-3-phosphate dehydrogenase (gap), located in the leading strand. The cluster at kb 3475 → 3482 ostensibly renders the main glycolysis genes highly efficient, putatively making it less important to express many respiration genes. All clusters are located in the leading strand. B. subtilis also has a 245-kb stretch devoid of PHX genes, at kb 35 to 280.
DISCUSSION
Gene expression can be evaluated in several ways. One currently popular way centers on DNA microarrays (DNA chips) aiming to dissect gene expression under varied physiological, clinical, and environmental conditions. These DNA chips have been applied to the monitoring of genes in different situations for the discovery of genes associated with diseases; for assessment of gene expression under inducements from drugs, chemicals, or toxins; for ascertainment of genes compensatory for knockout mutations; and for profiling of gene expression patterns in temporal and tissue-specific localizations. The current microarray methodology is restricted to discriminating transcription levels and not levels of translation or protein abundances (33, 42). Also, DNA chip hybridizations are generally unable to detect unambiguously low-abundance gene transcripts. Experimental evaluations of protein abundances under different cellular conditions can be assayed by 2D gel electrophoresis (reviewed in reference 46) supplemented by mass spectrometry (51), by antibody associations, and by biochemical tests. Also, correlations of 2D gel proteomes and microarray assessments of transcriptomes generally appear to be weak (13). However, Futcher and coworkers (11) reexamined these correlations in yeast and found generally good agreement.
Codon choice is presumably influenced by protein structure via evolutionary selection for the most accurately translated sequences at structurally important locations. Codon choices may be different at the beginning of a gene than at the central part of the gene (6). It has been suggested that translation pause sites, especially early in the coding sequence, can slow translation initiation (16). Accordingly, there appear to be conflicting selection pressures imposed by constraints on ribosomal binding for the rate of initiation, rate of elongation, and overall translation fidelities. In rapidly growing cells, where ribosomes are limiting for protein synthesis, a ribosome stalled at a rare codon is unavailable for the synthesis of other proteins, and the higher the molar abundance of the stalled protein, the greater the disruption of cellular growth (52). Protein structure may be correlated with codon usage (e.g., see references 30 and 44). Thanaraj and Argos (44) argue the rare-codon hypothesis for domains and secondary structures, in which repetition of rare codons reduces translation rates and introduces translation pauses, allowing time for protein domains and secondary structures to fold into native structural conformations.
Codon usage offers another way to evaluate gene expression with a different set of limitations. Our sequence methods are effectively complementary to the experimental procedures of 2D gel electrophoresis and DNA microarray analysis in assessing gene expression levels. By our methods, genes similar in codon frequencies to RP, TF, and CH genes but strongly deviant in codon usage from the average gene are identified as PHX. Our analyses and data support the hypothesis that each genome has evolved codon usage patterns indicating “optimal” gene expression levels for most situations of its habitat, energy sources, and lifestyle. The three protein families—ribosomal proteins, major translation/transcription processing factors, and chaperone/degradation proteins—are fundamental at many stages of the cell life in promoting growth and stability. Generally, PHX genes exploit favorable codon usages, tend to possess strong Shine-Dalgarno sequences, and putatively possess strong promoter sequences (cf. reference 21). Some limitations of our method result from an implicit assumption that the codon usage of a gene is not affected by its location in the genome, e.g., G+C-rich versus A+T-rich regions. The high variance of G+C composition (isochores) along mammalian genomes may be prohibitive with respect to predicting gene expression levels from codon usages. However, the nucleotide compositions of bacterial genomes are largely homogeneous. Some genes that deviate in G+C content (e.g., those for transposases or specialized pathogenicity islands) tend to be detected as “putative alien” genes (22, 28).
What does the expression level E(g) for a gene g reflect? Gene expression in prokaryotes is regulated at initiation, elongation, and termination of transcription and of translation, by different rates of transcription and translation, by differential mRNA stabilities, by segmental stability differences in polycistronic messages, by codon preferences, and by interactions with chaperone and other proteins. Expression is also influenced by lifestyle, habitat, and energy sources. The classes (RP, TF, and CH) of proteins that we have chosen to represent highly expressed genes are needed in high molar abundances when a high rate of protein synthesis is essential.
