

Abstract
Coronavirus disease 2019 (COVID‐19) remains a major public health concern, and vaccine unavailability, hesitancy, or failure underscore the need for discovery of efficacious antiviral drug therapies. Numerous approved drugs target protein kinases associated with viral life cycle and symptoms of infection. Repurposing of kinase inhibitors is appealing as they have been vetted for safety and are more accessible for COVID‐19 treatment. However, an understanding of drug mechanism is needed to improve our understanding of the factors involved in pathogenesis. We tested the in vitro activity of three kinase inhibitors against severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), including inhibitors of AXL kinase, a host cell factor that contributes to successful SARS‐CoV‐2 infection. Using multiple cell‐based assays and approaches, gilteritinib, nintedanib, and imatinib were thoroughly evaluated for activity against SARS‐CoV‐2 variants. Each drug exhibited antiviral activity, but with stark differences in potency, suggesting differences in host dependency for kinase targets. Importantly, for gilteritinib, the amount of compound needed to achieve 90% infection inhibition, at least in part involving blockade of spike protein‐mediated viral entry and at concentrations not inducing phospholipidosis (PLD), approached a clinically achievable concentration. Knockout of AXL, a target of gilteritinib and nintedanib, impaired SARS‐CoV‐2 variant infectivity, supporting a role for AXL in SARS‐CoV‐2 infection and supporting further investigation of drug‐mediated AXL inhibition as a COVID‐19 treatment. This study supports further evaluation of AXL‐targeting kinase inhibitors as potential antiviral agents and treatments for COVID‐19. Additional mechanistic studies are needed to determine underlying differences in virus response.
Keywords: antiviral therapy, COVID‐19, gilteritinib, imatinib, kinase inhibitor, nintedanib, SARS‐CoV‐2
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is a positive‐sense, single‐stranded, enveloped RNA virus belonging to the Beta coronavirus genus, first reported in Wuhan, China in 2019. SARS‐CoV‐2 shares 80% RNA sequence identity with SARS‐CoV, a Beta coronavirus that first appeared in Guangdong Province, China, in 2002, before spreading to 37 countries with a case fatality rate of nearly 10% and 8098 documented cases. 1 , 2 SARS‐CoV‐2 also shares around 50% sequence identity with Middle East respiratory syndrome coronavirus (MERS‐CoV), another member of this genus that causes respiratory disease, first reported in Saudi Arabia in 2012 before affecting 29 countries, with a fatality rate of around 34% and over 2521 documented cases. 2 At the time of this writing, in the United States there have been over 93 million cases and over 1 000 000 deaths. While MERS‐CoV and SARS‐CoV‐2 are in circulation, cases of SARS‐CoV have not been reported since 2004.
SARS‐CoV‐2 causes Coronavirus disease 2019 (COVID‐19), ranging in symptoms from asymptomatic, to fever and dry cough and loss of smell and taste. 3 , 4 While COVID‐19 is associated with a lower fatality rate than SARS‐CoV or MERS‐CoV, a portion of patients show severe symptoms with a 1% fatality rate, often due to the onset of a virus‐induced hyper‐inflammatory response characterized by diffuse alveolar damage preceding fibroproliferation and fibrosis over time. Patients undergoing respiratory failure resulting from acute respiratory distress syndrome require supportive therapy in the form of intubation and mechanical ventilation.
Since the appearance of SARS‐CoV‐2, variants have emerged that contain numerous mutations across the genome but that are often concentrated in the SARS‐CoV‐2 spike protein gene. Among these are the Alpha (lineage B.1.1.7) variant, first detected in the United Kingdom and characterized by a 45%–71% increased transmissibility, 5 , 6 and the Beta (B.1.351, B.1.351.2, B.1.351.3) variant, which first emerged in South Africa and is marked by a 55% increased transmissibility. 5 , 7 Both variants have waned with the emergence of the Delta (lineage B.1.617.2) variant, which was first detected in India and was the dominant strain in the United States and numerous other countries. The Delta variant has transmissibility of around 50% more than the Alpha variant, with diminished sensitivity (around 2‐fold) to antibody neutralization. 5 During the completion of this manuscript, a highly mutated and transmissible coronavirus variant, Omicron (B.1.1.529), harboring >30 mutations on the spike protein and first detected in South Africa, has emerged. 8 , 9 It is unclear how each of these variants differs in host factor dependence and how this may affect the actions of small molecule therapeutics.
All SARS‐CoV‐2 virus variants have a similar infection cycle to that of SARS‐CoV and MERS‐CoV. In brief, viral entry is initiated when the receptor binding domain (RBD) of the S1 subunit of the spike protein on the surface of SARS‐CoV‐2 physically interacts with the host cell surface ACE2 receptor. 10 Following clathrin‐dependent endocytosis of the virion and pH‐dependent cleavage of the S2 spike subunit, the viral envelope fuses with the endosome to release the viral nucleocapsid. 11 The viral RNA genome is then transcribed and replicated, leading to the assembly of new viral particles that are released from the host cell and go on to establish new cycles of infection. A recent report indicated that AXL kinase activity was required for uptake of the virus 12 suggesting that inhibitors, targeting AXL and related kinases, and used in the clinic, may offer a path to treatment for COVID‐19.
The SARS‐CoV‐2 outbreak has highlighted the need for identification and/or development of effective antiviral drug treatments, especially for those for whom vaccination is not an option due to immune deficiency, lack of availability, or personal choice. Preliminary data suggest efficacy of novel oral drug treatments, such as the main protease inhibitor, Paxlovid (Pfizer Inc.), 13 and the RNA‐dependent RNA polymerase inhibitor, molnupiravir (EIDD‐2801/MK‐4482) (Merck and Ridgeback Biotherapeutics). 14 , 15 Like many RNA viruses, SARS‐CoV‐2 has shown an ability to rapidly adapt to humans and new selective pressures and may become resistant to new therapies. Thus, there is a need for an armament of treatments as an attempt to override potential drug resistance. Repurposing of approved, targeted kinase inhibitors provides a clinical option to treatment of SARS‐CoV‐2 infection and COVID‐19, as an impressive number of cellular and viral protein kinases are implicated in viral infection or organ‐damaging symptoms caused by the infection. Importantly, some of these kinases can be targeted by FDA‐approved kinase inhibitors, and thus clinically available drugs can in theory be readily and speedily repurposed for treatment of infected, symptomatic patients.
