

Abstract
Mesenchymal chondrosarcoma is a rare, high‐grade, primitive mesenchymal tumor. It accounts for around 2–10% of all chondrosarcomas and mainly affects adolescents and young adults. We previously described the HEY1–NCOA2 as a recurrent gene fusion in mesenchymal chondrosarcoma, an important breakthrough for characterizing this disease; however, little study had been done to characterize the fusion protein functionally, in large part due to a lack of suitable models for evaluating the impact of HEY1–NCOA2 expression in the appropriate cellular context. We used iPSC‐derived mesenchymal stem cells (iPSC‐MSCs), which can differentiate into chondrocytes, and generated stable transduced iPSC‐MSCs with inducible expression of HEY1–NCOA2 fusion protein, wildtype HEY1 or wildtype NCOA2. We next comprehensively analyzed both the DNA binding properties and transcriptional impact of HEY1–NCOA2 expression by integrating genome‐wide chromatin immunoprecipitation sequencing (ChIP‐seq) and expression profiling (RNA‐seq). We demonstrated that HEY1–NCOA2 fusion protein preferentially binds to promoter regions of canonical HEY1 targets, resulting in transactivation of HEY1 targets, and significantly enhances cell proliferation. Intriguingly, we identified that both PDGFB and PDGFRA were directly targeted and upregulated by HEY1‐NCOA2; and the fusion protein, but not wildtype HEY1 or NCOA2, dramatically increased the level of phospho‐AKT (Ser473). Our findings provide a rationale for exploring PDGF/PI3K/AKT inhibition in treating mesenchymal chondrosarcoma. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: mesenchymal chondrosarcoma, HEY1–NCOA2 fusion, RNA‐seq, ChIP‐seq
Introduction
Gene fusions derived from chromosome rearrangements have been recognized as driver mutations in neoplasia. Especially in bone and soft‐tissue tumors, more than 200 different fusions have been reported [1, 2, 3, 4]. Most of these fusions can serve as ideal molecular diagnostic markers because they are strongly associated with particular histological subtypes. Furthermore, detailed studies of their functions have provided pivotal knowledge about pathogenetic mechanisms for targeted cancer therapy.
The HEY1–NCOA2 fusion was identified as a recurrent fusion in mesenchymal chondrosarcoma by Wang et al in 2012 [5]. This fusion has been used successfully as a molecular marker for the diagnosis of mesenchymal chondrosarcoma [6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22]. HEY1 (hairy/enhancer‐of‐split related with YRPW motif 1) is a member of the basic helix‐loop‐helix‐orange (bHLH‐O) family of transcriptional repressors and a downstream effector of Notch signaling [23]. The protein consists of a basic helix‐loop‐helix (bHLH) domain, the Orange domain and two conserved C‐terminal motifs of which the function remains to be elucidated (Figure 1A). HEY1 protein, via its basic domain, directly contacts DNA and preferentially binds E‐box sequences (CACGTG, CACGCG) in the promoter regions of target genes as a dimer, recruiting corepressors to repress the target genes of Notch signaling [24].
Figure 1.
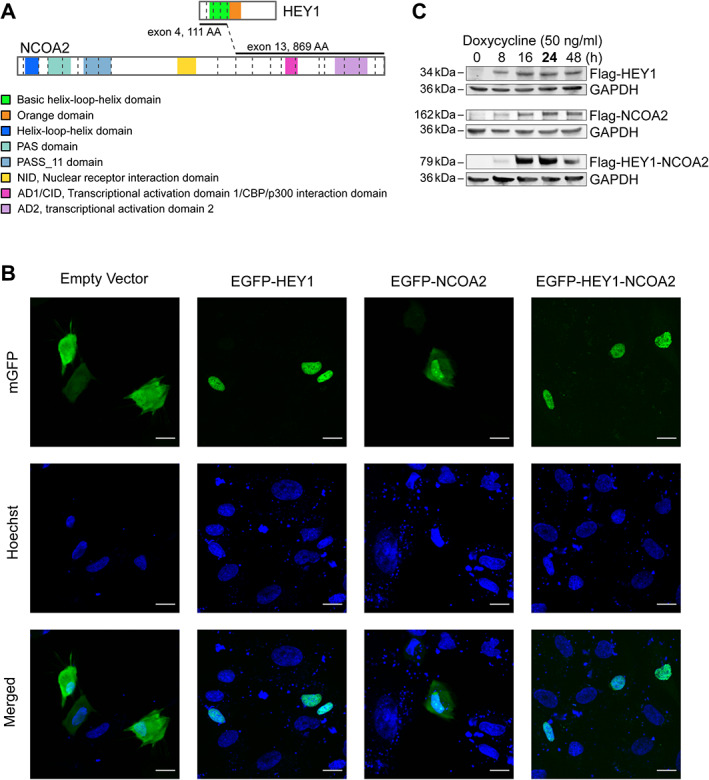
Schematic diagrams of HEY1, NCOA2, and HEY1–NCOA2 chimeric proteins and their intracellular localization, as well as the induced expression of FLAG‐tagged HEY1, NCOA2, and HEY1–NCOA2 in the iPSC‐MSCs cell models. (A) Schematic diagrams of HEY1, NCOA2, and HEY1–NCOA2 proteins. Dashed lines represent exon–exon borders. Only coding exons are presented. (B) The mEGFP‐HEY1, mEGFP‐NCOA2, and mEGFP‐HEY1–NCOA2 constructs as well as the empty vector were introduced into iPSC MSC cells transiently and photographed at 24 h. The first row shows the direct EGFP fluorescence, the second row shows Hoechst staining of DNA, and the third row shows the merged image indicating the intracellular localization. Bars, 20 μm. (C) Immunoblot using anti‐Flag antibody showed the induced expression of FLAG‐tagged HEY1–NCOA2, HEY1, and NCOA2 in stably transduced iPSC‐MSCs.
