

Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is known to present with pulmonary and extra‐pulmonary organ complications. In comparison with the 2009 pandemic (pH1N1), SARS‐CoV‐2 infection is likely to lead to more severe disease, with multi‐organ effects, including cardiovascular disease. SARS‐CoV‐2 has been associated with acute and long‐term cardiovascular disease, but the molecular changes that govern this remain unknown. In this study, we investigated the host transcriptome landscape of cardiac tissues collected at rapid autopsy from seven SARS‐CoV‐2, two pH1N1, and six control patients using targeted spatial transcriptomics approaches. Although SARS‐CoV‐2 was not detected in cardiac tissue, host transcriptomics showed upregulation of genes associated with DNA damage and repair, heat shock, and M1‐like macrophage infiltration in the cardiac tissues of COVID‐19 patients. The DNA damage present in the SARS‐CoV‐2 patient samples, were further confirmed by γ‐H2Ax immunohistochemistry. In comparison, pH1N1 showed upregulation of interferon‐stimulated genes, in particular interferon and complement pathways, when compared with COVID‐19 patients. These data demonstrate the emergence of distinct transcriptomic profiles in cardiac tissues of SARS‐CoV‐2 and pH1N1 influenza infection supporting the need for a greater understanding of the effects on extra‐pulmonary organs, including the cardiovascular system of COVID‐19 patients, to delineate the immunopathobiology of SARS‐CoV‐2 infection, and long term impact on health.
Keywords: cardiac, COVID‐19, SARS‐CoV‐2, spatial profiling, transcriptomic
Cardiac tissues collected at rapid autopsy from COVID‐19 and pandemic H1N1 patients were profiled using digital spatial profiling technology. The transcriptional profiles were compared and contrasted in the study to understand the impact of viral infection. Host transcriptomics showed distinct transcriptional profiles, such as DNA damage and repair which were upregulated in COVID‐19. The study highlights the need to understand the pathobiology of severe acute respiratory syndrome coronavirus 2 infection and the multi‐organ effects.
INTRODUCTION
Since 2019, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has led to the recognition of the emergence of broad‐spectrum multi‐organ disease with increasing prevalence of cardiac injury in hospitalized patients. SARS‐CoV‐2 has been associated with a wide range of cardiac complications. Specifically, the initial infection stage has been associated with acute myocardial injury [1, 2, 3] with abnormal echocardiography findings in both left and right ventricles [4], arrhythmias [1, 2], palpitations [5], myocarditis [6], heart failure [7] and other de novo problems [8, 9]. Paralleling these clinical symptoms is a growing body of evidence of increased cardiac complications in patients who have recovered from COVID‐19 [10]. Signs of persistent cardiac‐related issues among individuals who have suffered from severe disease during the acute infection appear to be relatively common. These include continued chest pain [11], palpitations [12], abnormal left ventricular function [13], myocardial infarction [13], late gadolinium enhancement (LGE) [13, 14] and ischaemia [13]. Ongoing myocardial inflammation and LGE [15] have also been reported even in patients who recovered from relatively mild, or even asymptomatic, COVID‐19. Individuals who have recovered from mild COVID‐19 suffer an increased burden of arrhythmias, chest pain, heart failure and vascular complications compared to uninfected patients, and this sequelae has been accompanied by excess use of drug therapies [16]. Currently, it remains unclear how long‐term cardiac complications from COVID‐19 will persist in convalescent patients. Studies have shown that the risk and 1‐year burden of cardiovascular disease is substantial in survivors of acute COVID‐19 [17].
Virus‐induced cardiac complications are not unique to SARS‐CoV‐2 infection. Influenza A virus (IAV) infection has been frequently associated with myocardial injury and infarction, endocarditis, tachycardia, ST segment echocardial changes and atrial fibrillation [18, 19], mostly resolving within a year of infection [19]. The mechanisms by which respiratory viruses may cause cardiac complications are manifold and are also not fully characterized [20]. Thus, it is currently unclear if SARS‐CoV‐2 and IAVs induce these complications via similar or distinct pathways.
Evidence of SARS‐CoV‐2 direct cardiac infection remains equivocal. In vitro studies show infection and replication within human pluripotent stem cell (hPSC)‐derived cardiomyocytes [21, 22], while hPSC‐smooth muscle cells remain uninfected [23]. Autopsies of 39 patients with COVID‐19 detected SARS‐CoV‐2 negative sense RNA indicative of active viral replication in the myocardium of only five patients with the highest viral loads [24]. In autopsies of a further 41 patients, SARS‐CoV‐2+ cells in the myocardium were rare despite viral RNA being detected in 30 hearts [25]. Furthermore, smaller studies of autopsy specimens from five and eight COVID‐19 patients failed to detect SARS‐CoV‐2 within the heart entirely, even when patients displayed severe myofibrillar anomalies [21, 26]. In IAV infection, it is equally uncertain whether early cardiomyocyte damage is linked primarily to virus presence or a secondary consequence of the immune response in IAV infection. IAV has been reported to replicate within hPSC‐cardiomyocytes [27], and IAV or antigens have been found directly in the heart in mice [27, 28] and humans [29, 30] in small case studies. In contrast, a larger US study of patients with acute viral myocarditis identified IAV RNA in cardiac samples from only 5 of 624 patients (0.8%) using polymerase chain reaction (PCR) analyses [31]. Therefore, the contribution of direct cardiac viral infection to SARS‐CoV‐2 and IAV‐induced cardiac complications remains both contentious and unclear.
One potential mechanistic disparity between influenza virus and SARS‐CoV‐2 induction of cardiac complications is the induction of an interferon (IFN) response in the heart. For example, 2 days post‐IAV infection, the expression of IFN stimulated genes within the heart of IAV‐infected mice was increased 50‐fold in infected compared with uninfected mice [32]. In contrast, several studies have found that both respiratory and extra‐respiratory type I IFN responses are significantly blunted during SARS‐CoV‐2 infection [33, 34]. For example, a Syrian hamster model of SARS‐CoV‐2 infection showed inhibited type I IFN responses in the respiratory tract despite a high burden of replicating virus, accompanied by inflammation in the heart, which also lacked type I IFN upregulation [35]. This may be attributable to SARS‐CoV‐2 production of proteins which suppress type I IFN release [36, 37]. The lack of efficient IFN induction by SARS‐CoV‐2 may be responsible for triggering the observed higher rate of cardiac complications than in more IFN‐stimulatory seasonal IAVs [38, 39]. Indeed, several studies have shown that type I IFNs play a protective role in the development of cardiovascular diseases such as pathological hypertrophy and virally induced left ventricular dysfunction [40, 41]. However, this hypothesis remains to be confirmed.
Transcriptomic analysis of patient myocardial tissue offers a unique opportunity to understand the mechanisms of SARS‐CoV‐2 and IAV‐induced cardiac complications. Specifically, spatial transcriptomics that consider intra‐organ heterogeneity [42, 43], provide a powerful tool for characterizing host responses to respiratory viral infections outside of the respiratory tract. Here, we use targeted spatial transcriptomic characterization of myocardial tissue to generate an in‐depth picture of the myocardial transcriptional landscape of COVID‐19, pandemic H1N1 influenza and uninfected control patients and shed light on the mechanisms that might drive these different clinical outcomes (Figure 1). Our study revealed that DNA damage pathways were enriched for in COVID‐19 tissues, whereas pH1N1 elicited a more inflammatory response in cardiac tissues.
FIGURE 1.