In a previous report, 16 we stratified a panel of kinase inhibitors according to their predicted ability to exhibit anti‐SARS‐CoV‐2 activity. Based on this study and the importance of AXL‐related kinases in virus entry into cells, we tested the activity of three potent kinase inhibitors using in vitro drug studies with models of SARS‐CoV‐2 infection in both Vero (African green monkey) and human lung adenocarcinoma (A549‐ACE2)‐cells. Here, we describe the anti‐SARS‐CoV‐2 efficacy of gilteritinib, nintedanib, and imatinib.
2. MATERIALS AND METHODS
Details are provided in the Supporting Information.
3. RESULTS
We previously predicted that a set of human kinase targets were associated with the coronavirus infection cycle and playing a role in development of symptoms associated with COVID‐19 (such as pneumonia, fibrosis, and inflammation) and prioritized pharmacokinetics that were tolerated at high doses over long periods of administration. 16 , 17 Of these we chose to examine three compounds, gilteritinib, nintedanib, and imatinib, which inhibit AXL or AXL‐related kinases, for activity against SARS‐CoV‐2. Each compound was evaluated using multipoint dose curves in Vero and A549‐ACE2 cells. Each cell line expresses the SARS‐CoV‐2 receptor, ACE2, either natively or after expression of recombinant protein respectively and so is susceptible to infection. For the A549‐ACE2 cells, infection was measured by immunofluorescent labeling of viral N‐protein and the proportion of N‐expressing cells was quantified from microscopy images (Figure 1A–C and Table 1). Vero cells were used in a cytopathic effect (CPE) protection assay, where compounds protect cells from infection and virus‐induced cytopathogenicity as measured by neutral red dye uptake into cells (Figure 1D,E and Table 1). Despite differences in approach, each assay gave similar outcomes with gilteritinib being the most potent, with an EC50 of 0.13 μM by CPE, and <0.1 μM by N staining assay (Figure 1 and Table 1). Imatinib was less potent with an EC50 of 2 μM by both assays. Gilteritinib is a potent and selective inhibitor of AXL (IC50 = 41 nM) 18 ; AXL is a target that has been reported as an important host factor for SARS‐CoV‐2 infection. 12 To assess whether the higher potency of gilteritinib could be associated specifically with AXL inhibition, we tested the triple angiokinase inhibitor nintedanib, which, according to KINOMEscan (https://lincs.hms.harvard.edu/kinomescan/) shows on‐target kinase activity against AXL (selected target kinase affinity [kd < 100 nM]) as well as other kinases (AAK1, ABL1, KIT, RET, YES1). Like gilteritinib, nintedanib proved to be a potent inhibitor of infection assay but unlike gilteritinib, caused little cytotoxicity in this assay suggesting a favorable selectivity index (SI) (Figure 1A–B, Table 1, and Supporting Information: Figure 2).
Figure 1.

Gilteritinib, imatinib, and nintedanib inhibit SARS‐CoV‐2 infection. (A–C) Representative dose–response curves showing effects of gilteritinib (A), nintedanib (B), and imatinib (C) on SARS‐CoV‐2 infection of A549‐ACE2 cells. One hour before infection cells were treated with a dilution series of each drug starting at 25 µM. Cells were infected with SARS‐CoV‐2 (USA‐WA1 strain) at a multiplicity of infection (MOI) ∼0.2 for 48 h. Cells were fixed and probed for SARS‐CoV‐2 infection using anti‐N protein antibody. Plates were imaged at 4x and infection was quantified using a Cell Profiler pipeline to count the number of N‐positive cells. Infection efficiency, defined as N‐positive cells divided by nuclei, was normalized to a DMSO negative control and dose–response curves and EC50 values were calculated using a four‐parameter variable‐slope model (GraphPad Prism). (D–E) Dose–response curves showing the toxicity of gilteritinib (D) and imatinib (E) on Vero 76 cells. An MOI of 0.001 was used for studies of SARS‐CoV‐2 in Vero 76 cells. The Neutral Red (cytopathic effect/toxicity) assay was carried out as a readout. DMSO, dimethylsulfoxide; ROI, reduction of infection; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
Table 1.
Investigation of a panel of kinase inhibitors for anti‐severe acute respiratory syndrome coronavirus 2 activity using a Vero 76 cell‐based assay and an ACE2‐A549 cell‐based assay
| Compound | Drug assay (Cytopathic effect/toxicity) | EC50 (μΜ) | CC50 (μΜ) | SI | |
|---|---|---|---|---|---|
| Test compounds (tyrosine kinase inhibitors) | Gilteritinib | Neutral red | 0.13 | 3.8 | 29 |
| NP stain | <0.1 | 1.2 | 12 | ||
| Imatinib | Neutral red | 2.0 | 24 | 12 | |
| NP stain | 2.0 | >20 | >10 | ||
| Nintedanib | Neutral red | nd | nd | nd | |
| NP stain | 1.0 | >20 | >20 |
Note: EC50 (µM), CC50 (µM). EC50=compound concentration that reduces viral replication by 50%. CC50=compound concentration that reduces cell viability by 50%. SI=CC50/EC50. Compounds with SI values > 10 are considered active. Nd indicates not done.
We further evaluated the antiviral activity of gilteritinib, nintedanib, and imatinib for inhibition of infection of the common SARS‐CoV‐2 variants, Alpha, Beta, and Delta. While the Alpha strain was similarly susceptible to the WA strain used in our initial work, the drugs were up to 10‐fold less potent against the Beta and Delta variants (Figure 2). This finding suggests a surprising difference in dependence on cellular kinases between viruses in the Alpha lineage versus other, later isolated variants.
