

Abstract
The nature and dynamics of mutations associated with the emergence, spread, and vanishing of SARS‐CoV‐2 variants causing successive waves are complex. We determined the kinetics of the most common French variant (“Marseille‐4”) for 10 months since its onset in July 2020. Here, we analyzed and classified into subvariants and lineages 7453 genomes obtained by next‐generation sequencing. We identified two subvariants, Marseille‐4A, which contains 22 different lineages of at least 50 genomes, and Marseille‐4B. Their average lifetime was 4.1 ± 1.4 months, during which 4.1 ± 2.6 mutations accumulated. Growth rate was 0.079 ± 0.045, varying from 0.010 to 0.173. Most of the lineages exhibited a bell‐shaped distribution. Several beneficial mutations at unpredicted sites initiated a new outbreak, while the accumulation of other mutations resulted in more viral heterogenicity, increased diversity and vanishing of the lineages. Marseille‐4B emerged when the other Marseille‐4 lineages vanished. Its ORF8 gene was knocked out by a stop codon, as reported in SARS‐CoV‐2 of mink and in the Alpha variant. This subvariant was associated with increased hospitalization and death rates, suggesting that ORF8 is a nonvirulence gene. We speculate that the observed heterogenicity of a lineage may predict the end of the outbreak.
Keywords: epidemic, Marseille‐4, ORF8, Pangolin B.1.1.160, SARS‐CoV‐2, variants
1. INTRODUCTION
The shape of epidemic curves of acute infectious diseases is the subject of several hypotheses and interpretations. The occurrence of successive waves of SARS‐CoV‐2 infections during the current pandemic was linked to the emergence of viral variants through various pathways of molecular changes, 1 , 2 , 3 , 4 , 5 , 6 while possible causes of the extinction of epidemics are viral load decrease, 7 herd immunity 8 (as hypothesized for influenza viruses), 9 the implementation of treatment or vaccination, 10 or outcompetition by another viral variant or lineage with higher fitness and transmissibility. 3 However, the factors and mechanisms involved in the rise and fall of SARS‐CoV‐2 variants have not been elucidated. In France, as of 20/07/2022, the number of SARS‐CoV‐2 cases was 33 375 449, with 152 019 deaths recorded (https://coronavirus.jhu.edu/map.html) 11 (https://coronavirus.jhu.edu/map.html, accessed 20/07/2022). We identified in July 2020 at the University Hospital Institute (IHU) Méditerranée Infection, Marseille, Southern France (which generated about 20% of the genomes deposited in GISAID (https://www.gisaid.org/) 12 by France as on December 16, 2021), a new SARS‐CoV‐2 variant, named Marseille‐4 (later classified as lineage 20A.EU2 and B.1.160 in Nextstrain 13 and Pangolin 14 classifications). 15 , 16 This variant is characterized by 20 mutations, including 13 specific compared with the Wuhan‐Hu‐1 isolate. 4 , 5 , 15 , 16 Seven mutations are nonsynonymous, including one in the spike glycoprotein (S477N). The Marseille‐4/B.1.160 variant was among the first variants that were detected during summer of 2020 and were described by us as early as during early September. 16 We defined it as a variant as its genomes harbored a set of more than 5 mutations absent in any other viral genomes and they were more than 30 in number. 5 This variant was the predominant one in France and our geographical area from August 2020 until January 2021 (Figure 1A,B). It was also retrospectively revealed as one of the major SARS‐CoV‐2 lineages that emerged in 2020. 1 , 4 In addition, the chronology of its incidence in France and genetic evidence strongly suggest that it originated from mink. 15 We analyzed the epidemiological source and features of this Marseille‐4 variant and accumulated genetic data through extensive SARS‐CoV‐2 genomic surveillance by next‐generation sequencing (NGS) from its onset until its disappearance 10 months later in April of 2021. Thus, we could study here the nature and dynamics of mutations associated with its emergence, spread, and vanishing.
Figure 1.
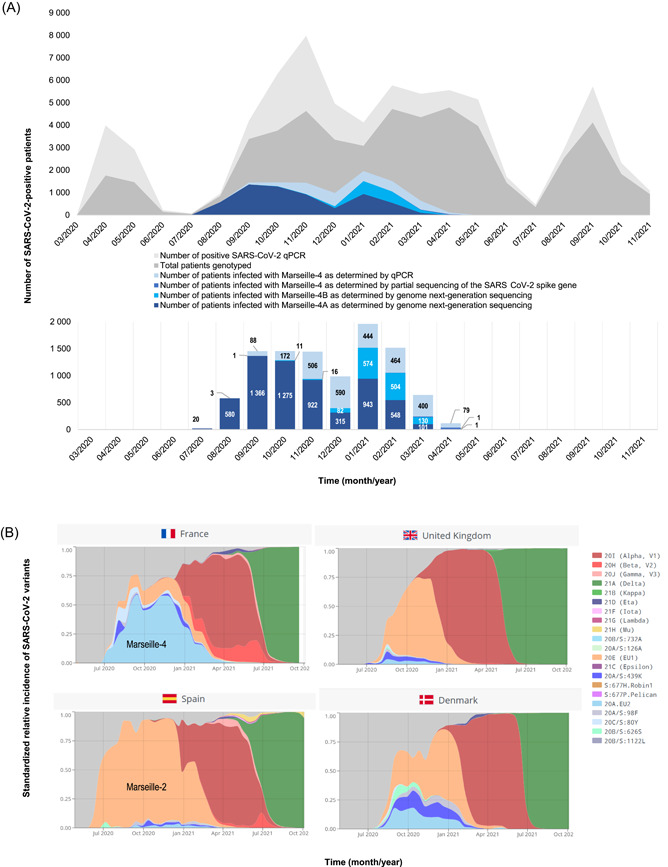
Weekly incidence of SARS‐CoV‐2 diagnoses at the IHU Méditerranée Infection, Marseille, France and incidence of the SARS‐CoV‐2 Marseille‐4 variant (A) and spread of the Marseille‐4 variant in France and three additional European countries (B). (B) Adapted from screenshots from the CoVariants website (https://covariants.org/). 17 IHU, University Hospital Institute; MRS, Marseille.
