

Abstract
Recent pandemic infection caused by SARS‐CoV‐2 (COVID‐19) led the scientific community to investigate the possible causes contributing to the physiopathology of this disease. In this context, analyses of the intestinal microbiota highlighted possible correlation between host‐associated bacterial communities and development of the COVID‐19. Nevertheless, a detailed investigation of the role of the human microbiota in the severity of the symptoms of this disease is still lacking. This study performed a comprehensive meta‐analysis of 323 faecal samples from public and novel Italian data sets based on the shotgun metagenomic approach. In detail, the comparative analyses revealed possible differences in the microbial biodiversity related to the individual health status, highlighting a species richness decrease in COVID‐19 patients with a severe prognosis. Moreover, healthy subjects resulted characterized by a higher abundance of protective and health‐supporting bacterial species, while patients affected by COVID‐19 disease displayed a significant increase of opportunistic pathogen bacteria involved in developing putrefactive dysbiosis. Furthermore, prediction of the microbiome functional capabilities suggested that individuals affected by COVID‐19 subsist in an unbalanced metabolism characterized by an overrepresentation of enzymes involved in the protein metabolism at the expense of carbohydrates oriented pathways, which can impact on disease severity and in excessive systemic inflammation.
INTRODUCTION
Coronavirus disease (COVID‐19) is an ongoing global pandemic emerged in late 2019 caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) (https://covid19.who.int/). To date, COVID‐19 pandemic involved more than 450 million cases worldwide (https://www.ecdc.europa.eu/en/covid-19), including asymptomatic as well as paucisymptomatic, and severely symptomatic individuals. The symptoms and the severity of COVID‐19 disease differ from individual to individual (Markovic et al., 2021; Vrotsou et al., 2021), and the underlying causes have not yet been fully clarified (Bohn et al., 2020).
Recent studies highlighted a possible correlation between COVID‐19 disease and intestinal and/or respiratory microbiota (Andrade et al., 2022; Sencio et al., 2022; Wang et al., 2021; Yeoh et al., 2021), suggesting that the resident microbial communities may serve as a novel therapeutic target for improving the prognosis of patients (McIlroy et al., 2020; Wu et al., 2020). In particular, the intestinal microbiota of subjects affected by COVID‐19 has been reported to be different to healthy individuals and it was mainly characterized by a low bacterial richness, diversity, and uniformity (Wang et al., 2021), along with a decrease of beneficial bacteria, such as Faecalibacterium, Roseburia, and Blautia, in favour of several opportunistic pathogen bacteria, such as Actinomyces, Rothia, Veillonella, and Streptococcus (Wang et al., 2021; Xu et al., 2021). In fact, the reduction of these short‐chain fatty acid‐producing bacteria could increase oxidative stress and ROS production that affect the regulation of the intestinal barrier and contribute to the systemic inflammation (Wang et al., 2021). These observations were mainly obtained through the 16S rRNA gene sequencing analysis (Chen et al., 2022; Mazzarelli et al., 2021; Romani et al., 2022; Wang et al., 2021; Xu et al., 2021), whose main limit is to allow a taxonomic profiling of the microbial communities down to genus level while being inaccurate at the species level and preventing the reconstruction of functional impact of the microbiome.
Moreover, currently published information regarding the possible correlations between microbiota composition and COVID‐19 disease are generally linked to a specific nationality (Britton et al., 2021; Liu et al., 2021; Mazzarelli et al., 2021; Zhang et al., 2022; Zhou et al., 2022; Zuo et al., 2020). For these reasons, we decided to perform an in‐depth meta‐analysis based on seven public shotgun metagenomics data sets corresponding to 291 faecal samples from North American and Chinese populations, obtained from healthy and COVID‐19 disease patients, for which metadata regarding disease severity was available. Moreover, in order to extend the variety of the populations collected in the meta‐analysis, we included a novel data set of faecal samples, which was collected and analysed in the framework of this study, encompassing by both healthy and COVID‐19‐affected Italian individuals. All the shotgun metagenomics data sets included in this study were employed to achieve a detail taxonomical profile at species level and to examine the genetic repertoire of the gut microbiome in relationship to COVID‐19 disease severity.
EXPERIMENTAL PROCEDURES
Selection and collection of samples included in the meta‐analysis
In this meta‐analysis‐based study, we retrieved six publicly available data sets from studies regarding the possible correlation between human gut microbiome and COVID‐19 disease, performed in accordance with the relevant guidelines and regulations. Moreover, in order to overcome the absence of American samples healthy control and to be able to exclude possible biases related to nationality, we retrieved 28 faecal samples from a publicly available data set. In particular, we selected only shotgun metagenomic datasets obtained by an Illumina sequencing platform to avoid the variability of the input data as much as possible. In detail, we selected shotgun metagenomic data sets from 291 samples from adult healthy or diseased individuals from China and North America (Table 1). In addition, we enrolled 32 Italian hospitalized patients with or without COVID‐19 disease in the framework of the project COVIDbiome from which faecal samples were collected (Table 1) and submitted to shallow metagenomics analyses. In detail, the fresh faecal samples obtained from the Italian hospitalized patients were immediately inactivated with DNA/RNA shield buffer (Zymo Research, USA) and subsequently submitted to the extraction of bacterial DNA using the protocol previously described (Mancabelli et al., 2021). The COVIDbiome study was approved as part of a larger project on the study of respiratory microbiome in COVID‐19 by the local Ethics Committee (Comitato Etico dell'Area Vasta Emilia Nord, Emilia‐Romagna Region, Italy), under the ID 1131/2020/TESS/UNIPR.
TABLE 1.