Figure 2.

Kinase inhibitors are less potent against the Beta and Delta strains compared to the Alpha strain. Representative dose–response curves of A549‐ACE2 cells treated with each compound and infected with SARS‐CoV‐2 Alpha, Beta, or Delta strains (A–C). Cells were treated with compounds 1 h before infection with each variant at an MOI ∼0.2. After 48 h of infection plates were fixed and probed for N‐protein. Infection efficiency was calculated by normalizing the proportion of N‐positive cells in each well to an untreated DMSO control. Concentrations listed for each variant are EC50s calculated using a four‐parameter variable slope model (GraphPad Prism). DMSO, dimethylsulfoxide; MOI, multiplicity of infection; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
As AXL is the main target of gilteritinib and one tyrosine kinase target of nintedanib, and has been reported to interact with the N‐terminus of the spike protein, 12 , 19 we evaluated the role of AXL in defining infection efficiency for the WA, Alpha, and Delta strains. AXL was knocked out by CRISPR‐targeted gene disruption and loss of AXL expression was confirmed by immunoblotting (Figure 3A) and flow cytometry (Figure 3B). For each, ACE2 overexpression, as assessed by immunoblotting, was not affected and was similar for each cell line. For clones A08 and D10, AXL expression was reduced by 50% and 90% respectively compared to E05, which was made using a nontargeting guide RNA and had wild‐type levels of AXL expression. Despite having residual AXL expression, significant reductions in infection efficiency of each virus variant were seen for the D10 clone (Figure 3C,D). For A08, infection consistently trended lower than E05 for all three strains, but statistical significance was not achieved in all repeats of assays. Overall, lower AXL expression was associated with reduced infection efficiency, which supports a role for AXL, the target of gilteritinib, in SARS‐CoV‐2 infection.
Figure 3.
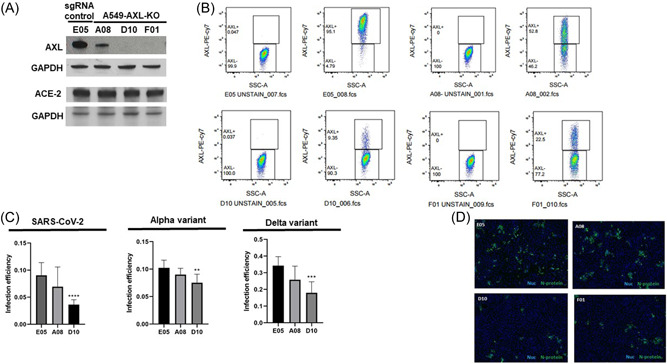
SARS‐CoV‐2 infection is reduced in AXL KO cells and correlates with AXL expression level. (A) AXL and ACE‐2 expression assessed by immunoblotting. (B) AXL expression assessed by flow cytometry. (C) A549‐ACE2 cells with AXL knocked out reduced infection efficiency of SARS‐CoV‐2 (left panel), the Alpha variant (middle panel), and the Delta variant (right panel), as compared to control cells (E05). The level of AXL expression correlated with infection inhibition (A08 53%, D10 9%). Graphed results for the Alpha and Delta variant are representative of three independent replicates, for which similar results were observed. AXL expression for the variant studies as measured by flow cytometry is shown in Supporting Information: Figure 1 (D) Representative images A549‐ACE2‐Cas9 cells with AXL knocked down stained with anti‐N protein antibody. Shown are cells infected with SARS‐CoV‐2 at an MOI ∼0.2 for 24 h. Green is N protein, blue is nuclei. Scale bar indicates 300 µM. A549‐ACE2‐Cas9 cells with AXL knocked down were infected with an MOI ∼0.2 for 24 h, fixed in formalin, and probed for infection using an anti‐N protein antibody. Plates were imaged and infection was quantified using a Cell Profiler pipeline to count the number of N‐positive cells. Infection efficiency, defined as N‐positive cells divided by nuclei, was normalized to a DMSO negative control and is shown for the indicated virus variants. Sidak's multiple comparison tests were used to test for significant differences in infection efficiency. Asterisks denote statistical differences versus E05, an AXL‐expressing control line. Data are representative of three or four biological replicates. DMSO, dimethylsulfoxide; MOI, multiplicity of infection; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
Since major differences for Alpha versus Beta and Delta variants are present in the virus glycoprotein gene (as many as 20 amino acid substitutions), and AXL is involved in virus entry into cells, a step controlled by the spike protein, it is possible that drug potency is related to changes in the spike protein and its interactions with the drug targets. To evaluate the contribution of differences in the spike protein to drug sensitivity, we challenged pretreated A549‐ACE2 cells with pseudotyped viruses (PVs) made using a lentiviral core and bearing spike proteins from each variant. Specifically, we tested the Wuhan (similar to WA) and Delta spike sequences, as well as Omicron. The VSV‐G protein served as a specificity control for drug activity. Consistent with the results from the live virus experiments, nintedanib and gilteritinib were more potent than imatinib, with gilteritinib treatments showing the greatest inhibition of PV infection (Figure 4A). Imatinib exhibited similar potency against the Wuhan and Delta PVs and higher potency against the Omicron PV (Figure 4A). Similarly, nintedanib was most potent against the Omicron PV and least potent against the Wuhan PV (Figure 4A). Gilteritinib only exhibited significant infection inhibition against Omicron PV compared to VSV‐G. There was a small decrease in Wuhan PV and Delta PV infection compared to VSV‐G, but this did not reach the level of statistical significance. This outcome suggests an effect on VSV‐G function or on expression of lentivirus infection reporter, or may be due to cytotoxicity caused by gilteritinib, as VSV‐G infection in gilteritinib‐treated cells was around 50% of the level attained in imatinib‐ or nintedanib‐treated cells. Indeed, cell viability as assessed by MTT assay was reduced by ∼30% after treatment with 0.63 µM gilteritinib (Supporting Information: Figure 2).