2. RESULTS
2.1. Kinetics of the Marseille‐4 variant infection
The identification of the Marseille‐4 variant in late July 2020 15 , 16 (Figure 1A) led to the design of a specific real‐time reverse transcription‐polymerase chain reaction (qPCR) assay to evaluate its incidence and 9616 cases were identified. It was the third most commonly diagnosed variant at our institution after variants Alpha/B.1.160 (n = 10 139) and Delta/B.1.617.2 (11 060). By contrast, it was rarely observed during this period in the United Kingdom or Spain, where the Marseille‐2/B.1.177 variant predominated (Figure 1B). 18 The curve of Marseille‐4 included several peaks, with a first in September (Week 37), a second in October (Week 43), and a third in January 2021 (Week 2) before the variant vanished in April 2021 (Figure 1A).
2.2. Marseille‐4 subvariants and lineages
NGS was performed when cycle threshold values (C t) of qPCR used for SARS‐CoV‐2 diagnosis was <30 and a genome was obtained from 7453 patients. Phylogenetic analysis and comparative genomics identified two subvariants, that is, new variants issued from a circulating variant (Marseille‐4A and Marseille‐4B). The Marseille‐4A subvariant contained 22 different lineages of at least 50 SARS‐CoV‐2 genomes harboring one to four hallmark nucleotide changes (Figure 2A; Supporting Information: Tables S1–S3). Interestingly, the single sequence reported from an infected mink farm in France was a Marseille‐4A variant. 15 The growth rate varied throughout the Marseille‐4 epidemic for each subvariant and lineage (Figure 2B–G; Supporting Information: Figure S1, Tables S2 and S3). It was 0.079 ± 0.045 on average and varied from 0.010 for the Marseille‐4A.17 lineage to 0.173 for the Marseille‐4A.15 lineage. Thus, we observed heterogeneous growth rates for Marseille‐4 subvariants and lineages, as indicated by a very high ratio of true heterogeneity to the total observed variation (I2 = 99%; p < 0.05; Supporting Information: Figure S2).
Figure 2.

Phylogenetic tree of SARS‐CoV‐2 Marseille‐4 genome sequences obtained from patients diagnosed with SARS‐CoV‐2 infection at IHU Méditerranée Infection (A) and time series of the number of additional mutations relatively to the root of subvariant or lineage along with the loess (locally estimated scatterplot smoothing) regression curve and its 95% confidence interval for the Marseille‐4 variant overall (B), for four Marseille‐4A lineages (C–F) and for the Marseille‐4B lineage (G). (A) The phylogenetic tree is adapted from a screenshot of the nextclade web application (https://clades.nextstrain.org). 13 IHU, University Hospital Institute.
2.3. ORF8 gene inactivation in the Marseille‐4B subvariant
The Marseille‐4B subvariant spread from September 2020 to March 2021, with an accepted homogeneous gamma distribution of the serial interval used to calculate the growth rate 19 (Figure 1A; Supporting Information: Tables S2 and S3). It expanded significantly in December 2020 during the vanishing of the Marseille‐4A lineages. It accumulated a mean number of 4.1 ± 1.6 mutations (range, 0–15; n = 1319 genomes). Interestingly, the ORF8 gene was knocked out by a stop codon (E64*) at the origin of the Marseille‐4B variant. This gene may play a key role in immune modulation and increases virus multiplication. 20 Its inactivation by a stop codon has been reported in SARS‐CoV‐2 of mink and in all genomes of the Alpha variant. 21
The dimeric structure of the ORF8 protein (wild‐type and mutant forms) is shown in Figure 3A–C. The dimer is stabilized by a covalent bond (a disulfide bridge) between two cysteine residues in the N‐terminal region of each subunit. 25 Mutation A65S harbored by lineage Marseille‐4A.02 does not induce major structural or electrostatic surface potential alterations (Figure 3B). Both the initial A65 and mutant 65S residues are well exposed to the solvent and occupy approximately the same volume. By contrast, the truncated 18‐63 form leads to a different protein, despite its sequence identity with the 18‐63 region of the initial ORF8 protein chains (Figure 3C). The electrostatic surface potential of the truncated protein is also significantly affected, with an increase in both neutral and electronegative surface areas. These structural data suggest a total loss of ORF8 function for the truncated 18‐63 form. Finally, mutation H17Y harbored by some genomes of the Marseille‐4A.02 lineage affects the C‐terminal residue of the signal peptide, so it is not present in the mature form of ORF8, as shown in Figure 3A–C.
Figure 3.