Data sets included in the meta‐analysis
| COVID‐19 | |||||||
|---|---|---|---|---|---|---|---|
| NCBI Bioproject | PMID | Nation | Number of samples | Healthy | Moderate | Severe | Total |
| This study | This study | Italy | 32 | 11 | 6 | 15 | 21 |
| PRJNA624223 | 32442562 | China | 30 | 15 | 10 | 5 | 15 |
| PRJNA660883 | 32909002 | North America | 31 | ‐ | 20 | 11 | 31 |
| PRJEB28543 | 31705027 | North America | 28 | ‐ | ‐ | ‐ | ‐ |
| PRJNA689961 | 34687739 | China | 136 | 70 | 66 | ‐ | 66 |
| PRJNA740067 | 35281785 | China | 26 | 13 | 12 | 1 | 13 |
| PRJNA762232 | 34926314 | China | 2 | 1 | 1 | ‐ | 1 |
| PRJNA792726 | ‐ | China | 38 | ‐ | 24 | 14 | 38 |
Shallow shotgun sequencing
According to the manufacturer's instructions, DNA library preparation was performed using the Nextera XT DNA sample preparation kit (Illumina, San Diego, CA, USA). First, 1 ng input DNA from each sample was used for the library preparation, which underwent fragmentation, adapter ligation, and amplification. Then, Illumina libraries were pooled equimolarly, denatured, and diluted to a concentration of 1.5 pM. Next, DNA sequencing was performed on a MiSeq instrument (Illumina) using a 2 × 250 bp Output sequencing Kit together with a deliberate spike‐in of 1% PhiX control library.
Taxonomic classification of sequence reads
Taxonomic profiling of sequenced reads was performed employing the METAnnotatorX2 bioinformatics platform (Milani et al., 2018, 2021). In detail, the downloaded fastq files were filtered to remove reads with quality of <25 and to retain reads with a length of >100 bp. Subsequently, a human host DNA filtering was performed through bowtie2 software (Langmead et al., 2019; Langmead & Salzberg, 2012), following the METAnnotatorX2 manual (Milani et al., 2021). Afterward, the taxonomic classification of 100,000 reads was achieved by means of MegaBLAST (Chen et al., 2015) employing a manually curated and pre‐processed database of genomes retrieved from the National Center for Biotechnology Information (NCBI), following the METAnnotatorX2 manual (Milani et al., 2021).
Functional prediction
Functional profiling of the sequenced reads was performed with the METAnnotatorX2 bioinformatics platform (Milani et al., 2018, 2021). Functional classification of reads was performed to reveal metabolic pathways based on the MetaCyc database (release 24.1) (Caspi et al., 2016) through RAPSearch2 software (Ye et al., 2011; Zhao et al., 2012).
Statistical analysis
ORIGIN 2021 (https://www.originlab.com/2021) and SPSS software (www.ibm.com/software/it/analytics/spss/) were used to compute statistical analyses, including HCL and Silhouette analyses. EMPeror tool was used to visualize principal coordinate analysis (PCoA) (Vazquez‐Baeza et al., 2013) calculated through ORIGIN 2021. PERMANOVA (Permutational analysis of variance) analyses were performed using 1000 permutations to estimate p values for differences among populations in PCoA. Furthermore, differential abundance of bacterial genera was tested by t‐test or ANOVA test analysis. Multiple comparison analyses were performed through Tukey's HSD (honestly significant difference) test.
RESULTS AND DISCUSSIONS
Dataset selection
In order to retrieve all the publicly available shotgun metagenomic data sets concerning the microbiota of COVID‐19 patients, an extensive scientific literature screening was performed. In detail, the scientific literature examination allowed us to collect data from six publicly available data sets (Britton et al., 2021; Liu et al., 2021; Ventura et al., 2019; Zhang et al., 2022; Zhou et al., 2022; Zuo et al., 2020), encompassing healthy and diseased human faecal samples based on Illumina shotgun metagenomic approaches (Table 1). Our database of metagenomic data about the gut microbiota of COVID‐19 patients included only those studies reporting metagenomic metadata about healthy from diseased individuals. Furthermore, healthy American samples from a single public bioproject (Ventura et al., 2019) were selected to overcome the absence of a control group for that nation (Table 1). Such database was extended by including metagenomic data of fresh faecal samples from 32 Italian hospitalized patients with or without COVID‐19 disease that were collected in the framework of a local project focusing on the exploration of the link between gut microbiota composition and severity of the symptoms named COVIDbiome (Table 1). Thus, the meta‐analysis included a total of 323 samples (Table 1 and Table S1). These include three groups, the group of healthy subjects that encompasses individuals resulted negative to SARS‐CoV‐2 infection, the group of asymptomatic or paucisymptomatic COVID‐19 patients, which includes individuals showing moderate COVID‐19 symptoms, and finally the group of severely symptomatic COVID‐19 patients, displaying severe COVID‐19 symptoms (Table 1 and Table S1).
Intra‐ and inter‐individual variability among healthy individuals and those affected by COVID‐19
The 323 stool samples analysed in this study, involving the 291 samples from public metagenomic datasets and the 32 samples collected in the framework of the COVIDbiome study, were used to assess the microbiota composition through the METAnnotatorX2 software (Milani et al., 2021) following the standard filtering parameters reported in the manual, comprising the Homo sapiens reads removal. All the fastq files analysed here were processed with the same bioinformatics pipeline to prevent biases, resulting in a total of 6,183,408,133 reads with an average per sample of 19,143,678 ± 15,020,002 after quality and human sequences filtering (Table S1). In order to optimize the taxonomical analysis, in accordance with the modern shallow shotgun metagenomics approach (Hillmann et al., 2018; Milani et al., 2021), we decided to analyse a subset of 100,000 reads for each sample, obtaining a total of 22,021,244 classified reads with an average per sample of 68,177 ± 10,074 (Table S1).