NCOA2 (Nuclear Receptor Coactivator 2) is a transcriptional coactivator for nuclear hormone receptors, including steroid, thyroid, retinoid, and vitamin D receptors. NCOA2 has three distinct regions, i.e. an N‐terminal bHLH‐PAS domain consisting of a bHLH sequence followed by a tandem PAS domain that mediates DNA binding and protein dimerization, a central area of nuclear receptor interaction domain (NID) with three LXLL motifs that drive NCOA2 interaction with nuclear hormone receptors, and a C‐terminal region that contains two transcriptional activation domains: AD1/CID (transcriptional activation domain 1/CBP/p300 interaction domain) and AD2 (transcriptional activation domain 2), which respectively recruit CBP/p300 and histone methyltransferases to facilitate transcription [25]. NCOA2 has been reported to be involved in translocations that results in fusions with multiple genes in various cancers [26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39]. Of note, in cancer‐associated NCOA2 fusions, only the C‐terminal AD1/CID and AD2 domains are consistently preserved in the fusion proteins. The involvement of the same functional domains of NCOA2 in various cancer gene fusions support a model in which the C‐terminal transcriptional activation domains (AD1/CID and AD2) of NCOA2 are aberrantly directed and utilized by the DNA‐binding domain contributed by the N‐terminal fusion partner.
The HEY1–NCOA2 fusion transcript identified in mesenchymal chondrosarcoma represents an in‐frame fusion of HEY1 5’‐exons (NM_012258, exons 1–4) to NCOA2 3’‐exons (NM_006540, exons 13–23) [5]. As such, it is predicted that the HEY1‐NCOA2 fusion protein replaces the C‐terminal portion of HEY1 with the NCOA2 transcriptional activation domains, AD1/CID and AD2, while retaining the HEY1 bHLH domain (Figure 1A). It was hypothesized that the HEY1‐NCOA2 fusion protein may act as a transcriptional activator by recruiting coactivators through its NCOA2 component to HEY1 target genes. In this study, we present a comprehensive analysis of both the DNA binding properties and transcriptional impact of HEY1‐NCOA2 expression. We demonstrate that the fusion protein preferentially binds to regions that are occupied by the wildtype HEY1 transcription factor and provide a correlation with its effect on gene expression.
Materials and methods
Cells and reagents
The human cell lines HEK293T (CRL3216) and iPSC‐derived Mesenchymal Stem Cells (iPSC‐MSCs, ACS‐7010) were purchased from the ATCC (Manassas, VA, USA) and maintained in the recommended medium and fetal bovine serum with 100 units/ml penicillin/streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. The polybrene infection/transfection reagent (TR‐1003), doxycycline hydrochloride, 3xFlag peptide and anti‐Flag M2 affinity gel (A2220) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Anti‐Flag M2 antibody (1:1,000, mouse monoclonal, F1804/M2) was from Sigma‐Aldrich. Anti‐CCND1 (1:1,000, rabbit monoclonal, 55506/E3P5S), anti‐HES1 (1:500, rabbit monoclonal, 11988/D6P2U), anti‐BCL2 (1:500, mouse monoclonal, 15071/124), anti‐phospho‐Akt (Ser473) (1:500, rabbit monoclonal, 4060/D9E), and anti‐GAPDH (1:1,000, rabbit monoclonal, 2118/14C10) were from Cell Signaling Technology (Danvers, MA, USA).
Construction of cell lines for ChIP‐seq, RNA‐seq and cell proliferation studies
Human HEY1 (CAT#: RC200257) and NCOA2 (CAT#: RC212235) cDNA clones, from OriGene Technologies (Rockville, MD, USA), were used to construct N‐terminal Flag‐tag full‐length coding sequences, i.e. FLAG‐HEY1, FLAG‐NCOA2, and FLAG‐HEY1‐NCOA2. In brief, the amplified coding sequence with Flag‐tag were cloned into the expression vector pINDUCER21 (#46948, Addgene, Watertown, MA, USA) using Gateway LR clonase II enzyme system (Invitrogen, Waltham, MA, USA). The selected clones were verified by Sanger sequencing. For the HEY1‐NCOA2 fusion construct, the sequence represents the fusion point as shown in supplementary material, Figure S1. For the generation of lentivirus, one million HEK293T cells were plated in each well of a 6‐well plate for 24 h, then transiently cotransfected with lentiviral packaging helper plasmids pHDM‐G, pCAGG‐HIVgpco, pCAG4‐RTR2 and the plasmid of interest at a 1:1 ratio using FuGene HD (Promega, Madison, WI, USA) according to the manufacturer's instructions. Cells were incubated for 48 h and the supernatant containing the virus was collected for cell transduction. The iPSC‐MSC cells were infected by the prepared lentivirus with 6.6 μg of polybrene for 72 h and GFP‐positive cells were selected by fluorescence‐activated cell sorting (FACS).
Intracellular localization of the HEY1–NCOA2 fusion protein
The coding sequences of HEY1, NCOA2, and HEY1–NCOA2 were cloned into the vector CL20‐mEGFP to generate fusions of EGFP with HEY1, NCOA2, or HEY1‐NCOA2, respectively. The iPSC‐MSC cells were cultured in μ‐Slide 4 Well (ibidi GmbH, Gräfelfing, Germany) and transiently transfected with the EGFP‐constructs for 24 h. Hoechst was directly added into the cultures and the subcellular localization of HEY1, NCOA2, and HEY1‐NCOA2 was observed using fluorescence microscopy. Images were capture using a Marianas 2 confocal microscope (Zeiss, White Plains, NY, USA).