Multifunctional proteins and PHX levels
A protein that belongs to a PHX class and that performs several functions might be expected to register higher E(g) values than the average PHX gene. We offer several examples.
Polynucleotide phosphorylase (Pnp) is fundamental in RNA processing and mRNA degradation, and the gene attains the highest E(g) value, 2.66, among all the E. coli genes. Pnp is also a component of the mRNA degradosome, which involves RNase E, DnaK, RhlB helicase, and enolase (27). RNase E is also PHX in E. coli, with an E(g) value of 1.22, but it is not PHX in H. influenzae and V. cholerae and it is missing from B. subtilis. As an important multifunctional protein, Pnp is expected to be PHX at an increased level. The Pnp gene also has the highest E(g) value among all the genes in B. burgdorferi. This gene is also significantly PHX in the genomes of H. influenzae, V. cholerae, Synechocystis, M. tuberculosis, T. pallidum, Chlamydia pneumoniae, A. aeolicus, and T. maritima.
Enolase obtains the very high E(g) values of 2.11 in E. coli, 1.93 in H. influenzae, 1.59 in V. cholerae, and 1.92 in B. subtilis. Again, enolase is multifunctional, acting in energy metabolism (glycolysis) and partly in RNA degradation.
The enzyme aconitate hydratase (aconitase) interconverts citrate and isocitrate in the TCA cycle. Aconitase also serves as a sensor, detecting changes in the redox state and assaying iron content within the cell (36). This protein can further function as a transcriptional activator that specifically regulates gene expression for the transferrin receptor and controls quantities of ferritin (2). At its iron sulfur center, aconitase can be inactivated by oxidative stress or iron deprivation. Aconitase has the highest E(g) value, 2.56, in D. radiodurans (see also reference 22), and its gene is PHX in many genomes.
Apart from structural roles in ribosome formation, several ribosomal proteins act in multifunctional capacities (50). For example, the S9 protein is an accessory protein functioning in DNA repair (49). The E(g) values for S9 in the four fast-growing bacteria studied here are all >1.50, particularly ≥1.90 in E. coli and V. cholerae, significantly higher than the average ribosomal protein E(g) value. The L25 ribosomal protein (93 aa in E. coli) is homologous to the general stress protein (Ctc). This protein achieves the very high E(g) values of 1.90 in E. coli and 1.89 in D. radiodurans. Ctc is PHX also in C. trachomatis, Campylobacter jejuni, H. pylori, T. maritima, and A. aeolicus, none of which carries the L25 gene. In contrast, Ctc is absent in E. coli, V. cholerae, and H. influenzae, but their genomes encode the L25 ribosomal protein. In almost all genomes, the Ctc and L25 protein genes are mutually exclusive. The large ribosomal protein S1 gene is almost always among the top levels of eubacterial PHX genes. We conjecture that the S1 protein (generally ≥500 aa) possesses multifunctional activity yet to be determined. Interestingly, the S1 protein is composed of repetitions of an 86-aa element, usually involving six or more copies.
Other multifunctional PHX proteins from many genomes include glyceraldehyde-3-phosphate dehydrogenase, acting primarily in the first step of the second phase of glycolysis. This protein is very promiscuous, showing uracil DNA glycosylase activity, and binds to tRNA and DNA and to proteins with glutamine repeats. In eukaryotes, it also structurally binds filaments of actin and microtubules (39).
The elongation factor EF-1α is an essential component of the translation apparatus and also has a major function in severing microtubules (38). Phosphoglycerate kinase also functions as a disulfide reductase (25). Many different metabolic proteins serve as crystalline components for the lenses of different animal eyes; these include PPIases, aldehyde dehydrogenase, arginosuccinate lyase, enolase, and aldose reductase.
Contrasts in PHX levels among genes involved in energy metabolism in E. coli and B. subtilis.