Figure 4.

Activity of kinase inhibitors against a panel of pseudotyped viruses. (A) Imatinib, nintedanib, and gilteritinib were tested for antiviral activity in vitro against pseudotyped lentiviruses expressing the spike protein from the SARS‐CoV‐2 (USA‐WA1), Delta, or Omicron strains. Vesicular stomatitis virus (VSV) G protein served as a specificity control. A549‐ACE2 cells were pretreated with the indicated dose of drug and infection was quantified by luciferase expression. Shown are results representative of two biological replicates. (B) The specificity of gilteritinib for SARS‐CoV‐2 and AXL inhibition was tested in vitro using a panel of recombinant VSVs (rVSVs) expressing the Ebola (EBOV), Lassa mammarenavrus (LASV), or VSV glycoproteins. A549‐ACE2 cells were pretreated with gilteritinib and infection was quantified by counting GPF + cells. Concentrations listed are EC50 values calculated using a four‐parameter variable slope model (GraphPad Prism). SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
The PV results, which show differences in infectivity of VSV‐G compared to the variants in response to drug treatment, suggest drug effects involve, at least in part, inhibition of spike protein‐mediated viral entry. However, differences in gilteritinib potency against the Delta versus Wuhan strains cannot be explained by spike protein mutations and suggest postentry events, such as replication, might be targeted by gilteritinib differently in the different strains.
Because Ebola virus (EBOV) is known to require AXL activity for infectivity, 20 we also tested whether gilteritinib could inhibit infection by a recombinant VSVΔG expressing the EBOV glycoprotein (rVSV‐EBOV). Consistent with this requirement, rVSV‐EBOV was inhibited by gilteritinib with an EC50 of 0.25 μM (Figure 4B).
Recently, it was proposed that many repurposed small molecules that require higher concentrations to work for SARS‐CoV‐2 inhibition than for their original indication could be acting by disrupting lipid metabolism, termed phospholipidosis (PLD). 21 PLD is a process by which a drug, often with amphipathic cationic properties, disrupts membrane fluidity and lipid metabolism in the cell. 22 The hallmark of this process is accumulation of lipids in intracellular vesicles overlapping with drug activity. We evaluated gilteritinib for PLD by measuring accumulation of a lipid‐based fluorescent dye. While gilteritinib did result in lipid accumulation, as do many small molecules, the difference between EC50 for antiviral and PLD activity was around 2–4‐fold (Figures 1 and 2, Supporting Information: Figure 3). Specifically, gilteritinib induced PLD in A549‐ACE2 cells with an EC50 of 390 nM, in comparison to an EC50 of <100 nM against SARS‐CoV‐2 in this cell‐based assay. Concentrations ≤100 nM were able to inhibit viral production, however these concentrations were not high enough to induce PLD. The observed margin suggests that PLD likely at most contributes to only a small degree to the antiviral activity of gilteritinib. Of note, the same cells (A549‐ACE2) used in antiviral assays were also used for the PLD assay, and thus the drug potency/membrane permeability and other otherwise influential factors associated with host cell sensitivity were comparable. In parallel with gilteritinib, we also tested nintedanib and imatinib for PLD. We observed that both nintedanib and imatinib induced as much PLD as amodiaquine (a known PLD inducer serving as a positive control), whereas gilteritinib induced only a small amount of PLD (maximum 15‐fold induction for gilteritinib vs. around 40‐fold induction for nintedanib and imatinib) (Figure 5). Taken together, it can be concluded that nintedanib and imatinib are strong PLD inducers, with the EC50s close to the EC50s against SARS‐CoV‐2 infection, whereas gilteritinib induces a small amount of PLD compared to amodiaquine at an EC50 above the EC50 for SARS‐CoV‐2 inhibition. However, PLD is still a possible additional mechanism of antiviral activity, and one that requires further study.
Figure 5.

Effects of gilteritinib, nintedanib, and imatinib on phospholipidosis (PLD) in ACE2‐A549 cells. Comparison of induction of PLD by gilteritinib, nintedanib, and imatinib versus the positive control amodiaquine. Cells were pretreated with NBD‐PE reagent for 2 h before compound addition. Compounds were added in dose curves and cells were incubated overnight. PLD was quantified using Cell Profiler to measure NBD‐PE signal intensity per nuclei, and values were normalized to the negative control DMSO. EC50 values were calculated in Prism using a variable‐slope four‐parameter model.
Finally, to show that observed antiviral effects of gilteritinib were due to effects on virus production in the target cells and not due to direct virucidal effects, we carried out a virucidal assay. Pretreating the SARS‐CoV‐2 Beta variant with 10 μM of gilteritinib did not greatly reduce viability compared to the untreated and post‐treated controls (Supporting Information: Figure 4).