The results obtained through METAnnotatorX2 software (Milani et al., 2021) were used to evaluate the microbial biodiversity of each faecal sample. In detail, we focused our interest on the comparison of the species richness between the three groups based on the individual health status, that is, healthy, moderate COVID‐19, and severe COVID‐19 (Figure 1A). The analysis revealed a decrease in bacterial species abundance related to the health status of the subjects included in the meta‐analysis, highlighting the smallest number of average bacterial species in the severe COVID‐19 samples (average of 59 ± 24, ANOVA and Tukey HSD post hoc p value < 0.05) (Figure 1A). Notably, the species richness values of the healthy and moderate COVID‐19 groups indicated a similar average number of species of 82 ± 19 and 78 ± 22, respectively (Tukey HSD post hoc p value = 0.278). These results suggested that the simplification of gut microbiota and consequently the possible presence of dysbiosis (Kriss et al., 2018; Mosca et al., 2016) could be associated with a COVID‐19 infections displaying severe disease symptoms (Wang et al., 2021). Moreover, the analysis of the species richness based on the geographical origin of the assayed faecal samples confirmed the higher microbial biodiversity in the group of healthy individuals compared to that including COVID‐19 subjects showing severe symptoms (Figure S1a), excluding a possible bias related to the geographical origin of the samples.
FIGURE 1.
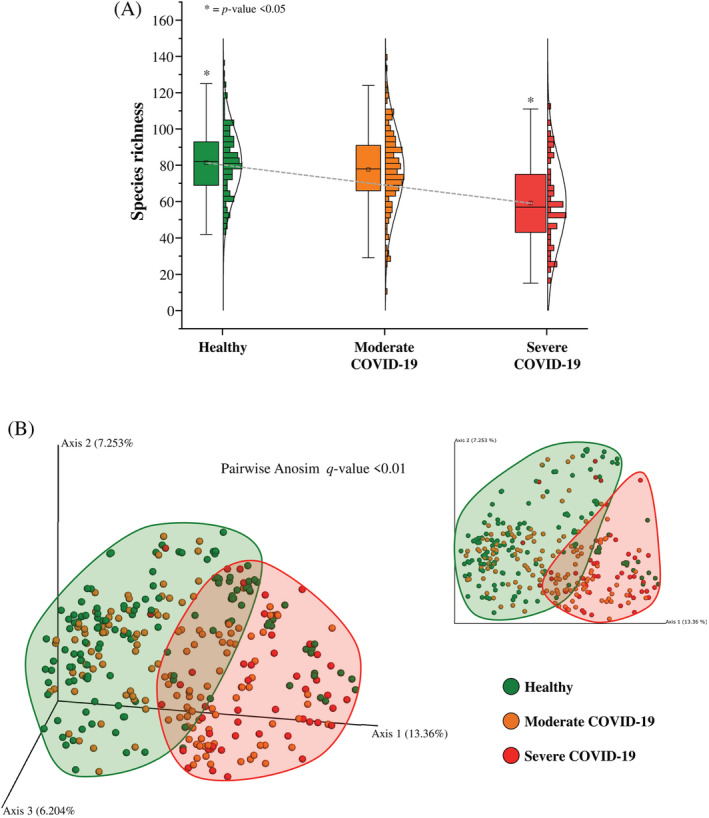
Evaluation of microbial biodiversity. Panel (A) displays the Whiskers plot representing the species richness identified from healthy, moderate and severe COVID‐19 subjects. The x‐axis represents the different groups, while the y‐axis indicates the number of species. The boxes are determined by the 25th and 75th percentiles. The whiskers are determined by 1.5 interquartile range (IQR). The line in the boxes represents the median, while the square represents the average. Panel (B) reports the principal coordinate analysis (PCoA) of the faecal samples included in the meta‐analysis, subdivided by health status.
Furthermore, the evaluation of the inter‐individual differences of the samples within each group (healthy, moderate, and severe COVID‐19 subjects) was assessed through the beta‐diversity calculated by Bray–Curtis dissimilarity and based on the taxonomical composition at species level. The beta‐diversity was reported through a three‐dimensional PCoA that showed a subdivision in clustering based on the individual health status (pairwise Anosim q‐value < 0.01; healthy vs. moderate COVID‐19 R = 0.039, healthy vs. severe COVID‐19 R = 0.407). Thus, suggesting taxonomic differences between the three groups (Figure 1B). Further PCoA based on individuals' health status and geographical origin revealed a partial subdivision correlated with the geographical origin, as expected but confirmed the separate cluster of severe COVID‐19 subjects (Figure S1b). Moreover, in order to identify possible correlations between the gut microbiota composition and SARS‐CoV‐2 vaccination (Brussow, 2021; Chen et al., 2021; Yarza et al., 2014), a PCoA was performed on the Italian samples (Figure S2). In fact, the absence of specific metadata regarding vaccination in the other public datasets did not allow to perform any comprehensive PCoA. In detail, the beta‐diversity analysis on Italian samples did not reveal any significant differences in composition between the vaccinated and unvaccinated individuals (pairwise Anosim q‐value < 0.197, R = 0.042) (Figure S2a). Similarly, the multiple comparisons between Italian samples based on health status and vaccination did not highlight any significant differences (pairwise Anosim p < 0.761, R = −0.052; pairwise PERMANOVA p value < 0.556, R = 0.125; pseudo‐F = 0.961) (Figure S2b), suggesting no correlation between vaccination and gut microbiota composition. However, the low number of samples included in this preliminary analysis and the low intra‐groups vaccination variability prevented to obtain statistically robust results, thus requiring further investigations to disentangle the identification of possible relationships between microbiota composition and vaccines against SARS‐CoV‐2.