ChIP‐seq
Stably transduced iPSC‐MSCs were treated with or without doxycycline (50 ng/ml) for 24 h, then subjected to the ChIP‐seq process following the protocol reported previously [40]. Immunoprecipitation was performed using anti‐Flag M2 affinity gel (Sigma, A2220) according to the manufacturer's protocol. Eluted samples were incubated with RNase A (50 μg/ml final concentration) at 37 °C for 30 min, overnight with proteinase K to a final concentration of 200 μg/ml at 65 °C, and cleaned up using a Monarch PCR & DNA Cleanup Kit (T1030; New England BioLabs, Ipswich, MA, USA). Two biological replicates were prepared and sequenced independently. In brief, DNA was quantified using the Quant‐iT PicoGreen ds DNA assay (ThermoFisher, Waltham, MA, USA). Libraries were prepared with the HyperPrep Library Preparation Kit (PN 07962363001, Roche, Indianapolis, IN, USA). Libraries were quantified using an Quant‐iT PicoGreen ds DNA assay (ThermoFisher) or by low‐pass sequencing with a MiSeq nano kit (Illumina, San Diego, CA, USA). Single or paired end 50 cycle sequencing was performed on a NovaSeq 6000 platform (Illumina) at the Hartwell Center for Biotechnology at the St. Jude Children's Research Hospital. Details for ChIP‐seq data analysis are presented in Supplementary materials and methods.
RNA‐seq and gene expression analysis
Stably transduced iPSC‐MSCs were treated with or without doxycycline (50 ng/ml) for 24 h, then subjected to RNA extraction using the Quick‐RNA Mini Prep kit (Zymo Research, Irvine, CA, USA). RNA‐sequencing was performed by the St. Jude Hartwell Center of Biotechnology. In brief, RNA was quantified using the Quant‐iT RiboGreen RNA assay (ThermoFisher) and quality checked using a 2100 Bioanalyzer RNA 6000 Nano assay (Agilent, Santa Clara, CA, USA) or 4200 TapeStation High Sensitivity RNA ScreenTape assay (Agilent) prior to library generation. Libraries were prepared from total RNA with a TruSeq Stranded mRNA Library Prep Kit according to the manufacturer's instructions (PN 20020595, Illumina). Paired end 100 cycle sequencing was performed using a NovaSeq 6000 (Illumina). Details for RNA‐seq data analysis as well as pathway and gene set enrichment analyses (GSEA) are described in Supplementary materials and methods.
Immunoblotting
Cell lysates were prepared with Laemmli buffer and separated on a 4–15% Mini‐Protean precast gels (Bio‐Rad, Hercules, CA, USA). The proteins were then transferred onto a nitrocellulose membrane and nonspecific binding was blocked by incubating with 5% nonfat milk in TBST buffer (0.01 m Tris–Cl, 0.15 m NaCl, 0.05% Tween‐20, pH 8.0) at room temperature for 1 h. The membrane was subjected to the indicated primary antibody with gentle agitation overnight at 4 °C and then horseradish peroxidase (HRP)‐conjugated secondary antibody at room temperature for 1.5 h. The proteins were detected using a ChemiDoc touch imaging system (Bio‐Rad) after the membrane was incubated in Clarity western ECL substrate (Bio‐Rad) for 5 min.
Real‐time quantitative polymerase chain reaction (RT‐qPCR)
Stably transduced iPSC‐MSCs were treated with or without doxycycline (50 ng/ml) for 24 h, then subjected to RNA extraction using the Quick‐RNA Mini Prep kit (Zymo Research). One μg of RNA was reverse‐transcribed into cDNA using a High‐Capacity RNA‐to‐cDNA kit (ThermoFisher, Cat#: 4387406), and quantitative polymerase chain reaction (qPCR) was performed in triplicate using iTaq Universal SYBR Green Supermix (Bio‐Rad, Cat#: 172‐5120) on the Applied Biosystems QuantStudio 7 Flex Real‐Time PCR platform. Expression of mRNA was normalized to GAPDH as a reference transcript, and the fold‐change of expression was calculated using the 2−ΔΔCt method. The sequences of PCR Primers used are listed in supplementary material, Table S1.
Cell proliferation assays
Stably transduced iPSC‐MSCs were seeded in a 6‐well plate (1 × 105 per well) for 24 h and then subjected to a cell proliferation study. Medium with or without doxycycline (50 ng/ml) respectively was used and replenished every 3 days. The cell number was counted using a Countess II FL Automated Cell Counter (Life Technologies, Waltham, MA, USA) at seeding and at day 6 and day 12 after the initial doxycycline treatment. Additionally, Click‐iT EdU flow cytometry cell proliferation assays (Invitrogen, C10634) were performed according to the manufacturer's protocol. In brief, cells were cultured with or without doxycycline (50 ng/ml) for 1 week, labeled with 10 μm EdU for 3 h, and then harvested, fixed, and stained using the Click‐iT Plus reaction cocktail with fluorescent picolyl azide and analyzed by flow cytometry using FlowJo software (v10.7.1, FlowJo, Ashland, OR, USA).
Three‐dimensional (3D) cell culture assay
3D cell cultures were performed in 96‐well, round black/clear bottom, ULA (Ultra‐Low Attachment surface) Corning spheroid microplates (Sigma, CLS4520). In each well, 1,500 stably transduced iPSC‐MSCs were cultured with or without doxycycline (50 ng/ml). Spheroid formation and growth were visualized microscopically at 24 h (day 1), 48 h (day 2), 72 h (day 3), 96 h (day 4), and day 7 after cell seeding. The sizes (radius) of spheroids were measured using TCapture imaging software (v5.1.1, Tucsen Photonics Co., Fuzhou, Fujian, PR China).
Results
Intracellular localization of HEY1–NCOA2 fusion protein
In order to evaluate the intracellular localization of HEY1–NCOA2, HEY1, and NCOA2, we generated EGFP‐ tagged constructs and monitored protein localization by fluorescence microscopy in transfected iPSC‐MSCs. We confirmed that HEY1–NCOA2, like wildtype HEY1, is localized to the nucleus. In contrast, wildtype NCOA2 is located in both the nucleus and cytoplasm (Figure 1B). In comparing the GFP signal patterns between EGFP‐HEY1‐NCOA2 and EGFP‐HEY1, our data showed similar intranuclear distribution of HEY1–NCOA2 and wildtype HEY1.