As indicated earlier, certain genes of energy metabolism are predominantly PHX in all four fast-growing bacteria and have high expression levels [E(g), often >2.00]. This is manifestly valid (Table 9) for glycolysis genes and for genes of pyruvate oxidation. Why should most of the TCA genes of E. coli be PHX but not those of B. subtilis? We suggest four possible contributing causes. (i) Perhaps B. subtilis makes less use of the TCA cycle for ATP production than E. coli. The principal glycolysis genes of B. subtilis, unlike those of E. coli (dispersed all over the E. coli genome), are encoded from a single cluster (gap, pgk, tpi, pgm, and eno); see our earlier discussion of PHX clusters. (ii) The TCA cycle has at least two main tasks: the first, aerobic energy (ATP) production, and the second, synthesis of carbon chain precursors to various essential metabolites, such as amino acids. Can many of these precursors be more easily acquired by other means in B. subtilis? B. subtilis, in marked contrast to E. coli, has four PHX flagellin genes (flagellin [hap], flagellar hook protein [flgE], flagellar hook basal body [fliE], and flagellin homolog [yvzB]), whereas a single flagellin gene of E. coli is PHX (21). Moreover, flagellar genes are strictly regulated and inducible in E. coli but constitutive in B. subtilis (40). Assuming that soil is the primary B. subtilis habitat and that the human gut is the primary habitat for E. coli, different metabolic patterns may be appropriate. The swimming movements of B. subtilis mediated by its PHX flagellar proteins may facilitate the acquisition of nutrients, such as amino acids, from an assortment of soil sources. B. subtilis also excretes many digestive enzymes in gathering macromolecular nutrients for possible predatory objectives (1). (iii) There are also differences between E. coli and B. subtilis in energy pathways, which can influence expression levels. For example, E. coli uses succinyl-CoA as a precursor in the biosynthesis of lysine and methionine, whereas B. subtilis uses acetyl-CoA for this objective. E. coli possesses isocitrate lyase (AceA) in competition with isocitrate dehydrogenase, the first enzyme of the glyoxylate shunt pathway, which is very effective for acquiring a net carbon gain in the metabolism of fatty acids, whereas B. subtilis lacks AceA. The early genes in the TCA cycle of B. subtilis, those for aconitase and isocitrate dehydrogenase, are PHX, whereas the remaining genes are only predicted moderately expressed. Apparently, the order of TCA genes can be important. (iv) B. subtilis and E. coli are both facultative aerobic organisms (29). For anaerobic respiration, B. subtilis relies exclusively on nitrate or nitrite as its terminal electron acceptor, whereas E. coli has many alternative acceptors.
Highly expressed genes under varying conditions.
Can our methods be applied in conjunction with microarray analysis? We cannot change the codon usage of a given gene, but we can change the gene class standards for discerning expression levels relative to these gene classes (see Materials and Methods). Here, the gene class standards are RP, TF, and CH. It is hypothesized that similarity of codon usages, as characterized in Materials and Methods, for two or more natural gene classes may identify new genes with similar properties, as in the defining gene classes. Effectively, codon usage patterns provide a means to correlate genes and functional categories (20). By using several gene classes as standards, a figure corresponding to Fig. 1 but in multiple dimensions, when coupled to a suitable clustering analysis, may discriminate additional genes highly expressed relative to the different gene class standards. For example, when we compare codon usages of genes with respect to the B. subtilis sporulation genes versus the class of all genes, the two coordinates plot a straight line. In another example, yeast mitochondrial genes feature a melange of PHX genes, putative alien genes, and average genes, and the genes for the ribosomal proteins functioning in the mitochondrion tend to show codon usages akin to average genes.
ACKNOWLEDGMENTS
We thank G. Miklos, F. Neidhardt, A. L. Sonenshein, and A. Spormann for valuable discussions on the manuscript.
This work was supported in part by NIH grants 5R01GM10452-35 and 5R01HG00335-12.
REFERENCES
- 1.Aizawa, S.-I., I. B. Zhulin, L. Marquez-Magana, and G. W. Ordal. Chemotaxis and motility in Bacillus subtilis. In A. L. Sonenshein (ed.), Bacillus subtilis, 2nd ed., in press. ASM Press, Washington, D.C.