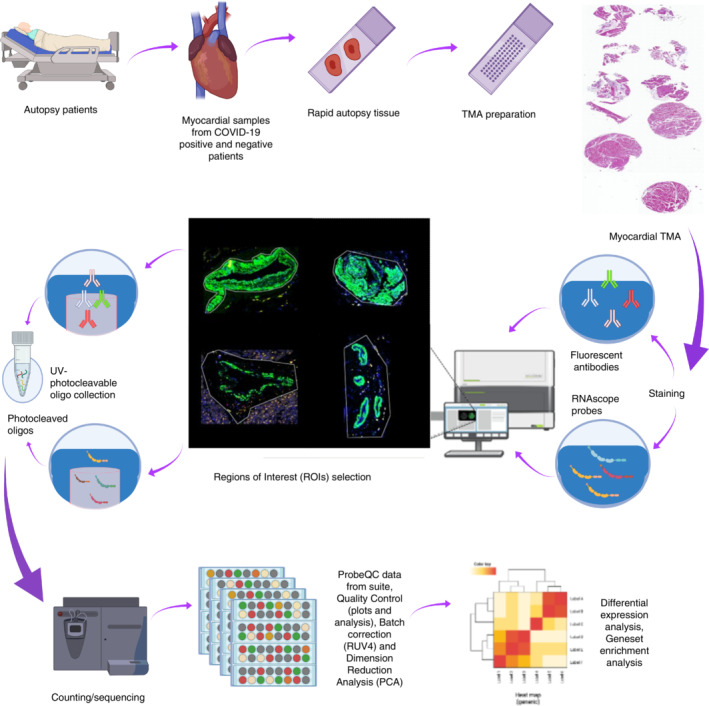
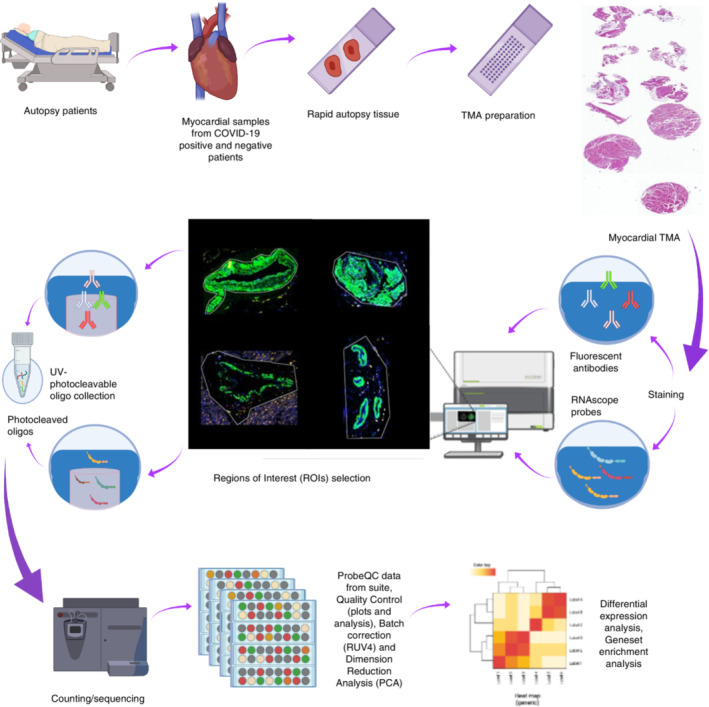
Study schema. Cardiac tissues were collected from COVID‐19 and pH1N1 patients at rapid autopsy. Samples were prepared onto tissue microarrays and profiled using targeted spatial transcriptomics (Immune Atlas Panel; NanoString technologies). Myocardial, blood vessels and mixed populations were captured using ‘region of interest (ROI)’ selection strategies to liberate the transcript data. These data were counted by next generation sequencing to obtain digital expression counts per ROI.
MATERIALS AND METHODS
Study design
Myocardial tissues were obtained from patients at the Pontificia Universidade Catolica do Parana PUCPR in accordance with the National Commission for Research Ethics (CONEP) under ethics approval numbers: protocol number 3.944.734/2020 (for COVID‐19 group), and 2.550.445/2018 (for pH1N1 and control group). Families permitted the post‐mortem biopsy of COVID‐19 and H1N1pdm09 samples and conventional autopsy for the cases of the control group. All SARS‐CoV‐2 and pH1N1 patients were confirmed for infection by RT‐qPCR of nasopharyngeal swab specimens. The control group did not have any indications of other infectious diseases. The study was ratified by The University of Queensland (UQ) Human Research Ethics Committee (HREC) (clearance number: 2020001792/30188020.7.1001.0020).
Tissue preparation and histopathology
Tissue microarray (TMA) was constructed of single cores from seven SARS‐CoV‐2, two pH1N1 and six control/healthy volunteer patients (Table 1) and cut onto positively charged slides (Bond Apex). Sections were stained by Fred Hutch pathology for haematoxylin and eosin and Mason's trichrome. Brightfield images were obtained using the Aperio (Leica Biosystems) slide scanner for histological assessment by a pathologist at The Prince Charles Hospital Pathology Laboratory.
TABLE 1.
Clinicopathological findings for the COVID‐19 patients.
| Patient 1—LN4 | Patient 2—LN6 | Patient 3—LN16 | Patient 4—LN17 | Patient 5—LN20 | Patient 6—LN21 | Patient 7—LN10 | |
|---|---|---|---|---|---|---|---|
| Gender, age (years) | Male, 70–75 | Male, 75–80 | Male, 55–60 | Female, 80–85 | Male, 70–75 | Male, 65–70 | Male, 45–50 |
| Underlying conditions |
Type 2 diabetes mellitus Chronic kidney disease Atrial fibrillation Coronary artery disease Heart failure Peripheral obstructive artery disease |
Arterial hypertension Coronary artery disease Heart failure Class III obesity |
Type 2 Diabetes mellitus Arterial hypertension Coronary artery disease Hepatic steatosis |
Type 2 diabetes mellitus Arterial hypertension Dyslipidemia |
Type 2 diabetes mellitus Arterial hypertension Dyslipidemia Hyperuricemia Coronary artery disease Myocardial infarction (April 2020) |
Type 2 diabetes mellitus Arterial hypertension Atrial fibrillation Interstitial pulmonary Fibrosis Pulmonary hypertension Former smoker |
Dyslipidemia |
| Length of stay on mechanical ventilation | 10 days | 21 days | 9 days | 14 days | 9 days | 15 days | 5 days |
| Chest computed tomography at admission | Diffuse and bilateral ‘opacities with ground‐glass attenuation’, suggestive of viral pulmonary infection | Diffuse and bilateral ‘opacities with ground‐glass attenuation’, suggestive of viral pulmonary infection | Peripheral, multifocal and bilateral ‘opacities with ground‐glass attenuation’, suggestive of viral pulmonary infection. Presence of bronchial thickening. | Diffuse and bilateral ‘opacities with ground‐glass attenuation’, thickening of the pulmonary septum, suggestive of viral pulmonary infection. | Diffuse and bilateral ‘opacities with ground‐glass attenuation’, thickening of the pulmonary septum, suggestive of viral pulmonary infection. Presence of bronchial thickening. Presence of diffuse bilateral bronchiectasis. Presence of parasseptal emphysema. | Peripheral, multifocal and bilateral ‘opacities with ground‐glass attenuation’, suggestive of viral pulmonary infection. Interstitial pulmonary fibrosis. Cardiomegaly. Increased pulmonary artery diameter (32 mm). | Peripheral, multifocal and bilateral ‘opacities with ground‐glass attenuation’, suggestive of viral pulmonary infection |
| Relevant initial laboratory tests |
C‐reactive protein = 83 mg/dl D‐dimer = 3436 μg/ml hs‐Troponin I = 12.6 pg/ml Creatinine = 7.45 mg/dl Globular volume = 25% Haemoglobin = 8.6 g/dl Leukocytes = 9200 |
C‐reactive protein = 52 mg/dl D‐dimer = 816 μg/ml hs‐Troponin I = 10.9 pg/dl Creatinine = 0.74 mg/dl Globular volume = 37.5% Haemoglobin = 12.8 g/dl Leukocytes = 4700 |
C‐reactive protein = 154.2 mg /dl D‐dimer = 628 μg/ml hs‐Troponin I = 3.9 pg/ml Creatinine = 0.82 mg/dl Globular volume = 38.3% Haemoglobin = 14 g/dl Leukocytes = 14 500 |
C‐reactive protein = 199.4 mg/dl D‐dimer = 83 143 μg/ml hs‐Troponin I = 42.1 pg/ml Creatinine = 1.33 mg/dl Globular volume = 41.3% Haemoglobin = 13.7 g/dl Leukocytes = 22 100 |
C‐reactive protein = 267 mg/dl D‐dimer = 152 174 μg/mL hs‐Troponin I = 13.3 pg/ml Creatinine = 1.61 mg/dl Globular volume = 43.1% Haemoglobin = 14.9 g/dl Leukocytes = 13 100 |
C‐reactive protein = 156.9 mg/dl D‐dimer = 1848 μg/ml hs‐Troponin I = 1750.2 pg/ml Creatinine = 1.37 mg/dl Globular volume = 52.8% Haemoglobin = 18.2 g/dl Leukocytes = 16 000 |
|
| Laboratory tests 24 h before death |
C‐reactive protein = 270 mg/dl D‐dimer = 4858 μg/ml hs‐Troponin I = 87.4 pg/dl Creatinine = 5.08 mg/dl Globular volume = 23% Haemoglobin = 8.0 g/dl Leukocytes = 22 000 |
C‐reactive protein = 407 mg/dl D‐dimer = 4507 μg/ml hs‐Troponin I = 32.7 pg/dl Creatinine = 1.81 mg/dl Globular volume = 29.4% Haemoglobin = 9.7 g/dl Leukocytes = 9400 |
C‐reactive protein = 267.2 mg/dl D‐dimer = 6571 μg/ml hs‐Troponin I = 19.9 pg/ml Creatinine = 2.43 mg/dl Globular volume = 27% Haemoglobin = 9 g/dl Leukocytes = 15 300 |
C‐reactive protein = 16.3 mg/dl D‐dimer = 19 137 μg/ml hs‐Troponin I = 324,7 pg/ml Creatinine = 1.06 mg/dl Globular volume = 29.2% Haemoglobin = 9.5 g/dl Leukocytes = 28 900 |
C‐reactive protein = 226.8 mg/dl Troponin = 21.2 μg/ml Creatinine = 2.14 mg/dl Globular volume = 19.7% Haemoglobin = 11 g/dl Leukocytes = 19 100 |
C‐reactive protein = 8.7 mg/dl Troponin = 245,2 pg/ml Creatinine = 1.66 mg/dl Globular volume = 32% Haemoglobin = 11.3 g/dl Leukocytes = 10 500 |
|
| Echocardiogram 24 h before death |
Ejection fraction = 43% Left ventricle = mild eccentric hypertrophy; akinesia of the infero‐lateral and basal lower walls. Right ventricle = increased basal dimension and normal systolic function. sPAP = 68 mmHg. |
Ejection fraction = 65% Left ventricle = preserved dimensions. Right ventricle = preserved dimensions and normal systolic function. sPAP = normal. |
Ejection fraction = 64% Left ventricle = preserved dimensions. Right ventricle = Increased dimensions and slightly reduced systolic function. sPAP = 51 mmHg. |
