

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the cause of the on‐going global pandemic of coronavirus disease 2019 (COVID‐19) that continues to pose a significant threat to public health worldwide. SARS‐CoV‐2 encodes four structural proteins namely membrane, nucleocapsid, spike, and envelope proteins that play essential roles in viral entry, fusion, and attachment to the host cell. Extensively glycosylated spike protein efficiently binds to the host angiotensin‐converting enzyme 2 initiating viral entry and pathogenesis. Reverse transcriptase polymerase chain reaction on nasopharyngeal swab is the preferred method of sample collection and viral detection because it is a rapid, specific, and high‐throughput technique. Alternate strategies such as proteomics and glycoproteomics‐based mass spectrometry enable a more detailed and holistic view of the viral proteins and host–pathogen interactions and help in detection of potential disease markers. In this review, we highlight the use of mass spectrometry methods to profile the SARS‐CoV‐2 proteome from clinical nasopharyngeal swab samples. We also highlight the necessity for a comprehensive glycoproteomics mapping of SARS‐CoV‐2 from biological complex matrices to identify potential COVID‐19 markers.
Keywords: COVID‐19, mass spectrometry, nasopharyngeal, proteomics, SARS‐CoV‐2
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
A
- sn
asparagine
- CCS
collisional cross section
- CDMS
charge detection mass spectrometry
- cIMS
cyclic ion mobility spectrometry
- COVID‐19
coronavirus disease 2019
- DDA
data‐dependent acquisition
- DIA
data‐independent acquisition
- E
envelope protein
- ESI
electrospray ionization
- EThcD
electron transfer/higher‐energy collision dissociation
- FAIMS
field asymmetric ion mobility spectrometry
- FT‐ICR
Fourier transform ion cyclotron resonance
- Fuc
fucose
- Gal
galactose
- GalNAc
N‐acetylgalactosamine
- Glc
glucose
- GlcNAc
N‐acetylglucosamine
- HPLC
high‐performance liquid chromatography
- IM‐MS
ion mobility mass spectrometry
- M
membrane protein
- MALDI
matrix‐assisted laser desorption/ionization
- Man
mannose
- MRM
multiple reaction monitoring
- MERS
Middle East respiratory syndrome
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- m/z
mass‐to‐charge
- N
nucleocapsid protein
- NeuAc
N‐acetylneuraminic acid
- NSP
non‐structural protein
- PASEF
parallel accumulation‐serial fragmentation
- PFU
plaque forming units
- PRM
parallel reaction monitoring
- PTM, post translational modification; QE
Quadrupole Exactive
- RBD
receptor‐binding domain
- RNA
ribonucleic acid
- RP
reversed phase
- RT‐PCR
reverse transcriptase polymerase chain reaction
- S
spike protein
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- Ser
serine
- SISCAPA
stable isotope standards and capture by anti‐peptide antibodies
- SWATH‐MS
sequential windowed acquisition of all theoretical fragment ion mass spectra
- Thr
threonine
- TIMS
trapped ion mobility spectrometry
- TOF
time‐of‐flight
- Tyr
tyrosine
- UPLC
ultra‐performance liquid chromatography
- ZIC‐HILIC
zwitterion hydrophilic interaction liquid chromatography
1. INTRODUCTION
The emergence of the pathogenic severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) in late 2019, is responsible for the on‐going global pandemic of coronavirus disease 2019 (COVID‐19) (Cui et al., 2019; Drosten et al., 2003; Xu et al., 2020). The first outbreak of SARS‐CoV‐1 in China in 2003, followed by the Middle East respiratory syndrome (MERS) epidemic 10 years later highlighted the danger of zoonotic transmission of Coronaviridae viruses (Zaki et al., 2012; Zhong et al., 2003). Detected in Wuhan, China in 2019, SARS‐CoV‐2 spread rapidly in the human population, has led to millions of deaths across the world, and has been identified as a disease with ambiguous etiology (Y. Huang et al., 2020; Meyerowitz et al., 2021). SARS‐CoV‐2 infections often show flu‐like symptoms but may also appear asymptomatically in individuals that further adds a layer of complexity and challenges, to diagnose and monitor the disease (Gao et al., 2021; Peiris et al., 2003).
SARS‐CoV‐2 is a positive‐sense single‐stranded ribonucleic acid (RNA) enveloped virus with viral particle size ranging from 60 to 140 nm (Alexandersen et al., 2020; Andersen et al., 2020; Chan et al., 2020; Masters, 2006; Naqvi et al., 2020). Researchers have deduced the molecular architecture and assembly of SARS‐CoV‐2 virus using cryo‐electron microscopy and tomography (Hardenbrook & Zhang, 2022; Kim et al., 2020; Yao et al., 2020). The SARS‐CoV‐2 genome (~30 kilobases) encodes four major structural proteins: membrane (M), nucleocapsid (N), envelope (E), and spike (S) (Arya et al., 2021; C. Cao et al., 2021), in addition to 16 nonstructural proteins (NSP1‐16) and 9 auxiliary proteins (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, ORF10) (Arya et al., 2021; Banerjee & Mukhopadhyay, 2016; Chan et al., 2020; D. J. Harvey, 2018; Redondo et al., 2021; Syed et al., 2021). These proteins function in viral replication, RNA binding, and packaging (Benton et al., 2020; de Haan & Rottier, 2005; Gordon et al., 2020; Smits et al., 2021). M‐protein (Uniprot ID: P0DTC5, ~25.2 kilodalton, 222 amino acids), the most abundant structural protein, defines the shape of the viral envelope. N‐protein (P0DTC9, ~45.6 kDa, 419 aa) is the next most abundant protein that can be detected as an early marker in the serum or nasopharyngeal aspirate of infected patients (Barlev‐Gross et al., 2021; Che et al., 2004; Shen et al., 2020). E‐protein (P0DTC4, ~8.4 kDa, 75 aa) forms only a small portion of the entire virion (Bar‐On et al., 2020). The trimeric glycosylated S‐protein (P0DTC2, ~141.2 kDa, 1273 aa), contributing to only ~5% of the total protein abundance on SARS‐CoV‐2 (Bezstarosti et al., 2021; B.‐Z. Zhang, Hu, et al., 2020) is composed of two subunits (S1 and S2). The virus enters the host cell via the widely known angiotensin‐converting enzyme 2 (ACE2), an important type‐I integral transmembrane metalloproteinase receptor (Q9BYF1, ~92.5 kDa, 805 aa) expressed on the human cell surface in the airway passage (W. T. Harvey et al., 2021; Ke et al., 2020; Oz et al., 2021). Trimming of the S‐protein by host cellular serine proteases (furin, TMPRSS2) is necessary for invasion of target cells (Hoffmann et al., 2020; Walls et al., 2020; Xia et al., 2020). Once inside the cell, the virus hijacks the host cell biosynthetic machinery to replicate and generate viral RNA. Apart from ACE2 receptor, studies have shown the presence of other factors such as extracellular vimentin that also facilitates the entry of SARS‐CoV‐2 into human endothelial cells further leading to vascular complications (Amraei et al., 2022). While the expression of ACE2 receptor is high in the upper respiratory tract (H. Zhang, Rostami, et al., 2020) it remains low in the lower respiratory tract that has enabled the identification of several other potential receptors such as neuropilin‐1, CD209L/L‐SIGN, and CD209/DC‐SIGN that can act as an entry point for SARS‐CoV‐2 in a cell‐dependent manner (Amraei et al., 2021; Lukassen et al., 2020; Sungnak et al., 2020). Interestingly, researchers have also made comparisons in the total number and mass of SARS‐CoV‐2 virions between samples with different viral load in a time‐dependent manner that demonstrated the genetic diversity of virions across many infected hosts (Sender et al., 2021). Although the trajectory of SARS‐CoV‐2 was unknown at the time of emergence, the virus was thought to have lower mutation rates when compared to other enveloped viruses (De Maio et al., 2021). We now know, tracked by the World Health Organization, the constant transmission of SARS‐CoV‐2 has generated variants of concern: Alpha (lineage B.1.1.7, United Kingdom), Beta (B.1.351, South Africa), Gamma (P.1, Brazil), Delta (B.1.617.2, India) and Omicron (B.1.1.529, South Africa) (Deng et al., 2021; L. Zhang, Jackson, et al., 2020). Recently, sequence variation analysis of SARS‐CoV‐2 isolates have identified several genetic modifications, such as D614G and L452R mutations in S‐protein that are shown to increase virion spike density and infectivity (Deng et al., 2021; Kuo et al., 2022; L. Zhang, Jackson, et al., 2020).
The highly contagious COVID‐19 disease requires an efficient and rapid testing system to isolate the infected individuals restricting escalation of SARS‐CoV‐2. It is estimated that an infected person carries 109−1011 virions during peak infection with a total mass in the range of 1 µg to 100 µg (Sender et al., 2021; Padhi & Tripathi, 2020). Reverse‐transcriptase polymerase chain reaction (RT‐PCR) has been the major tool for SARS‐CoV‐2 detection that identifies the viral RNA, primarily because it is minimally invasive and has a rapid turn‐around time (Corman et al., 2020; Freire‐Paspuel & Garcia‐Bereguiain, 2021; Studdert & Hall, 2020). While nasopharyngeal swab is the preferred sample collection method, (Basu et al., 2020; Pascarella et al., 2020), SARS‐CoV‐2 can also be detected from saliva (Costa et al., 2022; Hernandez et al., 2021; Takeuchi et al., 2020), sputum (Bezstarosti et al., 2021), gargle (Chivte et al., 2021; Ihling et al., 2020; Iles et al., 2020), blood (Li, Liu, et al., 2021; Peng et al., 2020), plasma (Lazari et al., 2021), serum (Shen et al., 2020; Yan et al., 2021), urine (Chavan et al., 2021; Peng et al., 2020), feces (Li, Liu, et al., 2021; Wang, Xu, et al., 2020) and breath (Ruszkiewicz et al., 2020; Steppert et al., 2021) samples. RT‐PCR lacks information on the infectious nature and host–pathogen interactions and cannot distinguish between viable and nonviable virus (Y. Chen et al., 2022; Healy et al., 2021). Alternative approaches to diagnose and test for SARS‐CoV‐2 include epitope mapping, enzyme‐linked immunoassays, and multi‐antigen serology assays (Haynes et al., 2021; Mazhari et al., 2021; Noy‐Porat et al., 2020; Roy et al., 2020).
Mass spectrometry (MS)‐based approaches have been employed in molecular pathology to study infectious diseases and other conditions (Aebersold & Mann, 2003; Mahmud & Garrett, 2020; Rybicka et al., 2021). In principle, MS analysis provides a holistic proteomic view of SARS‐CoV‐2 infected samples (Che et al., 2004; S.‐J. Cho et al., 2011; Kammila et al., 2008; Van Puyvelde et al., 2021) that complements RT‐PCR methods for the diagnosis of COVID‐19 (Lu et al., 2022; Overmyer et al., 2021; Wandernoth et al., 2020; Wörner et al., 2021).
Protein glycosylation, the addition of complex carbohydrates (hereby referred to as glycans) decorating polypeptide chains, is a post‐translational modification (PTM) that occurs to a wide range of intracellular and intercellular proteins. Quantification of the glycosylation changes is therefore an important aspect of mechanistic understanding of human health and disease processes (Kreisman & Cobb, 2012; Reily et al., 2019; Varki, 2017). Glycosylated spike proteins include the hemagglutinin glycoprotein of influenza (D. Chang et al., 2020; Khatri et al., 2016; Kobayashi & Suzuki, 2012), envelope glycoprotein of human immunodeficiency virus‐1 (L. Cao et al., 2018; Struwe et al., 2018), Ebola virus glycoprotein (Lee et al., 2008; Ritchie, Harvery, & Stroeher, et al., 2010), nipah virus glycoprotein (Z. Wang et al., 2022) and coronavirus S‐protein (D. Chang et al., 2021; Ritchie, Harvery, & Stroeher, et al., 2010; Walls et al., 2016). Viral protein glycosylation occurs by the host cell secretory pathway and is required for viral host cell invasion, budding and aggregation of virion particles, and immune evasion using glycan shield mechanisms (Bagdonaite & Wandall, 2018; Li, Liu, et al., 2021; Watanabe et al., 2019). The mechanistic aspects of SARS‐CoV‐2 S‐protein glycosylation is still emerging, and of particular interest is to characterize S‐protein glycosylation on the virions and its binding with the glycosylated ACE2 receptor that infect humans from nasal mucosal swabs. We summarize the progress in the field towards this goal. We highlight N‐linked (Asn x Ser/Thr, x ≠ Pro) (Breitling & Aebi, 2013) and mucin‐type O‐linked (Ser‐/Thr‐) protein glycosylation (Van den Steen et al., 1998) and their potential roles in the life cycles of enveloped viruses, particularly, of SARS‐CoV‐2. In animal cells, the N‐ and O‐glycoproteins are assembled using monosaccharide building blocks that include primarily mannose (Man), fucose (Fuc), galactose (Gal), glucose (Glc), N‐acetylgalactosamine (GalNAc), N‐acetylglucosamine (GlcNAc), and sialic acids (N‐acetylneuraminic acid, NeuAc or N‐glycolylneuraminic acid, NeuGc). Sulfate and phosphate modifications to glycans are also common. Incomplete biosynthetic reactions give rise to considerable macroheterogeneity and microheterogeneity of glycans. As a result, it remains a challenge to quantify changes in glycosylation that occur in biological processes.
In this review, we focus on the advantages and limitations of proteomics‐based MS methods for profiling SARS‐CoV‐2 from the nasopharyngeal swabs used to study the SARS‐CoV‐2 life cycle and understanding the host responses (Akgun et al., 2020; Crispin & Doores, 2015; Gordon et al., 2020; D. J. Harvey, 2018). We summarize state‐of‐the‐art MS‐based approaches to characterize the SARS‐CoV‐2 proteome and highlight the need for advanced glycoproteomics mapping of viral proteins from clinically relevant nasopharyngeal swab samples. Original contemporary research papers on SARS‐CoV‐2 published from 2018 onwards were considered for discussion in this review.
2. PROTEOMICS‐BASED MS OF SARS‐COV‐2‐INFECTED NASOPHARYNGEAL SWAB SAMPLES
Proteomics‐based MS, in the past decade, has renewed the growing interest in structural virology (Greco et al., 2014; Grenga & Armengaud, 2021; Grossegesse et al., 2020; Terracciano et al., 2021). A deep comparative and quantitative proteomics characterization often gives an overview of the viral and/or host protein changes during the infection stages. However, the hypervariable nature of viruses with immense heterogeneity across many variants can make the characterization more difficult. Developments in the proteomics workflows, using instruments with higher sensitivity and specificity, generating reliable spectral libraries from data‐dependent acquisition (DDA), to further improve on designing data‐independent acquisition (DIA) for identification of low abundant species have overcome some of the challenges and boosted viral research (Amiri‐Dashatan et al., 2022; SoRelle et al., 2020; Van Puyvelde & Dhaenens, 2021; Wörner et al., 2021; Zecha et al., 2020).
2.1. Virus sample workup
To characterize the proteome of SARS‐CoV‐2, a starting point for investigation has been the use of recombinantly‐expressed viral proteins in different expression systems such as HEK293, Vero E6, Caco‐2, or Calu‐3 cells. Creating plasmid constructs with desirable mutations generates either virus‐like particles or recombinant viral proteins that are then transfected into human cell culture for production (Renuse et al., 2021; Syed et al., 2021). Additionally, SARS‐CoV‐2 strains, specifically handled in a bio‐safety level 3‐guided laboratory have been also used to infect fresh cell cultures in vitro before conducting MS analysis. Often, studies have collected the cell lysates and performed in‐gel or in‐solution trypsin digestions that have generated a list of the major viral tryptic peptides. Some studies have found that cell culture supernatants with a titer value of at least 1 × 106 pfu/ml contain sufficient viral particles for MS detection of the viral proteins (Gouveia, Miotello, et al., 2020; Grenga et al., 2020). Finally, to identify the real‐time biological changes, researchers have attempted to characterize SARS‐CoV‐2 proteome from clinical nasopharyngeal swab samples from human donors. The following sections highlight in detail the present status of proteomics‐based MS methods to detect SARS‐CoV‐2 proteins using clinically available human nasopharyngeal swab samples. The number of nasopharyngeal samples, sample preparations, and LC‐MS conditions obtained from the existing literature is summarized in detail in Table 1.
Table 1.
Literature survey exploring proteomics workflows and mass spectrometry acquisitions to detect viral proteins from human nasopharyngeal swabs obtained from SARS‐CoV‐2 positive patients (negative samples = healthy controls).