- 2.Alén C, Sonenshein A L. Bacillus subtilis aconitase is an RNA-binding protein. Proc Natl Acad Sci USA. 1999;96:10412–10417. doi: 10.1073/pnas.96.18.10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antelmann H, Bernhardt J, Schmid R, Mach H, Volker U, Hecker M. First steps from two-dimensional protein index towards a response regulation map for Bacillus subtilis. Electrophoresis. 1997;18:1451–1463. doi: 10.1002/elps.1150180820. [DOI] [PubMed] [Google Scholar]
- 4.Bocharov E V, Gudkov A T, Budovskaya E V, Arseniev A S. Conformational independence of N- and C-domains in ribosomal protein L7/L12 and in the complex with protein L10. FEBS Lett. 1998;423:347–350. doi: 10.1016/s0014-5793(98)00121-5. [DOI] [PubMed] [Google Scholar]
- 5.Brocchieri L, Karlin S. A symmetric-iterated multiple alignment of protein sequences. J Mol Biol. 1998;276:249–264. doi: 10.1006/jmbi.1997.1527. [DOI] [PubMed] [Google Scholar]
- 6.Chen G T, Inouye M. Role of the AGA/AGG codons, the rarest codons in global gene expression in Escherichia coli. Genes Dev. 1994;8:2641–2652. doi: 10.1101/gad.8.21.2641. [DOI] [PubMed] [Google Scholar]
- 7.Dai Y, Wang C. A mutant truncated protein disulfide isomerase with no chaperone activity. J Biol Chem. 1997;272:27572–27576. doi: 10.1074/jbc.272.44.27572. [DOI] [PubMed] [Google Scholar]
- 8.Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- 9.Eisen J A, Hanawalt P C. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat Res. 1999;435:171–213. doi: 10.1016/s0921-8777(99)00050-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eyre-Walker A. Synonymous codon bias is related to gene length in Escherichia coli: selection for translational accuracy. Mol Biol Evol. 1996;13:864–872. doi: 10.1093/oxfordjournals.molbev.a025646. [DOI] [PubMed] [Google Scholar]
- 11.Futcher B, Latter G I, Monardo P, McLaughlin C S, Garrels J I. A sampling of the yeast proteome. Mol Cell Biol. 1999;19:7357–7368. doi: 10.1128/mcb.19.11.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez J C, Peariso K, Penner-Hahn J E, Matthews R G. Cobalamin-independent methionine synthase from Escherichia coli: a zinc metalloenzyme. Biochemistry. 1996;35:12228–12234. doi: 10.1021/bi9615452. [DOI] [PubMed] [Google Scholar]
- 13.Gygi S P, Rochon Y, Franza B R, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Handy J, Doolittle R F. An attempt to pinpoint the phylogenetic introduction of glutaminyl-tRNA synthetase among bacteria. J Mol Evol. 1999;49:709–715. doi: 10.1007/pl00006592. [DOI] [PubMed] [Google Scholar]
- 15.Herrmann J M, Malkus P, Schekman R. Out of the ER: outfitters, escorts and guides. Trends Cell Biol. 1999;9:5–7. doi: 10.1016/s0962-8924(98)01414-7. [DOI] [PubMed] [Google Scholar]
- 16.Irwin B, Heck J D, Hatfield G W. Codon pair utilization biases influence translational elongation step times. J Biol Chem. 1995;270:22801–22806. doi: 10.1074/jbc.270.39.22801. [DOI] [PubMed] [Google Scholar]
- 17.Jones C H, Danese P N, Pinkner J S, Silhavy T J, Hultgren S J. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J. 1997;16:6394–6406. doi: 10.1093/emboj/16.21.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlin S, Macken C. Some statistical problems in the assessment of inhomogeneities of DNA sequence data. J Am Stat Assoc. 1991;86:27–35. [Google Scholar]
- 19.Karlin S, Campbell A M, Mrázek J. Comparative DNA analysis across diverse genomes. Annu Rev Genet. 1998;32:185–225. doi: 10.1146/annurev.genet.32.1.185. [DOI] [PubMed] [Google Scholar]
- 20.Karlin S, Mrázek J, Campbell A M. Codon usages in different gene classes of the Escherichia coli genome. Mol Microbiol. 1998;29:1341–1355. doi: 10.1046/j.1365-2958.1998.01008.x. [DOI] [PubMed] [Google Scholar]