Ejection fraction = 45%. Left ventricle = mild eccentric hypertrophy; hypokinesia of the lower‐basal and inferoseptal walls. Right ventricle = preserved dimensions and normal systolic function. sPAP = 34 mmHg. |
Ejection fraction = 66% Left ventricle = preserved dimensions. Right ventricle = preserved dimensions and normal systolic function. sPAP = 28 mmHg. |
Ejection fraction = 57% Left ventricle = severe eccentric hypertrophy. Right ventricle = Increased dimensions and compromised systolic function. sPAP = 70 mmHg. |
Data not available |
| Therapeutic drugs |
Hydroxychloroquine Azithromycin Oseltamivir Metronidazole Meropenem Linezolid |
Hydroxychloroquine Azithromycin Oseltamivir Ceftriaxone |
Azithromycin Ceftriaxone Dexamethasone Enoxaparin Piperacillin + tazobactam Alteplase |
Azithromycin Ceftriaxone Oseltamivir Dexamethasone Enoxaparin Piperacillin + tazobactam |
Azithromycin Ceftriaxone Dexamethasone Enoxaparin |
Ceftriaxone Azithromycin Tocilizumabe Methylprednisolone Piperacillin + tazobactam Enoxaparin |
Azithromycin Dexamethasone |
| Invasive procedure | Haemodialysis three times a week | Tracheostomy | Chemical thrombolysis | Tracheostomy | Chest tube right (pneumothorax) | Tracheostomy | Data not available |
Note: Reference values: hs‐Troponin I < 19.8 pg/ml, D‐dimer < 500 μg/ml. The choice of the antibiotics was done according to the diagnosis and protocol for the patient's profile.
Abbreviation: sPAP, systolic pressure in pulmonary artery.
Immunohistochemistry and RNAscope (SARS‐CoV‐2)
Immunohistochemistry was performed on a Leica Bond RX autostainer (Leica Biosystems) with antibody targeting SARS‐CoV‐2 spike protein (ab272504; Abcam) at 2 μg/ml. Heat induced epitope retrieval was performed in buffer ER1 at 100°C for 20 min, and signal visualized with 3,3′‐diaminobenzidine substrate. Slides were imaged using a Zeiss Axioscanner (Carl Zeiss). RNAscope probes (ACDbio) targeting SARS‐CoV‐2 spike mRNA (nCoV2019, #848561‐C3), ACE2 host receptor mRNA (#848151‐C2) and host serine protease TMPRSS2 mRNA (#470341‐C1) were used as per manufacturer instructions for automation on Leica Bond RX. DNA was visualized with Syto13 (Thermo Fisher Scientific), channel 1 with Opal 570 (1:500), channel 2 with Opal 620 (1:1500) and channel 3 with Opal 690 (1:1500) (PerkinElmer). Fluorescent images were acquired with Nanostring Mars prototype DSP at ×20. Anti‐gamma H2A.X staining was performed as above on autostainer with antibody at 4 μg/ml (ab26350). Antigen retrieval was performed in buffer ER1 at 100°C for 10 min, and signal was visualized with Opal 520. Slides were scanned on Vectra Polaris (Akoya Biosciences) and unmixed in Inform (Akoya Biosciences). Image analysis was performed in QuPath [44] to generate H‐scores for γ‐H2AX nuclear staining.
Nanostring digital spatial profiling: COVID‐19 immune atlas panel
A serial section TMA slide, was freshly sectioned and prepared according to the Nanostring GeoMX Digital Spatial Profiler (DSP) slide preparation for RNA profiling (NanoString). Briefly, slides were baked 1 h at 60°C and then processed by Leica Bond RX autostainer. Slides were pre‐treated with proteinase K and then hybridized with mRNA probes in the COVID‐19 Immune Atlas panel with additional SARS‐CoV‐2 probe panel (https://nanostring.com/support-documents/geomx-cancer-transcriptome-atlas-rna-probe-list/). After incubation, slides were washed and then stained with αSMA, CD3, CD68 and Syto83 for 1 h then loaded into the NanoString GeoMX DSP instrument for scanning and region of interest (ROI) selection. ROI selection was guided by morphology markers to capture similar tissue structures across tissue cores where possible. Oligonucleotides linked to hybridized mRNA targets were cleaved and collected for counting using Illumina i5 and i7 dual indexing. PCR reactions were performed with 4 μl of a GeoMx DSP sample. AMPure XP beads (Beckman Coulter) were used at 1.2X bead‐to‐sample ratio for PCR product purification. Paired‐end sequencing (2 × 75) was performed using NextSeq550 up to 400M total aligned reads. Fastq files were processed by DND system and uploaded to GeoMX DSP system where raw and Q3 normalized counts of all targets were aligned with ROIs.
Transcriptomic data analysis
Data used in this study result from a mRNA assay conducted with the NanoString's GeoMx COVID‐19 Immune atlas panel using the GeoMX DSP. The data were measurements of RNA abundance of over 1800 genes, including 22 add‐in COVID‐19‐related genes, 4 SARS‐CoV‐2 specific genes and 2 negative control (SARS‐CoV‐2 Neg; NegProbe) genes and 32 internal reference genes. Transcriptomic measurements were made on regions of interests within each core. Control samples are COVID‐19‐free and pH1N1‐free. In total, 48 ROIs were analysed (16 COVID‐19, 4 pH1N1 and 28 control). Factors considered in this dataset include disease type (COVID‐19, pH1N1 and control), patient of origin, dominant tissue type (blood vessel, mixed vessel/myocardium, myocardium only).
Bioinformatics analyses
Data exploration and quality control
Data exploration and quality controls were conducted on negative probe‐QC count data generated from the Covid‐19 Immune atlas DSP mRNA assay. Data were first rescaled into log2‐transformed count per million (CPM) data to account for library size variation, followed by computing relative log expression (RLE) and principal component analysis (PCA) of the scaled count data to assess the overall distribution, factor variance from the experimental design and the presence of unwanted batch effects across all datasets.
Batch correction and normalization
The dataset comprise of two different experiments, leading to effects of batch between experiments that is required to be considered and eliminated. Batch correction and normalization requires negative control genes which were derived from RNA‐seq count data of atrial appendage, left ventricle and aorta tissues from the Genotype‐Tissue Expression (GTEx) project [45] (2017‐06‐05_v8_RNASeQCv1.1.9). Coefficient of variance (CV) was calculated for each gene after the transformation of raw count to log‐scaled CPM count to account for library size variation. Genes were then sorted based on a z‐score transformation. The top 500 heart‐related GTEx stable genes were intersected with the CTA panel genes used in this study, resulting in 32 overlapped genes (Table S1). The list of negative control genes was further curated with seven genes of potential biological relevance and three genes with a high CV (mean CV [log transformed] over 3.5) within each batch were removed from the list (Table S1 and Figure S1), leaving 22 genes as negative control genes for downstream analyses. RUV4 from the ruv R package [46] was then used to for the normalization using the 22 negative control genes and the factor k set to 3. The result of the normalization was then assessed by computing RLE and PCA of the normalized count data.
