

Abstract
Objectives
CYLD was a novel causative gene for frontotemporal dementia (FTD) and amyotrophic lateral sclerosis. Given the clinical and pathological overlap of FTD and Alzheimer's disease (AD), it is necessary to screen CYLD in AD patients and FTD patients in the Chinese population.
Methods
In our study, using a targeted sequencing panel, we sequenced the CYLD gene in a large cohort of 2485 participants in the Chinese population, including 1008 AD patients, 105 FTD patients, and 1372 controls.
Results
In the present study, the average onset age of AD and FTD patients was 66.84 ± 30.42 years old and 60 ± 10.00 years old, respectively. Our study reported three novel CYLD variants: p.Phe288Leu (patient No. 1, AD), p.Tyr485Phe (patients No. 6–9, all AD) and p.Thr951Ala (patient No. 10, AD), plus a previously reported variant: p.Arg397Ser (patient No. 2–5, AD and No. 11, FTD). These variants were absent in our in‐house controls and predicted to be deleterious according to the MutationTaster. The variant carriers were composed of 10 AD patients and one FTD patient, and the average onset age was 61.2 ± 10.9 years. The frequency of CYLD variants in AD was similar to that in FTD, which was 0.99% (10/1008) and 0.95% (1/105), respectively.
Interpretation
Our finding extended the genotype and phenotype of the CYLD gene and demonstrated that CYLD rare damaging variants may be implicated in AD and FTD pathogenesis.
Introduction
Dementia is a devastating disease and imposes a huge burden on patients and their caregivers. Currently, approximately 50 million people suffer from dementia and its prevalence is projected to triple by 2050. 1 Alzheimer's disease (AD) is the most prevalent dementia and accounts for 60%–80% of dementia cases. 2 Frontotemporal dementia (FTD) is the second most common dementia with a prevalence ranging from 3% to 26% in all dementia patients. 3 Clinically, both AD and FTD exhibit progressive impairments in different cognitive domains, including memory, language, behavior, and execution. Of note, atypical AD cases closely resemble FTD patients. 4 Aggregation of transactive response (TAR)‐DNA binding protein 43 (TDP‐43) and tau protein are shared pathological hallmarks in AD and FTD. 5 Additionally, FTD causative genes identified previously, such as MAPT, C9orf72, GRN, TARDBP, UBQLN1, HNRNPA1, and CHCHD10 accounted for a proportion of AD pathogenesis. 6 , 7 , 8 , 9 , 10 For example, abnormal C9orf72 expansions were detected in AD patients but absent from controls.
Dobson‐Stone et al. recently identified that CYLD was a novel causative gene for frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS). A missense variant, CYLD p.M719V mutation was segregated in a large family with FTD and ALS. Functional studies revealed that CYLD p.M719V was associated with increased TAR DNA‐binding protein 43 (TDP‐43) and decreased axonal length. Additionally, this variant was involved in the inhibition of nuclear factor‐kappa B (NF‐κB) and impaired autophagy. 11 Intriguingly, one of the most dominant symptoms in patients with CYLD variant is memory impairment and some were clinically diagnosed with Alzheimer's disease (AD). 12 Also, two rare CYLD variants were observed in two patients with marked memory loss in a Portuguese cohort. 13
Given the clinical and pathological overlap between AD and FTD, we hypothesized that CYLD may contribute to AD pathogenesis. Besides, to date, no studies have investigated the association of CYLD variants AD or FTD in the non‐Caucasian population. Consequently, it is necessary to screen CYLD variants in AD and FTD patients in the Chinese population. In this study, we sequenced the CYLD gene in a large cohort of 2485 individuals using targeted gene sequencing.
Methods
Subjects
Our study recruited 1008 AD patients and 105 FTD patients from Xiangya Hospital and 1372 cognitively normal controls from a community in Changsha. According to the National Institute on Aging‐Alzheimer's Association criteria for probable AD and diagnostic criteria for FTD, 14 , 15 patients were diagnosed with AD or FTD by two expert neurologists. Patients with causative mutations for AD and FTD, including APP, PSEN1, PSEN2, MAPT, GRN, C9orf72, CHCHD10, CHMP2B, VCP, TARDBP, TBK1, and FUS had been excluded by Targeted genes sequencing or repeat‐prime PCR (RP‐PCR) analysis. Other inclusion criteria for AD or FTD patients include (1) the disease duration is larger than 6 months; (2) neuropsychological questionnaires indicated the patient had dementia; (3) The modified Hachinski ischemia scale score was ≤4; (4) The patients or their legal guardians agreed to participate in this study. Additionally, the inclusion criteria for controls involved (1) no chief complaint with cognitive impairment; (2) neuropsychological questionnaires revealed no dementia; (3) they had no other neurological diseases. This study was approved by the Ethics Committee of Xiangya Hospital, Central South University, China.