Species‐level taxonomic profiling of the gut microbiota of COVID‐19 and healthy individuals
The METAnnotatorX2 software (Milani et al., 2021) allowed to obtain a detailed taxonomical profile at species level for each sample assayed, including 291 samples from public metagenomic datasets and the 32 samples collected in the framework of the COVIDbiome study (Table S2). In detail, the analysis revealed that the group of patients with severe COVID‐19 prognosis showed a higher abundance of species belonging to the Bacteroidetes phylum (52.79% ± 31.09%) compared to healthy (31.64% ± 29.35%) and moderate COVID‐19 (28.47% ± 22.24%) groups (Tukey HSD post hoc p value < 0.05), and a lower abundance of species belonging to Firmicutes phylum (33.95% ± 27.13%) compared to healthy (48.84% ± 23.31%) and moderate COVID‐19 (53.06% ± 21.08%) groups (Tukey HSD post hoc p value < 0.05).
A specific taxonomical analysis based on the prevalence and the abundance trend of each bacterial species calculated separately for the three groups, also confirmed by ANOVA statistical analysis, revealed possible taxonomical differences linked to the level of severity of COVID‐19 symptoms. In particular, focusing on the bacterial species with a prevalence higher than 50% in each group (Figure 2A), the healthy individuals were characterized by four taxa, that is, Faecalibacterium prausnitzii, Faecalibacterium unknown species, Gemmiger unknown species, and Ruminococcus unknown species, which resulted more abundant compared to patients with severe COVID‐19 disease (Figure 2A), suggesting a possible role of these microorganisms in the protection from the diseases and/or mitigation of the symptoms thanks to their well‐known immunomodulation features (Yeoh et al., 2021; Zuo et al., 2020). In fact, species belonging to Faecalibacterium and Gemmiger genera are bacteria that produce butyrate, an important anti‐inflammatory compound (Sang et al., 2022; Yeoh et al., 2021). Remarkably, our hypothesis is reinforced by the absence of significant differences for three of the four taxa between healthy and moderate COVID‐19 groups, highlighting the possible presence of an abundance gradient correlated with the disease severity (Figure 2A).
FIGURE 2.
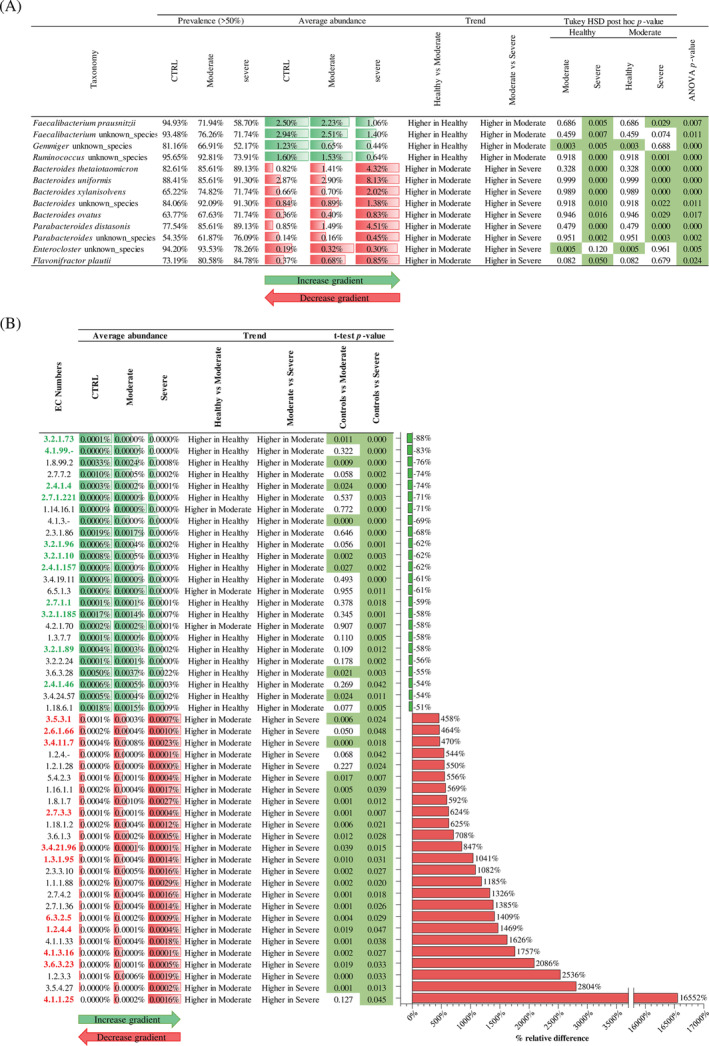
Taxonomical and functional comparison between healthy and COVID‐19 subjects. Panel (A) reports the significantly different taxa between groups, reporting prevalence, average abundance, trend, and ANOVA and Tukey HSD post hoc p value. Panel (B) indicates the EC numbers with an increase >400% and a decrease <50% in severe COVID‐19 subjects compared to healthy samples. In detail, the panel reports the average abundance of each group, the trend, the t‐test p value, and the bar plot representing the relative percentage difference. Moreover, EC numbers highlighted in green are related to the metabolism of complex carbohydrates, while EC numbers highlighted in red are related to the protein metabolism.