| # of Naso‐pharyngeal samplesa | Sample preparationb | Liquid chromatography and column informationb | Mass spectrometer and instrument parametersb | References |
|---|---|---|---|---|
| 237 samples | Direct spot of sample mixed in sinapinic acid matrix for MALDI‐TOF analysis | ‐ |
Bruker Autoflex maX MALDI‐TOF‐MS (combined with artificial Intelligence) Positive ion mode 20 laser shots at 2000 Hz frequency Total 1800 shots in 300 increments Mass range of 5000–20,000 Da |
Deulofeu et al. (2021) |
| 199 samples (107 positive, 92 negative) | Direct spot of sample mixed in α‐cyano‐4‐hydroxycinnamic acid matrix for MALDI‐TOF analysis | ‐ |
Shimadzu 8020 MALDI‐TOF‐MS (combined with machine learning) Positive ion mode 10 laser shots at 100 Hz frequency Total 1000 shots/well Mass range of 2000–20,000 Da |
Tran et al. (2021) |
| 223 samples (88 positive, 135 negative) | Direct spot of sample mixed in α‐cyano‐4‐hydroxycinnamic acid matrix for MALDI‐TOF analysis | ‐ |
MALDI‐TOF‐MS (Bruker Daltonics) Positive ion mode 240 laser shots collected in increments of 40 carried out at 40% of maximum laser energy Mass range of 2000–20,000 Da |
Rocca et al. (2020) |
|
23 samples (19 positive, 4 negative) |
Samples were concentrated using 300 kDa MWCO filter, in‐solution trypsin digestion, clean peptides were spotted mixed with 4‐chloro‐ α‐cyanocinnamic acid matrix for MALDI‐TOF analysis | ‐ |
MS‐S3000 SpiralTOF‐plus MS (JEOL) Positive ion mode Laser intensity 55% and frequency 250 Hz with 330 ns delay time Mass range of 700–3000 Da Both external and internal standards were used for mass calibration |
Yoshinari et al. (2022) |
| Unknown number of samples | Samples were concentrated using 300 kDa MWCO filter, in‐solution trypsin digestion, and cleaned up peptides were spotted mixed with α‐cyano‐4‐hydroxycinnamic acid matrix for MALDI‐TOF analysis | ‐ |
MALDI‐FT ICR‐MS (unknown company) No information given on the instrument company or parameters used for the experiments |
Dollman et al. (2020) |
|
44 samples (22 positive, 22 negative) |
RNA extraction (using chemagic viral DNA/RNA 300 Kit) and SARS‐CoV‐2 mass spectrometric multiplex assay performed on the samples | ‐ | MassARRAY MALDI‐TOF‐MS (Agena Bioscience) | Wandernoth et al. (2020) |
|
101 samples (71 positive, 30 negative) |
MassARRAY SARS‐CoV‐2 Panel manufacturer's protocol performed on the samples | ‐ |
MassARRAY Panel (Agena Bioscience), Cobas 6800/8800 System (Roche Molecular Diagnostic) |
Stelzl et al. (2021) |
|
244 samples (99 positive, 145 negative) |
Lipid extraction (using Bligh and Dyer method) performed on the samples | ‐ |
MasSpec Pen‐ESI system (LTQ‐Orbitrap XL and QE HF MS, Thermo Fisher Scientific) ESI voltage +3.3 kV, nitrogen gas 20 psi, and inlet temperature of 300°C Negative ion mode Mass range of m/z 100–1500 MS1 resolution 120,000 HCD fragmentation Acquisition: DDA mode |
Garza et al. (2021) |
|
350 samples (250 positive, 100 negative) |
Antibody‐based purification using anti‐nucleocapsid protein, in‐solution trypsin digestion on purified samples, clean up peptides on Evotips |
C18 column (Dr. Maisch) on preformed gradient LC system (EvoSep One) 1.9 µm, 4 cm × 150 µm analytical column (flow rate of 2 µl/min and gradient of 5.6 min) |
Orbitrap Exploris 480 MS (Thermo Fisher Scientific) Positive ion mode Mass range of m/z 560‐1000 MS1 resolution 60,000 AGC 5 ×104, injection time 118 ms HCD fragmentation (NCE 27%) Acquisition: PRM mode |
Maus et al. (2022) |
|
363 samples (204 positive, 159 negative) |
In‐solution trypsin digestion and SISCAPA enrichment of N‐peptides from nasal swab samples |
C18 column (PepMap100 and EasySpray, Thermo Fisher Scientific) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 2 cm × 75 µm trap column (flow rate of 20 µl/min) 1.9 µm, 100 Å, 50 cm × 75 µm analytical column (flow rate of 300 nl/min and gradient of 40 min) C18 column (Dr. Maisch) on preformed gradient LC system (EvoSep One) 1.9 µm, 100 Å, 4 cm × 150 µm analytical column (flow rate of 2 µl/min and gradient of 5.6 min) |
Orbitrap Eclipse MS (Thermo Fisher Scientific) ESI voltage +2.5 kV Positive ion mode Mass range of m/z 350‐1700 MS1 resolution 120,000 MS2 resolution 30,000 HCD fragmentation (NCE 28%) Acquisition: DDA mode Orbitrap Exploris 480 MS‐FAIMS Pro ion source (Thermo Fisher Scientific) Positive ion mode Mass range of m/z 560–1000 MS1 resolution 60,000 AGC 5 × 104, injection time 118 ms HCD fragmentation (NCE 27%) Acquisition: PRM mode |
Renuse et al. (2021) |
|
24 samples (18 positive, 6 negative) |
In‐solution trypsin/lys‐C digestion, tandem mass tag labeling, and high‐pH reversed‐phase fractionation, followed by phosphopeptide enrichment on nasal swab samples |
C18 column (PepMap100 and EasySpray; Thermo Fisher Scientific) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 2 cm × 75 µm trap column (flow rate of 20 µl/min) 50 cm × 75 µm analytical column (flow rate of 300 nl/min and gradient of over 2 h) |
Orbitrap Fusion Lumos MS (Thermo Fisher Scientific) Positive ion mode Mass range of m/z 370–1700 MS1 resolution 60,000 AGC 1 × 106, injection time 50 ms MS2 resolution 30,000 AGC 2 × 105, injection time 54 ms HCD fragmentation (NCE 35%) Acquisition: DDA mode Orbitrap Eclipse (Thermo Fisher Scientific) MS1 resolution 30,000 AGC 5 × 104, injection time 54 ms HCD fragmentation (NCE 28%) Acquisition: PRM mode |
Vanderboom et al. (2021) |
|
985 samples (540 positive, 445 negative) |
In‐solution trypsin digestion on samples |
C18 column (PepMap100, Thermo Fisher Scientific) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 5 µm, 0.3 ×5 mm trap column (flow rate of 150 µl/min) 2 µm, 15 cm x 150 µm analytical column (flow rate of 1.5 µl/min and gradient of 60 min) C18 column (Acquity BEH, Waters) on turbulent flow chromatography on Transcend TLX‐4 system (Thermo Fisher Scientific) 5 cm × 1.7 µm analytical column (flow rate of 1.2 ml/min) |
QE HF‐X MS (Thermo Fisher Scientific) ESI voltage +2.2 kV, capillary temperature 45°C and S‐lens RF level 50 Positive ion mode Mass range of m/z 350‐1650 MS1 resolution 120,000 AGC 3 ×106, injection time 60 ms MS2 resolution 15,000 AGC 2 ×105, injection time 60 ms HCD fragmentation (NCE 27%) Acquisition: DDA mode TSQ Altis Triple Quadrupole MS (Thermo Fisher Scientific) Acquisition: PRM mode |
Cardozo et al. (2020) |
|
71 samples (41 positive, 30 negative) |
In‐solution trypsin digestion and SISCAPA enrichment of N‐peptides from nasal swab samples |
C18 column (PepMap100 and PepSep, Thermo Fisher Scientific) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 2 cm × 100 µm trap column (flow rate of 20 µl/min) 1.9 µm, 100 Å analytical column (flow rate of 500 nl/min and gradient of 25 min) |
Orbitrap Eclipse MS (Thermo Fisher Scientific) ESI voltage +2.2 kV Positive mode Mass range of m/z 350–1700 MS1 resolution 120,000 AGC 3 × 104, injection time 200 ms MS2 resolution 30,000 HCD fragmentation (NCE 28%) Acquisition: PRM mode |
Mangalaparthi et al. (2021) |
|
9 samples (5 positive, 4 negative) |
In‐gel trypsin digestion on samples |
C18 column (PepMap100; Thermo Fisher Scientific) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 3 µm, 100 Å, 5 mm × 300 µm trap column 3 µm, 100 Å, 50 cm × 75 µm analytical column (flow rate of 0.2 µl/min and gradient of 90 min) |
QE HF MS (Thermo Fisher Scientific) Positive ion mode Mass range of m/z 350–1500 MS1 resolution 60,000 AGC 5 × 104 HCD fragmentation Acquisition: DDA mode |
Gouveia, Miotello, et al. (2020) |
|
40 samples (20 positive, 20 negative) |
In‐solution trypsin/Lys‐C digestion on samples | C18 column (PepMap100; Thermo Fisher Scientific) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) |
QE HF MS (Thermo Fisher Scientific) Positive ion mode Mass range of m/z 375–1650 MS1 resolution 60,000 AGC 3 × 106, injection time 100 ms MS2 resolution 15,000 AGC 2 × 105, injection time 25 ms HCD fragmentation (NCE 27%) Acquisition: DDA mode Multiple mass ranges between m/z 375–1650 MS1 resolution 60,000 AGC 1 × 106, injection time 55 ms MS2 resolution 30,000 AGC 1 × 106, injection time 55 ms HCD fragmentation (NCE 27%) Acquisition: DIA mode |
Liou et al. (2021) |
|
10 samples (5 positive, 5 negative) |
In‐gel trypsin digestion on samples |
C18 column (PepMap100; Thermo Fisher Scientific) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 3 µm, 2 cm × 75 µm trap column 2 µm, 100 Å, 50 cm × 75 µm analytical column (flow rate of 250 nl/min and gradient of 150 min) |
QE Plus (Q‐Orbitrap) MS (Thermo Fisher Scientific) ESI voltage +1.7 kV, capillary temperature 250°C and S‐lens RF level 50 Mass range of m/z 200‐2000 MS1 resolution 70,000 AGC 1 × 106, injection time 100 ms MS2 resolution 17,500 AGC 1 × 105, injection time 50 ms HCD fragmentation (stepped NCE 25%, 30%, 35%) Acquisition: DDA mode |
Rivera et al. (2020) |
|
19 samples (12 positive, 7 negative) |
In‐solution trypsin digestion on samples |
Aeris peptide XB‐C18 column (Phenomenex) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 1.7 µm, 100 Å, 15 cm × 2.1 mm analytical column (flow rate of 0.5 ml/min and gradient of 40 min) |
Quadrupole‐Orbitrap MS (Thermo Fisher Scientific) ESI voltage +4 kV, capillary temperature 320°C Positive ion mode Mass range of m/z 200–2000 MS1 resolution 70,000 AGC 1 × 106, injection time 250 ms MS2 resolution 17,500 HCD fragmentation (NCE 18%) Acquisition: DDA mode MS1 resolution 70,000 AGC 1 × 106, injection time 125 ms HCD fragmentation (NCE 15%–20%) Acquisition: PRM mode |
Saadi et al. (2021) |
|
16 samples (6 positive, 10 negative) |
In‐solution trypsin digestion on samples |
C18 column (Gemini) on 1290 HPLC system (Agilent) 3 µm, 10 cm × 2.1 mm analytical column (flow rate of 0.3 ml/min and gradient of 20 min) |
QE Plus Orbitrap MS (Thermo Fisher Scientific) ESI voltage +1.25 kV, capillary temperature 275°C, and S‐lens RF level 55 Positive ion mode Mass range of m/z 250–2000 MS1 resolution 140,000 AGC 3 × 106, injection time 100 ms MS2 resolution 35,000 AGC 2 × 105, injection time 100 ms HCD fragmentation (NCE 20%) Acquisition: DIA mode |
Schuster et al. (2021) |
|
26 samples (13 positive, 13 negative) |
Streptavidin‐magnetic based enrichment of SARS‐CoV‐2 from nasal swab samples followed by in‐solution trypsin digestion |
C18 column (Acquity, Waters) on Acquity UPLC‐I Class (SM‐FTN) system (Waters) 1.7 µm, 15 cm × 2.1 mm analytical column (flow rate of 0.4 ml/min and gradient of 10 min) |
Xevo Triple Q‐S MS (Waters) ESI voltage +0.6 kV and capillary temperature 150°C Positive ion mode Acquisition: MRM mode |
Schuster et al. (2022) |
|
103 samples (83 positive, 20 negative) |
In‐solution trypsin digestion on samples |
C18 column (ChromXP) on Eksigent nanoLC‐425 system 5 µm, 120 Å trap column 3 µm, 120 Å analytical column (flow rate of 5 µl/min and gradient of 40 min) C18 column (Acquity, Waters) on ExionLC UHPLC system (Sciex) 1.7 µm, 10 cm × 2.1 mm analytical column (flow rate of 600 µl/min and gradient of 2.3 min) |
TripleTOF 6600 (Sciex) ESI voltage +5.5 kV and capillary temperature 250°C Positive ion mode Mass range of m/z 400–1250 Acquisition: DDA mode QTrap 6500 + (Sciex) Acquisition: MRM mode No information given on the instrument parameters used for the experiments |
Singh et al. (2020) |
|
90 samples (45 positive, 45 negative) |
In‐solution trypsin digestion on samples |
C18 column (Xbridge) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 3.5 µm, 50 cm × 4.6 mm analytical column (flow rate of 300 nl/min and gradient of over 2 h) C18 column (IonOpticks) on nanoElute LC system (Bruker Daltonics) 2 cm × 100 µm trap column 1.6 µm, 25 cm × 75 µm analytical column (flow rate of 300 nl/min and gradient of over 2 h) |
TIMS‐TOF Pro (Bruker Daltonics) Acquisition: PASEF‐DDA mode Mass range of m/z 100–1700 100 ms of ramp time 100% duty cycle Ion mobility range 0.60–1.60 V s cm−2 10 PASEF MS/MS scans per cycle Isolation width of m/z 2 Stepwise NCE 20‐59% Acquisition: dia‐PASEF mode Mass range of m/z 400–1200 Ion mobility range 0.69–1.47 V s cm−2 Isolation width of m/z 25 Stepwise NCE 20%–59% |
Mun et al. (2021) |
|
8 samples (5 positive, 3 negative) |
In‐solution trypsin digestion and Rapigest digestion on samples |
C18 column (IonOpticks) on UltiMate 3000 RSLC nano system (Thermo Fisher Scientific) 1.6 µm, 25 cm × 75 µm analytical column (flow rate of 400 nl/min and gradient of 40 min) |
TIMS‐TOF Pro (Bruker Daltonics) Mass range of m/z 100–1700 Ion mobility range 0.60–1.60 V s cm−2 10 PASEF MS/MS scans per cycle Acquisition: dia‐PASEF mode |
Nikolaev et al. (2020) |
|
101 samples (81 positive, 20 negative) |
S‐Trap and in‐solution trypsin digestion on samples |
C18 column (Acquity, Waters) on Acquity UPLC‐I Class (SM‐FTN) system (Waters) 1.7 µm, 130 Å, 5 cm × 150 µm analytical column (flow rate of 3 µl/min and gradient of 40 min) |
Xevo Triple Q‐S MS (Waters) Positive ion mode Mass range of m/z 300–1000 MRM time window of 0.4–1 min/peptide Acquisition: MRM mode |
Pinto et al. (2021) |
|
356 samples (48 positive, 308 negative) |
In‐solution trypsin digestion and SISCAPA enrichment of N‐peptides from nasal swab samples |
C18 column (Acquity BEH, Waters) on Acquity UPLC‐I Class (SM‐FTN) system (Waters) 1.7 µm, 300 Å, 5 cm × 2.1 mm analytical column (flow rate of 0.6 ml/min and gradient of 5.7 min) |
Xevo Triple Q‐XS MS (Waters) ESI voltage +0.5 kV Source temperature 150°C and desolvation temperature 600°C Acquisition: MRM mode |
Hober et al. (2021) |
| Multiple cohorts of positive and negative samples | In‐solution trypsin digestion and SISCAPA enrichment of N‐peptides from nasal swab samples |
Luna Omega Polar C18 column (Phenomenex) on Eksigent nanoLC‐425 system (Sciex) Trap column (flow rate of 10 µl/min) 15 mm × 0.3 mm analytical column (flow rate of 5 µl/min and gradient of 60 min) |
TripleTOF 5600 and TripleTOF 6600 + (Sciex) Mass range of m/z 400‐1250 (MS1) and m/z 100–1500 (MS2) Acquisition: DDA, SWATH, narrow DIA, and MRM modes |
Van Puyvelde et al. (2021) |
| Unknown number of samples | In‐solution trypsin digestion and SP3 clean‐up of peptides from nasal swab samples |
C18 trap column (PepMap100; Thermo Fisher Scientific) C18 analytical column (Dr. Maisch) on EASY‐nLC 1200 system (Thermo Fisher Scientific) 1.9 µm, 50 cm × 75 µm column (flow rate of 250 nl/min and gradient of 60 or 90 min) |
Orbitrap Fusion Lumos MS (Thermo Fisher Scientific) Positive ion mode Mass range of m/z 375–1400 MS1 resolution 120,000 MS2 resolution 30,000 AGC 5 × 104, injection time 50 ms HCD fragmentation (NCE 30%) Acquisition: DDA mode Orbitrap Eclipse MS (Thermo Fisher Scientific) Positive ion mode Isolation width m/z 0.7 M2 resolution 30,000 HCD fragmentation (NCE 30%) Acquisition: PRM mode |
Bezstarosti et al. (2021) |
Abbreviations: LC‐MS, liquid chromatography–mass spectrometry; RT‐PCR, reverse‐transcription polymerase chain reaction.
Confirmation by RT‐PCR tests.
All samples were collected with consent from infected patients or healthy controls using nasopharyngeal or oropharyngeal swabs. The LC‐MS instrumentation and parameters listed in the table are taken directly from the main article and supplementary information provided in each of the respective references.
2.2. Nasopharyngeal swab sample preparation
Nasopharyngeal swab samples are collected in viral transport medium containing phosphate‐buffered saline and serum albumin, supplemented with antibiotics (McAuley et al., 2021; Radbel et al., 2020; K. P. Smith et al., 2020). For virus detection purposes, RT‐PCR‐focused studies have shown varied effects of long‐term nasal swab storage and usage conditions; for accurate results, swabs must be tested for viral RNA within 48–72 h (Yilmaz Gulec et al., 2021). The viral load, pH of transport media, and storage temperature can often alter the viral RNA and may potentially become contaminated with other agents leading to false PCR results. While RT‐PCR detects a precise target as the viral messenger RNA, MS analysis can measure many other biomolecules such as peptides and glycopeptides that remain stable for a longer time. Therefore, for MS investigations, the assumption is that proteins from swab samples can be analyzed even after long‐term storage at −80°C (Van Puyvelde et al., 2021). The samples are vortexed vigorously to release the virus into the solution before heat‐inactivation with or without detergents in the range of 56°C–70°C for at least 30 min. Depending on the research question, the specimens are either analyzed directly or grown in cell culture (e.g., Vero E6 or Calu‐3 cells) (Dollman et al., 2020; Hekman et al., 2020; Ng et al., 2003). Mainly used as standard controls, some studies also include recombinant SARS‐CoV‐2 N‐protein or S‐protein or generate virus‐like particles (Syed et al., 2021) that are either analyzed separately or spiked into negative nasal swab samples (Cardozo et al., 2020; Renuse et al., 2021; Schuster et al., 2021; Syed et al., 2021). For protein extraction, samples are reduced, alkylated, and digested in‐solution with one or more proteolytic enzymes such as trypsin, chymotrypsin, endoproteinase Lys‐C, serine protease Glu‐C, or alpha lytic proteases (Maus et al., 2022; Vanderboom et al., 2021; Wörner et al., 2021). Researchers have also used S‐Trap mini columns as an alternate approach for digestion of SARS‐CoV‐2 proteins (Cazares et al., 2020; Pinto et al., 2021). While sodium dodecyl sulfate is commonly used to denature proteins, it is avoided in proteomics sample preparations as MS analysis is very sensitive to detergents. However, S‐Trap approach has enabled researchers to incorporate the use of sodium dodecyl sulfate to denature nondigested proteins, induce enzymatic digestion and generate peptides for MS analysis in a rapid, spin‐column format. Immuno‐magnetic capture to enrich for SARS‐CoV‐2 virions was also attempted from the mucous‐rich nasal swab samples before digestion treatments which showed a 10‐fold increase in identifying markers when compared to samples without any enrichment (Schuster et al., 2022). Next, digested and cleaned up peptides can be directly analyzed or undergo further peptide (Cardozo et al., 2020; Hober et al., 2021; Schuster et al., 2021) and/or phosphopeptide (Vanderboom et al., 2021) enrichments to enhance sensitivity before MS analysis. A recent study employed the stable isotope standards and capture by antipeptide antibodies (SISCAPA), where the targeted proteolytically digested peptides of interest are enriched using specific antipeptide antibodies (Whiteaker et al., 2011). In this study, antibodies against the SARS‐CoV‐2 N‐peptides were generated and a semi‐automated SISCAPA‐based approach was used to detect the viral peptides from nasal swabs with very low viral loads thus improving the detection limit (Mangalaparthi et al., 2021).
2.3. Trends in MS instrumentation and acquisition modes
Electrospray ionization (ESI) and matrix‐assisted laser desorption/ionization (MALDI) are the most common soft ionization techniques used in MS‐based proteomics analyses. ESI operates in the presence of an electric field applied to the spray capillary to form charged ions and is often used as an on‐line LC detector (Mann, 2019). MALDI, on the other hand, employs a laser to vaporize analytes embedded in crystalline organic matrix on a target plate. MALDI is often coupled to TOF analyzers with CID‐type dissociation, either in the TOF‐TOF or in the hybrid quadrupole‐TOF formats (van den Boom et al., 2013) The use of LC interfaced to ESI‐MS facilitates the analysis of complex peptide mixtures due to the chromatographic separation of the analytes in time before entering the MS source. The LC step can also provide separation of isomeric analytes before the MS source. Although the LC step is time‐consuming, it serves to improve the dynamic range relative to the direct analysis of complex mixtures. Contrastingly, while MALDI‐TOF‐MS is not directly interfaced with LC, it provides rapid, high‐throughput and cost‐effective analysis and a nearly unlimited m/z range with resolution varying from 2000 to 100,000. MALDI‐MS is well suited to tissue imaging applications that help to determine the spatial arrangement of analytes (Nadler et al., 2017). As discussed below, investigation of SARS‐CoV‐2 using either of these ionization techniques have confidently advanced our understanding on this viral infection (Cardozo et al., 2020; Dollman et al., 2020; Grenga et al., 2020; Nachtigall et al., 2020; Preianò et al., 2021; Rybicka et al., 2021).
DDA selects precursor ions based on the top‐most abundant peptides detected at a given time. By contrast, DIA fragments all the ions in a specific m/z window and samples the entire m/z and retention time range without any bias towards a specific precursor ion (Bittremieux et al., 2021; Ludwig et al., 2018; F. Zhang, Ge, et al., 2020). While DDA cannot sample all the information present in a sample due to limited duty cycle, DIA acquires tandem mass spectra on all precursor ions but introduces complexity in terms of data analysis that requires appropriate tools for accurate interpretation (Bichmann et al., 2021). Both spectral library and library‐free methods have been developed for DIA data. Overall, DIA generates consistent, reproducible, and high‐precision data with optimal ability to quantify sample proteins (Meyer & Schilling, 2017; F. Zhang, Ge, et al., 2020). Various DIA‐based acquisition schemes have been developed over the years including sequential windowed acquisition of all theoretical fragment ion mass spectra (SWATH‐MS) (Ludwig et al., 2018), PulseDIA (Cai et al., 2021), and scanning quadrupole approach (SONAR) (Moseley et al., 2018). Recently, DIA‐parallel accumulation‐serial fragmentation (diaPASEF) (Meier et al., 2015; Meier et al., 2020) was developed to work in combination with a trapped ion mobility spectrometer (TIMS). Previously, the TIMS‐PASEF configuration was shown to have a 10‐fold increase in sequencing speed in DDA mode without any loss in sensitivity. Extending this observation, the authors investigated the PASEF principle in conjunction with DIA mode. This approach improved the proteome detection and coverage (as an example, >4000 proteins were identified from only 10 ng of HeLA mixture) due to the high ion sampling capability. Parallel reaction monitoring (PRM) and multiple reaction monitoring (MRM) methods are widely used methods for targeted quantitative proteomics analysis of SARS‐CoV‐2 (Meyer & Schilling, 2017; Zecha et al., 2020). Utilizing both DDA and DIA approaches to analyze SARS‐CoV‐2 nasopharyngeal samples have opened avenues to better understand the viral infection and its effect on the hosts (Liou et al., 2021; Saadi et al., 2021).