Identification of HEY1–NCOA2 DNA‐binding sites
The induced expression of FLAG‐tagged HEY1–NCOA2, HEY1, and NCOA2 proteins in stably transduced iPSC‐MSCs were verified by immunoblotting at 8, 16, 24, and 48 h after doxycycline induction (Figure 1C), and the genome‐wide binding profiles of HEY1‐NCOA2, HEY1, and NCOA2 were investigated through anti‐FLAG ChIP‐seq in each iPSC‐MSC cell model at 24 h after doxycycline (50 ng/ml) induction. By genome‐wide ChIP‐seq analyses, a total of 30,142 and 3,740 DNA‐binding peaks were identified for HEY1 and HEY1–NCOA2, respectively. No specific DNA‐binding peaks were identified with the induced expression of NCOA2 in comparison to controls, which is consistent with prior knowledge about NCOA2. NCOA2 is one of the members of the p160/steroid receptor coactivator (SRC) family. SRCs have not been shown to directly bind DNA, despite possessing the bHLH domain [41]. In reviewing the location of HEY1 and HEY1–NCOA2 binding sites in the genome, we observed remarkable overall similarity between those two (supplementary material, Figure S2A). Further comparison revealed that 92% (n = 3,445) of HEY1‐NCOA2 DNA‐binding sites were shared with HEY1 (Figure 2A). Interestingly, out of all those shared DNA‐binding targets, 88.5% are located within promoter regions (Figure 2A); in contrast, only 60.3% of HEY1‐only DNA‐binding peaks were located in promoter regions and HEY1–NCOA2‐only binding peaks (n = 295) exhibited a dramatically different binding profile across the genome (Figure 2A). The enrichment analysis of the ChIP‐seq data further demonstrated the remarkable similarity in the genome‐wide DNA binding feature between HEY1–NCOA2 fusion protein and wildtype HEY1 (Figure 2B), and suggested that the DNA regions bound by both HEY1 and HEY1–NCOA2 are enriched in high‐affinity binding targets of HEY1 (Figure 2C).
Figure 2.
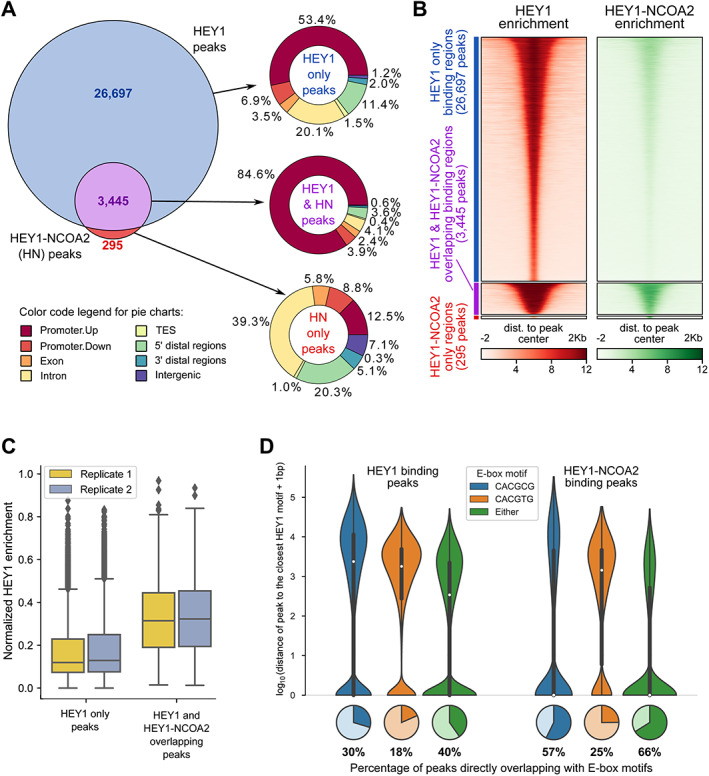
HEY1–NCOA2 fusion protein DNA‐binding pattern in the genome. (A) Genomic context of HEY1 and HEY1–NCOA2 binding peaks, with a breakdown into three groups: (1) Peaks that are overlapping between HEY1 and HEY1–NCOA2 fusion protein, (2) HEY1‐only binding peaks, and (3) HEY1–NCOA2‐only binding peaks. (B) Signal enrichment heatmap for the peaks specific for HEY1 only (blue bar), HEY1–NCOA2 only (red bar), and overlapping peaks (purple bar). (C) Normalized enrichment of HEY1‐binding targets, comparing HEY1‐only peaks and HEY1‐binding peaks overlapping with HEY1–NCOA2‐binding peaks. (D) E‐box sequences bound by the HEY1 and HEY1–NCOA2 fusion protein. Distribution of the distance between binding peaks and the closest E‐box motif(s); the distance equals 0 for the direct overlap between peak and motif, which fraction is visualized with pie plots.
The E‐box (CACGTG, CACGCG) DNA elements are HEY1‐preferred DNA binding motifs [24]. Therefore, we searched next for the E‐box sequences in our ChIP‐seq data. Indeed, as shown in Figure 2D, the E‐box sequences were identified in 12,157 out of 30,142 HEY1 binding peaks (40%) and 2,502 out of 3,740 HEY1–NCOA2 binding peaks (66%). Strikingly, about 98.8% (2,472 out of 2,502) of HEY1–NCOA2 binding peaks that contain E‐box sequences overlap with HEY1 binding peaks (supplementary material, Figure S2B).