Conversely, the microbiota of the group encompassing severe COVID‐19 patients revealed a significant increase (ANOVA and Tukey HSD post hoc p value < 0.05) of nine species belonging to Bacteroides, Parabacteroides, Enterocloster, and Flavonifractor genera compared to microbiota of the healthy subjects (Figure 2A). These bacteria were typical commensals of the human gastrointestinal tract involved in several metabolic activities, such as production of putrefactive compounds derived from protein fermentation (Mosele et al., 2015), and largely acknowledged as opportunistic pathogens (Zeng et al., 2017). Thus, the concomitance with an alteration of gut microbiota homeostasis, suggested by the decrease of the biodiversity, could promote an overgrowth of these opportunistic pathogen bacteria that could be responsible for a putrefactive dysbiosis. In detail, this typology of dysbiosis was usually correlated with an excessive intake of meat and saturated fats with a consequent increase of putrefying bacteria, such as species belonging to the Bacteroides genus (Baizabal‐Carvallo, 2021), involved in putrefaction and fermentation of proteins (Kaur et al., 2017). Therefore, the overgrowth of these bacteria and the concomitant expansion of protein metabolism could increase several disadvantageous compounds, such as ammonia and amines (Gagliardi et al., 2018), and further promote the susceptibility to pathogenic bacteria (Zeng et al., 2017). Intriguingly, also for these taxa, the moderate COVID‐19 subjects showed an intermediate microbiota and the possible presence of an abundance gradient correlated with the disease severity (Figure 2A), confirming a possible correlation between gut microbiota, its metabolism, and COVID‐19 symptoms and severity.
Functional prediction of gut microbiome
The taxonomical comparison between healthy and severe COVID‐19 subjects revealed specific differences in the bacterial communities, suggesting possible specific genetic repertoires. In order to explore the genetic features characterizing each faecal sample collected in this meta‐analysis, we performed a screening of metabolic enzymatic reactions based on the MetaCyc database (Caspi et al., 2016) and the Enzyme Commission (EC) classification.
Overall, the 306 enzymatic reactions that were identified as significantly different between healthy and severe COVID‐19 subjects display an interesting abundance gradient passing through the moderate group (Table S3). In fact, the relative abundance comparison between the healthy, moderate, and severe COVID‐19 groups highlighted that 86% of the enzymatic reactions presented an abundance gradient correlated with disease severity, probably indicating an intermediate microbiome of moderate COVID‐19 patients, and suggesting the important role of the metabolic capability of the bacterial communities on the host health status.
Furthermore, our analyses were focused on the main differences identified between healthy and severe COVID‐19 subjects, particularly on the enzymatic reactions with prevalence higher than 50% in at least one group. In detail, the analysis revealed a total of 306 enzymatic reactions significantly different between healthy and severe COVID‐19 patients (t‐test p value < 0.05) (Table S3). Among these, severe COVID‐19 subjects showed a total of 25 enzymatic reactions with an increase higher than 450% in the relative abundance, of which 11 are involved in protein metabolism (Figure 2B). This result is in accordance with the above taxonomical analysis and the increase of species related to putrefactive dysbiosis. Remarkably, several public studies highlighted the possible correlation between COVID‐19 disease severity and the alteration of systemic amino acid metabolism (Masoodi et al., 2022; Paez‐Franco et al., 2021; Rahnavard et al., 2022). In particular, the enrichment of specific amino acids, such as arginine and proline, could contribute to excessive systemic inflammation and a consequent increase in the severity of the disease (Rahnavard et al., 2022). Therefore, the increased protein metabolic capability of the intestinal microbiome observed in this study could suggest the intestinal bacterial communities' contribution to systemic inflammation.
Conversely, 24 enzymatic reactions showed a decrease lower than 50% in severe COVID‐19 subjects' relative abundance (Figure 2B) and the 46% of these resulted related to the metabolism of complex carbohydrate, such as hexokinase enzyme (EC 2.7.1.1), indicating a probably shift from a carbohydrate‐oriented to a protein‐oriented catabolism, which further support the onset of putrefactive gut microbiota dysbiosis in the patients with severe COVID‐19 symptoms. In this context, we could hypothesize a major role of the diet and lifestyle on microbiome composition and, consequently, a possible correlation with disease severity.
However, the absence of specific metadata regarding diet and subject's lifestyle prevented deeper correlation analyses, proposing the necessity for further specific analyses concerning the correlation between diet, microbiome, and COVID‐19 disease severity.
CONCLUSION
In order to investigate the impact of microbiome composition on the severity of the COVID‐19 disease, we performed an in‐depth meta‐analysis of healthy and diseased individuals from public metagenomic datasets based on 291 faecal samples and 32 new sampling of Italian subjects collected through the COVIDbiome project.
The meta‐analysis allowed to identify key differences in the microbial biodiversity between the gut microbiota of healthy and severe COVID‐19 disease patients, as well as detailed taxonomic differences that complete and corroborate recently published data, highlighting in the microbiota of COVID‐19 patients a decrease abundance of species belonging to Faecalibacterium and Gemmiger genera (Yeoh et al., 2021; Zuo et al., 2020) and an increase of several opportunistic pathogen bacteria (Patel & Roper, 2021). Moreover, our data clearly evidences the lack and/or high abundance of microbes that could be statistically associated with more serious symptoms and thus could be considered as key microbial markers associated with this disease. Notably, the high presence in diseased samples of microorganisms involved in the development of putrefactive dysbiosis may contribute to increased systemic inflammation by releasing compounds obtained through protein metabolism. Furthermore, the identification of a predominance of enzymatic reactions involved in protein metabolism in the gut microbiomes of individuals displaying severe symptoms could suggest that these functional variations together with a lower presence of anti‐inflammatory compounds, such as butyrate, could favour a putrefactive gut dysbiosis generating an exacerbation of systemic inflammation. Such findings clearly highlighted the important role exploited by gut microbiota in affecting the development of the COVID‐19 diseases, which might open novel avenues for the treatment of this disease, not only through the classical therapeutic protocols but also by the inclusion of new strategies aimed to modulate the gut microbiota through the establishment and/or enhancement of protective bacteria towards COVID‐19 disease. In this context, according to recent scientific publications (Kurian et al., 2021; Nguyen et al., 2022; Tang et al., 2021), the modulation of gut microbiota with specific probiotics supplementation could reduce the severity of COVID‐19 morbidity and mortality severity, improving the health of the patients.