Traditionally, the analysis of proteoforms is performed using either the bottom‐up (proteolytic protein) or top‐down (intact protein) proteomics approach. Bottom‐up, also known as shotgun proteomics, is widely used to identify peptides and quantify protein. By contrast, top‐down approaches use RP‐LC to first separate intact proteins without any digestion from biologically complex mixture which are then identified from the MS fragmentation patterns. Complexity of the data and other technical challenges impede the top‐down approach, but it can provide high‐quality protein information (L. M. Smith & Kelleher, 2013; L. M. Smith et al., 2021). In addition, native MS is a powerful tool to interrogate the structure of intact macromolecular assemblies in their near‐native state while preserving noncovalent protein‐protein or ligand interactions (Tamara et al., 2021). This complex technique can be employed in the analyses of glycan‐specific drug binding, in lectin recognition, and in glycoengineering (S. Chen et al., 2021). However, native MS of glycoproteins has achieved modest success, primarily because of the diversity and overlap of multiple glycosites and glycoforms on proteins that requires extensive orthogonal strategies such as glycomics (released glycans), bottom‐up and top‐down glycoproteomics for accurate analysis (Struwe & Robinson, 2019). Although limited, there have been reports that used native MS to study SARS‐CoV‐2 interactions which are discussed in Section 2.3.3.
2.3.1. DDA‐ and DIA‐based LC‐ESI‐MS/MS
In 2003, a preliminary report of MS characterization of cultured SARS‐CoV‐1 virus used a MALDI‐TOF‐MS approach to map virus proteolytic peptides release from gel bands. The authors identified the prominent ~46 kDa N‐protein and ~139 kDa S‐protein (Krokhin et al., 2003). Later, in early 2020, Grenga et al. performed shotgun proteomics on SARS‐CoV‐2 infected Vero E6 cells over a week at different multiplicities of infection with a virus titer of 1 × 107 pfu/ml. Virus‐infected cell lysates were heat‐inactivated, and proteins were precipitated with cold trichloroacetic acid before gel electrophoresis. The protein gel bands were excised and in‐gel trypsin digestion was done before LC analysis coupled to a Quadrupole Exactive (QE) HF MS. Correlations were made between the higher protein abundances found across the increasing time‐points spanning several peptides from the three structural viral proteins (M‐, N‐, and S‐) as well as other accessory proteins (Grenga et al., 2020). Using a similar shotgun proteomics approach, Gouveia et al. analyzed SARS‐CoV‐2 infected Vero E6 cell supernatants on a QE HF Orbitrap MS system. The study identified 101 peptides from six viral proteins including M‐, N‐, and S‐proteins (Gouveia, Grenga, et al., 2020). A shortlist of 14 peptides from the viral proteins were used for targeted MS method development and diagnostic studies of SARS‐CoV‐2 infection. The study was expanded into targeting the low abundance peptides of N‐protein from 9 positive nasal swabs, analyzed on a QE HF Orbitrap MS system, and detecting them in only a 3‐min MS window (Gouveia, Grenga, et al., 2020). In another study, also published in 2020, Cazares et al. prepared cultured kidney epithelial cells and in vitro‐derived mucus samples, both spiked with SARS‐CoV‐2 inactivated virus. Using DDA data, the authors developed a PRM assay on QE HF‐X Orbitrap MS with a spectral library containing 7 proteotypic peptides (4 from S‐protein and 3 from N‐protein). The PRM assay efficacy was evaluated, and it was estimated that 2 × 105 viral particles/ml sufficed for the detection of S‐ and N‐proteins of SARS‐CoV‐2. A limit of detection (~200 attomoles) and a limit of quantitation (~390 attomoles) was also formed for the developed PRM assay (Cazares et al., 2020). These findings showed that developing targeted or DIA approaches from DDA data could be useful to target low abundant or specific peptides from both pure protein and complex mixtures. Additionally, Bojkova et al. devised a cell‐culture model to perform proteomic analysis and determine SARS‐CoV‐2‐infections in Caco‐2 cells using a QE HF MS. The proteome profiling revealed modulation of core cellular pathways that occur after infection as well as identified several potential drugs that can inhibit viral replications such as ribavirin (Bojkova et al., 2020). Other studies have also used proteomics as the primary tool to identify dysregulated proteins after viral infection from 144 autopsy samples were obtained from seven organs (Nie et al., 2021) as well as reported on altered mechanisms involved in tissue fibrosis and autophagy in the host caused by SARS‐CoV‐2 (Stukalov et al., 2021).
Renuse et al. (2021) conducted preliminary discovery experiments on recombinant SARS‐CoV‐2 N‐protein on an Orbitrap Exploris 480 MS mass analyzer to generate a list of detectable viral peptides. To improve the sensitivity of detection, an automated mass spectrometric immuno assay‐based workflow specific to N‐proteins were performed on 116 SARS‐CoV‐2 positive and 71 SARS‐CoV‐2 negative nasal swab samples. The authors used reversed‐phase high‐performance liquid chromatography (RP‐HPLC) in conjunction with field asymmetric ion mobility spectrometry (FAIMS) separation for on‐line fractionation. FAIMS or differential IMS operates in alternating low and high electric field strengths that filters and separates the ions. This technique improves the linear dynamic range and the ion detection limits by filtering out background noise which enhances the resolution and sensitivity of targeted peaks in both MS and tandem MS levels (Swearingen & Moritz, 2012). In discovery analysis, the authors tested 17 different antibodies and identified 42 peptides belonging to N‐protein, some of which being low signal peptides, from a range of samples with both low and high viral loads. FAIMS compensation voltages were optimized for N‐protein peptides (Renuse et al., 2021). Based on these observations, a PRM method was optimized for the best‐performing peptides and the method was used to analyze 350 clinical samples (250 SARS‐CoV‐2 positive and 100 negative swabs) where four low‐abundant N‐peptides were observed (Maus et al., 2022). These studies together demonstrate that N‐protein peptides can be assayed using FAIMS‐PRM‐MS in a high‐throughput workflow.
FAIMS can be easily interfaced with an existing MS and is compatible with LC‐MS; however, due to the asymmetric waveform of the measured mobilities, it cannot provide the collisional cross‐section values (CCS) that correlate with the structure of an ion (Dodds & Baker, 2019). Contrastingly, TIMS operates by first accumulating and trapping ions in an opposing electric field, and then ejecting them sequentially based on their mobilities. The development of TIMS greatly increased the robustness, resolution, and sensitivity of ion mobility separation whereby isomeric peptides or lipids from very small sample amounts could be distinguished based on the CCS values of the observed ions (Aballo et al., 2021; Vasilopoulou et al., 2020). Application of diaPASEF acquisition mode on the TIMS‐TOF instrument was employed in two studies to determine SARS‐CoV‐2 viral proteins from positive nasopharyngeal swab samples. Mun et al. analyzed 45 positive and 45 negative samples where first the swab samples were heat‐inactivated at 70°C for 30 min, then the proteins were precipitated using methanol, before employing a proteomics workflow. They identified 7723 proteins using PASEF‐enabled DDA mode from which they built a spectral library to quantify 5023 proteins in diaPASEF mode. Upregulated proteins in positive samples were found to be involved in key biological processes of the innate immune system, viral protein assembly, and exocytosis (Mun et al., 2021). The use of PASEF in both DDA and DIA modes on the TIMS‐TOF was an advanced choice to study SARS‐CoV‐2. Using the same LC‐MS and acquisition setup, Nikolaev et al. investigated nasopharynx epithelial scrapings from 5 positive and 3 negative individuals using the standard proteomics workflow and an express preparation procedure (using RapiGest SF Surfactant) whereby the latter confidently identified N‐proteins even in the samples with the lowest viral loads. In addition, low abundant and unique phosphopeptides of the N‐protein were also detected using the diaPASEF approach (Nikolaev et al., 2020).
Interestingly, Schuster et al. developed a multistep procedure to obtain a set of reliable peptide markers for SARS‐CoV‐2 infection. RP‐HPLC coupled to a QE Plus Orbitrap MS was used to analyze SARS‐CoV‐2 infected Vero E6 cells. Recombinant N‐ and S‐proteins were codon‐optimized in Escherichia coli cells, virus stocks were propagated in the Vero E6 cells, and the viral titers were determined by serial dilutions of the cell cultures. The resultant protein fractions were reduced and alkylated before trypsin digestion. They identified almost all tryptic N‐ and S‐peptides in buffer samples spiked with cell‐cultured SARS‐CoV‐2 at 1 × 106 pfu/ml concentration. However, when they used this workflow to analyze viral proteins spiked into clinical nasal swab samples, the matrix background of the nasal swab samples interfered with identification of some of the viral peptides (Schuster et al., 2021). In a recent paper, the authors showed enrichment of SARS‐CoV‐2 from nasopharyngeal swab samples by first combining the biotinylated S‐protein antibodies with streptavidin‐coated magnetic beads and then enriching the virions. After enrichment and tryptic digestion for viral proteins, analysis was completed on an RP ultra‐performance liquid chromatography (UPLC) combined with Xevo Triple‐Q MS in MRM mode. After rounds of method optimization, they improved the sensitivity and selectivity of the workflow (Schuster et al., 2022). Given the complexity of nasal swab samples, enrichment steps to further isolate the SARS‐CoV‐2 proteome is recommended to improve the identification and quantitation of these proteins.
Similar viral N‐protein enrichment strategies were used in a study by Cardozo et al. where the authors analyzed 985 respiratory tract samples (540 positive and 445 negative samples, based on RT‐PCR results). Recombinant N‐protein was added as a standard and DDA acquisition was performed on QE HF‐X Orbitrap MS that identified 119 peptides from 8 SARS‐CoV‐2 viral proteins and 23.5% of the identified peptides belonged to N‐protein. A spectral library of 17 peptides (9 from N‐protein, 5 from S‐protein, 2 from M‐protein, and 1 from protein 3a) was then generated for PRM analysis that was performed on a turbulent flow on a Transcend TLX‐4 system consisting of four Dionex Ultimate 3000 quaternary and binary pumps coupled to a TSQ Altis Triple Quadrupole MS with heated ESI source that enabled processing of more than 500 samples per day. N‐peptides were found to exhibit 80‐fold higher relative intensities compared to the other peptides. Together, this workflow showed a high sample stability with 84% sensitivity and 97% specificity that could be considered for large‐scale SARS‐CoV‐2 testing (Cardozo et al., 2020).
Rivera et al. demonstrated proteomic analysis of oro‐ and nasopharyngeal samples from 5 SARS‐CoV‐2 positive and 5 negative samples analyzed on a nano‐LC coupled to a QE Plus Orbitrap MS. A total of 1177 proteins were observed in the DDA data where unique set of proteins were identified for each of the positive and negative cohorts, N‐protein being one of the top hits in the positive samples (Rivera et al., 2020). Alternatively, using an Orbitrap Fusion Lumos MS with multiplexed isobaric tagging and basic pH reversed‐phase chromatography fractionation, Vanderboom et al. identified 7582 proteins from a set of four SARS‐CoV‐2 positive and four negative nasal swabs. Tandem mass tags composed of an amine‐reactive NHS‐ester group, a spacer arm, and an MS/MS reporter generate low mass reporter ions that quantify the ratios of peptides in multiplexed samples. Using this strategy, significant upregulation of interferon‐related proteins and downregulation of proteasomal subunits in the infected samples were observed in this study. They also used immobilized metal affinity chromatography enrichment of nasopharyngeal swab tryptic phosphopeptides and identified more than 8500 phosphorylation sites, among which 194 were upregulated and 213 were downregulated. The alterations were primarily at the phosphorylation level, with the protein level unchanged (Vanderboom et al., 2021).
Bezstarosti et al. first assessed the limit of detection for SARS‐CoV‐2 N‐protein from infected Vero E6 cell lysates analyzed on an Orbitrap Eclipse MS with label‐free quantification. They then reported that 4% of the total proteome of the positive nasal swab samples corresponded to SARS‐CoV‐2 peptides, of which 88% corresponded to N‐protein. Next, they developed a PRM method for the viral proteins and applied it to 15 positive clinical patient samples with a stable isotope‐labeled N‐protein peptide as an internal control. They showed that the abundances of the N‐protein peptides correlated with the viral load for nasopharyngeal swab samples (Bezstarosti et al., 2021). Saadi et al. selected proteotypic peptides for the M‐, N‐, and proteins using untargeted proteomics. They then used stable isotope‐labeled synthetic peptides as absolute quantification standards corresponding to three viral peptides each from M‐, N‐, and S‐proteins of SARS‐CoV‐2 from 12 positive nasopharyngeal swabs (compared with 7 negative samples) using a microflow RP‐HPLC interfaced with a Q‐Orbitrap MS in PRM acquisition mode. Their LC‐PRM method confirmed 75% of samples that were positive by RT‐PCR assay (Saadi et al., 2021).
Finally, two studies used the MRM acquisition approach. Singh et al. identified 22 viral peptides covering three structural (N‐, E‐ and S‐) and one nonstructural (replicase polyprotein 1ab) proteins from 8 positive nasal swab samples on nanoflow RP‐HPLC coupled to a TripleTOF 6600 MS (Singh et al., 2020). Next, they developed a short‐scheduled MRM method using 8 proteotypic peptides from the four identified proteins. They reported detection of 2 peptides specific to SARS‐CoV‐2 with 100% and 90.5% specificity, respectively, compared to results from an RT‐PCR assay. Pinto et al. employed an S‐Trap mini spin column digestion and an LC‐MRM method to quantify 23 viral peptides spanning the 4 major structural and 2 accessory proteins from 81 positive samples on a Xevo Triple‐Q MS (Pinto et al., 2021). Hober et al. (2021) utilized the SISCAPA approach to enrich viral N‐proteins and identified 4 low‐abundance targeted peptides of N‐protein using a Xevo Triple‐Q MS instrument.
The MasSpec Pen is an automated and biocompatible MS handheld device that uses a discrete water droplet to extract biomolecules from ex vivo or in vivo tissue samples for direct transportation to an MS system (J. Zhang et al., 2017). This device has been used to study protein and lipid profiles directly from the tissues from various cancers (Sans et al., 2019; J. Zhang et al., 2017) and is compatible with different MS instruments. As a method for rapid screening of SARS‐CoV‐2 from mucous secretions, the MasSpec Pen was redesigned to incorporate a disposable sampling device for optimal analysis of swab tips via liquid extraction and direct connection to an ESI MS source. This system was used to analyze lipid profiles from 268 nasal swab samples. The samples were analyzed using two MS systems: LTQ‐Orbitrap XL and a QE HF Orbitrap MS with ESI in the negative ion mode. A set of 75 mass spectra were extracted and analyzed for each sample. SARS‐CoV‐2 positive and negative samples were determined using this tool which were in accordance to existing RT‐PCR results, and alterations in lipid profiles were observed between the samples (Garza et al., 2021).
2.3.2. MALDI‐MS
The MALDI‐TOF peptide mass fingerprinting method has been used to characterize the proteome of SARS‐CoV‐2 infected nasal swabs without any enrichments from large sample cohorts of 311 (Rocca et al., 2020), 237 patients (Deulofeu et al., 2021), and 107 (Tran et al., 2021), respectively, each of which detected peptides of N‐protein only. In the first study, MS testing of the nasal samples were performed on a 8020 MALDI‐TOF‐MS analyzer. The investigators used the Machine Intelligence Learning Optimizer (MILO) platform that consisted of a combination of supervised and unsupervised algorithms that identifies machine learning models with optimal performance (Tran et al., 2020). With the purpose of rapid screening of SARS‐CoV‐2, a selected subset of 82 samples (40 positive and 42 negative cases) was first used to validate the MILO models, which was then tested on the remaining 117 samples (67 positive and 50 negative cases). A total of 379,269 models were generated exhibiting high‐performance characteristics and two optimized machine learning models were identified with 98.3% and 96.6% accuracy respectively (Tran et al., 2021). Next, samples were analyzed on the Autoflex maX system and the data were used to build Extreme Gradient Boosting Trees and Support Vector Machines algorithms with hyper‐variable parameters. This differentiated SARS‐CoV‐2 positive samples from healthy controls with an accuracy, sensitivity, and specificity of more than 90%, promoting a rapid and economic diagnostic testing alternative for COVID‐19 disease. Finally, this study demonstrated the power of machine learning tools to generate models that correspond to distinct fingerprints of nasal swab samples in two different viral transfer media as well (Deulofeu et al., 2021).
Yoshinari et al. demonstrated purification of inactivated SARS‐CoV‐2 viral particles (focusing on N‐protein) first from control nasopharyngeal swabs inoculated with the virus, and then in clinically available samples (19 positive and 4 negative cases) using an ultrafiltration cartridge and anion exchange chromatography step before trypsin digestion. The N‐peptides were detected on a JMS‐S3000 SpiralTOF‐plus MS with a limit of detection of 106.7 viral copies and 7 N‐peptides were selected as the target molecules for the detection of SARS‐CoV‐2 (Yoshinari et al., 2022).
Dollman et al. investigated SARS‐CoV‐2 proteins from both nasal swabs and tryptic digests of whole virus cultured in Vero E6 cells by combining MALDI with Fourier transform ion cyclotron resonance (FT‐ICR) MS. The major advantage of the MALDI FT‐ICR MS setup is its ability in distinguishing isobars with a high resolving power (greater than 50,000) and mass accuracies (better than 5 ppm). In fact, this tool can provide confident peptide mass assignment from minimal samples without the need for tandem mass spectrometry (Spraggins et al., 2015). However, relatively data slow acquisition rates are a major limitation of this approach. Nasal swab samples were first washed in saline and water, and then passed through a 300 K molecular weight cut‐off filter. Cultured viral samples were chemically‐inactivated with 1% formalin, heat‐inactivated at 100°C for 15 min, filtered, and precipitated using polyethylene glycol, similar to an approach described previously (Fernandes et al., 2014). Cultured viral samples produced higher virus titers which was used for analysis to overcome potential false negative results associated with low viral loads in clinical samples. In determining the power of high‐resolution MS, despite low virus titers, peptides of M‐, N‐, and S‐proteins were detected with reduced signal‐to‐noise ratio in SARS‐CoV‐2 positive nasal samples. 5 distinct and reliable peptides were consistently identified in all clinical samples for proteotyping of SARS‐CoV‐2 using this approach (Dollman et al., 2020).
The recently developed MassARRAY system, a benchtop MS, is based on a combination of RT‐PCR and MALDI‐TOF‐MS customized for the detection of nucleic acid molecules (Jurinke et al., 2002). Two separate studies evaluated the sensitivity and reliability of this tool, one with 44 samples and another with a set of 101 nasopharyngeal or oropharyngeal swabs that were analyzed using the MassARRAY that effectively detected the viral nucleocapsid genes (Stelzl et al., 2021; Wandernoth et al., 2020).
2.3.3. Native MS
Native MS has been a particularly challenging tool for SARS‐CoV‐2 studies because of its extreme degree of heterogeneity and interactions with its host system. Additionally, the highly glycosylated nature of SARS‐CoV‐2 S‐protein and its interaction with the ACE2 receptor makes the analyses more complex, which is why there is a paucity of native MS studies on SARS‐CoV‐2. Two studies from a single group of researchers used recombinant forms of human ACE2 receptor and the S‐protein to perform native MS and molecular modeling. Using a combination of these advanced techniques, they first showed that heparin plays a major disruptor of the SARS‐CoV‐2 interaction with the host cell receptor (Yang, Hughes, et al., 2020) and then evaluated the interactions between S1 domain of S‐protein with the ACE2 ectodomain (Yang et al., 2021). In addition to this, another group of researchers characterized the functional relevance of SARS‐CoV‐2 N‐protein and main protease using native MS. In one study, Lutomski et al. identified multiple N‐terminal proteoforms of N‐protein that differentially interacted with the antibodies (IgM, IgG, and IgA) from convalescent plasma, whereas C‐terminal proteoforms showed increased reactivity with convalescent antibodies. N‐protein was also shown to bind to RNA preferentially via the GGG motifs which plays a role in packaging signals (Lutomski et al., 2021). In another study, El‐baba et al. investigated the SARS‐CoV‐2 main protease known to play a role in viral replication. Monomer/dimer equilibrium was analyzed in the presence of several small molecules that non‐covalently bound to the protease slowing down the substrate processing, thereby optimizing the use of antiviral compounds in this context (El‐Baba et al., 2020). Native and denaturing top‐down MS‐based approaches to determine the glycosylation of the SARS‐CoV‐2 S‐protein and its interaction with the ACE2 receptor is described in Section 3.4.