- 21.Karlin S, Mrázek J. Predicted highly expressed genes of diverse prokaryotic genomes. J Bacteriol. 2000;182:5238–5250. doi: 10.1128/jb.182.18.5238-5250.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karlin S, Mrázek J. Predicted highly expressed and putative alien genes of Deinococcus radiodurans and implications for resistance to ionizing radiation damage. Proc Natl Acad Sci USA. 2001;98:5240–5245. doi: 10.1073/pnas.081077598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ladenstein R, Schneider M, Huber R, Bartunik H D, Wilson K, Schott K, Bacher A. Heavy riboflavin synthase from Bacillus subtilis. Crystal structure analysis of the icosahedral beta 60 capsid at 3.3 A resolution. J Mol Biol. 1988;203:1045–1070. doi: 10.1016/0022-2836(88)90128-3. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence J G, Roth J R. Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics. 1996;143:1843–1860. doi: 10.1093/genetics/143.4.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lay A J, Jiang X M, Kisker O, Flynn E, Underwood A, Condron R, Hogg P J. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature. 2000;408:869–873. doi: 10.1038/35048596. [DOI] [PubMed] [Google Scholar]
- 26.Matin A, Baetens M, Pandza S, Park C H, Waggoner S. Survival strategies in stationary phase. In: Rosenberg E, editor. Microbial ecology and infectious diseases. Washington, D.C.: American Society for Microbiology; 1999. pp. 32–48. [Google Scholar]
- 27.Miczak A, Kaberdin V R, Wei C L, LinChao S. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci USA. 1996;93:3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mrázek J, Bhaya D, Grossman A R, Karlin S. Highly expressed and alien genes of the Synechocystis genome. Nucleic Acids Res. 2001;29:1590–1601. doi: 10.1093/nar/29.7.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano M M, Zuber P. Anaerobic growth of a “strict aerobe” (Bacillus subtilis) Annu Rev Microbiol. 1998;52:165–190. doi: 10.1146/annurev.micro.52.1.165. [DOI] [PubMed] [Google Scholar]
- 30.Netzer W J, Hartl F U. Protein folding in the cytosol: chaperonin-dependent and -independent mechanisms. Trends Biochem Sci. 1998;23:68–73. doi: 10.1016/s0968-0004(97)01171-7. [DOI] [PubMed] [Google Scholar]
- 31.Oberto J, Bonnefoy E, Mouray E, Pellegrini O, Wikstrom P M, Rouviere-Yaniv J. The Escherichia coli ribosomal protein S16 is an endonuclease. Mol Microbiol. 1996;19:1319–1330. doi: 10.1111/j.1365-2958.1996.tb02476.x. [DOI] [PubMed] [Google Scholar]
- 32.Raivio T L, Popkin D L, Silhavy T J. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J Bacteriol. 1999;181:5263–5272. doi: 10.1128/jb.181.17.5263-5272.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richmond C S, Glasner J D, Mau R, Jin H, Blattner F R. Genome-wide expression profiling in Escherichia coli K-12. Nucleic Acids Res. 1999;27:3821–3835. doi: 10.1093/nar/27.19.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rocha E P, Danchin A, Viari A. Translation in Bacillus subtilis: roles and trends of initiation and termination, insights from a genome analysis. Nucleic Acids Res. 1999;27:3567–3576. doi: 10.1093/nar/27.17.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rolland N, Janosi L, Block M A, Shuda M, Teyssier E, Miege C, Cheniclet C, Carde J P, Kaji A, Joyard J. Plant ribosome recycling factor homologue is a chloroplastic protein and is bactericidal in Escherichia coli carrying temperature-sensitive ribosome recycling factor. Proc Natl Acad Sci USA. 1999;96:5464–5469. doi: 10.1073/pnas.96.10.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rouault T A, Klausner R D. Iron-sulfur clusters as biosensors of oxidants and iron. Trends Biochem Sci. 1996;21:174–177. [PubMed] [Google Scholar]