AUTHOR CONTRIBUTIONS
Leonardo Mancabelli: processed the metagenomic data, conducted and interpreted the analyses, and wrote the manuscript. Christian Milani: participated in the study's design and contributed to the manuscript preparation. Federico Fontana: contributed to the statistical analyses. Gabriele Andrea Lugl: contributed to the metagenomic analyses. Chiara Tarracchini: contributed to the metagenomic analyses. Alice Viappiani: performed the sequencing process of the samples included in the study. Tecla Ciociola: performed the extraction of bacterial DNA. Andrea Ticinesi: contributed to patient selection and enrolment, clinical data collection, biological sample collection, communications with the Ethics Committee and official reporting of the study. Antonio Nouvenne: contributed to the coordination of patient selection and enrolment, clinical data collection and biological sample collection. Tiziana Meschi: contributed to the coordination of patient selection and enrolment, clinical data collection, biological sample collection, communications with the Ethics Committee and official reporting of the study. Francesca Turroni: contributed to the conception and analysis of the study. Marco Ventura: conceived the study, participated in its design and coordination and contributed to the manuscript preparation. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
Supporting information
Figure S1 Evaluation of microbial biodiversity based on health status and nationality. Panel a displays the Whiskers plot representing the species richness identified from healthy, moderate, and severe COVID‐19 subjects subdivided by nationality. The x‐axis represents the different groups, while the y‐axis indicates the number of species. The boxes are determined by the 25th and 75th percentiles. The whiskers are determined by 1.5 IQR (interquartile range). The line in the boxes represents the median, while the square represents the average.
Panel b reports the principal coordinate analysis (PCoA) of the faecal samples included in the meta‐analysis, subdivided by health status and nationality.
Figure S2 Evaluation of microbial biodiversity based on health status and vaccination against SARS‐CoV‐2. Panel a reports the principal coordinate analysis (PCoA) of the Italian faecal samples included in the meta‐analysis, subdivided according to the administration of the vaccine. Panel b shows the PCoA of the Italian faecal samples included in the meta‐analysis, divided according to the health status and vaccine treatment.
Table S1 Metadata and data relate to the samples sequenced in this study.
Table S2 Bacterial profile at species level of healthy, moderate, and severe COVID‐19 subjects.
Table S3 Functional prediction of healthy, moderate, and severe COVID‐19 subjects.
ACKNOWLEDGEMENTS
The authors thank GenProbio srl for financial support of the Laboratory of Probiogenomics. This study was supported by ‘Programma Operativo Nazionale Ricerca e Innovazione’ 2014‐2020 (PON ‘R&I’ 2014‐2020) (project ARS01_00530) and by the extraordinary call of the University of Parma 2020 for biomedical research projects in the SARS‐CoV‐2 and COVID‐19 field (project COVIDbiome). Francesca Turroni was supported by PROGETTO Ricerca Finalizzata, Ministero della Salute (RF GR‐2018‐12365988). Part of this research is conducted using the high performance computing (HPC) facility of the University of Parma. Open Access Funding provided by Universita degli Studi di Parma within the CRUI‐CARE Agreement.
Mancabelli, L. , Milani, C. , Fontana, F. , Lugli, G.A. , Tarracchini, C. , Viappiani, A. et al. (2022) Untangling the link between the human gut microbiota composition and the severity of the symptoms of the COVID‐19 infection. Environmental Microbiology, 1–10. Available from: 10.1111/1462-2920.16201
Funding information PROGETTO Ricerca Finalizzata, Ministero della Salute, Grant/Award Numbers: GR‐2018‐12365988, RF GR‐2018‐12365988; Programma Operativo Nazionale Ricerca e Innovazione, Grant/Award Number: PON “R&I” 2014‐ 2020; Project COVIDbiome; GenProbio srl
DATA AVAILABILITY STATEMENT
Raw sequences of the Italian shallow‐shotgun metagenomics data are accessible through SRA under study accession number PRJNA864754.