3. GLYCOPROTEOMICS‐BASED MS OF SARS‐COV‐2 VIRAL PROTEINS
Enveloped viruses are often decorated with surface glycoproteins that play vital roles in the viral life cycle and evasion of host immune responses to infection (H. Y. Huang et al., 2022; Tapper, 2006; Watanabe et al., 2019; Watanabe, Berndsen, et al., 2020). Viral evolution and mutation can alter the glycoproteome profile which contributes to the survival and transmissibility of the viruses (H. C. Huang et al., 2021; Zhao et al., 2020). For example, the glycosylated S‐protein of SARS‐CoV‐2 improves viral entry and binding efficacy with the host receptors that when inhibited has shown a reduction in the spread of the infection (Casas‐Sanchez et al., 2022; Yang, Hughes, et al., 2020). Understanding the glycoproteome alterations are also essential in identifying vaccine targets, particularly of the receptor binding domain (RBD) of the S‐protein (Gstöttner et al., 2021; Watanabe, Berndsen, et al., 2020; Yang, Hughes, et al., 2020). While several forms of protein glycosylation occur in nature, we will focus on N‐linked and mucin‐type O‐linked glycosylation of SARS‐CoV‐2.
N‐linked glycan biosynthesis originates in the endoplasmic reticulum where there is an en bloc transfer of the immature glucose‐capped precursors (Glc3Man9GlcNAc2) to an Asn residue in an NxS/T sequon via the oligosaccharyltransferase protein complex (Varki, 2017; Varki & Kornfeld, 2015). A series of glycan processing steps catalyzed by dozens of glycosyltransferases and glycoside hydrolases occur that generates four heterogeneous types of N‐glycans: oligomannosidic‐, paucimannosidic‐, hybrid‐, or complex‐types (Mikolajczyk et al., 2020; Nagae et al., 2020; Rini & Esko, 2015), Figure 1A. A common trimannosyl chitobiose core with or without Fuc (Man3GlcNAc2Fuc0‐1) is common for oligomannose, complex, and hybrid classes (Breitling & Aebi, 2013), whereas paucimannose type N‐glycans range between Man1‐3GlcNAc2Fuc0‐1 (Thaysen‐Andersen et al., 2015). While oligomannose and paucimannose type N‐glycans have only Man residues in the antennae, complex type N‐glycans contain GlcNAc‐Gal‐NeuAc antennae or structures with bisecting GlcNAc. Hybrid type N‐glycans have one Man‐containing arm whereas the other arm is occupied by saccharides other than mannose, including GlcNAc, Gal, NeuAc, and Fuc (D. J. Harvey, 2018). In addition to their original structures, some of these glycan types may have elongated Gal‐GlcNAc repeating units, phosphate, or sulfate groups (D. J. Harvey, 2018).
Figure 1.
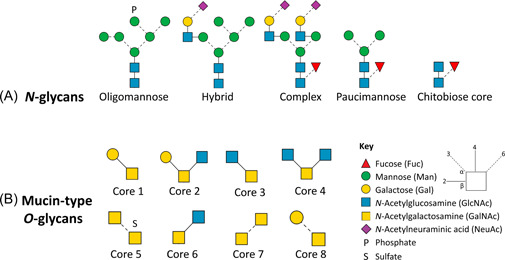
Summary of key human N‐ and O‐linked glycosylation. (A) N‐glycans are classified into oligomannosidic‐, hybrid‐, complex‐, paucimannosidic‐, and chitobiose core types. (B) Mucin‐type O‐glycans, also referred to as O‐GalNac, are divided into eight types, each with a common core.
Mucin‐type O‐glycans are simpler than N‐glycans, albeit they lack a single, uniform core structure. Generally, for mucin‐type O‐glycans, the amino acid residues (Ser, Thr, or Tyr) are covalently attached to a GalNAc residue by polypeptide‐N‐acetylgalactosaminyltransferases to form the Tn antigen (Pratt et al., 2004) which becomes modified by glycosyltransferases to form a total of 8 mucin‐type O‐glycan core‐structures, Figure 1B. Cores 1‐4 are common O‐GalNAc structures found in nature, where cores 1 and 2 are predominantly found in viruses that infect humans (Cipollo & Parsons, 2020).
Of the four coronaviruses major structural proteins, membrane (M), envelope (E), and spike (S) are known to be glycosylated whereas nucleocapsid (N) protein is heavily phosphorylated (Fung & Liu, 2018). While M‐ and E‐protein glycosylation have not been studied in detail, S‐protein glycosylation (derived from cell cultures or commercial recombinant sources) has been studied extensively because of its interaction with the crucial host ACE2 receptor that allows the first viral entry into the host cells (Gong et al., 2021; Shajahan, Archer‐Hartmann, et al., 2021).
3.1. Membrane, M‐protein
Membrane or M‐protein, which defines the viral shape and binds to all other structural proteins, is a transmembrane protein that becomes glycosylated in the host cell secretory pathway. It contains a short N‐terminal ectodomain, 3 hydrophobic transmembrane domains, and a large C‐terminal endodomain that play a role in viral particle assembly by interacting with the other structural proteins (Boson et al., 2021; Fu et al., 2021; Thomas, 2020). M‐protein resembles closely the semiSWEET family (Semi‐sugars Will Eventually be Exported Transporters) of prokaryotic transporters that catalyze diffusion of sugars through concentration gradients (Feng & Frommer, 2015). In silico experiments have predicted 8 N‐glycosylation sites: Asn5, Asn21, Asn41, Asn43, Asn117, Asn121, Asn203, and Asn216 (Dawood, 2021; Oostra et al., 2006; Thomas, 2020) but there is a gap in knowledge of site occupancy, glycoforms, and heterogeneity across all the sites. No O‐glycosylation has been reported for M‐protein (Figure 2A).
Figure 2.
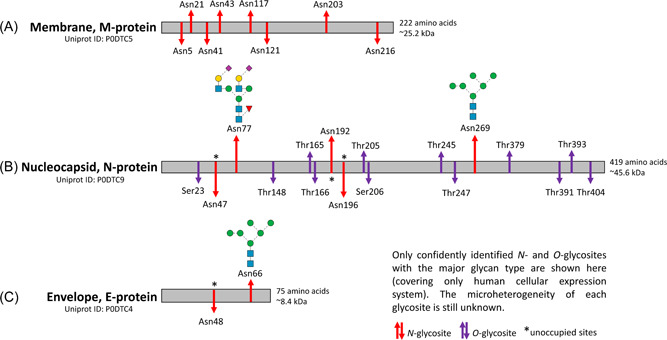
Overview of N‐ and O‐linked glycosites with an example of the major glycan class identified for (A) membrane protein, (B) nucleocapsid protein, and (C) envelope protein of SARS‐CoV‐2.
3.2. Nucleocapsid, N‐protein
Nucleocapsid or N‐protein constitutes the helical ribonucleocapsid that encapsulates the viral genome and interacts with the M protein (which is in a two‐dimensional lattice) during viral assembly (C. K. Chang et al., 2014; Kim et al., 2021; Zeng et al., 2020). Using an enzyme‐linked immunoassay‐based “bait and prey” system and recombinant fusion proteins, interactions between N‐ and S‐proteins were monitored and captured using monoclonal antibodies for N‐protein produced in SARS‐CoV‐2 infected Vero E6 cell cultures (Kim et al., 2021). N‐protein contains two major N‐ and C‐terminal domains that are both rich in Ser and Arg motifs (C. K. Chang et al., 2006). Since the N‐protein does not pass through the secretory pathway, the protein remains unglycosylated in the assembled virions but is known to be phosphorylated at Ser176 and Thr393 (observed on HEK293‐derived N‐protein). Supekar et al. compared the posttranslational modifications for N‐proteins derived from HEK293 cells, one prepared in‐house without a signal peptide sequence, and the other commercially obtained with a signal peptide and channeled through the secretory pathway (Supekar et al., 2021). N‐ and O‐glycans were released, purified and permethylated, and the samples were analyzed on the 5800 MALDI‐TOF/TOF‐MS and an Orbitrap Fusion MS in DDA mode. The commercial HEK293‐derived N‐protein with a signal peptide showed extensive N‐glycosylation (Asn47, Asn77, Asn192, Asn196, and Asn269), O‐glycosylation (Ser23, Thr148, Thr165, Thr166, Thr205, Ser206, Thr245, Thr247, Thr379, Thr391, Thr393, and Ser404) and phosphorylation at Thr393 (Figure 2B). Among the N‐glycosylation sites, Asn47 and Asn269 were decorated with complex and oligomannose‐type N‐glycans respectively, while other sites were found to be unoccupied. However, the native N‐protein without a signal peptide showed only a single O‐phosphorylation at Ser176 and no glycosylation, as would be expected for a protein that does not pass through the secretory pathway (Supekar et al., 2021). ADP‐ribosylation modifications have also been observed for N‐protein from SARS‐CoV‐1 virus‐infected Vero E6 cells (Grunewald et al., 2018). Sumoylation has been identified in SARS‐CoV‐1 N‐protein that was cloned and expressed in mammalian cells (F. Q. Li et al., 2005).
Another study by Sun et al. that used purified N‐protein highlights the dangers of using a secreted recombinant form of the protein to assess biologically relevant PTMs. HEK293 or Vero E6 cells were transfected with the plasmid‐encoded for SARS‐CoV‐2 N‐protein and after 40–60 h of incubation, the cells were lysed using immunoprecipitation buffer. Purified N‐protein fractions after multienzyme digestions using trypsin, chymotrypsin, and Lys‐C were enriched for N‐glycopeptides using zwitterion hydrophilic interaction liquid chromatography (ZIC‐HILIC) which were then separated on an RP‐HPLC connected to a QE HF‐X MS in DDA mode (Sun, Zheng, et al., 2021). While this study revealed multiple PTMs including acetylation, succinylation, ubiquitination, methylation, and phosphorylation on the secreted N‐protein, there is little evidence that these results are biologically relevant. A total of 27 phosphorylation sites on N‐protein were identified out of 80 potential sites (as reported by Group‐based Prediction System 5.0; Wang, Xu, et al., 2020). Intact N‐glycopeptide profiling confirmed the presence of N‐glycosylation on Asn77 (complex‐type N‐glycans) and Asn269 (oligomannose‐type N‐glycans), while no evidence was detected for the other sites. O‐glycosylation was not investigated in this study (Sun, Zheng, et al., 2021). Research showed presence of N‐protein in nasopharyngeal swabs from SARS‐CoV‐2 positive individuals, the detection of which was improved using an abbreviated workup without reduction and alkylation (Nikolaev et al., 2020).
3.3. Envelope, E‐protein
Envelope or E‐protein, the smallest of the four structural proteins, contains a short outer hydrophilic amino acid terminal domain, a single helix of hydrophobic transmembrane domain and a long inner hydrophilic carboxy‐terminal domain (Schoeman & Fielding, 2019). E‐protein interacts with M‐ and N‐proteins and the accessory proteins 3a and 7a (Boson et al., 2021) that plays critical roles in viral infectivity (Duart et al., 2020; Nieto‐Torres et al., 2014). Based on existing observations and sequence predictions, E‐protein of SARS‐CoV‐2 has two N‐glycosylation sites (Asn48 and Asn66) (Duart et al., 2020). Asn66 is known to be decorated with oligomannose‐type N‐glycans (Duart et al., 2020) whereas Asn48 is predicted to be a site which is difficult to be glycosylated due to its proximity of the residue to the membrane in the hydrophobic region (Schoeman & Fielding, 2019). No O‐glycosylation have been reported for E‐protein (Figure 2C).
3.4. Spike, S‐protein
Spike or S‐protein is a trimeric transmembrane protein made up of two subunits; S1 (~100–200 kDa, composed of 672 aa) with an N‐ and C‐terminal domain and two subdomains, and S2 (~80–150 kDa, 588 aa) with a hydrophobic N‐terminal domain, two heptad repeats, a transmembrane domain and a cytoplasmic tail (Du et al., 2009; Duan et al., 2020; Y. Huang et al., 2020; Riley et al., 2021; Wrapp et al., 2020). S‐protein binds to the ACE2 receptor of the host cells via the RBD (319–541 aa) of the S1 subunit, an interaction that determines the infectivity and transmissibility rate of SARS‐CoV‐2 (Gong et al., 2021; Walls et al., 2020). S2 subunit is involved in the fusion of the viral and host cellular membranes (Hoffmann et al., 2020; Shajahan, Archer‐Hartmann, et al., 2021). The heavy glycosylation of both S‐protein and ACE2 receptor may play a role in fostering binding mechanism to initiate infectivity (Campos et al., 2022; Gong et al., 2021; Shang et al., 2020; H. Zhang, Rostami, et al., 2020; Zhao et al., 2020). Interestingly, some studies have indicated that altering the host glycosylation can effectively inhibit viral binding to the host. Particularly, human milk oligosaccharides that are heavily decorated with α−2,6‐sialyl‐lactose are capable of preventing SARS‐CoV‐2 infection (Moore et al., 2021; Sheng et al., 2022). In‐depth characterization of the viral glycoproteome is therefore vital to understand the roles of SARS‐CoV‐2 in host‐pathogen mechanisms. A comparison of S‐protein from MERS and SARS‐CoV‐1 with the SARS‐CoV‐2, respectively, concluded that coronaviruses are extensively shielded (~40%) from antibody recognition due to the glycan decoration (Allen et al., 2021; B. G. Cho et al., 2021; Grant et al., 2020; Walls et al., 2019; Yang, Hughes, et al., 2020). Owing to these functions and its antigenicity, S‐protein is often the chosen target for vaccine and therapeutic developments (Du et al., 2009; Hussain et al., 2020; Papageorgiou & Mohsin, 2020; Samrat et al., 2020; Yang, Hughes, et al., 2020; Y. Zhang & Kutateladze, 2020; Zhao et al., 2021).
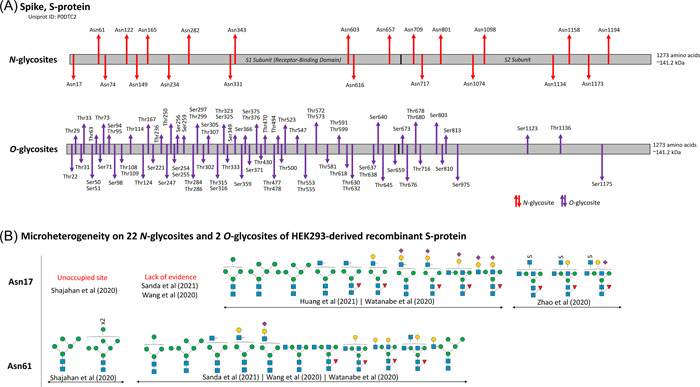
Previous studies on cryo‐based electron microscopy performed on trimeric S‐protein from SARS‐CoV‐1 and SARS‐CoV‐2 resolved at least 16 N‐glycosites with several glycoforms occupancy (Ke et al., 2020; Romeo et al., 2020; Walls et al., 2016; Walls et al., 2020; Wrapp et al., 2020; Yuan et al., 2017). In 2009, Ritchie et al. characterized N‐glycans of S‐protein from SARS‐CoV‐1 infected Vero E6 cells using a combination of normal‐phase HPLC with exoglycosidase digestion and MALDI‐TOF‐MS. Oligomannosylation was found to be the major class of N‐glycans, followed by complex‐type N‐glycans comprising of bi‐, tri‐, and tetra‐antennary structures with and without bisecting GlcNAc (Ritchie, Harvey, & Stroeher, 2010). Later, SARS‐CoV‐2 S‐protein derived recombinantly in the monomeric and trimeric forms, from cultured viral stocks and from virus‐infected cells were analyzed using site‐specific MS approaches, where a number of studies confirmed a total of 22 N‐glycosites with more than 95% occupancy and high macro‐ and micro‐heterogeneity (Antonopoulos et al., 2021; Bagdonaite et al., 2021; D. Chang et al., 2021; Duan et al., 2020; Sanda et al., 2021; Shajahan et al., 2020; Shajahan, Archer‐Hartmann, et al., 2021; Y. Zhang, Zhao, et al., 2021a; Zhao et al., 2021; Zhou et al., 2021). Apart from the canonical N‐glycosylation sequons, 3 other non‐canonical motifs of N‐glycosites with cysteine sequon are known including Asn164, Asn334, and Asn536, but no glycoforms have been identified on these sites (Gong et al., 2021; Y. Zhang, Zhao, et al., 2021a). S1 subunit of SARS‐CoV‐2 has 13 putative N‐glycosites: Asn17, Asn61, Asn74, Asn122, Asn149, Asn165, Asn234, Asn282, Asn331, Asn343, Asn603, Asn616, and Asn657, and 3 putative O‐glycosites: Thr323, Thr325, and Thr678. S2 subunit has 9 putative N‐glycosites: Asn709, Asn717, Asn801, Asn1074, Asn1098, Asn1134, Asn1158, Asn1173, and Asn1194. At least 60 O‐glycosites have been identified on S‐protein, of which many belong to the RBD region; however, the exact glycoforms and sites of occupancy are still under investigation (Figure 3A) (Ritchie, Harvey, & Stroeher, 2010; Roberts et al., 2021; Sanda et al., 2021; Shajahan et al., 2020; Shajahan, Archer‐Hartmann, et al., 2021; Tian et al., 2021; Y. Zhang, Zhao, et al., 2021a; S. Zhang, Go, et al., 2022; Zhao et al., 2021).
Figure 3.
Glycosylation of SARS‐CoV‐2 spike protein. (A) Overview of N‐ and O‐linked glycosites found on the S1 and S2 subunits of spike protein. (B) Site‐specific micro‐heterogeneity observed on 22 N‐glycosites and 2 O‐glycosites of recombinant spike protein derived from HEK293 cells.
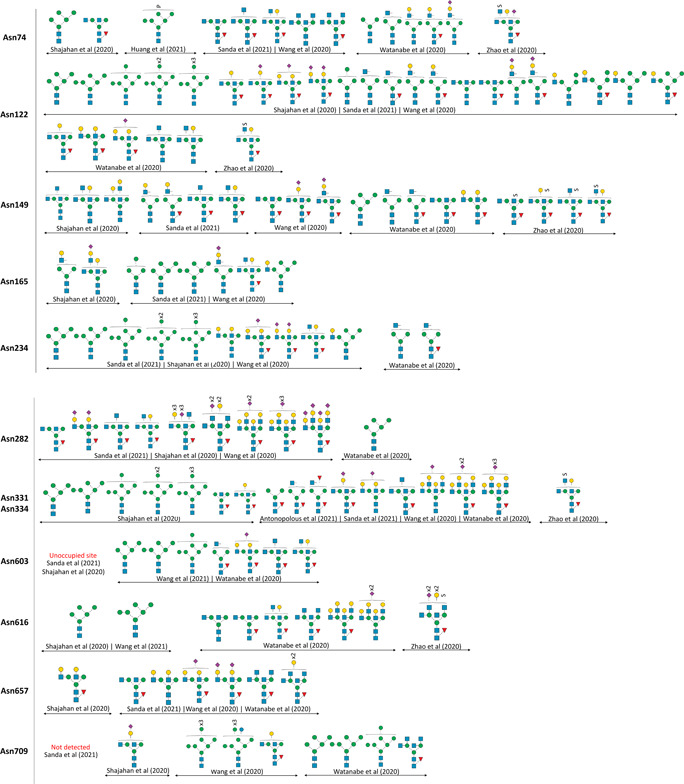
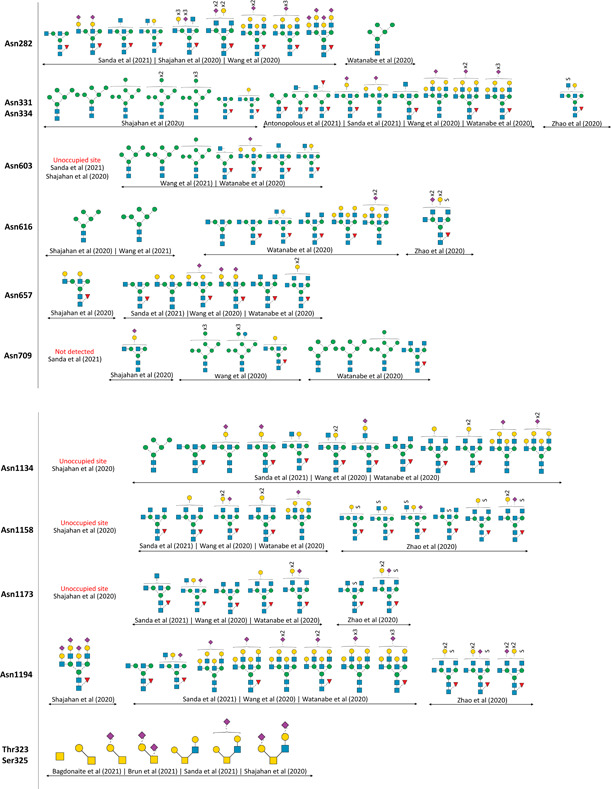
Glycosylation of recombinant trimeric S‐protein expressed in different systems have been studied by various groups around the world. While the identified N‐ and O‐glycosites are mostly consistent across different studies, the glycan compositions and occupancies on each site are unique depending on the analysis platform and expression system, respectively (Krishnan & Krishnan, 2021). As an example, the micro‐heterogeneity of N‐ and O‐glycoforms observed for only HEK293‐derived S‐protein from different research groups are depicted in Figure 3B.