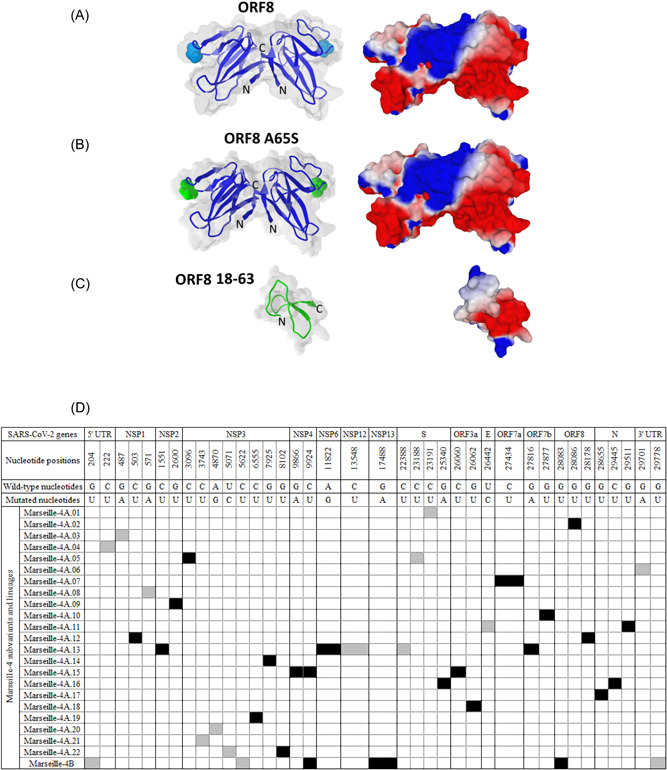
Structure of the SARS‐CoV‐2 ORF8 protein and its mutated and truncated forms. (A–C) Signature mutations in the genomes obtained for each of the Marseille‐4 subvariants and lineages (D). (A–C) The upper panel (A) shows the structure of dimeric SARS‐CoV‐2 ORF8 shown as superimposed surface and cartoon representations. The missing amino acids (65‐66 in chain A and 66‐68 in chain (B) were inserted with Swiss‐PdbViewer 22 in pdb file 7JTL, and the resulting model was minimized using the Polak–Ribiere algorithm of HyperChem, 23 as previously described. 24 Residue A65 in both chains is highlighted in cyan. The structure of the ORF8 mutant A65S (highlighted in green) was modeled using Swiss‐PdbViewer and HyperChem (middle panels) (B). The structure of truncated ORF8 18‐63 (bottom panels) was obtained using HyperChem (C). For all models, the surface potential of the protein is shown in the right panels (blue, positive; red, negative; white, neutral). (B) Synonymous nucleotide changes are indicated by a gray background. Nonsynonymous nucleotide changes are indicated by a black background.
2.4. Severity of the Marseille‐4B subvariant infections
We compared the characteristics of the first 181 patients identified as infected with Marseille‐4B and 1647 patients identified as infected with Marseille‐4A (Table 1a,b). Patients infected with Marseille‐4B were more likely to be female and older than those infected with Marseille‐4A. The mean cycle threshold (C t) values of the qPCR used for SARS‐CoV‐2 diagnosis did not differ between the two groups of patients. Higher hospitalization and death rates were observed in patients infected with Marseille‐4B. Multivariate analysis (Table 1b) confirmed that increased hospitalization rate was significantly associated with male sex, older age, a lower viral load (increased C t value) and Marseille‐4B infection. An increased rate of transfer to the intensive care unit was significantly associated with male sex and older age. An increased death rate was significantly associated with male sex, older age, and Marseille‐4B infection. Thus, we concluded that Marseille‐4B was more virulent and suspected that it is related to the knock‐out of ORF8.
Table 1.
Characteristics of 1828 patients infected with Marseille‐4A or Marseille‐4B (a) and risk factor analysis for hospitalization, transfer to the intensive care unit, and death in 1647 patients infected with Marseille‐4A or Marseille‐4B (b).
| a. Characteristics of 1828 patients infected with Marseille‐4A or Marseille‐4B | |||
|---|---|---|---|
| Epidemiological, clinical, and virological features | Marseille‐4A | Marseille‐4B | p Value |
| Male sex (%) | 790 (48.0) | 74 (40.9%) | 0.0710 |
| Mean age in years (standard deviation) | 51.3 (24.1) | 57.5 (21.5) | 0.0008 |
| Mean qPCR C t value (standard deviation) | 20.0 (3.5) | 20.1 (3.8) | 0.9246 |
| Hospitalization (%) | 195 (11.8) | 34 (18.8) | 0.0124 |
| Transfer to intensive care unit (%) | 59 (3.6) | 7 (3.9) | 0.8330 |
| Death (%) | 72 (4.4) | 17 (9.4) | 0.0058 |
| b. Risk factor analysis for hospitalization, transfer to the intensive care unit, and death in 1647 patients infected with Marseille‐4A or Marseille‐4B | |||
|---|---|---|---|
| Epidemiological, clinical, and virological features | OR (95% CI, p value) | ||
| Hospitalization | Transfer to intensive care unit | Death | |
| Male sex | 1.48 (1.08–2.03, p= 0.0142) | 4.70 (2.21–10, p< 0.0001) | 3.07 (1.85–5.1, p< 0.0001) |
| Age | 1.06 (1.05–1.06, p< 0.0001) | 1.03 (1.02–1.05, p< 0.0001) | 1.07 (1.06–1.09, p< 0.0001) |
| qPCR C t value | 1.09 (1.05–1.14, p< 0.0001) | 1.07 (0.98–1.16, p= 0.1368) | 0.97 (0.91–1.04, p= 0.3478) |
| Presence of the stop codon 64 in ORF8 | 2.24 (1.41–3.56, p= 0.0006) | 1.18 (0.41–3.43, p= 0.7625) | 2.60 (1.36–4.98, p= 0.0039) |
Abbreviations: CI, confidence interval; C t, cycle threshold value
2.5. Some mutations are associated with the expansion of Marseille‐4 lineages, others with their vanishing
Analyzing the kinetics of new variants without enough viral sequences leads to confused interpretation because of the superposition of different lineages in this clade. As for Marseille‐4, we identified nucleotide changes among the 22 Marseille‐4A lineages and the Marseille‐4B subvariant. Most of the Marseille‐4 subvariants and lineages showed a bell‐shaped distribution of cases, and their lifetime was 4.1 ± 1.4 months on average (Supporting Information: Table S3), during which 4.1 ± 2.6 mutations accumulated in viral genomes. RNA viruses have a high mutation rate 26 , 27 , 28 and SARS‐CoV‐2 accumulates approximately one mutation every 2 weeks. 29 We found signature mutations in each of these lineages, including in the NSP1, NSP2, NSP3, NSP4, NSP6, NSP12 (RNA‐dependent RNA polymerase), NSP13, S (spike), ORF3a, E (envelope), ORF7a, ORF7b, ORF8 and N (nucleocapsid) genes (Supporting Information: Table S1; Figure 3D). Interestingly, the accumulation of mutations resulting in an increasing genetic divergence correlated with a decreased incidence. Thus, the accumulation of nonlethal and nonfavoring mutations leads gradually to a genetic dispersion of the lineages, a decrease in viral fitness, and vanishing of the epidemic.