Differential expression analysis
Differential expression (DE) analysis was performed using R packages edgeR [47] (v3.34.0) and limma [48] (3.48.0). Briefly, DE was modelled using linear models with experimental factors as predictors. The variation in gene expression was modelled as the combination of a common dispersion that applies to all genes and a gene‐specific dispersion. To estimate the common and gene‐wise variation, the variation of each gene was modelled by borrowing information from all other genes using an empirical Bayes approach while treating the variation of patients as a random effect using DuplicateCorrelation in Limma. The linear model was then fitted to a given experimental design containing the biological factors of interest and the weight matrix from RUV4 normalization to account for unwanted variations. DE was performed for distinct contrasts of interest. The resulting statistic was an empirical Bayes moderated t‐statistic which was more robust than a t‐statistic from a classic t‐test. Multiple testing adjustment was carried out with the Benjamini–Hochberg procedure, adjusted p < 0.05 was used as the threshold to identify significant differentially expressed (DE) genes. In this study, the main factor of interest is disease with the main contrasts investigated as COVID19 versus control, pH1N1 versus control and COVID19 versus pH1N1.
Gene set enrichment analysis
Gene sets from the Molecular Signatures Database [49] (MsigDB, v7.2) Hallmarks, C2 (curated gene sets) and C5 (gene ontology terms) categories and KEGG pathways gene sets were obtained using the getMisgdb and appendKEGG functions from the msigdb R package (v1.1.5) [49]. Gene set enrichment analysis (GSEA) were performed using fry from the limma package. False discovery rate of 0.05 was set as the threshold to determine significantly enriched gene sets. Taking advantage of the fact that most gene sets are related and share genes in common, the results of the GSEA were interrogated and visualized by an unbiased approach using a novel network enrichment and visualization R package vissE [50]. The vissE approach exploits this geneset–geneset overlap relationship to enable interpretation of clusters of gene sets and further summarize results. This approach allows natural clustering of the gene sets to take place, so that significant perturbations can be better visualized while allowing the other less obvious, sensible but unexpected results to be highlighted. To inspect the concordance between the transcriptomic profile of each ROI and DNA damage‐related gene set, we used R package singscore [51].
RESULTS
Cardiac tissue was obtained from seven SARS‐CoV‐2 infected patients, two pH1N1 influenza patients and six control patients at rapid autopsy. Patients were confirmed as having SARS‐CoV‐2 infection by RT‐qPCR of nasopharyngeal swabs and confirmed ‘glass opacities’ characteristic of pulmonary infection. The SARS‐CoV‐2 cohort was composed of six male and one female with a mean age of 69 years (range 46–81) (Table 1). These patients had a number of comorbidities, including type II diabetes, cardiac disease and arterial hypertension. All patients underwent mechanical ventilation ranging from 5 to 21 days. Tissue immunohistochemistry showed oedema in all SARS‐CoV‐2‐infected patients and myocarditis in one patient. Pathology review of the myocardial histology samples were indifferent to patients of similar age and comorbidities. All COVID‐19 samples were found to have RNAscope SARS‐CoV‐2 near background levels and considered not to have viral RNA present.
Spatial transcriptomic analysis
NanoString DSP analysis was performed on (n = 7) SARS‐CoV‐2, (n = 2) pH1N1 and (n = 6) control patients across 2 TMAs. ROI selection was performed to enable the capture of targeted transcriptome from sufficient myocardial tissue (>200 cells) to generate robust count data. Regions selected included blood vessels (endothelial cells, αSMA+), myocardial muscle (striated tissue) and mixed regions containing both vessels and surrounding muscle tissue (Figure 2 and Table S2). Bar codes targeting these regions were collected to generate morphology marker informed compartments. The barcodes were sequenced, mapped, and counted by NGS readout as per manufacturer's instructions. Quality control QC was performed to remove outlying probes and collapse counts from five genes per probe to single gene measurements. The QC files were output for further bioinformatics analysis.
FIGURE 2.

Representative immunohistochemistry. (a) Haematoxylin and eosin staining of the tissue microarray. (b) Regions of interest selected for spatial profiling by the Nanostring GeoMX digital spatial profiler (DSP) assay. (c) Regions of interest for the blood vessel (top), myocardium (middle), and mixed vessel/myocardium (bottom). Morphology markers for CD3E (red, T‐cell marker), CD68 (yellow, macrophage marker) and ACTA2 (green, smooth muscle alpha‐2 actin) and nuclear (blue) shown here.
Data standardization and batch correction
The batch correction adjusted for effects from the two independent batches of TMA data. This can be seen from the PCA (Figure 3a,b) where control samples from two batches are not clustered until the batch correction was performed. Additional to batch effect, technical variations of the data were mostly removed after batch correction, which can be visualized in the RLE plots (Figure 3d,e), which are known to be sensitive to technical variation while insensitive to biological variation (REF). In terms of biological variance (Figure 3b), differences in disease types are mainly explained by the variance from PC1 and PC2, while consistent local clustering of different tissue types can be observed within each disease type cluster. However, slight patient effect can still be observed, especially in control samples (Figure S2), which is required to be account for in the downstream analysis.
FIGURE 3.
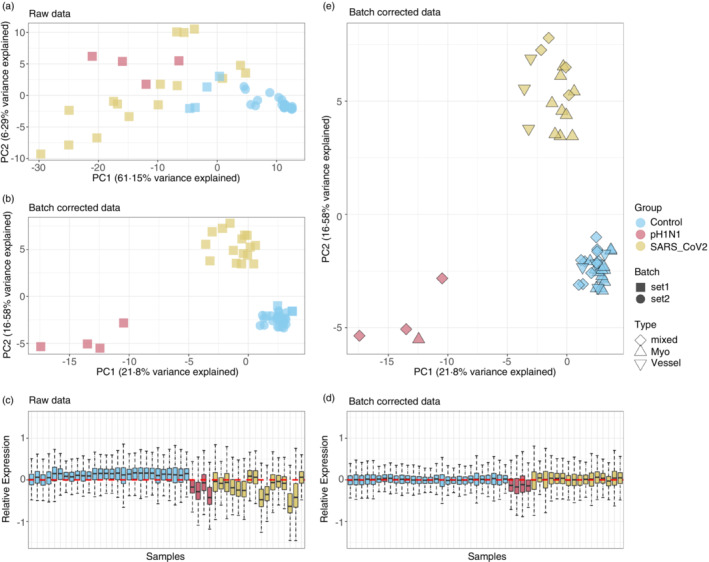
Batch correction and variability assessment of the spatial transcriptomic data. Principal component (PCs) analysis identifies the variability from batch effect in the transcriptomic data before (a) and after (b) batch correction. Relative log expression (RLE) plots (d and e) show the removal of technical variations after batch correction. Variabilities contributed by biological factors are visualized (c) in PC1 and PC2 of the batch‐corrected data.
DE analysis
To determine the virus‐induced effects on the cardiac tissues, we performed DE analysis of (a) COVID‐19 versus pH1N1, (b) COVID‐19 versus control and (c) pH1N1 versus control (Figure 4 and Table S3). Notably, there is an increased expression of IFN response gene expression in pH1N1 patients compared with COVID‐19 patients (Figure 4a). This is shown by an increase in IFN‐induced transmembrane proteins (IFITM1, IFITM2, IFIT3) and IFN stimulating genes (ISGs). In contrast, in COVID‐19, increased expression of chemokine ligands such as CCL15 (also known as macrophage inflammatory protein 5) were found, which has a chemotactic effect eliciting a transient increase of intracellular calcium are contributes towards inflow of monocytes/macrophages and neutrophils [52]. Moreover, CCL15 and SSX1 (a member of the synovial sarcoma X breakpoint protein family) were also significantly DE in COVID‐19 versus control tissues (Figure 4b). SSX1 has been previously found to be present in lung tissues with high amounts of SARS‐CoV‐2 RNA [53], and thought to be induced by SARS‐CoV‐2 as an adaptive response to IFN. HSPA1A, of the family of heat shock proteins, HSP70s, a classical molecular chaperone, was found to be higher expressed in COVID‐19 than control, and is thought to elicit a potent anti‐inflammatory effect [54]. In pH1N1 versus control (Figure 4c), a strong anti‐viral, IFN response was observed.