REFERENCES
- Andrade, B.G.N. , Cuadrat, R.R.C. , Tonetti, F.R. , Kitazawa, H. & Villena, J. (2022) The role of respiratory microbiota in the protection against viral diseases: respiratory commensal bacteria as next‐generation probiotics for COVID‐19. Bioscience of Microbiota, Food and Health, 41, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baizabal‐Carvallo, J.F. (2021) Gut microbiota: a potential therapeutic target for Parkinson's disease. Neural Regeneration Research, 16, 287–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn, M.K. , Hall, A. , Sepiashvili, L. , Jung, B. , Steele, S. & Adeli, K. (2020) Pathophysiology of COVID‐19: mechanisms underlying disease severity and progression. Physiology (Bethesda), 35, 288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton, G.J. , Chen‐Liaw, A. , Cossarini, F. , Livanos, A.E. , Spindler, M.P. , Plitt, T. et al. (2021) Limited intestinal inflammation despite diarrhea, fecal viral RNA and SARS‐CoV‐2‐specific IgA in patients with acute COVID‐19. Scientific Reports, 11, 13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussow, H. (2021) COVID‐19: vaccination problems. Environmental Microbiology, 23, 2878–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi, R. , Billington, R. , Ferrer, L. , Foerster, H. , Fulcher, C.A. , Keseler, I.M. et al. (2016) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Research, 44, D471–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Liu, X. , Liu, W. , Yang, C. , Jia, R. , Ke, Y. et al. (2022) Comparison of the respiratory tract microbiome in hospitalized COVID‐19 patients with different disease severity. Journal of Medical Virology, 94, 5284–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Vitetta, L. , Henson, J.D. & Hall, S. (2021) The intestinal microbiota and improving the efficacy of COVID‐19 vaccinations. Journal of Functional Foods, 87, 104850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Ye, W. , Zhang, Y. & Xu, Y. (2015) High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Research, 43, 7762–7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi, A. , Totino, V. , Cacciotti, F. , Iebba, V. , Neroni, B. , Bonfiglio, G. et al. (2018) Rebuilding the gut microbiota ecosystem. International Journal of Environmental Research and Public Health, 15, 1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmann, B. , Al‐Ghalith, G.A. , Shields‐Cutler, R.R. , Zhu, Q. , Gohl, D.M. , Beckman, K.B. et al. (2018) Evaluating the information content of shallow shotgun metagenomics. mSystems, 3, e00069–e00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur, H. , Das, C. & Mande, S.S. (2017) In silico analysis of putrefaction pathways in bacteria and its implication in colorectal cancer. Frontiers in Microbiology, 8, 2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriss, M. , Hazleton, K.Z. , Nusbacher, N.M. , Martin, C.G. & Lozupone, C.A. (2018) Low diversity gut microbiota dysbiosis: drivers, functional implications and recovery. Current Opinion in Microbiology, 44, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian, S.J. , Unnikrishnan, M.K. , Miraj, S.S. , Bagchi, D. , Banerjee, M. , Reddy, B.S. et al. (2021) Probiotics in prevention and treatment of COVID‐19: current perspective and future prospects. Archives of Medical Research, 52, 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Wilks, C. , Antonescu, V. & Charles, R. (2019) Scaling read aligners to hundreds of threads on general‐purpose processors. Bioinformatics, 35, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Zhang, H. , Tang, X. , Jiang, X. , Yan, X. , Liu, X. et al. (2021) Distinct metagenomic signatures in the SARS‐CoV‐2 infection. Frontiers in Cellular and Infection Microbiology, 11, 706970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancabelli, L. , Mancino, W. , Lugli, G.A. , Argentini, C. , Longhi, G. , Milani, C. et al. (2021) Amoxicillin‐clavulanic acid resistance in the genus Bifidobacterium . Applied and Environmental Microbiology, 87, e03137–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic, S. , Rodic, A. , Salom, I. , Milicevic, O. , Djordjevic, M. & Djordjevic, M. (2021) COVID‐19 severity determinants inferred through ecological and epidemiological modeling. One Health, 13, 100355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoodi, M. , Peschka, M. , Schmiedel, S. , Haddad, M. , Frye, M. , Maas, C. et al. (2022) Disturbed lipid and amino acid metabolisms in COVID‐19 patients. Journal of Molecular Medicine (Berlin, Germany), 100, 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarelli, A. , Giancola, M.L. , Farina, A. , Marchioni, L. , Rueca, M. , Gruber, C.E.M. et al. (2021) 16S rRNA gene sequencing of rectal swab in patients affected by COVID‐19. PLoS One, 16, e0247041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy, J.R. , Mullish, B.H. , Goldenberg, S.D. , Ianiro, G. & Marchesi, J.R. (2020) Intestinal microbiome transfer, a novel therapeutic strategy for COVID‐19 induced hyperinflammation? In reply to, 'COVID‐19: immunology and treatment options', Felsenstein, Herbert McNamara et al. 2020′. Clinical Immunology, 218, 108542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, C. , Casey, E. , Lugli, G.A. , Moore, R. , Kaczorowska, J. , Feehily, C. et al. (2018) Tracing mother‐infant transmission of bacteriophages by means of a novel analytical tool for shotgun metagenomic datasets: METAnnotatorX. Microbiome, 6, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, C. , Lugli, G.A. , Fontana, F. , Mancabelli, L. , Alessandri, G. , Longhi, G. et al. (2021) METAnnotatorX2: a comprehensive tool for deep and shallow metagenomic data set analyses. mSystems, 6, e0058321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca, A. , Leclerc, M. & Hugot, J.P. (2016) Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Frontiers in Microbiology, 7, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosele, J.I. , Macia, A. & Motilva, M.J. (2015) Metabolic and microbial modulation of the large intestine ecosystem by non‐absorbed diet phenolic compounds: a review. Molecules, 20, 17429–17468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, Q.V. , Chong, L.C. , Hor, Y.Y. , Lew, L.C. , Rather, I.A. & Choi, S.B. (2022) Role of probiotics in the management of COVID‐19: a computational perspective. Nutrients, 14, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez‐Franco, J.C. , Torres‐Ruiz, J. , Sosa‐Hernandez, V.A. , Cervantes‐Diaz, R. , Romero‐Ramirez, S. , Perez‐Fragoso, A. et