3. DISCUSSION
Here, we described the complete cycle of emergence, spread, and vanishing of the Marseille‐4 variant identified in France from mink 15 by analyzing 7453 genomes. The viral mutation rate leads to the accumulation of many random mutations, most presumably mildly deleterious, with little effect on fitness. Only 10−8 may be associated with a fitness gain. 26 , 30 , 31 Overall, the RNA virus fitness evolution includes an initial period of rapid multiplication possibly caused by a positive mutation followed by the decline of viral fitness caused by the accumulation of unfit mutations, as described for the vesicular stomatitis virus. 26 , 32
As we described in previous studies, 5 , 15 the Marseille‐4/B.1.160 variant appeared in July 2020 in our geographical area, 5 months before the Alpha/B.1.177 variant. In the second part of its lifetime, it co‐existed from December 2020 with the Alpha/B.1.177 variant, and vanished in April 2021. The same history was observed at our country scale (Figure 1B). We observed here a heterogeneity of the growth rates for the different Marseille‐4 subvariants and lineages, making it challenging to generalize the behavior of one SARS‐CoV‐2 subvariant or lineage to all of them. All Marseille‐4 lineages present a genetic signature, with mutations sometimes associated with the inactivation of ORF7a or ORF7b, as described. 33 None of these mutations, apart from those located in the spike gene, were predicted to be possibly associated with increased transmissibility.
Analysis of the Marseille‐4B subvariant that extended from December 2020 revealed the presence at its origin of a stop codon in ORF8, which was notably observed in the Alpha variant and viruses infecting mink and pangolin. 21 The ORF8 protein is 121 amino acids long and is one of the nine accessory proteins of SARS‐CoV‐2. It has been reported to be multifunctional, including inhibiting the presentation of viral antigens by the major histocompatibility complex of class I and suppressing the antiviral response mediated by type I interferon. 34 It has been also reported that ORF8 was one of the SARS‐CoV‐2 genes under positive selection 35 , 36 , 37 and that ORF8 of SARS‐CoV‐1 was under strong positive selection during animal‐to‐human transmission. 38 As expected, structural analysis performed here of the truncated form of ORF8 (ORF8 18‐63) confirms that the large deletion induced by the stop codon results in a distinct protein that does not retain any resemblance to the native ORF8 dimer. Nonetheless, Marseille‐4B may be more virulent than the other Marseille‐4 subvariant as suggested by clinical data, and it expanded at a time when the other Marseille‐4 lineages had vanished or were vanishing. These findings suggest that SARS‐CoV‐2 virulence may be associated with gene loss, as shown for some bacteria in which the decline in genomic content is associated with an increased host specificity and virulence, 39 , 40 and that ORF8 may be a so‐called “nonvirulence gene.” 40 Such nonvirulence or antivirulence genes were reported as genes whose inactivation or deletion is associated with improved fitness in a new host niche, with a novel lifestyle. 40 This concept of pathoadaptation by loss of gene function was initially described in Shigella spp. Interestingly, partial or complete deletions of the ORF8 gene were observed during the middle and late phases of the SARS‐CoV epidemic in 2002–2003. 41
The emergence of SARS‐CoV‐2 genomes harboring stop codons in ORF8 could be explained through at least two hypotheses: The first one is that the virus might benefit from not replicating and expressing useless genes. This can be related to the “use it or lose it” theory that proposes that the loss of a gene may provide a selective advantage when the function of a gene is dispensable, leading to the absence of correction of gene‐inactivating mutations. 42 The knock‐out of the ORF8 gene suggests that SARS‐CoV‐2 originates from a distinct animal reservoir, explaining why several of its genes are not essential and may be knocked out in humans, minks, and pangolins. Similarly, ORF8 truncation has been hypothesized to have occurred for HCoV‐229E in humans after zoonotic transmission from bats or intermediate hosts. 43 Functionally, ORF8 may help the virus to adapt to new hosts by facilitating immune evasion due to functional mimicry with immunological molecules. 44 This function may not remain critical as soon as the virus is well adapted to its new hosts. This first hypothesis also obeys what we named the “backpack rule,” which means that when we no longer need what is in our backpack it is better for us to put it down. Interestingly, loss of accessory genes was described as a major evolutionary pathway among poxviruses. 45 The second hypothesis can be related to the Red Queen theory, 46 which illustrates the arm race between competing biological entities. It has been reported that ORF8 protein was highly immunogenic and even elicited with ORF3a the strongest specific humoral responses. 47 , 48 Hence, the virus may benefit from the non‐expression of this protein as an immune escape strategy by getting rid of a major immune target. Overall, it appears that the fate of the Marseille‐4 variant, whether explained by one or both of these hypotheses, obeys the “Mistigri rule,” which we named in reference to the game of cards for which the winners are those who get rid of the Mistigri (jack of club, or of spades) card.
Finally, regarding the spread of Marseille‐4 lineages, their average detection duration was approximately 4 months, indicating that the accumulation of mutations beyond 8 was associated with a vanishing. This accumulation of mutations is associated with genetic heterogeneity and increased diversity. This leads to a funnel‐shaped evolution of the viral population. Thus, several beneficial mutations at unpredicted sites increase fitness, while the accumulation of other mutations results in decreased fitness, loss of clonality, and vanishing of the lineage. We speculate that the observed heterogeneity of a lineage may predict the end of the outbreak.