4. DISCUSSION
The kinase inhibitors investigated in our study were selected based on KINOMEscan biochemical kinase profiling assay data from the Harvard Medical School Library of Integrated Network‐based Cellular Signatures (LINCS) 17 and data derived from the ChEMBL database that identified target proteins implicated in SARS‐CoV, MERS‐CoV, and related virus infections. 16 Priority was also given to those with potential clinical benefits in the context of COVID‐19 disease symptoms, including anti‐inflammatory, antifibrotic or anticytokine activities, and pharmacokinetics for short‐term therapy. 16 Here, we focused on the tyrosine kinase inhibitors gilteritinib, nintedanib and imatinib. Both gilteritinib and nintedanib have overlapping activity against AXL while nintedanib and imatinib overlap in targeting c‐Kit. A functional relationship between c‐Kit and AXL is implicated: Within the C‐terminus cytoplasmic domain AXL shows 61% homology to c‐Kit, suggesting that both receptor tyrosine kinases may communicate through shared downstream mediating signaling factors. 23 In addition, AXL contributes to c‐Kit phosphorylation and thus may participate in its positive regulation by counteracting phosphatase activity that inhibits active c‐Kit. 23
We observed a significant and strong correlation between exogenous AXL levels and the degree of viral growth, consistent with previous reports. Of potential significance to SARS‐CoV‐2 infection, AXL has been demonstrated to be involved in SARS‐CoV‐2 infection of pulmonary cells and may act like a coreceptor promoting entry of the virus by binding the virus spike protein. 19 In this study by Wang and colleagues, AXL expression in HEK293 and pulmonary‐derived HT1299 (which do not normally express ACE2 at appreciable levels), was knocked out. For the HEK293 cells, restoration of AXL resulted in increased infection. SARS‐CoV‐2 binding, uptake and then infection was also reduced in each cell type. 19 Here, we show a similar effect on SARS‐CoV‐2 infection in human lung derived A549 cells but in the presence of ACE2 indicating that AXL still plays an important role in infection even when other receptors are present. Importantly, AXL was shown to physically interact with the N‐terminal domain (NTD) of the SARS‐CoV‐2 spike protein. 12 , 19 Collectively, these studies suggest that AXL is a target worthy of investigating for blocking SARS‐CoV‐2 entry and support the potential of an AXL‐targeting drug such as gilteritinib as a therapy for COVID‐19.
Gilteritinib exhibited the most potent activity, with an EC50 close to 100 nM, against SARS‐CoV‐2 infection. Gilteritinib is FDA‐approved for adult patients harboring mutated FLT3‐expressing refractory/relapsed AML, 18 but also has potent activity against AXL and to a lesser extent ALK. Gilteritinib was previously tested against SARS‐CoV‐2 in a study using a Vero cells. 24 In this earlier study, gilteritinib exhibited an EC50 of 6.75 uM and CC50 = 37.16 μM. The SI was 5.5, which is less than 10 and therefore would not be considered useful as a potential therapy. The same investigators later utilized a Calu‐3 cells (derived from human non‐small‐cell lung cancer) but showed no detectable activity. 25 Gilteritinib was also tested (Mount Sinai) for anti‐SARS‐CoV‐2 activity in a Vero cell‐based system, using an anti‐NP antibody, and was observed to exhibit an EC50 of around 0.8 μM with a SI < 10. 26 One other study evaluated gilteritinib using A549‐ACE2 cells similar to those used here and showed inhibition of virus replication down to the lowest concentration tested of 0.5 μM, with little effect on cell growth. 27 In another study an EC50 of 0.2 μM in Huh7 cells (derived from human hepatocellular carcinoma) was reported with an SI of 8.9 28 and worked regardless of whether drug was added pre‐ or postinfection, suggesting that gilteritinib inhibits postbinding events. Finally, gilteritinib was tested (Institut Pasteur) for anti‐SARS‐CoV‐2 activity in an A549‐ACE2 cell‐based system, using RT‐PCR and plaque assay and was observed to exhibit an EC50 of 0.06–0.12 μM with a SI > 10. 26 The results of these latter studies are consistent with our results using Vero and A549 cells. Differences compared to the earlier studies likely reflect cell types used and assay approaches, however the majority of work indicates that gilteritinib is a candidate for further testing in other systems that reflect virus infection in vivo, such as tissue explants. Such studies will be needed to advance this candidate. Of relevance, a successful remission with gilteritinib was reported in a FLT3‐mutant‐positive AML patient with severe COVID‐19. 29
In addition, our studies identified the kinase inhibitor, nintedanib, as displaying anti‐SARS‐CoV‐2 activity. Nintedanib, which is approved for idiopathic pulmonary fibrosis and exhibits anti‐inflammatory activity through TNF‐Alpha and IL‐6 inhibition, 30 is a potent inhibitor of AXL as well as several other kinases (AAK1, ABL1, KIT, RET, YES1). 16 Aside from AXL, the numb‐associated kinase family member, AAK1, a target of nintedanib, is involved in endocytosis of several RNA viruses, such as hepatitis C virus and dengue, which, like SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2, belong to the Baltimore Group IV classification of RNA viruses. 31 , 32 , 33 Knockdown of AXL, c‐Kit, and RET using siRNA revealed that each was important for infection by dengue viruses, and the SRC member, YES, was also shown to be important for infection. 34 , 35 The kinase profile of nintedanib, coupled with its well‐established antifibrotic activity, make nintedanib a candidate COVID‐19 therapy that could potentially reduce viral load in patients and also provide clinical benefit for the symptoms of the disease caused by SARS‐CoV‐2 infection.
Imatinib was previously tested for SARS‐CoV‐2 activity in a Vero E6 cell‐based assay system, with RNA analysis performed as a readout for virus levels. 36 In this former study, imatinib inhibited SARS‐CoV‐2 infection with an EC50 of 3–10 μM depending upon the multiplicity of infection used. 36 In contrast, our work yielded efficacy of 2 μM for both Vero and A549 cells with low cell toxicity evident in the Vero cell‐based assay. Like nintedanib, imatinib can mitigate some physical symptoms of COVID‐19. Case study reports and small clinical trial data are generally in favor of the anti‐inflammatory and antifibrotic effects of imatinib, although outcomes of such studies have been variable. 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45
Previous reports typically have not looked at sensitivity of different SARS‐CoV‐2 variants to drug treatments. When targeting the replication mechanism, such as RNA synthesis or virus protease activity required to process the virus polyproteins into mature subunits, there is little variation seen in each variant. Given tyrosine kinases often enable signal transduction from cell surface receptors, such as through AXL, changes in the virus spike protein could result in differences in susceptibility to treatment. Indeed, compared to the Alpha variant we observed infection efficiency of the WA and Delta variants to be more impaired by loss of AXL in A549 cells. However, this did not correlate to sensitivity to compounds, where Alpha and WA were consistently more sensitive than Delta (the latter being more similar in sensitivity to Beta). The Alpha, Beta, and Delta variants share the spike (D614G) mutation, the hallmark of all the variants located on the outside of the RBD, and which enhances the binding potential of ACE2 and consequently heightens the potential for viral infectivity. 5 , 7 , 46 However, changes in the NTD of the spike protein are thought to interact with AXL and other cell receptors.