- 37.Selmer M, Al-Karadaghi S, Hirokawa G, Kaji A, Liljas A. Crystal structure of Thermotoga maritima ribosome recycling factor: a tRNA mimic. Science. 1999;286:2349–2352. doi: 10.1126/science.286.5448.2349. [DOI] [PubMed] [Google Scholar]
- 38.Shiina N, Gotoh Y, Kubomura N, Iwamatsu A, Nishida E. Microtubule severing by elongation factor 1 alpha. Science. 1994;266:282–285. doi: 10.1126/science.7939665. [DOI] [PubMed] [Google Scholar]
- 39.Sirover M A. New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim Biophys Acta. 1999;1432:159–184. doi: 10.1016/s0167-4838(99)00119-3. [DOI] [PubMed] [Google Scholar]
- 40.Soutourina O, Kolb A, Krin E, Laurent-Winter C, Rimsky S, Danchin A, Bertin P. Multiple control of flagellum biosynthesis in Escherichia coli: role of H-NS protein and the cyclic AMP-catabolite activator protein complex in transcription of the flhDC master operon. J Bacteriol. 1999;181:7500–7508. doi: 10.1128/jb.181.24.7500-7508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takusagawa F, Kamitori S, Misaki S, Markham G D. Crystal structure of S-adenosylmethionine synthetase. J Biol Chem. 1996;271:136–147. [PubMed] [Google Scholar]
- 42.Tao H, Bausch C, Richmond C, Blattner F R, Conway T. Functional genomics: expression analysis of Escherichia coli growing on minimal and rich media. J Bacteriol. 1999;181:6425–6440. doi: 10.1128/jb.181.20.6425-6440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teter S A, Houry W A, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P, Georgopoulos C, Hartl F U. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell. 1999;97:755–765. doi: 10.1016/s0092-8674(00)80787-4. [DOI] [PubMed] [Google Scholar]
- 44.Thanaraj T A, Argos P. Ribosome-mediated translational pause and protein domain organization. Protein Sci. 1996;5:1594–1612. doi: 10.1002/pro.5560050814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.VanBogelen R A, Abshire K Z, Pertsemlidis A, Clark R L, Neidhardt F C. Gene-protein database of Escherichia coli K-12, edition 6. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C.: ASM Press; 1996. pp. 2067–2117. [Google Scholar]
- 46.VanBogelen R A, Schiller E E, Thomas J D, Neidhardt F C. Diagnosis of cellular states of microbial organisms using proteomics. Electrophoresis. 1999;20:2149–2159. doi: 10.1002/(SICI)1522-2683(19990801)20:11<2149::AID-ELPS2149>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 47.Wang C C, Tsou C L. Protein disulfide isomerase is both an enzyme and a chaperone. FASEB J. 1993;7:1515–1517. doi: 10.1096/fasebj.7.15.7903263. [DOI] [PubMed] [Google Scholar]
- 48.Warner J R. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 49.Woodgate R, Rajagopalan M, Lu C, Echols H. UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc Natl Acad Sci USA. 1989;86:7301–7305. doi: 10.1073/pnas.86.19.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wool I G. Extraribosomal functions of ribosomal proteins. Trends Biochem Sci. 1996;21:164–165. [PubMed] [Google Scholar]
- 51.Yates J R., III Mass spectrometry. From genomics to proteomics. Trends Genet. 2000;16:5–8. doi: 10.1016/s0168-9525(99)01879-x. [DOI] [PubMed] [Google Scholar]
- 52.Zahn K. Overexpression of an mRNA dependent on rare codons inhibits protein synthesis and cell growth. J Bacteriol. 1996;178:2926–2933. doi: 10.1128/jb.178.10.2926-2933.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]