4. METHODS
4.1. Patients
Patients included in the present study were those identified as infected with the SARS‐CoV‐2 Marseille‐4 variant. 5 , 15 The present study has been approved by the ethics committee of the IHU Méditerranée Infection (N°2022‐001). Epidemiological and clinical data were retrieved for patients registered in the Assistance Publique‐Hôpitaux de Marseille (APHM) information system. Access to the patients’ biological and registry data issued from this system was approved by the data protection committee of APHM and was recorded in the European General Data Protection Regulation registry under number RGPD/APHM 2019‐73. Statistical processes were performed using R software version 4.0.2 (https://cran.r-project.org/). A p < 0.05 was considered statistically significant.
4.2. SARS‐CoV‐2 genotyping
SARS‐CoV‐2 genotyping was performed on RNA extracts from nasopharyngeal samples tested between July 1, 2020, and April 30, 2021 (10 months) at the IHU Méditerranée Infection Institute (https://www.mediterranee-infection.com/). Viral RNA was extracted from 200 µl of nasopharyngeal swab fluid using the EZ1 Virus Mini Kit v2.0 and the EZ1 Advanced XL instrument (Qiagen, Courtaboeuf) or the KingFisher Flex system (Thermo Fisher Scientific) following the manufacturer's instructions. NGS was performed when the cycle threshold value (C t) of the qPCR used to diagnose SARS‐CoV‐2 infection was <30. This qPCR was performed using a previously described protocol targeting the envelope (E) gene 49 or the BGI real‐time fluorescent RT‐PCR (BGI Genomics, Shanghai Fosun Long March Medical Science Co., Ltd.). When the Ct was ≥30 and in any case in the absence of an available genome sequence, the genotype was determined for SARS‐CoV‐2‐positive specimens using a variant‐specific qPCR or a partial sequencing of the SARS CoV‐2 spike gene, as previously described. 5 , 15 , 50 SARS‐CoV‐2 genome sequences were obtained as follows: by NGS using Illumina technology, the Nextera XT paired‐end strategy, and the MiSeq instrument (Illumina Inc.) since February 2020, as previously described 5 ; using the Illumina COVID‐seq protocol and the NovaSeq 6000 instrument (Illumina Inc.) since April 2021; or using Oxford Nanopore technology (ONT) and the GridION instrument (Oxford Nanopore Technologies Ltd.), as previously described. 5 NGS with ONT was performed without or with (since March 2021) synthesized cDNA amplification using a multiplex PCR protocol with ARTIC nCoV‐2019 v3 Panel primers purchased from Integrated DNA Technologies (IDT) according to the ARTIC procedure (https://artic.network/), as previously described. 5 Postextraction, viral RNA was reverse‐transcribed using SuperScript IV (Thermo Fisher Scientific) before cDNA second strand synthesis with Klenow Fragment DNA polymerase (New England Biolabs) when performing NGS using the Illumina MiSeq instrument (Illumina Inc.), LunaScript RT SuperMix kit (New England Biolabs) when performing NGS with the ONT, or according to the COVIDSeq protocol (Illumina Inc.) following the manufacturer's recommendations. The generated cDNA was purified using Agencourt AMPure XP beads (Beckman Coulter) and quantified using a Qubit 2.0 fluorometer (Invitrogen).
4.3. Assembly and analyses of genome sequences
Genome sequences were assembled by mapping on the SARS‐CoV‐2 genome GenBank accession no. NC_045512.2 (Wuhan‐Hu‐1 isolate) using CLC Genomics workbench v.7 (with the following thresholds: 0.8 for coverage and 0.9 for similarity) (https://digitalinsights.qiagen.com/) as previously described 5 or Minimap2 (https://github.com/lh3/minimap2). 51 Samtools (https://www.htslib.org/) was used for soft clipping of Artic primers (https://artic.network/) and to remove sequence duplicates. 52 Consensus genomes were generated using CLC Genomics workbench v.7 and Sam2consensus (https://github.com/vbsreenu/Sam2Consensus) through a first in‐house script written in Python language (https://www.python.org/). Mutation detection was performed using the Nextclade tool (https://clades.nextstrain.org/) and freebayes (https://github.com/freebayes/freebayes) 53 using a mapping quality score of 20 and results filtered by the Python script based on major nucleotide frequencies ≥70% and nucleotide depths ≥10 (when sequence reads were generated on the NovaSeq Illumina instrument) or ≥5 (when sequence reads were generated on the MiSeq Illumina instrument). SARS‐CoV‐2 genotyping was performed using a second in‐house script written in Python language (https://www.python.org/) by comparing mutation patterns with those of our database of SARS‐CoV‐2 variants. Nextstrain clades and Pangolin lineages provided in the present study were determined using the Nextclade web application (https://clades.nextstrain.org/) 13 , 54 and the Pangolin web application (https://cov-lineages.org/pangolin.html), 14 respectively. The sequences described in the present study have been deposited in the IHU Marseille Infection website (https://www.mediterranee-infection.com/acces-ressources/donnees-pour-articles/marseille-4-evolution/) and have also been deposited in the GISAID sequence database (https://www.gisaid.org/) 12 and can be retrieved online using the GISAID online search tool with “IHU” as keyword and B.1.160 as lineage criteria or using GISAID identifiers provided in Supporting Information: Table S4.
4.4. Phylogenetic reconstruction and definition and naming of Marseille‐4 subvariants and lineages
Phylogenetic reconstruction based on SARS‐CoV‐2 genomes were performed for genome sequences >24 000 nucleotide‐long using the Nextstrain/ncov tool (https://github.com/nextstrain/ncov) and then were visualized using Auspice (https://docs.nextstrain.org/projects/auspice/en/stable/). Marseille‐4A lineages comprised at least 50 SARS‐CoV‐2 genomes harboring one to four hallmark nucleotide changes. The Marseille‐4B subvariant was a new variant issued from a circulating variant (Marseille‐4).