The NTD of the spike protein is proposed to be involved in adhesion to the host cell surface and therefore is associated with viral infectivity. 46 There are key structural amino acid interactions within the spike NTD that contribute to its stability and shape. Mutations in these regions that have been identified in strains with high transmissibility, including Ala67 and Asp89 mutations, His69/Val70 and Tyr144 deletions, and deletions in amino acids 241‐243 and 246‐252, could conceivably impact the ability of the NTD to interact with AXL. 47 Of note, the Ala67 and Asp80 residues in the NTD, which are important for the maintenance of interloop interactions in the NTD supersite, are mutated in the Beta variant characterized by the D80A mutation. 47 , 48 The Alpha variant harbors HV69‐70del and Y144del, A570D, T716I, S982A, D1118H, E484K, N501Y, the Beta variant harbors mutations R246I, D215G, D80A, L18F, A701V, L242H, L242‐244del, and the Delta variant harbors E154K, R158G, T19R, G142D, 156del, 157del, L452R, E484Q. 5 , 7 , 49 , 50 Unfortunately, none of these changes appears to account for the Beta and Delta vs WA and Alpha sensitivity difference to the compounds tested. Indeed, while the PVs each showed higher sensitivity to the compounds than the VSV‐G PVs, there appeared to be little difference in the sensitivity of PVs bearing Wuhan (similar to WA) or Delta spike proteins. In summary, our work suggests that the compounds affect SARS‐CoV‐2 virus entry into cells but there are likely other residues in the spike that influence sensitivity and will require further investigation to be fully understood.
The flurry of drug repurposing studies sparked by the COVID‐19 pandemic has identified numerous candidates for therapeutic use, yet the mechanism of action as it relates to SARS‐CoV‐2 specifically is unknown for many. One consideration is whether the activities of such drugs are specific and target‐based, or whether antiviral effects are confounded by common functional groups that broadly alter host cell function. Drug‐induced PLD, a lysosomal storage disorder marked by hyperaccumulation of tissue phospholipids, has been revealed to be a common and shared property underlying the in vitro antiviral activity of numerous repurposed drugs investigated for activity against SARS‐CoV‐2. 21 , 22 This process is dependent on physical drug characteristics, is independent of antiviral effects stemming from target specificity, and would instead lead to antiviral effects through altering the lipid trafficking pathways viruses rely on to complete infection cycles. Results presented in our study suggest that the antiviral effects for gilteritinib against the variants may be target‐specific, as the concentrations required to induce PLD in A549‐ACE2 cells are significantly greater than the concentrations inhibiting SARS‐CoV‐2 infectivity and that of the variants.
There are currently no ongoing COVID‐19 trials investigating gilteritinib as a therapeutic. There are additional kinase inhibitors that are either approved or that are in clinical trials with anti‐AXL activity, and our findings presented herein with gilteritinib warrant further investigation of their anti‐SARS‐CoV‐2 activity within this context. Imatinib has been of clinical interest as a therapeutic agent based on its potential antiviral properties (due to ABL targeting) as well as potential anti‐inflammatory and anti‐cytokine effects (NCT04394416, NCT04346147, NCT04794088, NCT05220280, NCT04422678, NCT05273242, NCT04357613, NCT04330690), and clinical investigation is underway for nintedanib as an antifibrotic agent for pulmonary fibrosis in COVID‐19 patients (NCT04338802; NCT04856111; NCT04619680). Of note, nintedanib is not being studied as an antiviral agent.
5. CONCLUSION
Overall, our findings support further investigation of targeted kinase inhibitors, such as gilteritinib, as antiviral agents and potential therapies that may offer the advantage of mitigating the symptoms of COVID‐19, which can be life‐threatening. The repurposing of approved therapeutics that block the function of proteins required by SARS‐CoV‐2 to enter host cells and replicate, and that also inhibit the activity of proteins implicated in disease symptoms caused by the virus, is an attractive option for the rapid implementation of efficacious treatments, readily available and vetted for safety, in the setting of a pandemic outbreak.
AUTHOR CONTRIBUTIONS
RuthMabel Boytz wrote the manuscript and carried out the viral assays. Mikołaj Słabicki, Radosław P. Nowak, and Charles Zou performed the AXL KO studies. Sita Ramaswamy, Rahm Gummuluru, Brett L. Hurst, and J. J. Patten performed the viral assays. Chengcheng Meng validated AXL KO. Jinhua Wang acquired and prepared drug stocks for the viral assays. Priscilla L. Yang, Martin Sattler, James D. Griffin, and Richard M. Stone provided valuable scientific feedback. Nathanael S. Gray initiated the study, provided the drug stocks, and provided valuable scientific feedback. Robert A. Davey and Ellen Weisberg wrote the paper and designed the research study.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
Supplementary information.
ACKNOWLEDGMENTS
President and fellows of Harvard University and Dana‐Farber Cancer Institute will be utilizing/has utilized the nonclinical and preclinical services program offered by the National Institute of Allergy and Infectious Diseases. Part funding for this study was supported by a grant from MassCPR to R. A. D., R. M. B., J. J. P., and R. A. D. were additionally supported in part by NIH R01 AI128364, UC7AI095321, and P01AI120943.