Genomic DNA isolation
We extracted genomic DNA from the peripheral blood leukocytes via phenol‐chloroform extraction and ethanol precipitation. 16 The DNA's quality and quantity were assessed using a NanoDrop spectrophotometer (Thermo Scientific). The DNA sample was diluted to 50–100 ng/μL.
Targeted gene sequencing
The targeted sequencing panel comprised CYLD and APOE genes. The genomic DNA was broken into 150–200 bp length fragments using Biorupter Pico. End‐repairing, A‐tailing, adaptor ligation, and pre‐capture PCR amplification were performed. The DNA was captured by a targeted panel and sequenced on Illumina NovaSeq 6000 platform. Using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), the low‐quality reads fastq data were removed. The reads were aligned to the human reference genome (hg19) via the BWA software (http://bio‐bwa.sourceforge.net). 17 Duplicate sequence reads were discarded with Picard (version 2.18.7, http://broadinstitute.github.io/picard/). Using the Genome Analysis Toolkit (version 3.2), 18 we performed the quality‐score recalibration, local realignments, and variant calling. Variants were annotated with ANNOVAR (https://hpc.nih.gov/apps/ANNOVAR.html). 19 According to minor allele frequencies (MAF), rare variants were defined by variants with MAF < 0.001. Furthermore, the pathogenicity of missense variants was predicted using MutationTaster (https://www.mutationtaster.org/), SIFT, and PolyPhen2.
Results
In the present study, the average onset age of AD and FTD patients was 66.84 ± 30.42 years old and 60 ± 10.00 years old, respectively. Besides, the average enrolled age was 67.98 ± 6.75 years old in controls. We identified three CYLD novel rare variants, including c.864 T > A, p.Phe288Leu, c.1454A > T, p.Tyr485Phe, and c.2851A > G, p.Thr951Ala, as well as one previously reported variant c.1189C > A:p.Arg397Ser (Fig. 1). 20 All these rare CYLD variants were verified by Sanger sequencing. These variants exhibited extremely low frequency (minor allele frequency < 0.001) in the Genome Aggregation Database (gnomAD) database and were predicted to be damaging using the MutationTaster (https://www.mutationtaster.org/). All these variants were absent in our in‐house controls. The variant carriers consisted of 10 AD patients (patients No.1‐No.10) and one FTD patient (patient No.11), who had an average onset age of 61.2 ± 10.9 years (range 46–79 years) (Table 1). The frequency of CYLD variants in AD was similar to that in FTD, which was 0.99% (10/1008) and 0.95% (1/105), respectively.
Figure 1.
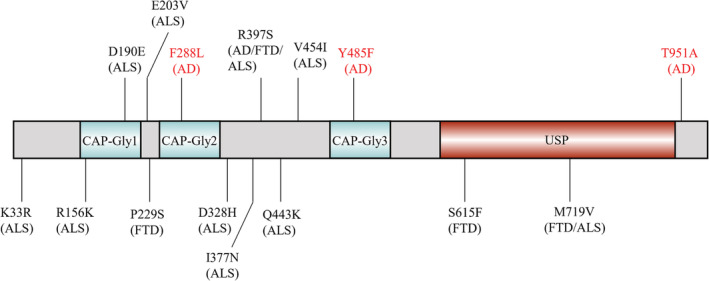
Schematic of rare damaging variants in the CYLD gene. AD, Alzheimer's disease; FTD, Frontotemporal dementia; ALS, Amyotrophic lateral sclerosis; CAP‐Gly, cytoskeleton‐associated protein‐glycine conserved domain; USP, ubiquitin‐specific protease domain; Variants firstly were identified in our study (red), and variants were previously reported (black). The corresponding diagnoses are in parentheses.
Table 1.