Gene expression profile associated with HEY1–NCOA2
Our ChIP‐data demonstrated that HEY1–NCOA2 fusion protein preferentially binds to promoter regions across the genome that are also high‐affinity binding targets of HEY1 with canonical E‐box binding motif, which confirmed our hypothesis based on the predicted structure of HEY1–NCOA2 fusion protein. We speculated further that the expression of those target genes, which are normally repressed by HEY1, may be transactivated in the presence of HEY1–NCOA2 fusion protein, given that the NCOA2 transcriptional activation domains replace the C‐terminal portion of HEY1. To verify this hypothesis, we performed RNA‐seq in stably transduced iPSC‐MSCs expressing exogenous HEY1, NCOA2, and HEY1–NCOA2 after 24 h doxycycline induction, which we will refer to as ‘MSC‐HEY1(+),’ ‘MSC‐NCOA2(+),’ and ‘MSC‐HEY1–NCOA2(+)’ respectively, in this article. Transduced iPSC‐MSCs that lacked doxycycline treatment, which we will refer to as ‘MSC‐HEY1ctrl,’ ‘MSC‐NCOA2ctrl’ and ‘MSC‐HEY1‐NCOA2ctrl,’ respectively, were similarly profiled as controls. Overall, MSC‐HEY1‐NCOA2(+), MSC‐HEY1(+), and MSC–NCOA2(+) cells can be robustly separated by gene expression profiling (supplementary material, Figure S3A,B). Differential gene expression (DGE) analysis was performed to compare gene expression values between MSC‐HEY1‐NCOA2(+) and MSC‐HEY1(+) cells and identified a total of 2,440 differentially expressed genes (fold change >2, false discovery rate [FDR] < 0.05), about two‐thirds of which (1,563; 64%) showed increased expression in MSC‐HEY1‐NCOA2(+) cells (Figure 3A, supplementary material, Table S2). In addition, DGE analysis was performed to compare gene expression values between MSC‐HEY1‐NCOA2(+) and MSC‐HEY1‐NCOA2ctrl, MSC‐HEY1(+) and MSC‐HEY1ctrl, as well as MSC‐NCOA2(+) and MSC‐NCOA2ctrl, respectively. Differentially expressed genes identified in each matched pair were subjected to Gene Set Enrichment Analysis (GSEA). As shown in Figure 3B, genes downregulated by HEY1 [MSC‐HEY1(+) versus MSC‐HEY1ctrl] were significantly enriched among HEY1‐NCOA2‐upregulated genes (NES = 1.8, p value ≈ 0, FDR ≈ 0). GSEA analysis showed no significant correlation in the expression between NCOA2‐regulated and HEY1–NCOA2‐regulated genes (data not shown). Further on, as shown in Figure 3C, the additional GSEA showed that genes that were directly targeted by both HEY1 and HEY1‐NCOA2 (binding peaks identified in their promoters by ChIP‐seq, n = 3,194), were significantly enriched in the gene set that were upregulated by HEY1–NCOA2 in comparison to HEY1 [MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1(+)]. The integrated genome‐wide ChIP‐seq and RNA‐seq expression profiling demonstrated that HEY1‐NCOA2 fusion protein activated the expression of canonical HEY1 target genes that were transcriptionally repressed by wildtype HEY1.
Figure 3.
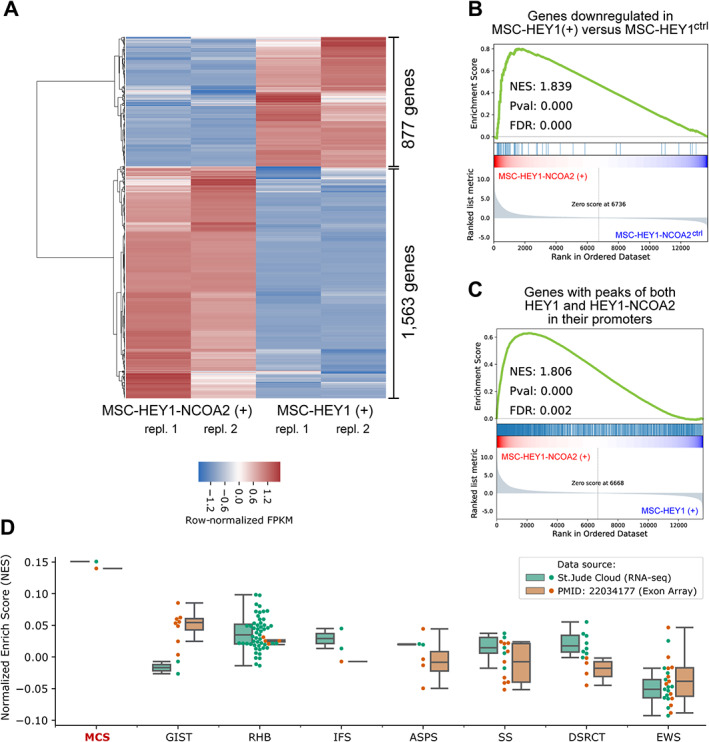
Gene expression profile associated with HEY1–NCOA2. (A) Heatmap showing row‐normalized expression level of genes differentially regulated by HEY1‐NCOA2 in comparison to wildtype HEY1 [MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1(+)]. (B) GSEA Enrichment plot of the expression of genes that were identified as downregulated by HEY1 in MSC‐HEY1(+) versus MSC‐HEY1ctrl. The gene rank was based on the log2(fold‐change) of the expression of genes in MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1‐NCOA2ctrl. (C) GSEA enrichment plot for the expression of genes of which their promoters had binding peaks of both HEY1 and HEY1–NCOA2. The gene rank was based on the log2(fold‐change) of the gene expression in MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1(+). (D) Boxplots visualizing the enrichment of the 674 HEY1–NCOA2 direct‐target‐and‐transactivating genes identified using the iPSC‐MSCs cell models in sarcoma tumor samples. The enrichment was visualized across patients' samples from a total of eight sarcoma entities. GSEA, Gene Set Enrichment Analysis; NES, Normalized Enrichment Score; FDR, False Discovery Rate; MCS, Mesenchymal Chondrosarcoma; GIST, Gastrointestinal Stromal tumor; RHB, Rhabdomyosarcoma; IFS, Infantile Fibrosarcoma; ASPS, Alveolar Soft Part Sarcoma; SS, Synovial Sarcoma; DSRCT, Desmoplastic Small Round Cell Tumors; EWS, Ewing's Sarcoma.