Shajahan et al. characterized the N‐ and O‐glycoproteomes of both S1 and S2 protein subunits expressed in HEK293‐derived cell culture supernatants on an Orbitrap Fusion MS in DDA mode. A total of 17 out of 22 N‐glycosylation sites were detected with heterogeneous glycoforms and 5 sites (Asn17, Asn603, Asn1134, Asn1158, and Asn1173) were found to remain unoccupied. Asn331 and Asn343 were heavily decorated with oligomannose‐type N‐glycans. The Asn234 and Asn282 sites (adjacent to RBD) contained highly sialylated complex structures that may influence virus binding to the ACE2 receptors. O‐glycosylation on sites Thr323 and Ser325 from a single O‐glycopeptide indicated the presence of core‐1 and core‐2 mucin‐type O‐glycans on Thr323, and a plausible core‐2 mucin‐type O‐glycan on Ser325 which needs further confirmatory evidence (Shajahan et al., 2020). By contrast, Sanda et al. analyzed N‐ and O‐glycoproteome of recombinant histidine‐tagged trimeric S‐protein expressed in HEK293 cells on an Orbitrap Fusion Lumos MS in DDA mode and on a cyclic ion mobility mass spectrometer (cIMS). Efforts to improve IM‐MS technology for high mobility resolution in separating isomers led to the development of cIMS instrument that operates on the principle of travelling wave ion mobility. Ions travel through the cyclic device where they get separated based on mobility through single or multiple passes that are detected on the TOF analyzer. An increased path length through the circular track greatly increases the mobility resolution (Giles et al., 2019). Data were analyzed using Byonic and Driftscope, respectively, and identified 17 out of 22 N‐glycosylation sites. While Asn17 and Asn603 remained unoccupied in accordance with Shajahan et al. (Shajahan et al., 2020); Asn1134, Asn1158, and Asn1173 were found to be glycosylated with more than 90% occupancy. Asn234 was exclusively occupied by oligomannose‐type N‐glycans whereas all other sites were predominantly decorated by complex‐type N‐glycans with many containing LacdiNAc and polylacNAc structural motifs (Sanda et al., 2021). 8 O‐glycopeptides occupied by core‐1 and core‐2 mucin‐type structures on Thr323 and Ser325 were identified, along with 13% occupancy of both O‐glycan types on Thr678 (Sanda et al., 2021).
Watanabe, Berndsen, et al. (2020) expressed and purified 3 biological replicates of the recombinant soluble stabilized trimeric S‐protein (with substitution at furin cleavage sites) from HEK293F cells, similar to the S‐protein obtained from cryo‐based electron microscopy studies (Wrapp et al., 2020). The majority of the sites and glycoforms identified in this study were in accordance with other publications; however, this study detected only low abundances of oligomannose‐type N‐glycans on Asn331 and Asn343, hybrid‐type N‐glycans on many sites, and did not report on the presence of LacdiNAc structures (Watanabe, Berndsen, et al., 2020). O‐glycosylation was not studied in this study.
Zhang et al. attempted to compare the N‐glycosylation profile of HEK293‐derived recombinant S‐protein and S‐protein S1 and RBD subunit expressed in High Five insect cells. Samples were enriched for intact N‐glycopeptides using ZIC‐HILIC and analyzed on an Orbitrap Fusion Lumos MS in DDA acquisition mode. Identifying all 22 N‐glycosites in both systems, HEK293‐derived S‐protein were dominated by complex‐type N‐glycans whereas insect‐derived S‐protein was oligomannose‐rich (Y. Zhang, Zhao, et al., 2021a) highlighting the differences in glycoforms based on different species‐specific glycosylation pathways. Similar observations of oligomannose‐rich N‐glycosylation on S‐protein secreted from baculovirus‐insect cells was made by Zhou et al. (2021).
O‐glycosylation profiling was conducted by Bagdonaite et al. between HEK293F and Drosophila melanogaster S2 cells‐derived S‐protein analyzed on both Orbitrap Fusion Lumos MS and QE HF‐X MS. A set of 25 O‐linked glycosites were confidently identified with similar pattern across both HEK293 and S2 cell‐types. Interestingly, more than 60% of the identified sites were located next to an unmodified N‐glycosite (Bagdonaite et al., 2021). These observations were also found consistent with a study where researchers analyzed the O‐glycoproteome landscape of recombinant S‐protein expressed in HEK293 cells and High Five baculovirus‐insect cells (Y. Zhang, Zhao, et al., 2021b). Research has also highlighted the importance of O‐linked glycosylation in the RBD region when expressed as a monomer where the Thr323 site is almost completely occupied, in contrast to the native S‐protein where Thr323 displays low O‐glycan occupancy (Eldrid et al., 2021).
Antonopoulos et al. (2021) performed glycoproteomics analysis of the RBD (Arg319‐Phe541) region of monomeric S1 subunit recombinantly expressed in HEK293 cells on a Xevo G2‐XS Q‐TOF MS system that identified two N‐glycosylation sites: Asn331 and Asn343. In contrast to the findings presented by Shajahan et al., both the sites contained exclusively complex‐type N‐glycans with 98% core‐fucosylation. They confirmed presence of LacdiNAc and polylacNAc structural motifs using MALDI‐TOF/TOF‐MS. A very small proportion of paucimannose‐type N‐glycan was also detected. A variety of O‐linked glycopeptides were observed with high site occupancy; for example, Thr323 site was found to be decorated with core‐1 di‐sialyl mucin‐type O‐glycans (Antonopoulos et al., 2021). Alternatively, nuclear magnetic resonance‐based studies of the RBD region identified similar epitopes of GalNAc and Fuc, as reported by Antonopoulos et al. (2021). Further, experiments were conducted to identify the interaction of S‐protein with relevant receptors and lectins such as DC‐SIGN, MGL, siglecs, and galectins (Lenza et al., 2020).
Brun et al. compared the site‐specific glycosylation changes among S‐protein derived from SARS‐CoV‐2 cultured virus propagated in Vero E6 and Calu‐3 cells, recombinant trimeric S‐protein expressed in HEK293F cells, expression constructs of S‐protein obtained from plasmids including nonstabilized S‐protein, recombinant trimeric soluble S‐protein and S1 subunit of S‐protein. Affinity purification was performed in the same manner for all the S‐protein types against CR3022 antibodies (recombinant monoclonal form expressed in HEK293F cells) and data were acquired using a QE HF‐X Orbitrap MS in DDA acquisition mode (Brun et al., 2021). Significant site‐specific N‐ and O‐glycome variations were found between the different S‐proteins which were suggested to appear due to the distinctive cellular secretion pathways resulting in altered glycosylation profiles. For example, S‐protein from viral cultures showed 79% complex‐type and 21% oligomannose‐type N‐glycans, whereas the recombinant trimeric form carried 89% complex‐type and only 11% oligomannose‐type N‐glycans. Byonic and Byologic software were used to obtain the area of each extracted ion chromatogram to subsequently quantify the site‐specific glycosylation on each site. Asn234 showed 100% complex‐type N‐glycans for recombinant S1 subunit which was completely absent from the recombinant trimeric S‐protein and S‐protein obtained from viral cultures. An O‐glycan site, Thr678, was identified on S‐protein from viral cultures and was found to be absent on the recombinant forms (Brun et al., 2021). Furthermore, Wang et al. investigated the differences in glycosylation of recombinant S‐protein S1 subunit obtained from different expression host systems including HEK293 and baculovirus‐insect cell cultures. N‐ and O‐glycans were detected on MALDI‐TOF/TOF‐MS and on Orbitrap Fusion Lumos MS with DDA acquisition (Y. Wang et al., 2021). A set of 12 and 13 N‐glycosites were identified on the S1 subunit derived from HEK293 and baculovirus systems respectively. Generally, HEK293‐derived S‐protein predominantly contained complex‐type N‐glycans, whereas insect‐derived S‐protein were found to be rich in oligomannose‐type N‐glycans. Site‐specific glycoform variations were also identified where for example Asn149 of HEK293 was decorated with only complex‐type N‐glycans, while insect‐derived Asn149 showed 50% hybrid, 30% oligomannose, and 20% complex‐type N‐glycans. Additionally, 9 and 15 O‐glycosites were identified on the S1 subunit derived from HEK293 and baculovirus systems respectively. The O‐glycosylation pattern was found to be more diverse and complex for the insect‐derived S‐protein when compared to HEK293 cells. Combining these observations indicated that the glycosylation of recombinant S‐protein may not be representative of the native S‐protein found in biological samples. This is an important consideration when selecting an expression system to develop relevant antibodies and vaccines for SARS‐CoV‐2.
Another major tropism determinant of SARS‐CoV‐2 spread is its interaction with the glycosylated ACE2 receptor commonly found on human host cells (Hoffmann et al., 2020; Mehdipour & Hummer, 2021; Oz et al., 2021; Shang et al., 2020). The N‐terminal domain of ACE2 has 7 putative N‐glycosylation sites: Asn53, Asn90, Asn103, Asn322, Asn432, Asn546, and Asn690 and several potential O‐glycosylation sites (Casalino et al., 2020; Gong et al., 2021; Shajahan, Archer‐Hartmann, et al., 2021; Sun, Ren, et al., 2021; Zhao et al., 2020) that potentially play vital roles in virus binding. Investigations have already elucidated that inhibition of glycosylation on specific sites (Asn122, Asn331, Asn334, Asn717, Asn801, and Asn1074) significantly reduces the viral infectivity rate (Wang, Xu, et al., 2020). Researchers continue to investigate the glycosylation profile of the ACE2 receptor to identify the features responsible for the virus‐receptor binding mechanisms.
Shajahan et al. characterized the N‐ and O‐glycosylation of recombinant histidine‐tagged human ACE2 expressed in HEK293 cells on an Orbitrap Fusion MS and on MALDI‐TOF/TOF‐MS (permethylated glycans). All 7 N‐glycosites on ACE2 were identified with more than 73% occupancy of complex‐type N‐glycans with high degree of sialylation and multiple antennae and primarily core‐fucosylated structures. Out of all the sites, the Asn432 and Asn690 sequons were found to be ~27% and ~1% unoccupied, respectively with equal proportion of sialylated and non‐sialylated structures. A single O‐glycosite, Thr730, was confidently assigned with almost 97% occupancy of core‐1 mucin‐type O‐glycans (Shajahan, Archer‐Hartmann, et al., 2021).
Subsequently, Zhao et al. used a glycomics‐informed glycoproteomics approach (Rosenbalm et al., 2020) with molecular dynamic simulations to assess the SARS‐CoV‐2 and ACE2 interactions (Zhao et al., 2020). This study used purified recombinant trimeric S‐protein and recombinant histidine‐tagged human ACE2, both expressed in HEK293F cells, applied different proteolytic digestions and released N‐ and O‐linked glycans, conducted extensive LC‐MS/MS acquisitions on an Orbitrap Lumos MS and performed analysis on multiple software for accurate data validations. All 22 N‐glycosites of S‐protein were detected showing a massive degree of micro‐heterogeneity in glycoforms spanning oligomannose‐, complex‐, and hybrid‐type N‐glycans. N‐glycans at sites Asn17, Asn74, Asn122, Asn149, Asn331, Asn616, Asn801, Asn1098, Asn1158, Asn1173, and Asn1194 were also found to be decorated with sulfated complex‐type N‐glycans. Sulfation on Asn74 was particularly in agreement with findings from Klein et al. where the authors re‐analyzed publicly available data (Watanabe, Berndsen, et al., 2020) that further reported the presence of sulfated N‐glycans on other sites such as Asn234 and Asn1074 (manuscript under review, doi 10.1101/2020.05.31.125302). O‐glycosylation on Thr323 was observed with only 11% occupancy of core‐1 and core‐2 mucin‐type O‐glycans. Using their systematic approach, the authors provided finer structural details of the S‐protein and highlighted the shielding effect of N‐glycans on the peptide backbone which was in agreement with other studies (Allen et al., 2021; Watanabe, Berndsen, et al., 2020). A total of 6 out of 7 N‐glycosylation sites on ACE2 was observed with more than 75% occupancy of complex‐type N‐glycans, and limited oligomannose‐ and hybrid‐type N‐glycans. No sulfated N‐linked glycans were detected. Additionally, the Ser155 O‐glycosite could be confidently assigned but several other O‐glycosites were observed in extremely low stoichiometry. Finally, glycan‐mediated interactions, specific to this system, were reported between the Asn74 and Asn149 sites of S‐protein and Asn90, Asn322, and Asn546 sites of ACE2 receptor (Zhao et al., 2020). In a molecular dynamics simulation‐focused study by Casalino et al., the authors revealed that the N‐glycans on Asn165 and Asn234 of the RBD region of S‐protein influenced ACE2 recognition. Using bio‐layer interferometry, deletion of glycans from these two N‐glycosites significantly reduced the binding affinity with ACE2 receptor (Casalino et al., 2020). However, the evolution in the S‐protein glycoforms across the different variants is still unknown.
Interestingly, Sun, Ren, et al. (2021) reported that the binding of recombinant S‐protein ectodomain expressed in insect cells and the recombinant extracellular domain of ACE2 receptor expressed in HEK 293 cells did not only depend on the N‐glycosylation. Using binding kinetics, the authors deduced that the ACE2 receptor bound to the deglycosylated S‐protein. The authors concluded based on proteomics database search results that methylation was present on lysine, arginine, and/or glutamic acid residues of both S‐protein and that ACE2 also contained hydroxylproline residues. While the study identified these PTMs through database searches, it will be important to confirm the presence of these PTMs and their biological significance using careful experiments to address if these modifications really play a role in the interactions between S‐protein and ACE2 receptor. S‐protein was expressed in Spodoptera frugiperda SF9 insect cells and analyzed on QE HF‐X MS in DDA mode. The binding evaluation was measured by bio‐layer interferometry on an Octet Re6E system. A total of 21 N‐glycosites were identified with glycoforms similar to other studies (Brun et al., 2021; Y. Wang et al., 2021). In another study, Liu et al. developed a novel MS‐based two‐step isotope labelling‐lysine reactivity profiling method to quantify the reactivity of lysine residues that are known to be crucial between the S1 subunit‐ACE2 receptor interactions. Recombinant S1 subunit and ACE2 receptor proteins were labelled, digested with Glu‐C or chymotrypsin and analyzed on an LTQ‐Orbitrap MS. Interestingly, significant alterations in lysine reactivities were observed particularly between Lys386‐Lys462 that was essential in the formation of the protein‐receptor complex (Liu et al., 2020). Recently, a study reported that a loss in glycosylation on Asn370 site of S‐protein fosters an enhanced infectivity for SARS‐CoV‐2. Previously observed on bat and pangolin‐derived coronavirus S‐protein trimers (S. Zhang et al., 2021), the glycosylation site at Asn370 was found to be lost in the S‐protein from SARS‐CoV‐2 because of the threonine‐to‐alanine mutation at that position that may have occurred over the viral evolution process. For this reason, the authors attempted to restore this N‐glycosite of S‐protein expressing it in different systems, and demonstrated through MS, surface plasmon resonance, and molecular dynamic simulation experiments that the absence of glycosylation on Asn370 facilitated a more efficient binding to the ACE2 receptor thereby providing a higher capacity for infection (S. Zhang, Liang, et al., 2022).
Researchers have also investigated enrichment and trigger methods to detect low‐abundant glycopeptides of S‐protein. In one study, dual‐functionalized titanium (IV)‐immobilized metal affinity chromatography approach was employed on HEK293‐derived recombinant S‐protein to separate neutral and sialyl glycopeptides analyzed on an Orbitrap Fusion Lumos MS (H. C. Huang et al., 2021). A total of 19 of 22 potential N‐glycosites with 398 unique glycoforms were profiled. Comparisons were drawn with HILIC‐MS analysis which identified 18 N‐glycosites with only 247 unique glycoforms. This approach also identified a mannose‐6‐phosphate glycan on Asn74 site and an O‐linked glycosylation site at Thr323 with three glycoforms. This approach assisted in eliminating signal suppression from neutral glycopeptides to increase detection of sialylated glycopeptides thereby increasing the overall glycoproteome coverage of S‐protein (H. C. Huang et al., 2021). In another study, W. Wang, Xu, et al. (2020) analyzed S‐protein from HEK293 cells and SF9 insect cells on an Orbitrap Eclipse MS using a signature ion‐triggered electron transfer/higher‐energy collision dissociation (EThcD) method. While HCD, the commonly used fragmentation mode on Q‐Orbitrap instruments generates efficient dissociation, EThcD fragmentation provides more abundant peptide backbone fragments that are useful for complete structural elucidation of glycopeptides. Glycan oxonium fragments of m/z 138.0545 (HexNAc fragment), m/z 204.0867 (HexNAc), and m/z 366.1396 (HexNAcHex) were included to trigger the ETD fragmentation. A set of 21 N‐glycosites with the known glycoforms were identified, and the presence of paucimannosylated N‐glycans were also confirmed in this approach. The authors also investigated the S1 subunit of S‐protein which primarily contained complex‐type N‐glycans. Many truncated N‐glycans with core‐fucosylation were observed on the S‐protein expressed in insect cells (Wang, Xu, et al., 2020). Glycosylation machinery in insects support a hexosaminidase pathway that can expectedly generate abundant paucimannosidic N‐glycans (Tjondro et al., 2019). While paucimannosylation is known to play important roles in human cancers, inflammation, and stemness (Chatterjee et al., 2019), its exact involvement in the context of SARS‐CoV‐2 infection is yet to be determined. While DDA‐MS acquisition has frequently and successfully been used by research groups, Chang et al. analyzed N‐glycosites of recombinant S‐protein without the furin cleavage site from HEK293 cells using both DDA and DIA approaches. Samples were separated on a nano RP‐UPLC system before acquiring the data on a QE HF‐MS instrument. This study identified 22 N‐glycosites using DDA approach with the glycoforms in agreement with other findings, whereas DIA approach identified 20 N‐glycosites with many more low‐abundant glycoforms. The study indicated that to survey the evolving viral glycoproteins, DIA mode can provide a higher sensitivity, selectivity, and reproducibility across the identified glycopeptides (D. Chang et al., 2021).
Finally, investigators have also explored top‐down MS‐based methods of SARS‐CoV‐2 S‐protein with its glycoforms. Using charge detection mass spectrometry (CDMS) approach, Miller et al. attempted to investigate the heterogeneity of N‐ and O‐glycosylation of S‐protein from HEK293, HEK293S, CHO and insect cell systems comparing it with existing glycoproteomics studies. CDMS measures both m/z and charge of individual ions making it a suitable approach to analyze large protein complexes or other heterogeneous macromolecules. Top‐down CDMS measurements indicated that the glycans were 35%–47% larger than the typical bottom‐up glycoproteomics approaches and some trimers were more heavily processed than the other ones (Miller et al., 2021). Additionally, in another study, structural O‐glycoform heterogeneity on S‐protein RBD expressed in HEK293 cells was explored using native and denaturing top‐down MS employing TIMS‐TOF MS and FT‐ICR MS, respectively. The authors exploited the exclusive features of these different mass analyzers to delineate a complete structural information of intact O‐glycoforms on S‐protein RBD. The ion mobility separation capacity of TIMS‐TOF was used to identify native protein conformers of the RBD region whereas the ultrahigh resolving power of 12 T on FT‐ICR MS instrument was used to provide an in‐depth characterization of the RBD O‐glycoforms. A total of eight O‐glycoforms spanning core‐1 and core‐2 type O‐glycans with their abundances and occupancies on RBD were reported using this approach. N‐terminal acetylation and sulfonation found on the glycans were not previously reported, thus overcoming the challenges of structural elucidation of O‐glycan proteoforms using bottom‐up glycoproteomics approaches (Roberts et al., 2021). Similarly, Gstöttner et al. also determined both N‐ and O‐glycosylation abundances on S‐protein RBD expressed in CHO and HEK293 cells using a combination of bottom‐up and top‐down approaches (Gstöttner et al., 2021). In a recent study by Wilson et al., (2022) top‐down MS‐based analysis of S‐protein RBD was greatly enhanced by using HILIC separation method that identified several low abundant forms showing the presence of more than 200 glycoforms only on the RBD. An in‐depth characterization of the S‐protein with its two subunits including the RBD region as well as the ACE2 receptor has provided important information on the N‐ and O‐glycome profile of this viral protein and human host receptor that forms the first key step in understanding the glycobiology of viral pathogenesis. However, it should also be noted that these observations are largely dependent on factors including the recombinant nature and the expression system of the proteins, employing enrichment or purification methods for the peptides or glycopeptides, MS instruments, parameters, and acquisitions modes as well as the data analyses tools (Hackett & Zaia, 2021). Hence, even though these essential preliminary findings advance our understanding on the glycosylation pattern of S‐protein and ACE2 receptor, they may not necessarily be representative of the actual biological scenario.