CYLD rare damaging variants identified in AD and FTD patients.
| Patient | Gender | Age at onset (years) | Age (years) | Disease duration (years) | Initial symptoms | Family history | MMSE | CDR | MTA | PET | APOE | Diagnosis | Variant | Allele and total read depth | GnomAD East Asian non‐neuro MAF | Damaging Prediction (MutationTaster/SIFT/PolyPhen2) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No 1 | M | 79 | 81 | 2 | Memory decline | + | 8 | 2 | 2 | Aβ deposition | 3/4 | AD | c.864 T > A:p.Phe288Leu | 40/58 | 0 | D/T/B |
| No 2 | F | 49 | 52 | 3 | Memory decline | − | 12 | 1 | 3 | NA | 3/3 | AD | c.1189C > A:p.Arg397Ser | 24/63 | 3.43 × 10−4 (5/14558) | D/T/B |
| No 3 | F | 46 | 51 | 5 | Memory decline | − | 17 | 1 | 2 | NA | 3/3 | AD | c.1189C > A:p.Arg397Ser | 38/69 | 3.43 × 10−4 (5/14558) | D/T/B |
| No 4 | F | 46 | 48 | 2 | Memory decline | − | 9 | 2 | 2 | Aβ deposition | 3/3 | AD | c.1189C > A:p.Arg397Ser | 38/72 | 3.43 × 10−4 (5/14558) | D/T/B |
| No 5 | F | 68 | 70 | 2 | Memory decline | + | 10 | 2 | 3 | NA | 3/4 | AD | c.1189C > A:p.Arg397Ser | 29/60 | 3.43 × 10−4 (5/14558) | D/T/B |
| No 6 | F | 74 | 80 | 6 | Memory decline | − | 4 | 3 | 3 | NA | 3/3 | AD | c.1454A > T:p.Tyr485Phe | 19/33 | 0 | D/T/B |
| No 7 | M | 58 | 60 | 2 | Memory decline | − | 7 | 2 | 2 | NA | 3/3 | AD | c.1454A > T:p.Tyr485Phe | 19/37 | 0 | D/T/B |
| No 8 | M | 68 | 72 | 4 | Memory decline | − | 7 | 2 | 3 | Aβ deposition | 3/3 | AD | c.1454A > T:p.Tyr485Phe | 11/21 | 0 | D/T/B |
| No 9 | M | 59 | 67 | 8 | Memory decline | + | 6 | 3 | 2 | NA | 4/4 | AD | c.1454A > T:p.Tyr485Phe | 45/91 | 0 | D/T/B |
| No 10 | M | 70 | 71 | 1 | Memory decline | + | 16 | 1 | 3 | Aβ deposition | 3/3 | AD | c.2851A > G:p.Thr951Ala | 23/45 | 0 | D/D/B |
| No 11 | M | 56 | 59 | 3 | Abnormal behavior | − | 13 | 1 | 1 |
Decreased metabolism in the bilateral frontal lobe No Aβ deposition |
3/4 | FTD | c.1189C > A:p.Arg397Ser | 17/33 | 3.43 × 10−4 (5/14558) | D/T/B |
AD, Alzheimer's disease; FTD, frontotemporal dementia; M, male; F, female; MMSE, Mini‐mental State Examination; MoCA, Montreal Cognitive Assessment; CDR, Clinical Dementia Rating; MTA, medial temporal lobe atrophy; PET, positron emission tomography; Aβ, amyloid β; MAF, minor allele frequency; P‐tau, phosphorylated tau; T‐tau, total tau; NA, not available; D, damaging; T, tolerable; B, benign.