Correlation between HEY1 and HEY1–NCOA2 binding profiles and gene expression
Cross‐comparison between ChIP‐seq and RNA‐seq expression data revealed that 674 out of 1,563 genes (43%) upregulated by HEY1–NCOA2 [MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1(+)] had binding peaks for HEY1–NCOA2 fusion protein as well as HEY1 in their promoter regions (the list of 674 genes, which we will refer to as ‘HEY1–NCOA2 key‐direct targets’ in this article, is available in supplementary material, Table S3). This finding indicates that almost half of the genes upregulated by HEY1–NCOA2 may result from direct transcriptional activation by the fusion protein. The 674 HEY1–NCOA2 key‐direct targets include previously identified targets of HEY1, such as HES1, KLF10, BMP2, and FOXC1 [42], as well as a variety of novel findings, including PDGFB, PDGFRA, SOX4, etc. HEY1–NCOA2 and HEY1 binding peaks in the promoter regions of HES1, PDGFB, PDGFRA, and SOX4, demonstrated by ChIP‐seq analysis, are presented in supplementary material, Figure S4.
Validation of the data derived from the in vitro iPSC‐MSC models on human mesenchymal chondrosarcoma tumor samples
To validate if the transcriptional dysregulation profile that were identified in the in vitro iPSC‐MSC‐HEY1‐NCOA2 model recapitulate the molecular phenotype of human mesenchymal chondrosarcoma, we performed single‐sample GSEA (ssGSEA) analysis to assess the expression of the 674 HEY1–NCOA2 key‐direct targets in a set of RNA‐seq data derived from 107 sarcoma patients' samples (RNA‐seq data downloaded from St. Jude Cloud, https://platform.stjude.cloud/), including one mesenchymal chondrosarcoma sample as well as 106 samples from eight other sarcoma entities, i.e. Gastrointestinal Stromal Tumor (GIST), Rhabdomyosarcoma (RHB), Infantile fibrosarcoma (IFS), Alveolar Soft Part Sarcoma (ASPS), Synovial Sarcoma (SS), Desmoplastic Small Round Cell Tumor (DSRCT), Ewing's Sarcoma (EWS), and Osteosarcoma (OS). The same ssGSEA strategy was also applied to the analysis of Exon‐Array data of 57 sarcoma samples published by Wang et al [5], in which the HEY1–NCOA2 fusion was identified as a novel and recurrent genetic alteration in mesenchymal chondrosarcoma (Exon‐Array data downloaded from http://cbio.mskcc.org/Public/sarcoma_array_data/). The detailed information about all samples accrued for the ssGSEA analysis are presented in supplementary material, Table S4. Our ssGSEA analysis, both in the RNA‐seq dataset and in the Exon‐array dataset, demonstrated that the 674 HEY1–NCOA2 key‐direct targets are highly enriched in the expression profile of mesenchymal chondrosarcoma tumor samples. As shown in Figure 3D, the Normalized Enrichment Scores (NESs) of the 674 genes in mesenchymal chondrosarcoma tumor samples are around 1.5; in contrast, the mean NESs in other sarcoma cases is between −0.5 and 0.5. Of note, samples of tumor types that are available in both the St. Jude cloud RNA‐seq dataset and the Exon‐array dataset are presented in Figure 3D. The ssGSEA NESs of all 164 samples are available in supplementary material, Table S5, and the corresponding box plots of NESs are presented in supplementary material, Figure S5. These results, although limited by the data availability, suggest that the key HEY1–NCOA2‐upregulated genes identified in our in vitro iPSC‐MSC cell models indeed represent the genes highly expressed in mesenchymal chondrosarcoma patients' tumors.
Functional classification of genes upregulated by the HEY–NCOA2 fusion
Functional classification of the 1,563 HEY1–NCOA2 upregulated genes was performed by pathway enrichment analysis. The top 10 significantly enriched pathways in KEGG are presented in Figure 4A, including cell cycle pathway, Hedgehog and WNT signaling pathways, as well as the PI3K–Akt signaling pathway, all of which were linked with promoting cell proliferation in cancer (refer to supplementary material, Table S6 for all KEGG pathways significantly enriched). The enrichment of these pathways was further supported by GSEA from the comparisons: (1) MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1‐NCOA2ctrl and (2) MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1(+) (Figure 4B, first two columns); whereas, in the direct comparison of MSC‐HEY1(+) versus MSC‐HEY1ctrl, GSEA demonstrated downregulation of the gene signatures related to the cell cycle in MSC‐HEY1(+) (Figure 4B, last column). The original GSEA enrichment plots are available in supplementary material, Figure S6.
Figure 4.
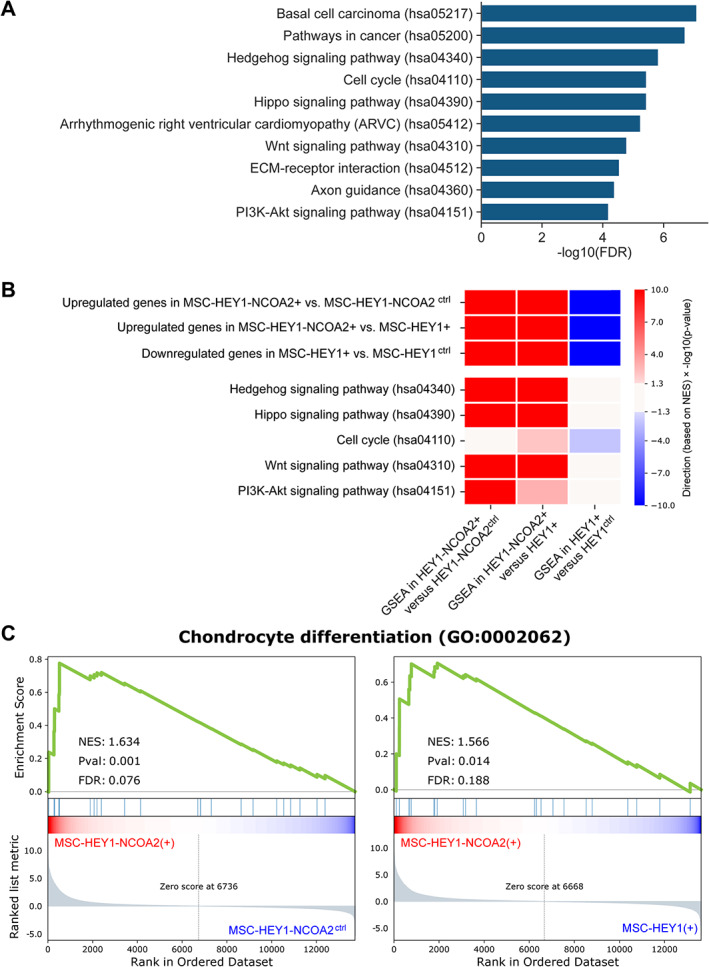
Functional pathways enriched in HEY1–NCOA2 upregulated genes. (A) Bar plot showing the −log10(FDR) of the top‐10 enriched KEGG pathways, computed for 1,563 HEY1–NCOA2 genes upregulated by HEY1–NCOA2 as compared with HEY1 [MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1(+)]. (B) Heatmap summarizing the GSEA (Gene Set Enrichment Analysis) of selected gene sets. Each row represents one gene set. Row #4–#8, gene signatures were selected based on the KEGG pathway enrichment analysis. The values in the heatmap indicate the −log10 of p value, multiplied by −1 if the NES score was indicating the enrichment biased toward the negative phenotype. (C) GSEA enrichment plot of MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1‐NCOA2ctrl and MSC‐HEY1‐NCOA2(+) versus MSC‐HEY1(+). The gene set ‘Chondrocyte Differentiation’ is from Gene Ontology (GO). The false discovery rate (FDR), nominal p value and normalized enrichment score were calculated by GSEA.