FIGURE 4.

Differential expression analysis. Distribution of differentially expressed (DE) genes as a function of the average transcript expression (log2) and fold change (log2) identified in the following comparisons were visualized: (a) COVID‐19 samples versus pH1N1 samples, (b) COVID‐19 samples versus control samples and (c) pH1N1 samples versus control. Green triangles indicate upregulated, blue triangles downregulated, and black dots indicate non‐DE genes. Differential expression genes were derived using voom‐limma pipeline with limma: Duplication correlations and false discovery rate threshold with Benjamini–Hochberg adjusted p < 0.05. Venn diagram (d and e) is used to visualize the intersection of DE genes from each comparison. Heatmap (f) is used to show the fold change (log2) of the DE genes that are distinctly upregulated or downregulated in Covid‐19 samples (the 16 and 24 genes showed in e).
Comparison of the overlapping DE genes between these comparisons (see Venn diagrams, in Figure 4d,e) suggested 16 and 24 unique DE genes are upregulated and downregulated, respectively, in COVID‐19 tissues compared with both pH1N1 and control samples. These 40 genes were not found to be DE between pH1N1 versus control samples, suggesting these as (Figure 4f) distinctly regulated for COVID‐19 patients. Of these COVID‐19 specific genes, the inflammatory response‐related gene NFAM1 and TNF receptor gene TNFRSF10A are found to be upregulated in concordance with previous reports suggesting higher expression in the throat swab samples [55] and in T cells [56] of COVID‐19 patients, respectively. Of the downregulated genes, of particular interest is IL1RL1 (which encodes the ST2 protein and is involved in the IL‐33/ST2 signalling pathway with its receptor cytokine IL‐33). This gene has been known to be cardioprotective for myocardial functions and has been implicated with increased cardiomyocyte hypertrophy and ventricular fibrosis upon germline deletion of ST2 [57] in mouse models. Furthermore, ST2 has also been tolled as a promising prognostic biomarker for COVID‐19 [58] with ST2 serum level suggested to be significantly increased in COVID‐19 patients [59], as opposed to the downregulation of IL1RL1 in COVID‐19 heart samples.
Gene set enrichment analysis
We performed GSEA on DE genes of each patient groupings (COVID19, pH1N1 and control) and visualized gene set enrichment by computing a similarity network from lists of curated gene sets using vissE [50] (Figure 5). For the comparison of COVID‐19 against pH1N1 samples, the gene set similarity network was visualized as a network (Figure 5b). We then identified higher‐order phenotypic changes associated with IFN responses in the clustering of gene sets (Figure 5a). Specifically for IFN responses, cluster 15 (Figure 5a,b, blue boundary) represents gene sets such as reactome IFN alpha beta signalling, IFN response and IFN‐induced anti‐viral module that are downregulated in SARS‐CoV‐2 samples relative to pH1N1 samples (Table S4). Within this cluster, IFN‐related genes including IFI27, IFIT3 and IFITM1 (Figure 5c, cluster 14) were all found to be downregulated.
FIGURE 5.
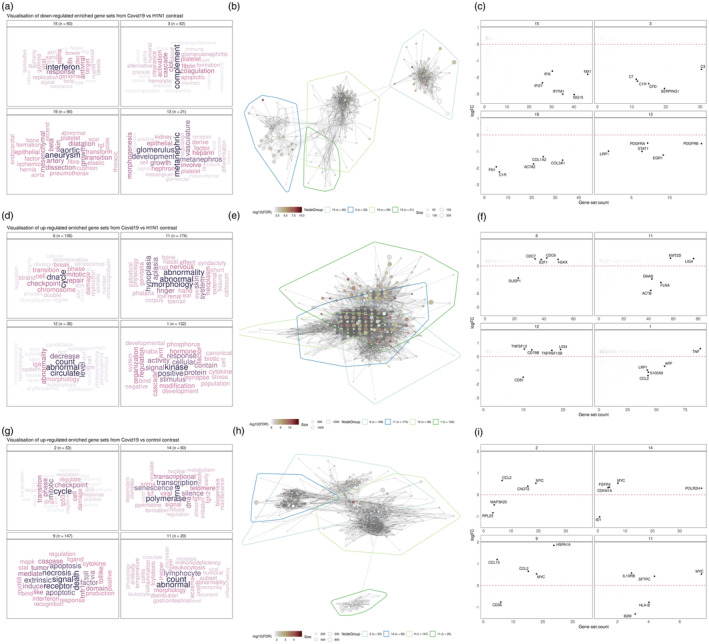
Visualization of significantly enriched gene sets different comparisons. (a, d and g) Cluster annotations based on text‐mining analysis of gene set names. Nine gene set clusters representing biological themes of each comparison are displayed. (b, e and h) Gene set overlap graphs of gene sets enriched in up/downregulated DE genes in different comparisons with nodes representing gene sets and edges representing overlaps based on the Jaccard index. Nodes are coloured based on the significance of enrichment. (c, f and i) Fold change (log2‐scaled) for genes belonging to gene sets in the cluster plot against the number of gene sets in the cluster the gene belongs to.
Visualizing downregulated gene sets comparing COVID‐19 to pH1N1, identified complement activation pathway was downregulated in cluster 3 (Figure 5a–c) for COVID‐19, reflected by initiator complement factor C1 complex, enzymatic mediator C3, membrane attach complex C7, among other genes in this pathway (Figure 5a–c, cluster 3). SERPING1 which encodes the C1 inhibitor (C1I) which inhibits C1r and C1s of the first complement component, is strongly downregulated in COVID‐19 compared with pH1N1 infection. Mutations of C1 are associated with dysregulation of the complement pathway and angioedema. The complement system is made up of about 40 heat‐stable plasma proteins which are involved in seven functional components, and cross‐talk in a catalytic cascade. Activation of the complement pathway is critical for immune homeostasis and deficiency can lead to life‐threatening infections [60].
The most predominant gene sets upregulated in COVID‐19 when compared with pH1N1 (Figure 5d–f), were DNA break, damage and repair, cellular abnormality, and cell cycle, including checkpoint and signalling (Figure 5d–f). This trend in cellular damage was seen in the COVID‐19 compared with normal cardiac tissue and showed enrichment of cell death and senescence gene sets (Figure 5g–i). In addition, leukocyte and myeloid cell gene sets were also enriched for in COVID‐19 patients compared with pH1N1 and control cardiac tissues, reflecting the higher number of neutrophils and leukocytes observed in severe COVID‐19 patients [2].
To further investigate and visualize the GSEA outcome of our transcriptomic data, staining for γ‐H2AX was performed [61], an established marker of DNA damage. Two of five COVID‐19 tissues exhibited significant nuclear γ‐H2AX signal, indicating the detection of DNA damage in these tissues. The degree of positive staining across these samples also coincided with the enrichment of GSEA scores of DSP regions for six DNA damage gene sets (Figure S3).
DISCUSSION
Pandemic H1N1 influenza drives a cytokine storm of generalized inflammation disrupting the heart which presents with fever, tachycardia, and arrhythmias. In contrast, COVID‐19 drives a now recognized syndrome which can result in acute myocardial infarction, myocardial injury, heart failure, disseminated thrombosis, hypotension, arrhythmias and sudden cardiac death [17].
In COVID‐19 patients with adverse outcomes, cardiac troponin I and brain‐type natriuretic peptide are elevated in ICU admission patients [62]. However, while biomarker changes are indicative of tissue damage, the mechanisms involved in cardiac injury have not been fully established. Recent studies have shown that myocarditis is prevalent in COVID‐19, however, evidence for subclinical cardiac inflammation or mechanisms regulating this process has been limited.
In influenza, viral binding to host cells induces a type I and III IFN response, including inflammatory cytokines (IL‐6 and TNF‐α) and chemokines. The binding of the type I and III IFNs to their receptors results in activation of the JAK/STAT pathways for the induction of ISGs such as IFI27. This can lead to acute myocarditis, a common complication of influenza infection [63]. In contrast to influenza infection, induction of these pathways in COVID‐19 is low [33], a feature supported by recent autopsy studies in which virus was not detected as a cause of myocarditis [64]. It was previously observed that excessive signalling induced by type 1 IFN induces inflammation‐driven myocardial infarction and this can be triggered by self‐DNA release and activation of the cGAS–STING–IRF3 pathway. This pathway is also connected to inflammation‐induced DNA damage, which was suggested by others and by our current dataset [65].