Patient No.1 was male and had a family history. At 79 years old, he presented memory decline and loss of orientation. He often forgot what others just said and where things were placed. His mother developed dementia at 80 years and died in her 90s. Brain magnetic resonance imaging (MRI) showed bilateral temporal lobe atrophy. 11C‐PiB‐positron emission tomography (PET) showed that amyloid β (Aβ) accumulated in the cerebral cortex. He carried the CYLD p.Phe288Leu variant. Patient No.2 was a 52‐year‐old woman with an onset of cognitive impairment at 49 years old, carrying the CYLD p.Arg397Ser variant. She was mainly presented with progressive episodic memory loss. Brain MRI revealed moderate hippocampal atrophy. Patient No.3 was a female with dementia starting at 46 years old, and also had CYLD p.Arg397Ser variant. She displayed symptoms such as forgetting what just happened and miscalculating money. Gradually, she could not tell the direction of home and got lost. She had no family history. MRI showed whole brain atrophy. Similarly, patient No. 4 was a female with an age of onset of 46 years old and carried CYLD p.Arg397Ser variant. She had typical episodic memory impairment. MRI showed bilateral hippocampal atrophy. 11C‐PiB‐PET revealed that there was diffuse amyloid β (Aβ) deposition in the whole cerebral cortex. Meanwhile, reduced Aβ42 and elevated total‐tau and phosphorylated tau were observed in the cerebrospinal fluid (CSF) test. Patient No.5 was a female who started dementia at 68 years and carried CYLD p.Arg397Ser variant. She showed progressive memory loss, such as forgetting put salt in dishes when cooking. Also, she had decreased execution ability. Her brother and mother both presented dementia in their 70s and died several years later. Brain MRI showed bilateral hippocampal atrophy. Patient No.6 was an 80‐year‐old female who developed dementia at 74 years old and carried CYLD p.Tyr485Phe variant. She displayed symptoms such as forgetting what just happened. MRI indicated whole brain atrophy. Patient No.7 was a male with an onset age of 58 years old. He often forgot what others just said and could not recognize his relatives. MRI showed medial temporal atrophy. He finally carried CYLD p.Tyr485Phe variant. Patient No. 8 presented at our hospital because of memory loss and loss of orientation. His symptoms began at age of 68 years. Temporal lobes atrophy was observed in brain MRI. PiB‐PET showed diffuse Aβ deposition. The genetic screening revealed that he carried CYLD p.Tyr485Phe variant. Patient No. 9 was a male with an onset age of 59 years. His father had similar symptoms and died in his 60s. He presented episodic memory impairment, and MRI indicated bilateral temporal and parietal atrophy. He also carried CYLD p.Tyr485Phe variant. Patient No. 10 was a 71‐year‐old patient with an onset age of 70 years, carrying a CYLD p.Thr951Ala variant. His sister also had similar symptoms. He presented with marked memory loss, disorientation, and deterioration of visuospatial skills. Brain MRI revealed global brain atrophy and mild leukodystrophy. PiB‐PET revealed showed that Aβ was deposited in the temporal lobe and parietal lobe. Patient No.11 was a male with dementia starting at age of 56 years with a negative family history. He visited our hospital because of abnormal behaviors, such as picking up the garbage and stealing money. Also, he became irritable and often lost his temper for no reason, while his memory remains intact during the disease progression. MRI showed moderate bilateral frontal lobe atrophy, and FDG‐PET indicated decreased metabolism in the bilateral frontal lobe while PiB‐PET revealed no Aβ deposits in the cortex. This patient had no signs of amyotrophic lateral sclerosis (ALS). He was finally diagnosed with behavioral variant FTD (bvFTD) and carried the CYLD p.Arg397Ser variant.
Discussion
In our study, we identified three novel CYLD rare damaging variants (p.Phe288Leu, p.Tyr485Phe, and p.Thr951Ala) and one variant previously reported (p.Arg397Ser) in ten AD patients and one FTD patient. This is the first study of screening CYLD variants in AD patients. Also, we are the first to sequence the CYLD gene in FTD cases in the Chinese population.
CYLD gene is located in the chromosome 16q12.1 region, encoding a protein including three cytoskeleton‐associated protein glycine‐rich (CAP‐Gly) domains and a ubiquitin‐specific protease (USP) domain. 13 In our study, CYLD rare variants were predicted to be damaging by MutationTaster but not by SIFT and PolyPhen2. Although the predictions of these variants were not consistent, they may still exert effects on AD or FTD development. Firstly, MutationTaster had the best prediction performance when compared to PolyPhen2, Provean, SIFT, and MutationAssessor. 21 Additionally, in silico tools only predict variants' pathogenicity, and functional studies are warranted to determine their roles in AD or FTD pathogenesis. CYLD is a deubiquitinating enzyme targeting ubiquitin chains and regulating the NF‐κB pathway or autophagy. 