3.5. Accessory proteins
Out of the other SARS‐CoV‐2 accessory viral proteins, ORF3a (P0DTC3, ~31.2 kDa, 274 aa), a nonstructural accessory protein localized on the surface is essential for viral replication and transcription. ORF3a is known to contain an N‐terminal ectodomain, three transmembrane domains, and a C‐terminal endodomain (Tan et al., 2004). ORF3a protein was found to localize in the Golgi compartment as well as on the cell surface, from which it is presumably endocytosed (Tan et al., 2004; Yu et al., 2004). According to hydrophobicity analyses and topology studies, ORF3a has four O‐linked glycosites (Ser27, Thr28, Thr32, and Thr34) but there is no information on the O‐glycoforms (Oostra et al., 2006; Yadav et al., 2021). An absence of N‐glycosylation has been confirmed in ORF3a (Majumdar & Niyogi, 2020).
4. CONCLUSION AND FUTURE DIRECTIONS
In this review, we first summarized the current state‐of‐the‐art MS‐based proteomics approaches used to characterize the SARS‐CoV‐2 proteome obtained from clinical human nasopharyngeal nasal swabs. Based on the research question, a number of different sample preparation workflows and LC‐MS instrumentation and methods were used to detect SARS‐CoV‐2 proteins. Many of the studies in the early 2019–2020 used DDA‐based workflows that revealed several upregulated and downregulated proteins caused by COVID‐19 infection. The use of enrichment tools was shown to improve the detection of proteolytic peptides primarily of SARS‐CoV‐2 M‐, N‐, and S‐proteins from infected cell cultures or biological samples with an abundance of proteins from both host and virus. Multi‐enzyme digestion prior or in parallel to enrichments also allow increased peptide detection. Alternatively, peptides of interest may be isolated using appropriate SWATH‐ or DIA‐based MS methods. As discussed in Section 2.3, each MS analyzer came with its own benefits and limitations, but the experimental choices including the acquisition compatibility with instruments were heavily dependent on the aims of the study. Only a few studies reported on the presence of the less abundant E‐protein and accessory proteins which are still under investigation.
Next, we recapitulated the known glycosylation pattern of SARS‐CoV‐2 viral proteins from commercial or expression sources, highlighting the different methods and glycoproteomics‐based MS approaches used to profile the N‐ and O‐glycoproteome, particularly of recombinantly expressed S‐protein and ACE2 receptor. The macro‐ and micro‐heterogeneity of protein glycosylation make it harder to accurately identify glycopeptides with their multiple glycoforms, but with the advancement of MS techniques and bioinformatics, observations have shed light on the potential glycosites and glycoforms that are now known to play significant roles in viral binding to the host receptors. Generally, the studies have emphasized on the identification of the glycosites, glycopeptides, and glycan compositions using high‐throughput MS, but only few have deduced the isomers and linkages and attempted to quantify the detected glycans. Notably, many N‐ and O‐ glycosites are lacking information on the extensive isomeric glycoforms and occupancies that still needs to be investigated further to get a complete glycoproteome picture of SARS‐CoV‐2. Glycopeptide enrichment using suitable HILIC resin is a common step for glycopeptide detection because analysis of the enriched fraction, devoid of the unmodified peptides, allows better separation and fragmentation of glycopeptides. While HILIC is popularly used for enrichment before LC‐MS, the chromatography separation is often performed on the C18‐RP setup. RP‐LC is robust in separating glycopeptides based on the hydrophobic nature of the peptide part; but produces only a small degree of separation based on the glycopeptide glycan composition. While a few reports have suggested that HILIC is capable of resolving some glycan isomers (Huang et al., 2016; van der Burgt et al., 2020), particularly for top‐down MS‐based characterization of SARS‐CoV‐2 (Wilson et al., 2022), this has yet to be tested on the viral protein glycopeptides.
While there has been much focus on studying S‐protein for its interaction with the ACE2 receptor, glycoprofiling of other structural proteins such as M‐, N‐, and E‐proteins remain under investigation. There is also a considerable gap in understanding of the protein glycosylation alterations of the SARS‐CoV‐2 structural viral proteins from biologically relevant samples (naso‐ or oro‐pharyngeal swabs, or other biological fluids), what mutations occur, how the glycosylation profile changes across the different variants of concern, and if new methods and bioinformatics approaches are needed to accurately quantify site‐specific glycosylation changes. Together, these findings will form essential components for sensitive vaccine and therapeutic developments against SARS‐CoV‐2.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors acknowledge funding by National Institute of Health (NIH) grant R01AI155975.
Biographies
Dr. Sayantani Chatterjee is a Post‐Doctoral Associate in the Zaia research group in the Department of Biochemistry, Boston University School of Medicine, Boston, USA. She is currently investigating viral glycoproteomics and host‐pathogen alterations, particularly of influenza and SARS‐CoV‐2 viruses. Previously, she completed her MRes (2017) and received her PhD (2021) in Molecular Sciences from the Analytical Glycoimmunology Group in the Department of Molecular Sciences, Macquarie University, Sydney, Australia. Her research focused on characterizing the structural and functional diversity of mannose‐terminating glycoproteins in cancer, innate immunity and pathogenic infections. Earlier, she received her BSc in Microbiology (Honors) in 2015 from St. Xavier's College (Autonomous), India.

Prof. Joseph Zaia is Professor of Biochemistry at Boston University School of Medicine. His research group focuses on analytical biochemistry of glycoproteins and proteoglycans. They have developed methods for combined glycomics, proteomics and glycoproteomics from tissue slides. Their major goals are to track changes in protein glycosylation during biological processes. Specific projects include (i) mapping changes in extracellular matrix protein glycosylation during brain disease mechanisms and (ii) analysis of the roles of viral spike protein glycosylation in evolution of pathogenic enveloped viruses. Dr. Zaia has published more than 150 articles in peer‐reviewed scientific journals and has an H‐index of 54. He is an editor of Analytical and Bioanalytical Chemistry.

Chatterjee S, Zaia J. (2022). Proteomics‐based mass spectrometry profiling of SARS‐CoV‐2 infection from human nasopharyngeal samples. Mass Spectrometry Reviews e21813. 10.1002/mas.21813
REFERENCES
- Aballo TJ, Roberts DS, Melby JA, Buck KM, Brown KA, Ge Y. 2021. Ultrafast and reproducible proteomics from small amounts of heart tissue enabled by Azo and timsTOF Pro. Journal of Proteome Research 20(8):4203‐4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R, Mann M. 2003. Mass spectrometry‐based proteomics. Nature 422(6928):198‐207. [DOI] [PubMed] [Google Scholar]
- Akgun E, Tuzuner MB, Sahin B, et al. 2020. Proteins associated with neutrophil degranulation are upregulated in nasopharyngeal swabs from SARS‐CoV‐2 patients. PLoS One 15(10):e0240012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandersen S, Chamings A, Bhatta TR. 2020. SARS‐CoV‐2 genomic and subgenomic RNAs in diagnostic samples are not an indicator of active replication. Nature Communications 11(1):6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JD, Chawla H, Samsudin F, et al. 2021. Site‐specific steric control of SARS‐CoV‐2 spike glycosylation. Biochemistry 60(27):2153‐2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri‐Dashatan N, Koushki M, Rezaei‐Tavirani M. 2022. Mass spectrometry‐based proteomics research to fight COVID‐19: an expert review on hopes and challenges. OMICS: A Journal of Integrative Biology 26(1):19‐34. [DOI] [PubMed] [Google Scholar]
- Amraei R, Yin W, Napoleon MA, et al. 2021. CD209L/L‐SIGN and CD209/DC‐SIGN act as receptors for SARS‐CoV‐2. ACS Central Science 7(7):1156‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amraei R, Xia C, Olejnik J, et al. 2022. Extracellular vimentin is an attachment factor that facilitates SARS‐CoV‐2 entry into human endothelial cells. Proceedings of the National Academy of Sciences of the United States of America 119(6):e2113874119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. 2020. The proximal origin of SARS‐CoV‐2. Nature Medicine 26(4):450‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonopoulos A, Broome S, Sharov V, et al. 2021. Site‐specific characterization of SARS‐CoV‐2 spike glycoprotein receptor‐binding domain. Glycobiology 31(3):181‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya R, Kumari S, Pandey B, et al. 2021. Structural insights into SARS‐CoV‐2 proteins. Journal of Molecular Biology 433(2):166725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdonaite I, Wandall HH. 2018. Global aspects of viral glycosylation. Glycobiology 28(7):443‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdonaite I, Thompson AJ, Wang X, et al. 2021. Site‐specific O‐glycosylation analysis of SARS‐CoV‐2 spike protein produced in insect and human cells. Viruses 13(4):551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee N, Mukhopadhyay S. 2016. Viral glycoproteins: biological role and application in diagnosis. Virus Disease 27(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐On YM, Flamholz A, Phillips R, Milo R. 2020. SARS‐CoV‐2 (COVID‐19) by the numbers. eLife 9:e57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev‐Gross M, Weiss S, Ben‐Shmuel A, et al. 2021. Spike vs nucleocapsid SARS‐CoV‐2 antigen detection: application in nasopharyngeal swab specimens. Analytical and Bioanalytical Chemistry 413(13):3501‐3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Zinger T, Inglima K, et al. 2020. Performance of Abbott ID Now COVID‐19 rapid nucleic acid amplification test using nasopharyngeal swabs transported in viral transport media and dry nasal swabs in a New York City Academic Institution. Journal of Clinical Microbiology 58(8):e01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton DJ, Wrobel AG, Xu P, et al. 2020. Receptor binding and priming of the spike protein of SARS‐CoV‐2 for membrane fusion. Nature 588(7837):327‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezstarosti K, Lamers MM, Doff WAS, et al. 2021. Targeted proteomics as a tool to detect SARS‐CoV‐2 proteins in clinical specimens. PLoS One 16(11):e0259165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichmann L, Gupta S, Rosenberger G, et al. 2021. DIAproteomics: a multifunctional data analysis pipeline for data‐independent acquisition proteomics and peptidomics. Journal of Proteome Research 20(7):3758‐3766. [DOI] [PubMed] [Google Scholar]
- Bittremieux W, Adams C, Laukens K, Dorrestein PC, Bandeira N. 2021. Open science resources for the mass spectrometry‐based analysis of SARS‐CoV‐2. Journal of Proteome Research 20(3):1464‐1475. [DOI] [PubMed] [Google Scholar]
- Bojkova D, Klann K, Koch B, et al. 2020. Proteomics of SARS‐CoV‐2‐infected host cells reveals therapy targets. Nature 583(7816):469‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boson B, Legros V, Zhou B, et al. 2021. The SARS‐CoV‐2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus‐like particles. The Journal of Biological Chemistry 296:100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling J, Aebi M. 2013. N‐linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harbor Perspectives in Biology 5(8):a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun J, Vasiljevic S, Gangadharan B, et al. 2021. Assessing antigen structural integrity through glycosylation analysis of the SARS‐CoV‐2 viral spike. ACS Central Science 7(4):586‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Ge W, Yi X, et al. 2021. PulseDIA: data‐independent acquisition mass spectrometry using multi‐injection pulsed gas‐phase fractionation. Journal of Proteome Research 20(1):279‐288. [DOI] [PubMed] [Google Scholar]
- Campos D, Girgis M, Sanda M. 2022. Site‐specific glycosylation of SARS‐CoV‐2: Big challenges in mass spectrometry analysis. Proteomics 22(15‐16):2100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Cai Z, Xiao X, et al. 2021. The architecture of the SARS‐CoV‐2 RNA genome inside virion. Nature Communications 12(1):3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Pauthner M, Andrabi R, et al. 2018. Differential processing of HIV envelope glycans on the virus and soluble recombinant trimer. Nature Communications 9(1):3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo KHM, Lebkuchen A, Okai GG, et al. 2020. Establishing a mass spectrometry‐based system for rapid detection of SARS‐CoV‐2 in large clinical sample cohorts. Nature Communications 11(1):6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L, Gaieb Z, Goldsmith JA, et al. 2020. Beyond shielding: the roles of glycans in the SARS‐CoV‐2 spike protein. ACS Central Science 6(10):1722‐1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas‐Sanchez A, Romero‐Ramirez A, Hargreaves E, et al. 2022. Inhibition of protein N‐glycosylation blocks SARS‐CoV‐2 infection. mBio 13(1):e0371821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazares LH, Chaerkady R, Samuel Weng SH, et al. 2020. Development of a parallel reaction monitoring mass spectrometry assay for the detection of SARS‐CoV‐2 spike glycoprotein and nucleoprotein. Analytical Chemistry 92(20):13813‐13821. [DOI] [PubMed] [Google Scholar]
- Chan JF, Kok KH, Zhu Z, et al. 2020. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging Microbes & Infections 9(1):221‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CK, Sue SC, Yu TH, et al. 2006. Modular organization of SARS coronavirus nucleocapsid protein. Journal of Biomedical Science 13(1):59‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CK, Hou MH, Chang CF, Hsiao CD, Huang TH. 2014. The SARS coronavirus nucleocapsid protein‐‐forms and functions. Antiviral Research 103:39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Hackett WE, Zhong L, Wan XF, Zaia J. 2020. Measuring site‐specific glycosylation similarity between influenza a virus variants with statistical certainty. Molecular & Cellular Proteomics 19(9):1533‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Klein JA, Nalehua MR, Hackett WE, Zaia J. 2021. Data‐independent acquisition mass spectrometry for site‐specific glycoproteomics characterization of SARS‐CoV‐2 spike protein. Analytical and Bioanalytical Chemistry 413(29):7305‐7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Lee LY, Kawahara R, et al. 2019. Protein paucimannosylation is an enriched N‐glycosylation signature of human cancers. Proteomics 19(21‐22):e1900010. [DOI] [PubMed] [Google Scholar]
- Chavan S, Mangalaparthi KK, Singh S, et al. 2021. Mass spectrometric analysis of urine from COVID‐19 patients for detection of SARS‐CoV‐2 viral antigen and to study host response. Journal of Proteome Research 20(7):3404‐3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che XY, Hao W, Wang Y, et al. 2004. Nucleocapsid protein as early diagnostic marker for SARS. Emerging Infectious Diseases 10(11):1947‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu D, Robinson CV, Struwe WB. 2021. Native mass spectrometry meets glycomics: resolving structural detail and occupancy of glycans on intact glycoproteins. Analytical Chemistry 93(30):10435‐10443. [DOI] [PubMed] [Google Scholar]
- Chen Y, Huang S, Zhou L, Wang X, Yang H, Li W. 2022. Coronavirus disease 2019 (COVID‐19): emerging detection technologies and auxiliary analysis. Journal of Clinical Laboratory Analysis 36(1):e24152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivte P, LaCasse Z, Seethi VDR, et al. 2021. MALDI‐ToF protein profiling as a potential rapid diagnostic platform for COVID‐19. Journal of Mass Spectrometry and Advances in the Clinical Lab 21:31‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho BG, Gautam S, Peng W, Huang Y, Goli M, Mechref Y. 2021. Direct comparison of N‐glycans and their isomers derived from spike glycoprotein 1 of MERS‐CoV, SARS‐CoV‐1, and SARS‐CoV‐2. Journal of Proteome Research 20(9):4357‐4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S‐J, Woo H‐M, Kim K‐S, Oh J‐W, Jeong Y‐J. 2011. Novel system for detecting SARS coronavirus nucleocapsid protein using an ssDNA aptamer. Journal of Bioscience and Bioengineering 112(6):535‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipollo JF, Parsons LM. 2020. Glycomics and glycoproteomics of viruses: mass spectrometry applications and insights toward structure‐function relationships. Mass Spectrometry Reviews 39(4):371‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman VM, Landt O, Kaiser M, et al. 2020. Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Euro surveillance: bulletin Europeen sur les maladies transmissibles = European Communicable Disease Bulletin 25(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MM, Martin H, Estellon B, et al. 2022. Exploratory study on application of MALDI‐TOF‐MS to detect SARS‐CoV‐2 infection in human saliva. Journal of Clinical Medicine 11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispin M, Doores KJ. 2015. Targeting host‐derived glycans on enveloped viruses for antibody‐based vaccine design. Current Opinion in Virology 11:63‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li F, Shi ZL. 2019. Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology 17(3):181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood AA. 2021. Glycosylation, ligand binding sites and antigenic variations between membrane glycoprotein of COVID‐19 and related coronaviruses. Vacunas 22(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan CAM, Rottier PJM. Molecular interactions in the assembly of coronaviruses. In: Advances in Virus Research. 64. Academic Press; 2005:165‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio N, Walker CR, Turakhia Y, Lanfear R, Corbett‐Detig R, Goldman N. 2021. Mutation rates and selection on synonymous mutations in SARS‐CoV‐2. Genome Biology and Evolution 13(5):evab087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Garcia‐Knight MA, Khalid MM, et al. 2021. Transmission, infectivity, and neutralization of a spike L452R SARS‐CoV‐2 variant. Cell 184(13):3426‐3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deulofeu M, García‐Cuesta E, Peña‐Méndez EM, et al. 2021. Detection of SARS‐CoV‐2 Infection in Human Nasopharyngeal Samples by Combining MALDI‐TOF MS and Artificial Intelligence. Frontiers in Medicine 8:661358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds JN, Baker ES. 2019. Ion mobility spectrometry: fundamental concepts, instrumentation, applications, and the road ahead. Journal of the American Society for Mass Spectrometry 30(11):2185‐2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollman NL, Griffin JH, Downard KM. 2020. Detection, mapping, and proteotyping of SARS‐CoV‐2 coronavirus with high resolution mass spectrometry. ACS Infectious Diseases 6(12):3269‐3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C, Günther S, Preiser W, et al. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. The New England Journal of Medicine 348(20):1967‐1976. [DOI] [PubMed] [Google Scholar]
- Du L, He Y, Zhou Y, Liu S, Zheng BJ, Jiang S. 2009. The spike protein of SARS‐CoV—a target for vaccine and therapeutic development. Nature Reviews Microbiology 7(3):226‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, Wang H. 2020. The SARS‐CoV‐2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike‐based vaccine immunogens. Frontiers in Immunology 11:576622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duart G, García‐Murria MJ, Grau B, Acosta‐Cáceres JM, Martínez‐Gil L, Mingarro I. 2020. SARS‐CoV‐2 envelope protein topology in eukaryotic membranes. Open Biology 10(9):200209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Baba TJ, Lutomski CA, Kantsadi AL, et al. 2020. Allosteric inhibition of the SARS‐CoV‐2 main protease: insights from mass spectrometry based assays**. Angewandte Chemie 59(52):23544‐23548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldrid CFS, Allen JD, Newby ML, Crispin M. 2021. Suppression of O‐linked glycosylation of the SARS‐CoV‐2 spike by quaternary structural restraints. Analytical Chemistry 93(43):14392‐14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Frommer WB. 2015. Structure and function of SemiSWEET and SWEET sugar transporters. Trends in Biochemical Sciences 40(8):480‐486. [DOI] [PubMed] [Google Scholar]
- Fernandes ND, Downard KM, Caliendo AM. 2014. Incorporation of a proteotyping approach using mass spectrometry for surveillance of influenza virus in cell‐cultured strains. Journal of Clinical Microbiology 52(3):725‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire‐Paspuel B, Garcia‐Bereguiain MA. 2021. Clinical performance and analytical sensitivity of three SARS‐CoV‐2 nucleic acid diagnostic tests. The American Journal of Tropical Medicine and Hygiene 104(4):1516‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YZ, Wang SY, Zheng ZQ, et al. 2021. SARS‐CoV‐2 membrane glycoprotein M antagonizes the MAVS‐mediated innate antiviral response. Cellular & Molecular Immunology 18(3):613‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung TS, Liu DX. 2018. Post‐translational modifications of coronavirus proteins: roles and function. Future Virology 13(6):405‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Xu Y, Sun C, et al. 2021. A systematic review of asymptomatic infections with COVID‐19. Journal of Microbiology, Immunology, and Infection = Wei mian yu gan ran za zhi 54(1):12‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza KY, Silva AAR, Rosa JR, et al. 2021. Rapid screening of COVID‐19 directly from clinical nasopharyngeal swabs using the MasSpec Pen. Analytical Chemistry 93(37):12582‐12593. [DOI] [PubMed] [Google Scholar]