4.5. Structural analysis of the untruncated and truncated ORF8 protein
A structural model of the ORF8 protein was generated from pdb file 7JTL. 25 The gaps in the crystal structure were fixed by incorporating the missing amino acids with the Robetta protein structure prediction tool, 55 followed by energy minimization with the Polak–Ribière algorithm as previously reported. 22 Mutant and truncated proteins were then generated with Swiss‐PdbViewer 21 and submitted to several rounds of energy minimization as described. 56
4.6. Evolution of Marseille‐4 subvariants and lineages and time dynamics of SARS‐CoV‐2 mutation accumulation
The duration of circulation of the different subvariants and lineages was calculated using the differences between the 5th and 95th percentiles of sampling dates; this allowed considering the time periods during which the subvariants and lineages had a significant incidence. Time dynamics of mutation accumulation were analyzed using locally estimated scatterplot smoothing (loess) for regression fitting. Latent structural changes in mutation distributions were retrieved using change point analysis of mean and variance with binary segmentation method and Schwartz information criterion penalty associated with a penalty threshold of 0.05. 57 We assessed the epidemiological capabilities of subvariants using the early stages of each epidemic curve. During this exponential phase, the size of the susceptible population may be considered as constant, and the cumulated number of cases exponentially increases at an approximately constant rate that was defined as the growth rate. After logarithmic transformation, the cumulated number of cases followed a linear model as follows: ln(It) = ln(I0) + Λt, where It is the cumulated incidence at time t, I0 is the initial number of cases, and Λ is the regression slope and the growth rate. From Λ, the reproduction rate, R, may be easily retrieved using the following equation 58 : R = (I + ΛD)(1 + ΛD′), where D and D′ are the average infectious and pre‐infectious periods (according to the SEIR model). Here, we set D at 9.3 59 and D′ at 3.3. 60 To calculate the growth rate, we used Chow's F test to determine the inflexion point of the logarithm of cumulated number of cases, which corresponded to the end of the exponential phase. Then, we applied a linear model for this phase to obtain the regression slope (i.e., the growth rate) and its 95% confidence interval. Statistical processes were performed using R software version 4.0.2 (https://cran.r-project.org/). All statistical conclusions were made using a 0.05 threshold.
AUTHOR CONTRIBUTIONS
Didier Raoult, Philippe Colson, Bernard La Scola, Pierre‐Edouard Fournier, and Philippe Gautret designed the study. Philippe Colson, Philippe Gautret, Jeremy Delerce, Hervé Chaudet, Marielle Bedotto, Léa Delorme, Wahiba Bader, Anthony Levasseur, Jean‐Christophe Lagier, Matthieu Million, Nouara Yahi, Jacques Fantini, and Pierre‐Edouard Fournier provided materials, data or analysis tools. Didier Raoult, Philippe Colson, Hervé Chaudet, Jeremy Delerce, Marielle Bedotto, Léa Delorme, Wahiba Bader, Nouara Yahi, Jacques Fantini, Bernard La Scola, and Pierre‐Edouard Fournier analyzed the data. Philippe Colson, Didier Raoult and Philippe Gautret wrote the first draft of the manuscript. Didier Raoult, Philippe Colson, Philippe Gautret, Pierre Pontarotti, Patrick Forterre, Jacques Fantini, Bernard La Scola, and Pierre‐edouard Fournier critically reviewed and revised the manuscript. All authors approved the final manuscript.
CONFLICTS OF INTEREST
Didier Raoult was a consultant for the Hitachi High‐Technologies Corporation, Tokyo, Japan, from 2018 to 2020. He is a scientific board member of the Eurofins company and a founder of a microbial culture company (Culture Top). The remaining authors declare that there are no conflict of interest.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
We are thankful to Mamadou Beye, Emilie Burel, Elsa Prudent, Céline Gazin, Sofiane Bakour, Laurence Thomas, Séverine Guillon, Véronique Filosa, Ludivine Bréchard, and Claudia Andrieu for their technical help. This work was supported by the French Government under the “Investments for the Future” program managed by the National Agency for Research (ANR) (Méditerranée‐Infection 10‐IAHU‐03); by the Région Provence Alpes Côte d'Azur and European funding FEDER PRIMMI (Fonds Européen de Développement Régional‐Plateformes de Recherche et d'Innovation Mutualisées Méditerranée Infection) (FEDER PA 0000320 PRIMMI); and by the French Ministry of Higher Education, Research and Innovation (Ministère de l'Enseignement supérieur, de la Recherche et de l'Innovation) and the French Ministry of Solidarity and Health (Ministère des Solidarités et de la Santé). Funding sources had no role in the design and conduct of the study, the collection, management, analysis, and interpretation of the data, and the preparation, review, or approval of the manuscript.
Colson P, Gautret P, Delerce J, et al. The emergence, spread and vanishing of a French SARS‐CoV‐2 variant exemplifies the fate of RNA virus epidemics and obeys the Mistigri rule. J Med Virol. 2022;1‐11. 10.1002/jmv.28102
DATA AVAILABILITY STATEMENT
Viral genomes analyzed in the present study are available from the IHU Méditerranée Infection website (https://www.mediterranee-infection.com/acces-ressources/donnees-pour-articles/marseille-4-evolution/) and have also been deposited in the GISAID sequence database (https://www.gisaid.org/) 12 and can be retrieved online using the GISAID online search tool with “IHU” as keyword and B.1.160 as lineage criteria or using GISAID identifiers provided in Supporting Information: Table S4.