Boytz R, Słabicki M, Ramaswamy S, et al. Anti‐SARS‐CoV‐2 activity of targeted kinase inhibitors: repurposing clinically available drugs for COVID‐19 therapy. J Med Virol. 2022;1‐10. 10.1002/jmv.28157
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Morse JS, Lalonde T, Xu S, Liu WR. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019‐nCoV. ChemBioChem. 2020;21(5):730‐738. 10.1002/cbic.202000047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi H, Han X, Jiang N, et al. Radiological findings from 81 patients with COVID‐19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. 2020;20(4):425‐434. 10.1016/S1473-3099(20)30086-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gautier JF, Ravussin Y. A new symptom of COVID‐19: loss of taste and smell. Obesity. 2020;28(5):848. 10.1002/oby.22809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jacob JJ, John Fletcher G, Monisha Priya T, Veeraraghavan B, Mutreja A. Relevance of immune response and vaccination strategies of SARS‐CoV‐2 in the phase of viral red queen dynamics. Indian J Med Microbiol. 2021;39(4):417‐422. 10.1016/j.ijmmb.2021.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies NG, Abbott S, Barnard RC, et al. Estimated transmissibility and impact of SARS‐CoV‐2 lineage B.1.1.7 in England. Science. 2021;372(6538):eabg3055. 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winger A, Caspari T. The spike of concern‐the novel variants of SARS‐CoV‐2. Viruses. 2021;13(6):1002. 10.3390/v13061002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vaughan A. Omicron emerges. New Sci. 2021;252(3363):7. 10.1016/S0262-4079(21)02140-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Daria S, Bhuiyan MA, Islam MR. Detection of highly muted coronavirus variant Omicron (B.1.1.529) is triggering the alarm for South Asian countries: associated risk factors and preventive actions. J Med Virol. 2021;94:1267‐1268. 10.1002/jmv.27503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao YC, Deng QX, Dai SX. Remdesivir for severe acute respiratory syndrome coronavirus 2 causing COVID‐19: an evaluation of the evidence. Travel Med Infect Dis. 2020;35:101647. 10.1016/j.tmaid.2020.101647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koch J, Uckeley ZM, Doldan P, Stanifer M, Boulant S, Lozach PY. TMPRSS2 expression dictates the entry route used by SARS‐CoV‐2 to infect host cells. EMBO J. 2021;40(16):e107821. 10.15252/embj.2021107821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bohan D, Ert HV, Ruggio N. Phosphatidylserine Receptors Enhance SARS‐CoV‐2 Infection: AXL as a Therapeutic Target for COVID‐19. bioRxiv. 2021;17(11):e1009743. 10.1101/2021.06.15.448419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahase E. Covid‐19: Pfizer's paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ. 2021;375:n2713. 10.1136/bmj.n2713 [DOI] [PubMed] [Google Scholar]
- 14. Painter WP, Holman W, Bush JA, et al. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad‐spectrum oral antiviral agent with activity against SARS‐CoV‐2. Antimicrob Agents Chemother. 2021;65(5):e02428‐20. 10.1128/AAC.02428-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hashemian SMR, Pourhanifeh MH, Hamblin MR, Shahrzad MK, Mirzaei H. RdRp inhibitors and COVID‐19: is molnupiravir a good option? Biomed Pharmacother. 2021;146:112517. 10.1016/j.biopha.2021.112517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weisberg E, Parent A, Yang PL, et al. Repurposing of kinase inhibitors for treatment of COVID‐19. Pharm Res. 2020;37(9):167. 10.1007/s11095-020-02851-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rouillard AD, Gundersen GW, Fernandez NF, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database. 2016;2016:baw100. 10.1093/database/baw100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee LY, Hernandez D, Rajkhowa T, et al. Preclinical studies of gilteritinib, a next‐generation FLT3 inhibitor. Blood. 2017;129(2):257‐260. 10.1182/blood-2016-10-745133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang S, Qiu Z, Hou Y, et al. AXL is a candidate receptor for SARS‐CoV‐2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021;31(2):126‐140. 10.1038/s41422-020-00460-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimojima M, Ikeda Y, Kawaoka Y. The mechanism of Axl‐mediated Ebola virus infection. J Infect Dis. 2007;196(suppl 2):S259‐S263. 10.1086/520594 [DOI] [PubMed] [Google Scholar]
- 21. Tummino TA, Rezelj VV, Fischer B, et al. Drug‐induced phospholipidosis confounds drug repurposing for SARS‐CoV‐2. Science. 2021;373(6554):541‐547. 10.1126/science.abi4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Breiden B, Sandhoff K. Emerging mechanisms of drug‐induced phospholipidosis. Biol Chem. 2019;401(1):31‐46. 10.1515/hsz-2019-0270 [DOI] [PubMed] [Google Scholar]
- 23. Park IK, Giovenzana C, Hughes TL, Yu J, Trotta R, Caligiuri MA. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood. 2009;113(11):2470‐2477. 10.1182/blood-2008-05-157073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeon S, Ko M, Lee J, et al. Identification of antiviral drug candidates against SARS‐CoV‐2 from FDA‐approved drugs. Antimicrob Agents Chemother. 2020;64(7):e00819‐20. 10.1128/AAC.00819-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ko M, Jeon S, Ryu WS, Kim S. Comparative analysis of antiviral efficacy of FDA‐approved drugs against SARS‐CoV‐2 in human lung cells. J Med Virol. 2021;93(3):1403‐1408. 10.1002/jmv.26397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bouhaddou M, Memon D, Meyer B, et al. The global phosphorylation landscape of SARS‐CoV‐2 infection. Cell. 2020;182(3):685‐712. 10.1016/j.cell.2020.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stukalov A, Girault V, Grass V, et al. Multilevel proteomics reveals host perturbations by SARS‐CoV‐2 and SARS‐CoV. Nature. 2021;594:246‐252. 10.1038/s41586-021-03493-4 [DOI] [PubMed] [Google Scholar]