22 In our cohort, two rare variants, including p.Phe288Leu and p.Tyr485Phe, were located within the CAP‐Gly domains (residues 232–303 and residues 472–540). Additionally, one rare variant (p.Arg397Ser) is located near the CAP‐Gly domain while p.Thr951Ala is near the USP domain (residues 589–947). Cap‐Gly domains play important roles in the interaction of CYLD and microtubule‐associated proteins, such as proteins encoded by MAPT. 23 Disruption of the USP domain was associated with loss of CYLD catalytic activity. 24 Therefore, CYLD p.Thr951Ala may regulate CYLD catalytic activity. Also, it was speculated that the USP domain accounts for autophagy. 13 Thus, CYLD rare damaging variants we identified may be involved in AD pathogenesis via modulating CYLD catalytic activity or its interaction with other microtubule‐associated proteins. Our study firstly revealed that CYLD rare damaging variants were involved in AD pathogenesis. Interestingly, it is reported that CYLD can interact with protein phosphatase 2A (PP2A) and increase PP2A's ability to bind and dephosphorylate Aurora‐B. 25 PP2A is able to regulate tau phosphorylation, 26 therefore, although there are no studies that CYLD promotes PP2A's capacity to regulate the phosphorylation level of tau, it is plausible to suppose that CYLD may be associated with tau's phosphorylation. Furthermore, neuroinflammation is a key pathologic event in AD. Elevated levels of tumor necrosis factor‐α (TNF‐α) induced necroptosis in HT‐22 hippocampal neuronal cells, which was mediated by a signaling pathway that CYLD is involved. 27
To date, no studies have shown that CYLD is implicated in AD pathogenesis. Our study is the first to reveal that the frequency of CYLD rare variants was common in a Chinese AD cohort, in whom approximating 1% of AD patients carried these variants. Our finding indicated that the CYLD rare variants probably play a role in AD pathogenesis, and further functional studies are needed to investigate whether CYLD interacts with the proteins encoded by AD pathogenic genes. Furthermore, Dobson‐Stone et al. identified that 0.54% (6/1105) of bvFTD patients carried CYLD rare variants in Australia, North America, and Europe, while Tábuas‐Pereira et al. showed that CYLD rare variants accounted for 3.08% (2/65) of FTD patients in a Portuguese cohort. 11 , 13 Compared to the two aforementioned studies in the Caucasian population, in our cohort, the frequency of CYLD rare variants in FTD patients (1/105, 0.95%) was intermediate, suggesting that population differences may exist.
Some limitations exist in our study. First, some AD patients were diagnosed with probable AD, pathogenic evidence, such as CSF biomarkers or PiB‐PET scan, is needed to ensure the diagnosis. Second, the pathogenicity of rare variants we found was based on bioinformatic analysis, and the functional studies are warranted to confirm their exact roles. Thirdly, only the variants identified in AD or FTD patients were sequenced in controls. Further systematic studies of screening CYLD rare variants between patients and controls are needed.
Taken together, we identified four rare damaging CYLD variants in 10 AD patients and one FTD patient. We are the first to reveal that CYLD rare variants may be implicated in the pathogenesis of AD, which extended the genotype and phenotype of CYLD. Additionally, our study further supported the potential role of CYLD in FTD pathogenesis.
Conflict of Interest
Nothing to report.
Author Contributions
Study concept: XX, BJ, and LS. Design: XX, TX, HL, and XL. Acquisition of the data: XL, YZ, LZ, XW, YZ, QY, XH, YL, HJ, and JG. Analysis, or interpretation of data: XX, JW, BT, and JL. Statistical analysis: XX and JL. Drafting of the manuscript: XX. Critical revision of the manuscript for important intellectual content: all authors.
Acknowledgements
The authors are grateful to all subjects for participation in our study. This study was supported by the National Key R&D Program of China (No. 2020YFC2008500), the National Major Projects in Brain Science and Brain‐like Research (No.2021ZD0201803), the National Natural Science Foundation of China (No.81971029, 82071216, 81901171), Hunan Innovative Province Construction Project (No.2019SK2335), the Youth Program of Science Foundation of Xiangya Hospital (No.2018Q017, 2018Q020) and Hu‐Xiang Youth Project (No. 2021RC3028).
Funding Information This study was supported by the National Key R&D Program of China (No. 2020YFC2008500), the National Major Projects in Brain Science and Brain‐like Research (No. 2021ZD0201803), the National Natural Science Foundation of China (No. 81971029, 82071216, 81901171), Hunan Innovative Province Construction Project (No. 2019SK2335), the Youth Program of Science Foundation of Xiangya Hospital (No. 2018Q017, 2018Q020) and Hu‐Xiang Youth Project (No. 2021RC3028).
Funding Statement
This work was funded by Hunan Innovative Province Construction Project grant 2019SK2335; Hu‐Xiang Youth Project grant 2021RC3028; National Major Projects in Brain Science and Brain‐like Research grant 2021ZD0201803; the National Key R&D Program of China grant 2020YFC2008500; the National Natural Science Foundation of China grants 81971029, 82071216, and 81901171; Youth Program of Science Foundation of Xiangya Hospital grants 2018Q017 and 2018Q020.