REFERENCES
- 1. Lemey P, Ruktanonchai N, Hong SL, et al. Untangling introductions and persistence in COVID‐19 resurgence in Europe. Nature. 2021;595:713‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li J, Lai S, Gao GF, Shi W. The emergence, genomic diversity and global spread of SARS‐CoV‐2. Nature. 2021;600:408‐418. [DOI] [PubMed] [Google Scholar]
- 3. Vöhringer HS, Sanderson T, Sinnott M, et al. Genomic reconstruction of the SARS‐CoV‐2 epidemic in England. Nature. 2021;600:506‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rochman ND, Wolf YI, Faure G, Mutz P, Zhang F, Koonin EV. Ongoing global and regional adaptive evolution of SARS‐CoV‐2. Proc Natl Acad Sci USA. 2021;118:e2104241118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colson P, Fournier P‐E, Chaudet H, et al. Analysis of SARS‐CoV‐2 variants from 24,181 patients exemplifies the role of globalisation and zoonosis in pandemics. Front Microbiol. 2022;12:786233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tomaszewski T, DeVries RS, Dong M, et al. New pathways of mutational change in SARS‐CoV‐2 proteomes involve regions of intrinsic disorder important for virus replication and release. Evol Bioinform Online. 2020;16:1176934320965149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hay JA, Kennedy‐Shaffer L, Kanjilal S, et al. Estimating epidemiologic dynamics from cross‐sectional viral load distributions. Science. 2021;373:eabh0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Omer SB, Yildirim I, Forman HP. Herd immunity and implications for SARS‐CoV‐2 control. JAMA. 2020;324:2095‐2096. [DOI] [PubMed] [Google Scholar]
- 9. Palese P, Wang TT. Why do influenza virus subtypes die out? A hypothesis. mBio. 2011;2:e00150‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pegu A, O'connell SE, Schmidt SD, et al. Durability of mRNA‐1273 vaccine‐induced antibodies against SARS‐CoV‐2 variants. Science. 2021;373:1372‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dong E, Du H, Gardner L. An interactive web‐based dashboard to track COVID‐19 in real time. Lancet Infect Dis. 2020;20(5):533‐534. 10.1016/S1473-3099(20)30120-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elbe S, Buckland‐Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. 2017;1:33‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aksamentov I, Roemer C, Hodcroft EB, Neher RA. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J Open Source Softw. 2021;6:3773. [Google Scholar]
- 14. Rambaut A, Holmes EC, O'toole Á, et al. A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fournier PE, Colson P, Levasseur A, et al. Emergence and outcomes of the SARS‐CoV‐2 ‘Marseille‐4' variant. Int J Infect Dis. 2021;106:228‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colson P, Levasseur A, Delerce J, et al. Dramatic increase in the SARS‐CoV‐2 mutation rate and low mortality rate during the second epidemic in summer in Marseille. IHU Preprints . 2020. https://www.mediterranee-infection.com/dramatic-increase-in-the-sars-cov-2-mutation-rate-and-low-mortality-rate-during-the-second-epidemic-in-summer-in-marseille/. 10.35088/68c3-ew82 [DOI]
- 17. Hodcroft E. CoVariants: SARS‐CoV‐2 mutations and variants of interest; 2021. Available from https://covariants.org/
- 18. Hodcroft EB, Zuber M, Nadeau S, et al. Spread of a SARS‐CoV‐2 variant through Europe in the summer of 2020. Nature. 2021;595:707‐712. [DOI] [PubMed] [Google Scholar]
- 19. Bi Q, Wu Y, Mei S, et al. Epidemiology and transmission of COVID‐19 in 391 cases and 1286 of their close contacts in Shenzhen, China: a retrospective cohort study. Lancet Infect Dis. 2020;20:911‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prates ET, Garvin MR, Pavicic M, et al. Potential pathogenicity determinants identified from structural proteomics of SARS‐CoV and SARS‐CoV‐2. Mol Biol Evol. 2021;38:702‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pereira F. SARS‐CoV‐2 variants lacking ORF8 occurred in farmed mink and pangolin. Gene. 2021;784:145596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guex N, Peitsch MC. SWISS‐MODEL and the Swiss‐PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714‐2723. [DOI] [PubMed] [Google Scholar]
- 23. Froimowitz M. 1HyperChem: a software package for computational chemistry and molecular modeling. Biotechniques. 1993;14:1010‐1013. [PubMed] [Google Scholar]
- 24. Fantini J, Yahi N, Azzaz F, Chahinian H. Structural dynamics of SARS‐CoV‐2 variants: a health monitoring strategy for anticipating Covid‐19 outbreaks. J Infect. 2021;83:197‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flower TG, Buffalo CZ, Hooy RM, Allaire M, Ren X, Hurley JH. Structure of SARS‐CoV‐2 ORF8, a rapidly evolving immune evasion protein. Proc Natl Acad Sci USA. 2021;118:e2021785118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elena SF, Miralles R, Cuevas JM, Turner PE, Moya A. The two faces of mutation: extinction and adaptation in RNA viruses. IUBMB Life. 2000;49:5‐9. [DOI] [PubMed] [Google Scholar]
- 27. Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 1997;51:151‐178. [DOI] [PubMed] [Google Scholar]
- 28. Domingo E. Virus as Population. 1st ed. Elsevier, Academic Press; 2016:1‐412. [Google Scholar]
- 29. van Dorp L, Richard D, Tan CCS, Shaw LP, Acman M, Balloux F. No evidence for increased transmissibility from recurrent mutations in SARS‐CoV‐2. Nat Commun. 2020;11:5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miralles R, Gerrish PJ, Moya A, Elena SF. Clonal interference and the evolution of RNA viruses. Science. 1999;285:1745‐1747. [DOI] [PubMed] [Google Scholar]
- 31. Muller HJ. The relation of recombination to mutational advance. Mutat Res. 1964;106:2‐9. [DOI] [PubMed] [Google Scholar]