- Giles K, Ujma J, Wildgoose J, et al. 2019. A cyclic ion mobility‐mass spectrometry system. Analytical Chemistry 91(13):8564‐8573. [DOI] [PubMed] [Google Scholar]
- Gong Y, Qin S, Dai L, Tian Z. 2021. The glycosylation in SARS‐CoV‐2 and its receptor ACE2. Signal Transduction and Targeted Therapy 6(1):396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, et al. 2020. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature 583(7816):459‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia D, Grenga L, Gaillard JC, et al. 2020. Shortlisting SARS‐CoV‐2 peptides for targeted studies from experimental data‐dependent acquisition tandem mass spectrometry data. Proteomics 20(14):e2000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia D, Miotello G, Gallais F, et al. 2020. Proteotyping SARS‐CoV‐2 virus from nasopharyngeal swabs: a proof‐of‐concept focused on a 3 min mass spectrometry window. Journal of Proteome Research 19(11):4407‐4416. [DOI] [PubMed] [Google Scholar]
- Grant OC, Montgomery D, Ito K, Woods RJ. 2020. Analysis of the SARS‐CoV‐2 spike protein glycan shield reveals implications for immune recognition. Scientific Reports 10(1):14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco TM, Diner BA, Cristea IM. 2014. The impact of mass spectrometry‐based proteomics on fundamental discoveries in virology. Annual Review of Virology 1(1):581‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenga L, Gallais F, Pible O, et al. 2020. Shotgun proteomics analysis of SARS‐CoV‐2‐infected cells and how it can optimize whole viral particle antigen production for vaccines. Emerging Microbes & Infections 9(1):1712‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenga L, Armengaud J. 2021. Proteomics in the COVID‐19 Battlefield: first semester check‐up. Proteomics 21(1):2000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossegesse M, Hartkopf F, Nitsche A, Schaade L, Doellinger J, Muth T. 2020. Perspective on proteomics for virus detection in clinical samples. Journal of Proteome Research 19(11):4380‐4388. [DOI] [PubMed] [Google Scholar]
- Grunewald ME, Fehr AR, Athmer J, Perlman S. 2018. The coronavirus nucleocapsid protein is ADP‐ribosylated. Virology 517:62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gstöttner C, Zhang T, Resemann A, et al. 2021. Structural and functional characterization of SARS‐CoV‐2 RBD domains produced in mammalian cells. Analytical Chemistry 93(17):6839‐6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett WE, Zaia J. 2021. The need for community standards to enable accurate comparison of glycoproteomics algorithm performance. Molecules (Basel, Switzerland) 26(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardenbrook NJ, Zhang P. 2022. A structural view of the SARS‐CoV‐2 virus and its assembly. Current Opinion in Virology 52:123‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey DJ. 2018. Mass spectrometric analysis of glycosylated viral proteins. Expert Review of Proteomics 15(5):391‐412. [DOI] [PubMed] [Google Scholar]
- Harvey WT, Carabelli AM, Jackson B, et al. 2021. SARS‐CoV‐2 variants, spike mutations and immune escape. Nature Reviews Microbiology 19(7):409‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WA, Kamath K, Bozekowski J, et al. 2021. High‐resolution epitope mapping and characterization of SARS‐CoV‐2 antibodies in large cohorts of subjects with COVID‐19. Communications Biology 4(1):1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy B, Khan A, Metezai H, Blyth I, Asad H. 2021. The impact of false positive COVID‐19 results in an area of low prevalence. Clinical Medicine 21(1):e54‐e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman RM, Hume AJ, Goel RK, et al. 2020. Actionable cytopathogenic host responses of human alveolar type 2 cells to SARS‐CoV‐2. Molecular Cell 80(6):1104‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez MM, Banu R, Shrestha P, et al. 2021. RT‐PCR/MALDI‐TOF mass spectrometry‐based detection of SARS‐CoV‐2 in saliva specimens. Journal of Medical Virology 93(9):5481‐5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hober A, Tran‐Minh KH, Foley D, et al. 2021. Rapid and sensitive detection of SARS‐CoV‐2 infection using quantitative peptide enrichment LC‐MS analysis. eLife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Schroeder S, et al. 2020. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Lai YJ, Liao CC, et al. 2021. Targeting conserved N‐glycosylation blocks SARS‐CoV‐2 variant infection in vitro. EBioMedicine 74:103712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Liao HY, Chen X, et al. 2022. Vaccination with SARS‐CoV‐2 spike protein lacking glycan shields elicits enhanced protective responses in animal models. Science Translational Medicine 14(639):eabm0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Nie Y, Boyes B, Orlando R. 2016. Resolving isomeric glycopeptide glycoforms with hydrophilic interaction chromatography (HILIC). The Journal of Biomolecular Techniques 27(3):98‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yang C, Xu XF, Xu W, Liu SW. 2020. Structural and functional properties of SARS‐CoV‐2 spike protein: potential antivirus drug development for COVID‐19. Acta Pharmacologica Sinica 41(9):1141‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A, Hasan A, Nejadi Babadaei MM, et al. 2020. Targeting SARS‐CoV2 spike protein receptor binding domain by therapeutic antibodies. Biomedicine & Pharmacotherapy = Biomedecine & pharmacotherapie 130:110559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihling C, Tänzler D, Hagemann S, et al. 2020. Mass spectrometric identification of SARS‐CoV‐2 proteins from gargle solution samples of COVID‐19 patients. Journal of Proteome Research 19(11):4389‐4392. [DOI] [PubMed] [Google Scholar]
- Iles RK, Zmuidinaite R, Iles JK, Carnell G, Sampson A, Heeney JL. 2020. Development of a clinical MALDI‐ToF mass spectrometry assay for SARS‐CoV‐2: rational design and multi‐disciplinary team work. Diagnostics (Basel, Switzerland) 10(10):746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurinke C, van den Boom D, Cantor CR, Köster H. 2002. The use of MassARRAY technology for high throughput genotyping. Advances in Biochemical Engineering/Biotechnology 77:57‐74. [DOI] [PubMed] [Google Scholar]
- Kammila S, Das D, Bhatnagar PK, et al. 2008. A rapid point of care immunoswab assay for SARS‐CoV detection. Journal of Virological Methods 152(1):77‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z, Oton J, Qu K, et al. 2020. Structures and distributions of SARS‐CoV‐2 spike proteins on intact virions. Nature 588(7838):498‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri K, Klein JA, White MR, et al. 2016. Integrated omics and computational glycobiology reveal structural basis for influenza a virus glycan microheterogeneity and host interactions. Molecular & Cellular Proteomics 15(6):1895‐1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Lee J‐Y, Yang J‐S, Kim JW, Kim VN, Chang H. 2020. The architecture of SARS‐CoV‐2 transcriptome. Cell 181(4):914‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim J, Park S, et al. 2021. Production of SARS‐CoV‐2 N protein‐specific monoclonal antibody and its application in an ELISA‐based detection system and targeting the interaction between the spike C‐terminal domain and N protein. Frontiers in Microbiology 12:726231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Suzuki Y. 2012. Evidence for N‐glycan shielding of antigenic sites during evolution of human influenza A virus hemagglutinin. Journal of Virology 86(7):3446‐3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisman LS, Cobb BA. 2012. Infection, inflammation and host carbohydrates: a Glyco‐Evasion hypothesis. Glycobiology 22(8):1019‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S, Krishnan GP. 2021. N‐Glycosylation network construction and analysis to modify glycans on the spike (S) glycoprotein of SARS‐CoV‐2. Frontiers in Bioinformatics 1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokhin O, Li Y, Andonov A, et al. 2003. Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Molecular & Cellular Proteomics 2(5):346‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CW, Yang TJ, Chien YC, Yu PY, Hsu SD, Khoo KH. 2022. Distinct shifts in site‐specific glycosylation pattern of SARS‐CoV‐2 spike proteins associated with arising mutations in the D614G and Alpha variants. Glycobiology 32(1):60‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazari LC, Ghilardi FR, Rosa‐Fernandes L, et al. 2021. Prognostic accuracy of MALDI‐TOF mass spectrometric analysis of plasma in COVID‐19. Life Science Alliance 4(8):e202000946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454(7201):177‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenza MP, Oyenarte I, Diercks T, et al. 2020. Structural characterization of N‐linked glycans in the receptor binding domain of the SARS‐CoV‐2 spike protein and their interactions with human lectins. Angewandte Chemie (International ed in English) 59(52):23763‐23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FQ, Xiao H, Tam JP, Liu DX. 2005. Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome coronavirus. FEBS Letters 579(11):2387‐2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Tan C, Zeng J, et al. 2021. Analysis of viral load in different specimen types and serum antibody levels of COVID‐19 patients. Journal of Translational Medicine 19(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu D, Wang Y, Su W, Liu G, Dong W. 2021. The importance of glycans of viral and host proteins in enveloped virus Infection 12:638573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou TG, Adler FR, Cahill BC, et al. 2021. SARS‐CoV‐2 innate effector associations and viral load in early nasopharyngeal infection. Physiological Reports 9(4):e14761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhang W, Sun B, et al. 2020. Probing conformational hotspots for the recognition and intervention of protein complexes by lysine reactivity profiling. Chemical Science 12(4):1451‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Wang Y, Guo T. 2022. Multi‐omics in COVID‐19: seeing the unseen but overlooked in the clinic. Cell Reports Medicine 3(3):100580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R. 2018. Data‐independent acquisition‐based SWATH‐MS for quantitative proteomics: a tutorial. Molecular Systems Biology 14(8):e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukassen S, Chua RL, Trefzer T, et al. 2020. SARS‐CoV‐2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. The EMBO Journal 39(10):e105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutomski CA, El‐Baba TJ, Bolla JR, Robinson CV. 2021. Multiple roles of SARS‐CoV‐2 N protein facilitated by proteoform‐specific interactions with RNA, host proteins, and convalescent antibodies. JACS Au 1(8):1147‐1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud I, Garrett TJ. 2020. Mass spectrometry techniques in emerging pathogens studies: COVID‐19 perspectives. Journal of the American Society for Mass Spectrometry 31(10):2013‐2024. [DOI] [PubMed] [Google Scholar]
- Majumdar P, Niyogi S. 2020. ORF3a mutation associated with higher mortality rate in SARS‐CoV‐2 infection. Epidemiology and Infection 148:e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangalaparthi KK, Chavan S, Madugundu AK, et al. 2021. A SISCAPA‐based approach for detection of SARS‐CoV‐2 viral antigens from clinical samples. Clinical Proteomics 18(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M. 2019. The ever expanding scope of electrospray mass spectrometry—a 30 year journey. Nature Communications 10(1):3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters PS. 2006. The molecular biology of coronaviruses. Advances in Virus Research 66:193‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus A, Renuse S, Kemp J, et al. 2022. Targeted detection of SARS‐CoV‐2 nucleocapsid sequence variants by mass spectrometric analysis of tryptic peptides. Journal of Proteome Research 21(1):142‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhari R, Ruybal‐Pesántez S, Angrisano F, et al. 2021. SARS‐CoV‐2 multi‐antigen serology assay. Methods and Protocols 4(4), 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley J, Fraser C, Paraskeva E, et al. 2021. Optimal preparation of SARS‐CoV‐2 viral transport medium for culture. Virology Journal 18(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdipour AR, Hummer G. 2021. Dual nature of human ACE2 glycosylation in binding to SARS‐CoV‐2 spike. Proceedings of the National Academy of Sciences 118(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier F, Beck S, Grassl N, et al. 2015. Parallel accumulation–serial fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. Journal of Proteome Research 14(12):5378‐5387. [DOI] [PubMed] [Google Scholar]
- Meier F, Brunner A‐D, Frank M, et al. 2020. diaPASEF: parallel accumulation–serial fragmentation combined with data‐independent acquisition. Nature Methods 17(12):1229‐1236. [DOI] [PubMed] [Google Scholar]
- Meyer JG, Schilling B. 2017. Clinical applications of quantitative proteomics using targeted and untargeted data‐independent acquisition techniques. Expert Review of Proteomics 14(5):419‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerowitz EA, Richterman A, Gandhi RT, Sax PE. 2021. Transmission of SARS‐CoV‐2: a review of viral, host, and environmental factors. Annals of Internal Medicine 174(1):69‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolajczyk K, Kaczmarek R, Czerwinski M. 2020. How glycosylation affects glycosylation: the role of N‐glycans in glycosyltransferase activity. Glycobiology 30(12):941‐969. [DOI] [PubMed] [Google Scholar]
- Miller LM, Barnes LF, Raab SA, et al. 2021. Heterogeneity of glycan processing on trimeric SARS‐CoV‐2 spike protein revealed by charge detection mass spectrometry. Journal of the American Chemical Society 143(10):3959‐3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RE, Xu LL, Townsend SD. 2021. Prospecting human milk oligosaccharides as a defense against viral infections. ACS Infectious Diseases 7(2):254‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley MA, Hughes CJ, Juvvadi PR, et al. 2018. Scanning quadrupole data‐independent acquisition, part A: qualitative and quantitative characterization. Journal of Proteome Research 17(2):770‐779. [DOI] [PubMed] [Google Scholar]
- Mun DG, Vanderboom PM, Madugundu AK, et al. 2021. DIA‐based proteome profiling of nasopharyngeal swabs from COVID‐19 patients. Journal of Proteome Research 20(8):4165‐4175. [DOI] [PubMed] [Google Scholar]
- Nachtigall FM, Pereira A, Trofymchuk OS, Santos LS. 2020. Detection of SARS‐CoV‐2 in nasal swabs using MALDI‐MS. Nature Biotechnology 38(10):1168‐1173. [DOI] [PubMed] [Google Scholar]
- Nadler WM, Waidelich D, Kerner A, et al. 2017. MALDI versus ESI: the impact of the ion source on peptide identification. Journal of Proteome Research 16(3):1207‐1215. [DOI] [PubMed] [Google Scholar]
- Nagae M, Yamaguchi Y, Taniguchi N, Kizuka Y. 2020. 3D Structure and function of glycosyltransferases involved in N‐glycan maturation. International Journal of Molecular Sciences 21(2):437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi AAT, Fatima K, Mohammad T, et al. 2020. Insights into SARS‐CoV‐2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease 1866(10):165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ML, Tan SH, See EE, Ooi EE, Ling AE. 2003. Proliferative growth of SARS coronavirus in Vero E6 cells. The Journal of General Virology 84(Pt 12):3291‐3303. [DOI] [PubMed] [Google Scholar]
- Nie X, Qian L, Sun R, et al. 2021. Multi‐organ proteomic landscape of COVID‐19 autopsies. Cell 184(3):775‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto‐Torres JL, DeDiego ML, Verdiá‐Báguena C, et al. 2014. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathogens 10(5):e1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev EN, Indeykina MI, Brzhozovskiy AG, et al. 2020. Mass‐spectrometric detection of SARS‐CoV‐2 virus in scrapings of the epithelium of the nasopharynx of infected patients via nucleocapsid n protein. Journal of Proteome Research 19(11):4393‐4397. [DOI] [PubMed] [Google Scholar]
- Noy‐Porat T, Makdasi E, Alcalay R, et al. 2020. A panel of human neutralizing mAbs targeting SARS‐CoV‐2 spike at multiple epitopes. Nature Communications 11(1):4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra M, de Haan CA, de Groot RJ, Rottier PJ. 2006. Glycosylation of the severe acute respiratory syndrome coronavirus triple‐spanning membrane proteins 3a and M. Journal of Virology 80(5):2326‐2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmyer KA, Shishkova E, Miller IJ, et al. 2021. Large‐scale multi‐omic analysis of COVID‐19 severity. Cell Systems 12(1):23‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz M, Lorke DE, Kabbani N. 2021. A comprehensive guide to the pharmacologic regulation of angiotensin converting enzyme 2 (ACE2), the SARS‐CoV‐2 entry receptor. Pharmacology & Therapeutics 221:107750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi AK, Tripathi T. 2020. Can SARS‐CoV‐2 accumulate mutations in the S‐protein to increase pathogenicity? ACS Pharmacology & Translational Science 3(5):1023‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou AC, Mohsin I. 2020. The SARS‐CoV‐2 spike glycoprotein as a drug and vaccine target: structural insights into its complexes with ACE2 and antibodies. Cells 9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascarella G, Strumia A, Piliego C, et al. 2020. COVID‐19 diagnosis and management: a comprehensive review. Journal of Internal Medicine 288(2):192‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris JS, Yuen KY, Osterhaus AD, Stöhr K. 2003. The severe acute respiratory syndrome. The New England Journal of Medicine 349(25):2431‐2441. [DOI] [PubMed] [Google Scholar]
- Peng L, Liu J, Xu W, et al. 2020. SARS‐CoV‐2 can be detected in urine, blood, anal swabs, and oropharyngeal swabs specimens. Journal of Medical Virology 92(9):1676‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto G, Illiano A, Ferrucci V, et al. 2021. Identification of SARS‐CoV‐2 proteins from nasopharyngeal swabs probed by multiple reaction monitoring tandem mass spectrometry. ACS Omega 6(50):34945‐34953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt MR, Hang HC, Ten Hagen KG, et al. 2004. Deconvoluting the functions of polypeptide N‐α‐acetylgalactosaminyltransferase family members by glycopeptide substrate profiling. Chemistry & Biology 11(7):1009‐1016. [DOI] [PubMed] [Google Scholar]
- Preianò M, Correnti S, Pelaia C, Savino R, Terracciano R. 2021. MALDI MS‐based investigations for SARS‐CoV‐2 detection. BioChem 1(3):250‐278. [Google Scholar]
- Radbel J, Jagpal S, Roy J, et al. 2020. Detection of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is comparable in clinical samples preserved in saline or viral transport medium. The Journal of Molecular Diagnostics 22(7):871‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo N, Zaldívar‐López S, Garrido JJ, Montoya M. 2021. SARS‐CoV‐2 accessory proteins in viral pathogenesis: knowns and unknowns. Frontiers in Immunology 12:708264. [DOI] [PMC free article] [PubMed]
- Reily C, Stewart TJ, Renfrow MB, Novak J. 2019. Glycosylation in health and disease. Nature Reviews Nephrology 15(6):346‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renuse S, Vanderboom PM, Maus AD, et al. 2021. A mass spectrometry‐based targeted assay for detection of SARS‐CoV‐2 antigen from clinical specimens. EBioMedicine 69:103465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley TP, Chou H‐T, Hu R, et al. 2021. Enhancing the prefusion conformational stability of SARS‐CoV‐2 spike protein through structure‐guided design. Frontiers in Immunology 12:660198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini JM, Esko JD. 2015. Glycosyltransferases and glycan‐processing enzymes. In: Varki A, Cummings RD, Esko JD, et al., eds. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- Ritchie G, Harvey DJ, Feldmann F, et al. 2010. Identification of N‐linked carbohydrates from severe acute respiratory syndrome (SARS) spike glycoprotein. Virology 399(2):257‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie G, Harvey DJ, Stroeher U, et al. 2010. Identification of N‐glycans from Ebola virus glycoproteins by matrix‐assisted laser desorption/ionisation time‐of‐flight and negative ion electrospray tandem mass spectrometry. Rapid Communications in Mass Spectrometry 24(5):571‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera B, Leyva A, Portela MM, et al. 2020. Quantitative proteomic dataset from oro‐ and naso‐pharyngeal swabs used for COVID‐19 diagnosis: detection of viral proteins and host's biological processes altered by the infection. Data Brief 32:106121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DS, Mann M, Melby JA, et al. 2021. Structural O‐glycoform heterogeneity of the SARS‐CoV‐2 spike protein receptor‐binding domain revealed by top‐down mass spectrometry. Journal of the American Chemical Society 143(31):12014‐12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca MF, Zintgraff JC, Dattero ME, et al. 2020. A combined approach of MALDI‐TOF mass spectrometry and multivariate analysis as a potential tool for the detection of SARS‐CoV‐2 virus in nasopharyngeal swabs. Journal of Virological Methods 286:113991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo A, Iacovelli F, Falconi M. 2020. Targeting the SARS‐CoV‐2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Research 286:198068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbalm KE, Tiemeyer M, Wells L, Aoki K, Zhao P. 2020. Glycomics‐informed glycoproteomic analysis of site‐specific glycosylation for SARS‐CoV‐2 spike protein. STAR Protocols 1(3):100214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy V, Fischinger S, Atyeo C, et al. 2020. SARS‐CoV‐2‐specific ELISA development. Journal of Immunological Methods 484‐485:112832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruszkiewicz DM, Sanders D, O′Brien R, et al. 2020. Diagnosis of COVID‐19 by analysis of breath with gas chromatography‐ion mobility spectrometry—a feasibility study. EClinicalMedicine 29:100609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybicka M, Miłosz E, Bielawski KP. 2021. Superiority of MALDI‐TOF mass spectrometry over real‐time PCR for SARS‐CoV‐2 RNA detection. Viruses 13(5):730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadi J, Oueslati S, Bellanger L, et al. 2021. Quantitative assessment of SARS‐CoV‐2 virus in nasopharyngeal swabs stored in transport medium by a straightforward LC‐MS/MS assay targeting nucleocapsid, membrane, and spike proteins. Journal of Proteome Research 20(2):1434‐1443. [DOI] [PubMed] [Google Scholar]