DNA damage response and repair mechanisms have been involved in the pathogenesis of chronic conditions such as diabetes and cardiovascular disease [66]. However, the role of SARS‐CoV‐2 in inducing genome instability has not been fully ascertained. In vitro studies have shown that the SARS‐CoV‐2 spike protein inhibits DNA damage repair by impeding the recruitment of DNA damage repair checkpoint proteins, a pre‐requisite for V(D)J recombination in adaptive immunity. This has further been confirmed in spike protein over expressed cells by upregulation of DNA damage marker γ‐H2AX [67].
Key clusters of genes impacted were uniquely altered by SARS‐CoV‐2 infection and were distinct from pH1N1. These focus on DNA damage and repair pathways and the consequent cell cycle arrest pathways. Notably we observed upregulation of LIG4, an ATP‐dependent DNA ligase which acts to repair DNA double‐strand breaks via the non‐homologous enjoining pathway [68]. LIG4 expression is known to be enhanced following DNA damage and by Wnt/β‐catenin signalling [69], suggesting that COVID‐19‐induced DNA damage might be responsible for induction of LIG4 in cardiac tissue. While this remains to be determined, the helicase NSP13 protein expressed by the related SARS‐CoV‐1 is known to induce DNA damage and replication fork stress by interacting directly with DNA polymerase δ [70]. Given the NSP13 protein shares 99.8% sequence homology between SARS‐CoV and SARS‐CoV‐2, it is possible that infection may induce DNA damage within myocardial tissue. However, SARS‐CoV‐2 infection has been observed, at least in vitro, to induce telomere shortening [71]. This feature is attributed with senescence which aligns with the upregulation of this gene set pathway in COVID‐19 myocardial tissues in our study. Interestingly, telomere stability is controlled by the DNA damage response proteins, as a telomere resembles a DNA break. Shortened telomeres result in a persistent DNA damage response, although at this point the function of these foci are unknown [72].
In cardiac tissues, we also observed that COVID‐19 induced downregulation of gene clusters involved in in mitochondrial function and metabolic regulation. Mitochondrial dysfunction is linked with COVID‐19 whereby SARS‐CoV‐2 viral proteins interact with host mitochondrial proteins [73]. For example, viral open reading frame 9c interacts with NDUFAF1 and NDUFAB1 [73, 74], genes we identified in our study that are required for cellular bioenergetics as part of Complex I. Indeed, SARS‐CoV‐2 manipulation of mitochondrial activity is likely to enable evasion of mitochondrial‐mediated innate immunity [74, 75].
Dysfunctional mitochondria are also associated with myocarditis [76], and persistent inflammation causing irreversible myocardium damage [77, 78]. Damage to the myocardium is triggered by danger‐associated molecular patterns (DAMPs), which are recognized by toll‐like receptors (TLR) that are expressed on immune and heart parenchymal cells [78, 79]. Consistently, we observe upregulation of gene clusters associated with TLR signalling in heart tissue. Crucially, mitochondrial lipid, peptides and circulating mitochondrial DNA (mtDNA) are a source of DAMPs [76]. For example, increased circulating mtDNA is detected following myocardial infarction [80] and can cause TLR‐induced cardiomyocyte death [81]. Pertinently, antibodies against a key mitochondrial lipid, cardiolipin, have also been reported following serological testing of a critically ill COVID‐19 patient exhibiting thrombocytopenia and coagulopathy [82]. Indeed, the pathways and gene sets identified in our study point to a key role for SARS‐CoV‐2‐induced cardiac injury. However, further work is warranted to discern whether direct SARS‐CoV‐2 infection of cardiac tissue or other physiological events are responsible for the cardiac injury observed in our cohort.
Our study provides a comprehensive complex cellular blueprint across the full composition of cardiac tissues responding to SARS‐CoV2 and H1N1 influenza using highly sophisticated spatio‐temporal analyses. This study is limited by the number of samples for each cohort, in particular for the pH1N1 group and unequal sex distribution. Targeted transcriptome panels were used which limited the number of genes profiled in the study. In addition, this analysis was restricted to autopsy samples which are unlikely to reflect the full spectrum of COVID‐19 disease.
More comprehensive assessments of post‐acute sequelae are needed to determine the short and long‐term impacts of SARS‐CoV‐2 infection. It is known that DNA damage and impaired repair mechanisms foster genome instability and are involved in several chronic diseases. Long‐term studies are needed to identify new onset heart disease from the early, and even subclinical, lesions as time post‐infection transpires.
AUTHOR CONTRIBUTIONS
Idea/conceptualisation: Arutha Kulasinghe, Gabrielle T. Belz, Kirsty R. Short, Melissa J. Davis, Fernando Souza‐Fonseca Guimaraes, John F. Fraser. Experimental: Arutha Kulasinghe, Ning Liu, Chin Wee Tan, James Monkman, Jane E. Sinclair, Dharmesh D. Bhuva, David Godbolt, Liuliu Pan, Andy Nam, Habib Sadeghirad, Kei Sato, Gianluigi Li Bassi, Ken O'Byrne, Camila Hartmann, Anna Flavia Ribeiro dos Santos Miggiolaro, Gustavo Lenci Marques, Lidia Zytynski Moura, Derek Richard, Mark Adams, Lucia de Noronha, Cristina Pellegrino Baena, Jacky Y. Suen, Rakesh Arora. Analysis: Arutha Kulasinghe, Ning Liu, Chin Wee Tan, James Monkman, Dharmesh D. Bhuva, Liuliu Pan, AN, Gabrielle T. Belz, Kirsty R. Short, Melissa J. Davis. Writing and review: all authors.
CONFLICTS OF INTEREST
Liuliu Pan and Andy Nam are employed by Nanostring Technologies. Kirsty R. Short is a consultant for Sanofi, Roche and NovoNordisk. Other authors have no conflicts of interest.
Supporting information
Figure S1 The identification of negative control genes from the dataset. Coefficient of variance (log‐transformed) of all genes within each batch are computed, genes that are identified as GTEx stable genes are highlighted.
{kind=link}
Figure S2 Pair‐wise PCA plot of batch‐corrected data. Data are coloured by disease group (SARS_CoV2, pH1N1 and Control) and shaped by tissue cores, patient ID which are start with “LN” indicates SARS_CoV2 tissue cores, “N09” indicates pH1N1 tissue cores and both “Control” and “Norm” indicates healthy tissue cores.
{kind=link}
Figure S3 Gamma H2AX staining corresponds with GSEA scores of DSP regions. Two COVID‐19 cores, LN16, LN20 (coloured), exhibited positive staining. DSP regions within these cores were scored for enrichment of DNA damage GSEA gene sets using singscore. High levels of gamma‐H2AX in LN20 corresponded with a positive shift in score in GO DNA synthesis involved and in DNA damage/repair pathways.
{kind=link}
Table S1 GTEx heart‐specific stable genes and the negative control genes.
Table S2 GeoMx DSP data sample information.
Table S3 Differential expression analysis.
Table S4 Gene set enrichment analysis.
ACKNOWLEDGEMENTS
The authors thank Miki Haraguchi and Stephanie Weaver (Fred Hutchinson Cancer Research Pathology) and WEHI (previously known as the Walter and Eliza Hall Institute) Histology core for their assistance. This research was carried out at the Translational Research Institute, which is supported by a grant from the Australian Government. This study was funded by The Common Good (an initiative of The Prince Charles Hospital Foundation) and the Australian Academy of Sciences (AAS): Regional Collaborations Programme COVID‐19 Digital Grants scheme for Chin Wee Tan and Arutha Kulasinghe. Arutha Kulasinghe is supported by a fellowship from the NHMRC 1157741. Kirsty R. Short is supported by an NHMRC investigator grant 2007919. Gabrielle T. Belz is supported by fellowships from the NHMRC (SPRF, 1135898; Investigator, 2008542).
Kulasinghe A, Liu N, Tan CW, Monkman J, Sinclair JE, Bhuva DD, et al. Transcriptomic profiling of cardiac tissues from SARS‐CoV‐2 patients identifies DNA damage. Immunology. 2022. 10.1111/imm.13577
Arutha Kulasinghe and Ning Liu are co‐first authors.
Fernando Souza‐Fonseca Guimaraes and John F. Fraser are co‐senior authors.