Using the GSEA approach, we also demonstrated that chondrocyte differentiation genes (GO:0002062) are significantly enriched among HEY1–NCOA2‐upregulated genes (Figure 4C) and core enriched genes included WNT10B, WNT7A, MEF2C, etc. Mesenchymal chondrosarcoma is a neoplasm of very early prechondrogenic cells [43], so we reasoned that it would be crucial to model this fusion in the appropriate cellular context. As mesenchymal stem cells are multipotent stem cells that are capable of self‐renewing and differentiating into functional cell types, including chondrocytes [44], the iPSC‐derived mesenchymal stem cells (iPSC‐MSCs) were used in our study to generate the in vitro cell models. The findings from GSEA of chondrocyte differentiation genes not only validated the use of our HEY1–NCOA2‐expressing iPSC‐MSC cell model for studying mesenchymal chondrosarcoma, but also suggested that the HEY1‐NCOA2 fusion may drive the differentiation of mesenchymal stem cells toward chondrogenic lineage; unfortunately, as revealed by the overall functional characterization of HEY1‐NCOA2 upregulated genes, the fusion protein also rewires the transcriptional network during the differentiation, which may result in the development of mesenchymal chondrosarcoma.
Verification of the expression of selected targets upregulated by HEY1–NCOA2
The expression of 13 selected HEY1–NCOA2‐upregulated genes was verified by RT‐qPCR in an independent set of RNAs. As shown in Figure 5A, the significantly increased expression of these genes in HEY1–NCOA2‐expressing cells [MSC‐HEY1‐NCOA2(+)] was verified by RT‐qPCR.
Figure 5.
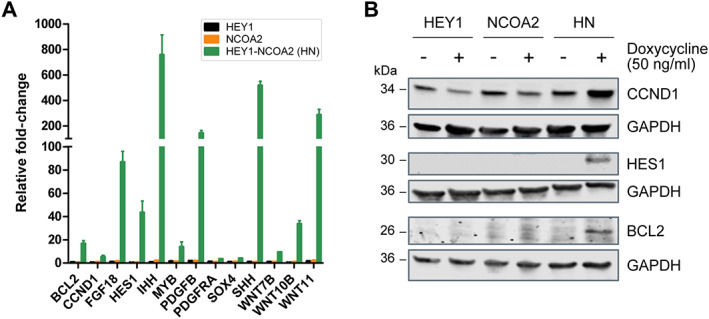
HEY1–NCOA2 target gene expression validation. (A) RT‐qPCR validation of genes regulation by inducible HEY1, NCOA2, or HEY1–NCOA2 expression in iPSC MSC. The graph represents the fold change between the induced and uninduced condition. (B) Immunoblotting was performed to further confirm the expression of BCL2, CCND1, and HES1 after HEY1–NCOA2 induction in stably transduced iPSC‐MSCs. GAPDH was used as a loading control.
All selected genes except CCND1 are among the 674 HEY1–NCOA2 key‐direct targets (supplementary material, Table S3). According to RNA‐seq, 10 out of the 12 genes barely had a detectable expression [Transcripts Per Million (TPM) << 1)] in control cells (MSC‐HEY1ctrl, MSC‐NCOA2ctrl, and MSC‐HEY1‐NCOA2ctrl). As for the remaining two genes, SOX4 and PDGFRA, RNA‐seq revealed ~4 and ~8 transcripts per million RNA molecules in control cells, respectively. In contrast, RNA‐seq revealed relatively high expression of CCND1 in control cells (~245 TPM). Moreover, the differential gene expression analysis demonstrated that HEY1 significantly downregulates CCND1 expression (>2‐fold), while HEY1–NCOA2 dramatically upregulates its expression (>4‐fold) in our iPSC‐MSC cell models (supplementary material, Table S2). In addition to verifying the expression of CCND1 by RT‐qPCR, immunoblotting was performed that demonstrated decent CCND1 expression in controls cells, decreased CCND1 expression in MSC‐HEY1(+), and dramatically increased CCND1 expression in MSC‐HEY1‐NCOA2(+) (Figure 5B). In addition, as shown in Figure 5B, there was no detectable expression of HES1 or BCL2 by immunoblotting in cells without HEY1–NCOA2 expression, which is compatible with the transcription expression data (TPM) obtained from RNA‐seq. Overall, the analysis of the expression of selected genes by RT‐qPCR and immunoblotting, including 12 HEY1–NCOA2 direct targets and the indirect‐dysregulated gene CCND1, confirmed the RNA‐seq results.