- Samrat SK, Tharappel AM, Li Z, Li H. 2020. Prospect of SARS‐CoV‐2 spike protein: potential role in vaccine and therapeutic development. Virus Research 288:198141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda M, Morrison L, Goldman R. 2021. N‐ and O‐Glycosylation of the SARS‐CoV‐2 spike protein. Analytical Chemistry 93(4):2003‐2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans M, Zhang J, Lin JQ, et al. 2019. Performance of the MasSpec Pen for rapid diagnosis of ovarian cancer. Clinical Chemistry 65(5):674‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeman D, Fielding BC. 2019. Coronavirus envelope protein: current knowledge. Virology Journal 16(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster O, Zvi A, Rosen O, et al. 2021. Specific and rapid SARS‐CoV‐2 identification based on LC‐MS/MS analysis. ACS Omega 6(5):3525‐3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster O, Atiya‐Nasagi Y, Rosen O, et al. 2022. Coupling immuno‐magnetic capture with LC‐MS/MS(MRM) as a sensitive, reliable, and specific assay for SARS‐CoV‐2 identification from clinical samples. Analytical and Bioanalytical Chemistry 414(5):1949‐1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Bar‐On YM, Gleizer S, et al. 2021. The total number and mass of SARS‐CoV‐2 virions. Proceedings of the National Academy of Sciences 118(25):e2024815118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A, Supekar NT, Gleinich AS, Azadi P. 2020. Deducing the N‐ and O‐glycosylation profile of the spike protein of novel coronavirus SARS‐CoV‐2. Glycobiology 30(12):981‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A, Archer‐Hartmann S, Supekar NT, Gleinich AS, Heiss C, Azadi P. 2021. Comprehensive characterization of N‐ and O‐glycosylation of SARS‐CoV‐2 human receptor angiotensin converting enzyme 2. Glycobiology 31(4):410‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A, Pepi LE, Rouhani DS, Heiss C, Azadi P. 2021. Glycosylation of SARS‐CoV‐2: structural and functional insights. Analytical and Bioanalytical Chemistry 413(29):7179‐7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Ye G, Shi K, et al. 2020. Structural basis of receptor recognition by SARS‐CoV‐2. Nature 581(7807):221‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Yi X, Sun Y, et al. 2020. Proteomic and metabolomic characterization of COVID‐19 patient sera. Cell 182(1):59‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Y, Vinjamuri A, Alvarez MRS, et al. 2022. Host cell glycocalyx remodeling reveals SARS‐CoV‐2 spike protein glycomic binding sites. Frontiers in Molecular Biosciences 9:799703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Chakraborty R, Marwal R, et al. 2020. A rapid and sensitive method to detect SARS‐CoV‐2 virus using targeted‐mass spectrometry. Journal of Proteins and Proteomics 11(3):159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KP, Cheng A, Chopelas A, et al. 2020. Large‐scale, in‐house production of viral transport media to support SARS‐CoV‐2 PCR testing in a multihospital health care network during the COVID‐19 pandemic. Journal of Clinical Microbiology 58(8):e00913‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Kelleher NL. 2013. Proteoform: a single term describing protein complexity. Nature Methods 10(3):186‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Agar JN, Chamot‐Rooke J, et al. 2021. The Human Proteoform Project: defining the human proteome. Science Advances 7(46):eabk0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits VAJ, Hernández‐Carralero E, Paz‐Cabrera MC, et al. 2021. The nucleocapsid protein triggers the main humoral immune response in COVID‐19 patients. Biochemical and Biophysical Research Communications 543:45‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SoRelle JA, Patel K, Filkins L, Park JY. 2020. Mass spectrometry for COVID‐19. Clinical Chemistry 66(11):1367‐1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spraggins JM, Rizzo DG, Moore JL, et al. 2015. MALDI FTICR IMS of intact proteins: using mass accuracy to link protein images with proteomics data. Journal of the American Society for Mass Spectrometry 26(6):974‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl E, Kessler HH, Mustafa HG, et al. 2021. Alternative detection of SARS‐CoV‐2 RNA by a new assay based on mass spectrometry. Clinical Chemistry and Laboratory Medicine 59(12):1998‐2002. [DOI] [PubMed] [Google Scholar]
- Steppert C, Steppert I, Sterlacci W, Bollinger T. 2021. Rapid detection of SARS‐CoV‐2 infection by multicapillary column coupled ion mobility spectrometry (MCC‐IMS) of breath. A proof of concept study. Journal of Breath Research 15(2):27105. [DOI] [PubMed] [Google Scholar]
- Struwe WB, Chertova E, Allen JD, et al. 2018. Site‐specific glycosylation of virion‐derived HIV‐1 Env Is mimicked by a soluble trimeric immunogen. Cell Reports 24(8):1958‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struwe WB, Robinson CV. 2019. Relating glycoprotein structural heterogeneity to function—insights from native mass spectrometry. Current Opinion in Structural Biology 58:241‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studdert DM, Hall MA. 2020. Disease control, civil liberties, and mass testing—calibrating restrictions during the Covid‐19 pandemic. The New England Journal of Medicine 383(2):102‐104. [DOI] [PubMed] [Google Scholar]
- Stukalov A, Girault V, Grass V, et al. 2021. Multilevel proteomics reveals host perturbations by SARS‐CoV‐2 and SARS‐CoV. Nature 594(7862):246‐252. [DOI] [PubMed] [Google Scholar]
- Sun Z, Ren K, Zhang X, et al. 2021. Mass spectrometry analysis of newly emerging coronavirus HCoV‐19 spike protein and human ACE2 reveals camouflaging glycans and unique post‐translational modifications. Engineering (Beijing, China) 7(10):1441‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Zheng X, Ji F, et al. 2021. Mass spectrometry analysis of SARS‐CoV‐2 nucleocapsid protein reveals camouflaging glycans and unique post‐translational modifications. Infectious Microbes & Diseases 3(3):149‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sungnak W, Huang N, Bécavin C, et al. 2020. SARS‐CoV‐2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nature Medicine 26(5):681‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supekar NT, Shajahan A, Gleinich AS, et al. 2021. Variable posttranslational modifications of severe acute respiratory syndrome coronavirus 2 nucleocapsid protein. Glycobiology 31(9):1080‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swearingen KE, Moritz RL. 2012. High‐field asymmetric waveform ion mobility spectrometry for mass spectrometry‐based proteomics. Expert Review of Proteomics 9(5):505‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed AM, Taha TY, Tabata T, et al. 2021. Rapid assessment of SARS‐CoV‐2‐evolved variants using virus‐like particles. Science (New York, NY) 374(6575):1626‐1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Furuchi M, Kamimoto A, Honda K, Matsumura H, Kobayashi R. 2020. Saliva‐based PCR tests for SARS‐CoV‐2 detection. Journal of Oral Science 62(3):350‐351. [DOI] [PubMed] [Google Scholar]
- Tamara S, den Boer MA, Heck AJR. 2021. High‐resolution native mass spectrometry. Chemical Reviews 122(8):7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YJ, Teng E, Shen S, et al. 2004. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. Journal of Virology 78(13):6723‐6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper ML. 2006. Emerging viral diseases and infectious disease risks. Haemophilia 12(Suppl 1):3‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano R, Preianò M, Fregola A, Pelaia C, Montalcini T, Savino R. 2021. Mapping the SARS‐CoV‐2‐host protein‐protein interactome by affinity purification mass spectrometry and proximity‐dependent biotin labeling: a rational and straightforward route to discover host‐directed anti‐SARS‐CoV‐2 therapeutics. International Journal of Molecular Sciences 22(2):532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaysen‐Andersen M, Venkatakrishnan V, Loke I, et al. 2015. Human neutrophils secrete bioactive paucimannosidic proteins from azurophilic granules into pathogen‐infected sputum. The Journal of biological chemistry 290(14):8789‐8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. 2020. The structure of the membrane protein of SARS‐CoV‐2 resembles the sugar transporter SemiSWEET. Pathogens & Immunity 5(1):342‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Li D, Zhang N, et al. 2021. O‐glycosylation pattern of the SARS‐CoV‐2 spike protein reveals an “O‐Follow‐N” rule. Cell Research 31(10):1123‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjondro HC, Loke I, Chatterjee S, Thaysen‐Andersen M. 2019. Human protein paucimannosylation: cues from the eukaryotic kingdoms. Biological Reviews of the Cambridge Philosophical Society 94(6):2068‐2100. [DOI] [PubMed] [Google Scholar]
- Tran NK, Albahra S, Pham TN, et al. 2020. Novel application of an automated‐machine learning development tool for predicting burn sepsis: proof of concept. Scientific Reports 10(1):12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NK, Howard T, Walsh R, et al. 2021. Novel application of automated machine learning with MALDI‐TOF‐MS for rapid high‐throughput screening of COVID‐19: a proof of concept. Scientific Reports 11(1):8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom D, Wjst M, Everts RE. 2013. MALDI‐TOF mass spectrometry. Methods in Molecular Biology (Clifton, NJ) 1015:71‐85. [DOI] [PubMed] [Google Scholar]
- Van den Steen P, Rudd PM, Dwek RA, Opdenakker G. 1998. Concepts and principles of O‐linked glycosylation. Critical Reviews in Biochemistry and Molecular Biology 33(3):151‐208. [DOI] [PubMed] [Google Scholar]
- van der Burgt YEM, Siliakus KM, Cobbaert CM, Ruhaak LR. 2020. HILIC–MRM–MS for linkage‐specific separation of sialylated glycopeptides to quantify prostate‐specific antigen proteoforms. Journal of Proteome Research 19(7):2708‐2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Puyvelde B, Dhaenens M. 2021. Add mass spectrometry to the pandemic toolbox. eLife 10:e75471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Puyvelde B, Van Uytfanghe K, Tytgat O, et al. 2021. Cov‐MS: a community‐based template assay for mass‐spectrometry‐based protein detection in SARS‐CoV‐2 patients. JACS Au 1(6):750‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderboom PM, Mun DG, Madugundu AK, et al. 2021. Proteomic signature of host response to SARS‐CoV‐2 infection in the nasopharynx. Molecular & Cellular Proteomics 20:100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Kornfeld S. 2015. Historical background and overview. In: Varki A, Cummings RD, Esko JD, et al., eds. Essentials of glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH, editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015–2017. [PubMed]
- Varki A. 2017. Biological roles of glycans. Glycobiology 27(1):3‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilopoulou CG, Sulek K, Brunner A‐D, et al. 2020. Trapped ion mobility spectrometry and PASEF enable in‐depth lipidomics from minimal sample amounts. Nature Communications 11(1):331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AC, Tortorici MA, Frenz B, et al. 2016. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo‐electron microscopy. Nature Structural & Molecular Biology 23(10):899‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AC, Xiong X, Park YJ, et al. 2019. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 176(5):1026‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AC, Park Y‐J, Tortorici MA, Wall A, McGuire AT, Veesler D. 2020. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 181(2):281‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandernoth P, Kriegsmann K, Groh‐Mohanu C, et al. 2020. Detection of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) by mass spectrometry. Viruses 12(8):849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xu H, Lin S, et al. 2020. GPS 5.0: an update on the prediction of kinase‐specific phosphorylation sites in proteins. Genomics, Proteomics & Bioinformatics 18(1):72‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Baudys J, Bundy JL, Solano M, Keppel T, Barr JR. 2020. Comprehensive analysis of the glycan complement of SARS‐CoV‐2 spike proteins using signature ions‐triggered electron‐transfer/higher‐energy collisional dissociation (EThcD) mass spectrometry. Analytical Chemistry 92(21):14730‐14739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu Y, Gao R, et al. 2020. Detection of SARS‐CoV‐2 in different types of clinical specimens. Jama 323(18):1843‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu Z, Hu W, Hao P, Yang S. 2021. Impact of expressing cells on glycosylation and glycan of the SARS‐CoV‐2 spike glycoprotein. ACS Omega 6(24):15988‐15999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Amaya M, Addetia A, et al. 2022. Architecture and antigenicity of the Nipah virus attachment glycoprotein. Science (New York, NY) 375(6587):1373‐1378. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Bowden TA, Wilson IA, Crispin M. 2019. Exploitation of glycosylation in enveloped virus pathobiology. Biochimica et Biophysica Acta General Subjects 1863(10):1480‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. 2020. Site‐specific glycan analysis of the SARS‐CoV‐2 spike. Science (New York, NY) 369(6501):330‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Berndsen ZT, Raghwani J, et al. 2020. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nature Communications 11(1):2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker JR, Zhao L, Abbatiello SE, et al. 2011. Evaluation of large scale quantitative proteomic assay development using peptide affinity‐based mass spectrometry. Molecular & Cellular Proteomics 10(4):M110.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JW, Bilbao A, Wang J, et al. 2022. Online hydrophilic interaction chromatography (HILIC) enhanced top‐down mass spectrometry characterization of the SARS‐CoV‐2 spike receptor‐binding domain. Analytical Chemistry 94(15):5909‐5917. [DOI] [PubMed] [Google Scholar]
- Wörner TP, Shamorkina TM, Snijder J, Heck AJR. 2021. Mass spectrometry‐based structural virology. Analytical Chemistry 93(1):620‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D, Wang N, Corbett KS, et al. 2020. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science (New York, NY) 367(6483):1260‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S, Lan Q, Su S, et al. 2020. The role of furin cleavage site in SARS‐CoV‐2 spike protein‐mediated membrane fusion in the presence or absence of trypsin. Signal Transduction and Targeted Therapy 5(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Chen P, Wang J, et al. 2020. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Science China Life Sciences 63(3):457‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Chaudhary JK, Jain N, et al. 2021. Role of structural and non‐structural proteins and therapeutic targets of SARS‐CoV‐2 for COVID‐19. Cells 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Yi J, Huang C, et al. 2021. Rapid detection of COVID‐19 using MALDI‐TOF‐based serum peptidome profiling. Analytical Chemistry 93(11):4782‐4787. [DOI] [PubMed] [Google Scholar]
- Yang J, Wang W, Chen Z, et al. 2020. A vaccine targeting the RBD of the S protein of SARS‐CoV‐2 induces protective immunity. Nature 586(7830):572‐577. [DOI] [PubMed] [Google Scholar]
- Yang Q, Hughes TA, Kelkar A, et al. 2020. Inhibition of SARS‐CoV‐2 viral entry upon blocking N‐ and O‐glycan elaboration. eLife 9:e61552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Du Y, Kaltashov IA. 2020. The utility of native MS for understanding the mechanism of action of repurposed therapeutics in COVID‐19: heparin as a disruptor of the SARS‐CoV‐2 interaction with its host cell receptor. Analytical Chemistry 92(16):10930‐10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ivanov DG, Kaltashov IA. 2021. The challenge of structural heterogeneity in the native mass spectrometry studies of the SARS‐CoV‐2 spike protein interactions with its host cell‐surface receptor. Analytical and Bioanalytical Chemistry 413(29):7205‐7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Song Y, Chen Y, et al. 2020. Molecular architecture of the SARS‐CoV‐2 virus. Cell 183(3):730‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz Gulec E, Cesur NP, Yesilyurt Fazlioğlu G, Kazezoğlu C. 2021. Effect of different storage conditions on COVID‐19 RT‐PCR results. Journal of Medical Virology 93(12):6575‐6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinari T, Hayashi K, Hirose S, et al. 2022. Matrix‐assisted laser desorption and ionization time‐of‐flight mass spectrometry analysis for the direct detection of SARS‐CoV‐2 in nasopharyngeal swabs. Analytical Chemistry 94(10):4218‐4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CJ, Chen YC, Hsiao CH, et al. 2004. Identification of a novel protein 3a from severe acute respiratory syndrome coronavirus. FEBS Letters 565(1‐3):111‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Cao D, Zhang Y, et al. 2017. Cryo‐EM structures of MERS‐CoV and SARS‐CoV spike glycoproteins reveal the dynamic receptor binding domains. Nature Communications 8:15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. The New England Journal of Medicine 367(19):1814‐1820. [DOI] [PubMed] [Google Scholar]
- Zecha J, Lee CY, Bayer FP, et al. 2020. Data, reagents, assays and merits of proteomics for SARS‐CoV‐2 research and testing. Molecular & Cellular Proteomics 19(9):1503‐1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Liu G, Ma H, et al. 2020. Biochemical characterization of SARS‐CoV‐2 nucleocapsid protein. Biochemical and Biophysical Research Communications 527(3):618‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B‐Z, Hu Y‐F, Chen L‐L, et al. 2020. Mining of epitopes on spike protein of SARS‐CoV‐2 from COVID‐19 patients. Cell Research 30(8):702‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ge W, Ruan G, Cai X, Guo T. 2020. Data‐independent acquisition mass spectrometry‐based proteomics and software tools: a glimpse in 2020. Proteomics 20(17‐18):e1900276. [DOI] [PubMed] [Google Scholar]
- Zhang H, Rostami MR, Leopold PL, et al. 2020. Expression of the SARS‐CoV‐2 ACE2 receptor in the human airway epithelium. American Journal of Respiratory and Critical Care Medicine 202(2):219‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Rector J, Lin JQ, et al. 2017. Nondestructive tissue analysis for ex vivo and in vivo cancer diagnosis using a handheld mass spectrometry system. Science Translational Medicine 9(406):eaan3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jackson CB, Mou H, et al. 2020. SARS‐CoV‐2 spike‐protein D614G mutation increases virion spike density and infectivity. Nature Communications 11(1):6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Qiao S, Yu J, et al. 2021. Bat and pangolin coronavirus spike glycoprotein structures provide insights into SARS‐CoV‐2 evolution. Nature Communications 12(1):1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Go EP, Ding H, et al. 2022. Analysis of glycosylation and disulfide bonding of wild‐type SARS‐CoV‐2 spike glycoprotein. Journal of Virology 96(3):e0162621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Liang Q, He X, et al. 2022. Loss of spike N370 glycosylation as an important evolutionary event for the enhanced infectivity of SARS‐CoV‐2. Cell Research 32(3):315‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kutateladze TG. 2020. Molecular structure analyses suggest strategies to therapeutically target SARS‐CoV‐2. Nature Communications 11(1):2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhao W, Mao Y, et al. 2021a. Site‐specific N‐glycosylation characterization of recombinant SARS‐CoV‐2 spike proteins. Molecular & Cellular Proteomics 20:100058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhao W, Mao Y, et al. 2021b. O‐Glycosylation landscapes of SARS‐CoV‐2 spike proteins. Frontiers in Chemistry 9:689521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Praissman JL, Grant OC, et al. 2020. Virus‐receptor interactions of glycosylated SARS‐CoV‐2 spike and human ACE2 receptor. Cell Host & Microbe 28(4):586‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Chen H, Wang H. 2021. Glycans of SARS‐CoV‐2 spike protein in virus infection and antibody production. Frontiers in Molecular Biosciences 8:629873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong NS, Zheng BJ, Li YM, et al. 2003. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet (London, England) 362(9393):1353‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Tian X, Qi R, Peng C, Zhang W. 2021. Identification of 22 N‐glycosites on spike glycoprotein of SARS‐CoV‐2 and accessible surface glycopeptide motifs: implications for vaccination and antibody therapeutics. Glycobiology 31(1):69‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]