Funding information Australian Academy of Sciences; NHMRC, Grant/Award Numbers: 1157741, 2007919, 1135898, 2008542; The Prince Charles Hospital Foundation (The Common Good)
DATA AVAILABILITY STATEMENT
The data generated in this study is available at Geo accession GSE212119.
REFERENCES
- 1. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID‐19). JAMA Cardiol. 2020;5(7):811–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giustino G, Croft LB, Stefanini GG, Bragato R, Silbiger JJ, Vicenzi M, et al. Characterization of myocardial injury in patients with COVID‐19. J Am Coll Cardiol. 2020;76(18):2043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dweck MR, Bularga A, Hahn RT, Bing R, Lee KK, Chapman AR, et al. Global evaluation of echocardiography in patients with COVID‐19. Eur Heart J Cardiovasc Imaging. 2020;21(9):949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu K, Fang YY, Deng Y, Liu W, Wang MF, Ma JP, et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin Med J (Engl). 2020;133(9):1025–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li N, Zhu L, Sun L, Shao G. The effects of novel coronavirus (SARS‐CoV‐2) infection on cardiovascular diseases and cardiopulmonary injuries. Stem Cell Res. 2021;51:102168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tersalvi G, Vicenzi M, Calabretta D, Biasco L, Pedrazzini G, Winterton D. Elevated troponin in patients with coronavirus disease 2019: possible mechanisms. J Card Fail. 2020;26(6):470–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. COVID‐19 and cardiovascular disease. Circulation. 2020;141(20):1648–55. [DOI] [PubMed] [Google Scholar]
- 9. Zheng YY, Ma YT, Zhang JY, Xie X. COVID‐19 and the cardiovascular system. Nat Rev Cardiol. 2020;17(5):259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Satterfield BA, Bhatt DL, Gersh BJ. Cardiac involvement in the long‐term implications of COVID‐19. Nat Rev Cardiol. 2021;19:332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carfi A, Bernabei R, Landi F, Gemelli Against COVID‐19 Post‐Acute Care Study Group . Persistent symptoms in patients after acute COVID‐19. JAMA. 2020;324(6):603–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6‐month consequences of COVID‐19 in patients discharged from hospital: a cohort study. Lancet. 2021;397(10270):220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kotecha T, Knight DS, Razvi Y, Kumar K, Vimalesvaran K, Thornton G, et al. Patterns of myocardial injury in recovered troponin‐positive COVID‐19 patients assessed by cardiovascular magnetic resonance. Eur Heart J. 2021;42(19):1866–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang L, Zhao P, Tang D, Zhu T, Han R, Zhan C, et al. Cardiac involvement in patients recovered from COVID‐2019 identified using magnetic resonance imaging. JACC Cardiovasc Imaging. 2020;13(11):2330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rajpal S, Tong MS, Borchers J, Zareba KM, Obarski TP, Simonetti OP, et al. Cardiovascular magnetic resonance findings in competitive athletes recovering from COVID‐19 infection. JAMA Cardiol. 2021;6(1):116–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Al‐Aly Z, Xie Y, Bowe B. High‐dimensional characterization of post‐acute sequelae of COVID‐19. Nature. 2021;594(7862):259–64. [DOI] [PubMed] [Google Scholar]
- 17. Xie Y, Xu E, Bowe B, Al‐Aly Z. Long‐term cardiovascular outcomes of COVID‐19. Nat Med. 2022;28(3):583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kwong JC, Schwartz KL, Campitelli MA, Chung H, Crowcroft NS, Karnauchow T, et al. Acute myocardial infarction after laboratory‐confirmed influenza infection. N Engl J Med. 2018;378(4):345–53. [DOI] [PubMed] [Google Scholar]
- 19. Wang J, Xu H, Yang X, Zhao D, Liu S, Sun X, et al. Cardiac complications associated with the influenza viruses a subtype H7N9 or pandemic H1N1 in critically ill patients under intensive care. Braz J Infect Dis. 2017;21(1):12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sato K, Sinclair JE, Sadeghirad H, Fraser JF, Short KR, Kulasinghe A. Cardiovascular disease in SARS‐CoV‐2 infection. Clin Transl Immunology. 2021;10(9):e1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perez‐Bermejo JA, Kang S, Rockwood SJ, Simoneau CR, Joy DA, Silva AC, et al. SARS‐CoV‐2 infection of human iPSC‐derived cardiac cells reflects cytopathic features in hearts of patients with COVID‐19. Sci Transl Med. 2021;13(590):eabf7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bailey AL, Dmytrenko O, Greenberg L, Bredemeyer AL, Ma P, Liu J, et al. SARS‐CoV‐2 infects human engineered heart tissues and models COVID‐19 myocarditis. JACC Basic Transl Sci. 2021;6(4):331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marchiano S, Hsiang TY, Khanna A, Higashi T, Whitmore LS, Bargehr J, et al. SARS‐CoV‐2 infects human pluripotent stem cell‐derived cardiomyocytes, impairing electrical and mechanical function. Stem Cell Reports. 2021;16(3):478–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lindner D, Fitzek A, Brauninger H, Aleshcheva G, Edler C, Meissner K, et al. Association of cardiac infection with SARS‐CoV‐2 in confirmed COVID‐19 autopsy cases. JAMA Cardiol. 2020;5(11):1281–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bearse M, Hung YP, Krauson AJ, Bonanno L, Boyraz B, Harris CK, et al. Factors associated with myocardial SARS‐CoV‐2 infection, myocarditis, and cardiac inflammation in patients with COVID‐19. Mod Pathol. 2021;34(7):1345–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Massoth LR, Desai N, Szabolcs A, Harris CK, Neyaz A, Crotty R, et al. Comparison of RNA in situ hybridization and immunohistochemistry techniques for the detection and localization of SARS‐CoV‐2 in human tissues. Am J Surg Pathol. 2021;45(1):14–24. [DOI] [PubMed] [Google Scholar]
- 27. Filgueiras‐Rama D, Vasilijevic J, Jalife J, Noujaim SF, Alfonso JM, Nicolas‐Avila JA, et al. Human influenza A virus causes myocardial and cardiac‐specific conduction system infections associated with early inflammation and premature death. Cardiovasc Res. 2021;117(3):876–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan HY, Yamada H, Chida J, Wang S, Yano M, Yao M, et al. Up‐regulation of ectopic trypsins in the myocardium by influenza A virus infection triggers acute myocarditis. Cardiovasc Res. 2011;89(3):595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ray CG, Icenogle TB, Minnich LL, Copeland JG, Grogan TM. The use of intravenous ribavirin to treat influenza virus‐associated acute myocarditis. J Infect Dis. 1989;159(5):829–36. [DOI] [PubMed] [Google Scholar]
- 30. Davoudi AR, Maleki AR, Beykmohammadi AR, Tayebi A. Fulminant myopericarditis in an immunocompetent adult due to pandemic 2009 (H1N1) influenza A virus infection. Scand J Infect Dis. 2012;44(6):470–2. [DOI] [PubMed] [Google Scholar]
- 31. Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R, et al. Detection of viruses in myocardial tissues by polymerase chain reaction: evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. 2003;42(3):466–72. [DOI] [PubMed] [Google Scholar]
- 32. Siegers JY, Novakovic B, Hulme KD, Marshall RJ, Bloxham CJ, Thomas WG, et al. A high‐fat diet increases influenza A virus‐associated cardiovascular damage. J Infect Dis. 2020;222(5):820–31. [DOI] [PubMed] [Google Scholar]
- 33. Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181(5):1036–1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee JS, Shin EC. The type I interferon response in COVID‐19: implications for treatment. Nat Rev Immunol. 2020;20(10):585–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Francis ME, Goncin U, Kroeker A, Swan C, Ralph R, Lu Y, et al. SARS‐CoV‐2 infection in the Syrian hamster model causes inflammation as well as type I interferon dysregulation in both respiratory and non‐respiratory tissues including the heart and kidney. PLoS Pathog. 2021;17(7):e1009705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuen CK, Lam JY, Wong WM, Mak LF, Wang X, Chu H, et al. SARS‐CoV‐2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9(1):1418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) membrane (M) protein inhibits type I and III interferon production by targeting RIG‐I/MDA‐5 signaling. Signal Transduct Target Ther. 2020;5(1):299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xie Y, Bowe B, Maddukuri G, Al‐Aly Z. Comparative evaluation of clinical manifestations and risk of death in patients admitted to hospital with covid‐19 and seasonal influenza: cohort study. BMJ. 2020;371:m4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng MP, Cau A, Lee TC, Brodie D, Slutsky A, Marshall J, et al. Acute cardiac injury in coronavirus disease 2019 and other viral infections‐a systematic review and meta‐analysis. Crit Care Med. 2021;49(9):1558–66. [DOI] [PubMed] [Google Scholar]