al. (2021) Metabolomics analysis reveals a modified amino acid metabolism that correlates with altered oxygen homeostasis in COVID‐19 patients. Scientific Reports, 11, 6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, P. & Roper, J. (2021) Gut microbiome composition is associated with COVID‐19 disease severity. Gastroenterology, 161, 722–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnavard, A. , Mann, B. , Giri, A. , Chatterjee, R. & Crandall, K.A. (2022) Metabolite, protein, and tissue dysfunction associated with COVID‐19 disease severity. Scientific Reports, 12, 12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani, L. , Del Chierico, F. , Macari, G. , Pane, S. , Ristori, M.V. , Guarrasi, V. et al. (2022) The relationship between pediatric gut microbiota and SARS‐CoV‐2 infection. Frontiers in Cellular and Infection Microbiology, 12, 908492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang, J. , Zhuang, D. , Zhang, T. , Wu, Q. , Yu, J. & Zhang, Z. (2022) Convergent and divergent age patterning of gut microbiota diversity in humans and nonhuman primates. mSystems, 7, e0151221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sencio, V. , Benech, N. , Robil, C. , Deruyter, L. , Heumel, S. , Machelart, A. et al. (2022) Alteration of the gut microbiota's composition and metabolic output correlates with COVID‐19‐like severity in obese NASH hamsters. Gut Microbes, 14, 2100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Bohannon, L. , Lew, M. , Jensen, D. , Jung, S.H. , Zhao, A. et al. (2021) Randomised, double‐blind, placebo‐controlled trial of probiotics to eliminate COVID‐19 transmission in exposed household contacts (PROTECT‐EHC): a clinical trial protocol. BMJ Open, 11, e047069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez‐Baeza, Y. , Pirrung, M. , Gonzalez, A. & Knight, R. (2013) EMPeror: a tool for visualizing high‐throughput microbial community data. Gigascience, 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura, R.E. , Iizumi, T. , Battaglia, T. , Liu, M. , Perez‐Perez, G.I. , Herbert, J. et al. (2019) Gut microbiome of treatment‐naive MS patients of different ethnicities early in disease course. Scientific Reports, 9, 16396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrotsou, K. , Rotaeche, R. , Mateo‐Abad, M. , Machon, M. & Vergara, I. (2021) Variables associated with COVID‐19 severity: an observational study of non‐paediatric confirmed cases from the general population of the Basque Country, Spain. BMJ Open, 11, e049066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Wang, H. , Sun, Y. , Ren, Z. , Zhu, W. , Li, A. et al. (2021) Potential associations between microbiome and COVID‐19. Frontiers in Medicine, 8, 785496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Zhao, S. , Yu, B. , Chen, Y.M. , Wang, W. , Song, Z.G. et al. (2020) A new coronavirus associated with human respiratory disease in China. Nature, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, R. , Lu, R. , Zhang, T. , Wu, Q. , Cai, W. , Han, X. et al. (2021) Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID‐19 in adults. Communications Biology, 4, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarza, P. , Yilmaz, P. , Pruesse, E. , Glockner, F.O. , Ludwig, W. , Schleifer, K.H. et al. (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nature Reviews Microbiology, 12, 635–645. [DOI] [PubMed] [Google Scholar]
- Ye, Y. , Choi, J.H. & Tang, H. (2011) RAPSearch: a fast protein similarity search tool for short reads. BMC Bioinformatics, 12, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoh, Y.K. , Zuo, T. , Lui, G.C. , Zhang, F. , Liu, Q. , Li, A.Y. et al. (2021) Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID‐19. Gut, 70, 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, M.Y. , Inohara, N. & Nunez, G. (2017) Mechanisms of inflammation‐driven bacterial dysbiosis in the gut. Mucosal Immunology, 10, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Wan, Y. , Zuo, T. , Yeoh, Y.K. , Liu, Q. , Zhang, L. et al. (2022) Prolonged impairment of short‐chain fatty acid and L‐isoleucine biosynthesis in gut microbiome in patients with COVID‐19. Gastroenterology, 162(548–561), e544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Tang, H. & Ye, Y. (2012) RAPSearch2: a fast and memory‐efficient protein similarity search tool for next‐generation sequencing data. Bioinformatics, 28, 125–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, T. , Wu, J. , Zeng, Y. , Li, J. , Yan, J. , Meng, W. et al. (2022) SARS‐CoV‐2 triggered oxidative stress and abnormal energy metabolism in gut microbiota. MedComm, 3, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, T. , Zhang, F. , Lui, G.C.Y. , Yeoh, Y.K. , Li, A.Y.L. , Zhan, H. et al. (2020) Alterations in gut microbiota of patients with COVID‐19 during time of hospitalization. Gastroenterology, 159(944–955), e948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Evaluation of microbial biodiversity based on health status and nationality. Panel a displays the Whiskers plot representing the species richness identified from healthy, moderate, and severe COVID‐19 subjects subdivided by nationality. The x‐axis represents the different groups, while the y‐axis indicates the number of species. The boxes are determined by the 25th and 75th percentiles. The whiskers are determined by 1.5 IQR (interquartile range). The line in the boxes represents the median, while the square represents the average.
Panel b reports the principal coordinate analysis (PCoA) of the faecal samples included in the meta‐analysis, subdivided by health status and nationality.
Figure S2 Evaluation of microbial biodiversity based on health status and vaccination against SARS‐CoV‐2. Panel a reports the principal coordinate analysis (PCoA) of the Italian faecal samples included in the meta‐analysis, subdivided according to the administration of the vaccine. Panel b shows the PCoA of the Italian faecal samples included in the meta‐analysis, divided according to the health status and vaccine treatment.
Table S1 Metadata and data relate to the samples sequenced in this study.
Table S2 Bacterial profile at species level of healthy, moderate, and severe COVID‐19 subjects.
Table S3 Functional prediction of healthy, moderate, and severe COVID‐19 subjects.
Data Availability Statement
Raw sequences of the Italian shallow‐shotgun metagenomics data are accessible through SRA under study accession number PRJNA864754.