References
- 1. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397(10284):1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang T, Ma S, Lv J, et al. The emerging role of exosomes in Alzheimer's disease. Ageing Res Rev. 2021;68:101321. [DOI] [PubMed] [Google Scholar]
- 3. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672‐1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Erkkinen MG, Kim MO, Geschwind MD. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2018;10(4):a033118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Montalbano M, McAllen S, Cascio FL, et al. TDP‐43 and tau oligomers in Alzheimer's disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol Dis. 2020;146:105130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kohli MA, John‐Williams K, Rajbhandary R, et al. Repeat expansions in the C9ORF72 gene contribute to Alzheimer's disease in Caucasians. Neurobiol Aging. 2013;34(5):1519.e5‐1519.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brouwers N, Bettens K, Gijselinck I, et al. Contribution of TARDBP to Alzheimer's disease genetic etiology. J Alzheimers Dis. 2010;21(2):423‐430. [DOI] [PubMed] [Google Scholar]
- 8. Xiao T, Jiao B, Zhang W, et al. Identification of CHCHD10 mutation in Chinese patients with Alzheimer disease. Mol Neurobiol. 2017;54(7):5243‐5247. [DOI] [PubMed] [Google Scholar]
- 9. Xiao X, Yuan Z, Guo L, et al. The role of frontotemporal dementia associated genes in patients with Alzheimer's disease. Neurobiol Aging. 2021;107:153‐158. [DOI] [PubMed] [Google Scholar]
- 10. Kämäläinen A, Viswanathan J, Natunen T, et al. GRN variant rs5848 reduces plasma and brain levels of granulin in Alzheimer's disease patients. J Alzheimers Dis. 2013;33(1):23‐27. [DOI] [PubMed] [Google Scholar]
- 11. Dobson‐Stone C, Hallupp M, Shahheydari H, et al. CYLD is a causative gene for frontotemporal dementia ‐ amyotrophic lateral sclerosis. Brain. 2020;143(3):783‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dobson‐Stone C, Luty AA, Thompson EM, et al. Frontotemporal dementia‐amyotrophic lateral sclerosis syndrome locus on chromosome 16p12.1‐q12.2: genetic, clinical and neuropathological analysis. Acta Neuropathol. 2013;125(4):523‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tábuas‐Pereira M, Santana I, Kun‐Rodrigues C, Bras J, Guerreiro R. CYLD variants in frontotemporal dementia associated with severe memory impairment in a Portuguese cohort. Brain. 2020;143(8):e67. [DOI] [PubMed] [Google Scholar]
- 14. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456‐2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di Pietro F, Ortenzi F, Tilio M, Concetti F, Napolioni V. Genomic DNA extraction from whole blood stored from 15‐ to 30‐years at −20 °C by rapid phenol‐chloroform protocol: a useful tool for genetic epidemiology studies. Mol Cell Probes. 2011;25(1):44‐48. [DOI] [PubMed] [Google Scholar]
- 17. Li H, Durbin R. Fast and accurate long‐read alignment with burrows‐wheeler transform. Bioinformatics. 2010;26(5):589‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20(9):1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu X, Chen Y, Wei Q, et al. Rare CYLD variants in Chinese patients with amyotrophic lateral sclerosis. Front Genet. 2021;12:740052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Q, Dai C, Zhang Q, Du J, Li W. Evaluation of performance of five bioinformatics software for the prediction of missense mutations. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2016;33(5):625‐628. [DOI] [PubMed] [Google Scholar]
- 22. Yamashita S, Matsuo Y, Tawara N, et al. CYLD dysregulation in pathogenesis of sporadic inclusion body myositis. Sci Rep. 2019;9(1):11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF‐kappaB activation and diverse biological processes. Cell Death Differ. 2010;17(1):25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Komander D, Lord CJ, Scheel H, et al. The structure of the CYLD USP domain explains its specificity for Lys63‐linked polyubiquitin and reveals a B box module. Mol Cell. 2008;29(4):451‐464. [DOI] [PubMed] [Google Scholar]
- 25. Sun L, Gao J, Huo L, et al. Tumour suppressor CYLD is a negative regulator of the mitotic kinase Aurora‐B. J Pathol. 2010;221(4):425‐432. [DOI] [PubMed] [Google Scholar]
- 26. Qian W, Shi J, Yin X, et al. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK‐3beta. J Alzheimers Dis. 2010;19(4):1221‐1229. [DOI] [PubMed] [Google Scholar]
- 27. Liu S, Wang X, Li Y, Xu L, Yu X, Ge L, Li J, Zhu Y, He S Necroptosis mediates TNF‐induced toxicity of hippocampal neurons. Biomed Res Int 2014;2014:290182, 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]