- 40. Jiang DS, Liu Y, Zhou H, Zhang Y, Zhang XD, Zhang XF, et al. Interferon regulatory factor 7 functions as a novel negative regulator of pathological cardiac hypertrophy. Hypertension. 2014;63(4):713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang XJ, Jiang DS, Li H. The interferon regulatory factors as novel potential targets in the treatment of cardiovascular diseases. Br J Pharmacol. 2015;172(23):5457–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Monkman J, Taheri T, Ebrahimi Warkiani M, O'Leary C, Ladwa R, Richard D, et al. High‐plex and high‐throughput digital spatial profiling of non‐small‐cell lung cancer (NSCLC). Cancers (Basel). 2020;12(12):3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kulasinghe A, Tan CW, Dos Santos Miggiolaro AFR, Monkman J, SadeghiRad H, Bhuva DD, et al. Profiling of lung SARS‐CoV‐2 and influenza virus infection dissects virus‐specific host responses and gene signatures. Eur Respir J. 2022;59:2101881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. GTEx Consortium . The GTEx consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gagnon‐Bartsch JA, Jacob L, Speed TP. Removing unwanted variation from high dimensional data with negative controls (Tech Reports 820). Berkeley: Dep Stat Univ California; 2013. [Google Scholar]
- 47. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bhuva DD, Tan CW, Liu N, Whitfield HJ, Papachristos N, Lee S, et al. vissE: a versatile tool to identify and visualise higher‐order molecular phenotypes from functional enrichment analysis. bioRxiv. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Foroutan M, Bhuva DD, Lyu R, Horan K, Cursons J, Davis MJ. Single sample scoring of molecular phenotypes. BMC Bioinformatics. 2018;19(1):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shimizu Y, Dobashi K. CC‐chemokine CCL15 expression and possible implications for the pathogenesis of IgE‐related severe asthma. Mediators Inflamm. 2012;2012:475253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Desai N, Neyaz A, Szabolcs A, Shih AR, Chen JH, Thapar V, et al. Temporal and spatial heterogeneity of host response to SARS‐CoV‐2 pulmonary infection. Nat Commun. 2020;11(1):6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krause M, Gerchman F, Friedman R. Coronavirus infection (SARS‐CoV‐2) in obesity and diabetes comorbidities: is heat shock response determinant for the disease complications? Diabetol Metab Syndr. 2020;12:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu B, Yan Y, Dong L, Han L, Liu Y, Yu J, et al. Integrated characterization of SARS‐CoV‐2 genome, microbiome, antibiotic resistance and host response from single throat swabs. Cell Discovery. 2021;7(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhu L, Yang P, Zhao Y, Zhuang Z, Wang Z, Song R, et al. Single‐cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID‐19 and influenza patients. Immunity. 2020;53(3):685–696.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kakkar R, Lee RT. The IL‐33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7(10):827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ragusa R, Basta G, Del Turco S, Caselli C. A possible role for ST2 as prognostic biomarker for COVID‐19. Vascul Pharmacol. 2021;138:106857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zeng Z, Hong X‐Y, Li Y, Chen W, Ye G, Li Y, et al. Serum‐soluble ST2 as a novel biomarker reflecting inflammatory status and illness severity in patients with COVID‐19. Biomark Med. 2020;14(17):1619–29. [DOI] [PubMed] [Google Scholar]
- 60. Fodil S, Annane D. Complement inhibition and COVID‐19: the story so far. Immunotargets Ther. 2021;10:273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mah LJ, El‐Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24(4):679–86. [DOI] [PubMed] [Google Scholar]
- 62. Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, et al. Characteristics and outcomes of 21 critically ill patients with COVID‐19 in Washington State. JAMA. 2020;323(16):1612–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ukimura A, Satomi H, Ooi Y, Kanzaki Y. Myocarditis associated with influenza a H1N1pdm2009. Influenza Res Treat. 2012;2012:351979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abbasi J. Researchers investigate what COVID‐19 does to the heart. JAMA. 2021;325(9):808–11. [DOI] [PubMed] [Google Scholar]
- 65. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23(12):1481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pánico P, Ostrosky‐Wegman P, Salazar AM. The potential role of COVID‐19 in the induction of DNA damage. Mutat Res Rev Mutat Res. 2022;789:108411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jiang H, Mei YF. SARS‐CoV‐2 spike impairs DNA damage repair and inhibits V(D)J recombination in vitro. Viruses. 2021;13(10):2056. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68. Williams GJ, Hammel M, Radhakrishnan SK, Ramsden D, Lees‐Miller SP, Tainer JA. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst). 2014;17:110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jun S, Jung YS, Suh HN, Wang W, Kim MJ, Oh YS, et al. LIG4 mediates Wnt signalling‐induced radioresistance. Nat Commun. 2016;7:10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xu LH, Huang M, Fang SG, Liu DX. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase delta. J Biol Chem. 2011;286(45):39546–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Victor J, Deutsch J, Whitaker A, Lamkin EN, March A, Zhou P, et al. SARS‐CoV‐2 triggers DNA damage response in Vero E6 cells. Biochem Biophys Res Commun. 2021;579:141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hewitt G, Jurk D, Marques FDM, Correia‐Melo C, Hardy T, Gackowska A, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress‐induced senescence. Nat Commun. 2012;3(1):708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Singh KK, Chaubey G, Chen JY, Suravajhala P. Decoding SARS‐CoV‐2 hijacking of host mitochondria in COVID‐19 pathogenesis. Am J Physiol Cell Physiol. 2020;319(2):C258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Srinivasan K, Pandey AK, Livingston A, Venkatesh S. Roles of host mitochondria in the development of COVID‐19 pathology: could mitochondria be a potential therapeutic target? Mol Biomed. 2021;2(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage‐associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32(4):157–64. [DOI] [PubMed] [Google Scholar]
- 77. Saleh J, Peyssonnaux C, Singh KK, Edeas M. Mitochondria and microbiota dysfunction in COVID‐19 pathogenesis. Mitochondrion. 2020;54:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. 2008;99:95–114. [DOI] [PubMed] [Google Scholar]
- 79. Hofmann U, Ertl G, Frantz S. Toll‐like receptors as potential therapeutic targets in cardiac dysfunction. Expert Opin Ther Targets. 2011;15(6):753–65. [DOI] [PubMed] [Google Scholar]
- 80. Bliksoen M, Mariero LH, Ohm IK, Haugen F, Yndestad A, Solheim S, et al. Increased circulating mitochondrial DNA after myocardial infarction. Int J Cardiol. 2012;158(1):132–4. [DOI] [PubMed] [Google Scholar]
- 81. Bliksoen M, Mariero LH, Torp MK, Baysa A, Ytrehus K, Haugen F, et al. Extracellular mtDNA activates NF‐κB via toll‐like receptor 9 and induces cell death in cardiomyocytes. Basic Res Cardiol. 2016;111(4):42. [DOI] [PubMed] [Google Scholar]
- 82. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid‐19. N Engl J Med. 2020;382(17):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The identification of negative control genes from the dataset. Coefficient of variance (log‐transformed) of all genes within each batch are computed, genes that are identified as GTEx stable genes are highlighted.
Figure S2 Pair‐wise PCA plot of batch‐corrected data. Data are coloured by disease group (SARS_CoV2, pH1N1 and Control) and shaped by tissue cores, patient ID which are start with “LN” indicates SARS_CoV2 tissue cores, “N09” indicates pH1N1 tissue cores and both “Control” and “Norm” indicates healthy tissue cores.
Figure S3 Gamma H2AX staining corresponds with GSEA scores of DSP regions. Two COVID‐19 cores, LN16, LN20 (coloured), exhibited positive staining. DSP regions within these cores were scored for enrichment of DNA damage GSEA gene sets using singscore. High levels of gamma‐H2AX in LN20 corresponded with a positive shift in score in GO DNA synthesis involved and in DNA damage/repair pathways.
Table S1 GTEx heart‐specific stable genes and the negative control genes.
Table S2 GeoMx DSP data sample information.
Table S3 Differential expression analysis.
Table S4 Gene set enrichment analysis.
Data Availability Statement
The data generated in this study is available at Geo accession GSE212119.