- 32. Elena SF, Moya A. Rate of deleterious mutation and the distribution of its effects on fitness in vesicular stomatitis virus. J Evol Biol. 1999;12:1078‐1088. [Google Scholar]
- 33. Nemudryi A, Nemudraia A, Wiegand T, et al. SARS‐CoV‐2 genomic surveillance identifies naturally occurring truncation of ORF7a that limits immune suppression. Cell Rep. 2021;35:109197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zinzula L. Lost in deletion: the enigmatic ORF8 protein of SARS‐CoV‐2. Biochem Biophys Res Commun. 2021;538:116‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Velazquez‐Salinas L, Zarate S, Eberl S, Gladue DP, Novella I, Borca MV. Positive selection of ORF1ab, ORF3a, and ORF8 genes drives the early evolutionary trends of SARS‐CoV‐2 during the 2020 COVID‐19 pandemic. Front Microbiol. 2020;11:550674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lo Presti A, Rezza G, Stefanelli P. Selective pressure on SARS‐CoV‐2 protein coding genes and glycosylation site prediction. Heliyon. 2020;6:e05001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng L, Han X, Zhu Z, Qi C, Wang P, Zhang X. Functional alterations caused by mutations reflect evolutionary trends of SARS‐CoV‐2. Brief Bioinform. 2021;22:1442‐1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lau SK, Feng Y, Chen H, et al. Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS‐Related coronavirus from greater horseshoe bats through recombination. J Virol. 2015;89:10532‐10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Georgiades K, Merhej V, El Karkouri K, Raoult D, Pontarotti P. Gene gain and loss events in rickettsia and orientia species. Biol Direct. 2011;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maurelli AT. Black holes, antivirulence genes, and gene inactivation in the evolution of bacterial pathogens. FEMS Microbiol Lett. 2007;267:1‐8. [DOI] [PubMed] [Google Scholar]
- 41. Chinese SARS Molecular Epidemiology Consortium . Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666‐1669. [DOI] [PubMed] [Google Scholar]
- 42. Moran NA. Microbial minimalism: genome reduction in bacterial pathogens. Cell. 2002;108:583‐586. [DOI] [PubMed] [Google Scholar]
- 43. Corman VM, Baldwin HJ, Tateno AF, et al. Evidence for an ancestral association of human coronavirus 229E with bats. J Virol. 2015;89:11858‐11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valcarcel A, Bensussen A, Álvarez‐Buylla ER, Díaz J. Structural analysis of SARS‐CoV‐2 ORF8 protein: pathogenic and therapeutic implications. Front Genet. 2021;12:693227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Senkevich TG, Yutin N, Wolf YI, Koonin EV, Moss B. Ancient gene capture and recent gene loss shape the evolution of orthopoxvirus‐host interaction genes. mBio. 2021;12:e0149521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Valen L. A new evolutionary law. Evol Theory. 1973;1:1‐30. [Google Scholar]
- 47. Hachim A, Kavian N, Cohen CA, et al. ORF8 and ORF3b antibodies are accurate serological markers of early and late SARS‐CoV‐2 infection. Nat Immunol. 2020;21:1293‐1301. [DOI] [PubMed] [Google Scholar]
- 48. Wang X, Lam JY, Chen L, et al. Mining of linear B cell epitopes of SARS‐CoV‐2 ORF8 protein from COVID‐19 patients. Emerg Microbes Infect. 2021;10:1016‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Amrane S, Tissot‐Dupont H, Doudier B, et al. Rapid viral diagnosis and ambulatory management of suspected COVID‐19 cases presenting at the infectious diseases referral hospital in Marseille, France, ‐January 31st to March 1st, 2020: a respiratory virus snapshot. Travel Med Infect Dis. 2020;36:101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bedotto M, Fournier PE, Houhamdi L, et al. Implementation of an in‐house real‐time reverse transcription‐PCR assay for the rapid detection of the SARS‐CoV‐2 Marseille‐4 variant. J Clin Virol. 2021;139:104814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094‐3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garrison E, Marth G. Haplotype‐based variant detection from short‐read sequencing. arXiv. 2012;arXiv:1207.3907. [Google Scholar]
- 54. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real‐time tracking of pathogen evolution. Bioinformatics. 2018;34:4121‐4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526‐W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Di Scala C, Fantini J. Hybrid in silico/In vitro approaches for the identification of functional cholesterol‐binding domains in membrane proteins. Methods Mol Biol. 2017;1583:7‐19. [DOI] [PubMed] [Google Scholar]
- 57. Chen J, Gupta AK. Parametric Statistical Change Point Analysis with Applications to Genetics, Medicine, and Finance. 2nd ed. Birkhauser; 2012. [Google Scholar]
- 58. White R, Vynnycky E. An Introduction to Infectious Disease Modelling. OUP Oxford; 2016. [Google Scholar]
- 59. Zhao S, Tang B, Musa SS, et al. Estimating the generation interval and inferring the latent period of COVID‐19 from the contact tracing data. Epidemics. 2021;36:100482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Byrne AW, McEvoy D, Collins AB, et al. Inferred duration of infectious period of SARS‐CoV‐2: rapid scoping review and analysis of available evidence for asymptomatic and symptomatic COVID‐19 cases. BMJ Open. 2020;10:e039856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
Viral genomes analyzed in the present study are available from the IHU Méditerranée Infection website (https://www.mediterranee-infection.com/acces-ressources/donnees-pour-articles/marseille-4-evolution/) and have also been deposited in the GISAID sequence database (https://www.gisaid.org/) 12 and can be retrieved online using the GISAID online search tool with “IHU” as keyword and B.1.160 as lineage criteria or using GISAID identifiers provided in Supporting Information: Table S4.