- 28. Mirabelli C, Wotring JW, Zhang CJ, et al. Morphological cell profiling of SARS‐CoV‐2 infection identifies drug repurposing candidates for COVID‐19. Proc Natl Acad Sci USA. 2021;118(36):e2105815118. 10.1073/pnas.2105815118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilson AJ, Troy‐Barnes E, Subhan M, et al. Successful remission induction therapy with gilteritinib in a patient with de novo FLT3‐mutated acute myeloid leukaemia and severe COVID‐19. Br J Haematol. 2020;190(4):e189‐e191. 10.1111/bjh.16962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu F, Bayliss G, Zhuang S. Application of nintedanib and other potential anti‐fibrotic agents in fibrotic diseases. Clin Sci (Lond). 2019;133(12):1309‐1320. 10.1042/CS20190249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Conner SD, Schmid SL. Identification of an adaptor‐associated kinase, AAK1, as a regulator of clathrin‐mediated endocytosis. J Cell Biol. 2002;156(5):921‐929. 10.1083/jcb.200108123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neveu G, Ziv‐Av A, Barouch‐Bentov R, Berkerman E, Mulholland J, Einav S. AP‐2‐associated protein kinase 1 and cyclin G‐associated kinase regulate hepatitis C virus entry and are potential drug targets. J Virol. 2015;89(8):4387‐4404. 10.1128/JVI.02705-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pu SY, Xiao F, Schor S, et al. Feasibility and biological rationale of repurposing sunitinib and erlotinib for dengue treatment. Antiviral Res. 2018;155:67‐75. 10.1016/j.antiviral.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bekerman E, Neveu G, Shulla A, et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad‐spectrum antiviral effects. J Clin Invest. 2017;127(4):1338‐1352. 10.1172/JCI89857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumar R, Agrawal T, Khan NA, Nakayama Y, Medigeshi GR. Identification and characterization of the role of c‐terminal Src kinase in dengue virus replication. Sci Rep. 2016;6:30490. 10.1038/srep30490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weston S, Coleman CM, Haupt R, et al. Broad anti‐coronavirus activity of food and drug administration‐approved drugs against SARS‐CoV‐2 in vitro and SARS‐CoV in vivo. J Virol. 2020;94(21):e01218‐20. 10.1128/JVI.01218-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Daniels CE, Wilkes MC, Edens M, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF‐beta and prevents bleomycin‐mediated lung fibrosis. J Clin Invest. 2004;114(9):1308‐1316. 10.1172/JCI19603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miyachi K, Ihara A, Hankins RW, Murai R, Maehiro S, Miyashita H. Efficacy of imatinib mesylate (STI571) treatment for a patient with rheumatoid arthritis developing chronic myelogenous leukemia. Clin Rheumatol. 2003;22(4‐5):329‐332. 10.1007/s10067-003-0716-3 [DOI] [PubMed] [Google Scholar]
- 39. Eklund KK, Joensuu H. Treatment of rheumatoid arthritis with imatinib mesylate: clinical improvement in three refractory cases. Ann Med. 2003;35(5):362‐367. 10.1080/07853890310001339 [DOI] [PubMed] [Google Scholar]
- 40. Ciarcia R, Vitiello MT, Galdiero M, et al. Imatinib treatment inhibit IL‐6, IL‐8, NF‐KB and AP‐1 production and modulate intracellular calcium in CML patients. J Cell Physiol. 2012;227(6):2798‐2803. 10.1002/jcp.23029 [DOI] [PubMed] [Google Scholar]
- 41. Kay J, High WA. Imatinib mesylate treatment of nephrogenic systemic fibrosis. Arthritis Rheum. 2008;58(8):2543‐2548. 10.1002/art.23696 [DOI] [PubMed] [Google Scholar]
- 42. Distler JH, Manger B, Spriewald BM, Schett G, Distler O. Treatment of pulmonary fibrosis for twenty weeks with imatinib mesylate in a patient with mixed connective tissue disease. Arthritis Rheum. 2008;58(8):2538‐2542. 10.1002/art.23694 [DOI] [PubMed] [Google Scholar]
- 43. Carnevale‐Schianca F, Gallo S, Rota‐Scalabrini D, et al. Complete resolution of life‐threatening bleomycin‐induced pneumonitis after treatment with imatinib mesylate in a patient with Hodgkin's lymphoma: hope for severe chemotherapy‐induced toxicity? J Clin Oncol. 2011;29(24):e691‐e693. 10.1200/JCO.2011.35.6733 [DOI] [PubMed] [Google Scholar]
- 44. Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo‐controlled trial results. Am J Respir Crit Care Med. 2010;181(6):604‐610. 10.1164/rccm.200906-0964OC [DOI] [PubMed] [Google Scholar]
- 45. Banakh I, Lam A, Tiruvoipati R, Carney I, Botha J. Imatinib for bleomycin induced pulmonary toxicity: a case report and evidence‐base review. Clin Case Rep. 2016;4(5):486‐490. 10.1002/ccr3.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Awasthi M, Gulati S, Sarkar DP, et al. The sialoside‐binding pocket of SARS‐CoV‐2 spike glycoprotein structurally resembles MERS‐CoV. Viruses. 2020;12(9):909. 10.3390/v12090909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klinakis A, Cournia Z, Rampias T. N‐terminal domain mutations of the spike protein are structurally implicated in epitope recognition in emerging SARS‐CoV‐2 strains. Comput Struct Biotechnol J. 2021. 10.1016/j.csbj.2021.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cai Y, Zhang J, Xiao T, et al. Structural basis for enhanced infectivity and immune evasion of SARS‐CoV‐2 variants. Science. 2021;373(6555):642‐648. 10.1126/science.abi9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aleem A, Akbar Samad AB, Slenker AK. Emerging variants of SARS‐CoV‐2 and novel therapeutics against coronavirus (COVID‐19). In: StatPearls [Internet]. 2022. Accessed StatPearls Publishing. [PubMed]
- 50. Greaney AJ, Loes AN, Crawford KHD, et al. Comprehensive mapping of mutations in the SARS‐CoV‐2 receptor‐binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021;29(3):463‐476. 10.1016/j.chom.2021.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.