HEY1–NCOA2 fusion enhances cell proliferation in iPSC MSC
RNA‐seq followed by pathway enrichment analysis revealed that the cell cycle pathway was significantly upregulated by the HEY1–NCOA2 fusion and downregulated by wildtype HEY1. We verified the effects of the HEY1–NCOA2 fusion, as well as wildtype HEY1 and NCOA2, on cell growth in stably transduced iPSC‐MSCs. As shown in Figure 6A, HEY1–NCOA2 expressing iPSC‐MSCs displayed significantly accelerated proliferation when compared with matched control cells. In contrast, cell proliferation was slightly inhibited with the induced expression of wildtype HEY1, while the induced expression of exogenous NCOA2 had no significant impact on cell growth. We next assessed cell cycle transit by an EdU incorporation assay. Compared to control cells, the proportion of HEY1–NCOA2‐expressing cells in S‐phase was increased by 8.2% (p = 0.0328) (Figure 6B), demonstrating a significant increase in cell cycle transit as a result of HEY1–NCOA2 expression.
Figure 6.
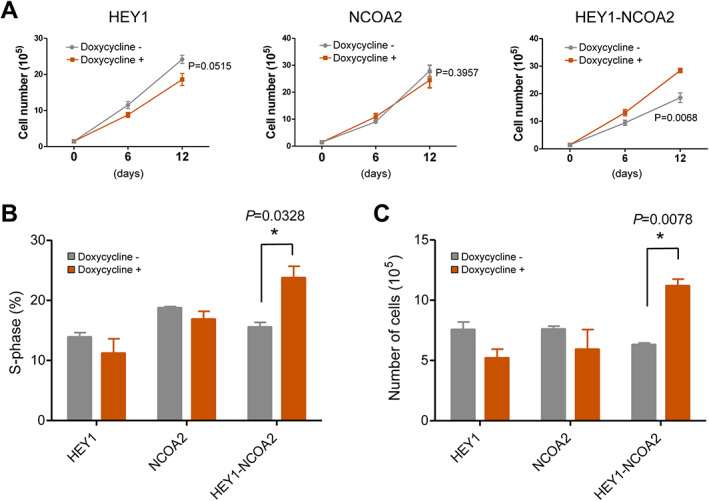
HEY1‐NCOA2 significantly increases cell proliferation in iPSC‐MSC. (A) 1 × 105 of FACS sorted GFP+ iPSC‐MSCs were seeded with or without doxycycline (50 ng/ml). Cell numbers were counted on days 6 and 12; the graph shows the mean ± SD (n = 3). (B) GFP+ iPSC‐MSCs were cultured with or without doxycycline (50 ng/ml) for 7 days and labeled with 10 μM of EdU for 3 h. Cells were then harvested, fixed, and stained with fluorescent dye 647 picolyl azide. EdU was detected by flow cytometry and the percentage of cells in S‐phase was analyzed using FlowJo software. The graph shows the mean ± SD (n = 2). (C) FACS‐sorted GFP+ iPSC‐MSCs were cultured as in panel A except without addition of rh‐FGF basic and rh‐IGF‐1 to the culture medium. Cell numbers were counted on day 7; the graph shows the mean ± SD (n = 3).
Human iPSC‐MSCs require the addition of rhFGF basic (recombinant human fibroblast growth factor, basic) and rhIGF‐1 (recombinant human insulin‐like growth factor‐1) growth factors to the growth medium as per the ATCC cell culture instruction. Since our RNA‐seq data revealed several FGF and IGF family genes were upregulated by HEY1–NCOA2 (supplementary material, Table S2), we tested whether HEY1–NCOA2 expression can induce cytokine‐independent growth. Indeed, as shown in Figure 6C, only HEY1–NCOA2 expressing iPSC‐MSCs displayed significantly accelerated proliferation in the absence of rhFGF basic and rhIGF‐1 growth factors in the culture medium (p = 0.0078).
HEY1–NCOA2 fusion increases cell growth in a 3D model
As 3D spheroid cell culture more closely resembles in vivo tissue in terms of cell–cell and cell–matrix interactions [45], we used a scaffold‐free technique to evaluate whether the fusion protein can also affect cell growth in a 3D model. FACS sorted iPSC‐MSCs stably transduced for inducible expression of HEY1, NCOA2 or HEY1–NCOA2 were seeded (1,500 cells per well, 96‐well plates) in wells with a round bottom and ultralow attachment surface. Doxycycline (50 ng/ml) was added into selected wells to induce the expression of transduced constructs, and no doxycycline in control cells' wells. After 24 h, cells of each well formed a single spheroid with a clear boundary. In addition, in the MSC‐HEY1‐NCOA2(+) wells, cell satellites around the main spheroid were visible (supplementary material, Figure S7B). As the culture continued, the spheroids shrunk in all wells except those in MSC‐HEY1‐NCOA2(+) wells. The size (radius) of each spheroid was measured at 24, 48, 72, and 96 h and 7 days. The graph in supplementary material, Figure S7A presents the mean value of radius ± SD (n = 10). Representative 3D cell growth images taken at 24 h and 72 h after seeding are shown in supplementary material, Figure S7B. The 3D culture experiment demonstrated the dramatic difference in size and morphology of spheroids between HEY1–NCOA2‐expressing iPSC‐MSCs [MSC‐HEY1‐NCOA2(+)] and wildtype protein expression cells [MSC‐HEY1(+) and MSC‐NCOA2(+)].
Discussion
Mesenchymal chondrosarcoma is a rare malignant tumor. It arises most commonly in the second and third decades of life, and the outcome for these patients appears to be poor [46]. In a recent study, the overall 5‐year and 10‐year survival rate for mesenchymal chondrosarcoma patients was 51% and 43%, respectively [47]. The discovery of the HEY1–NCOA2 fusion as a molecular marker for mesenchymal chondrosarcoma was an important breakthrough for characterizing this disease. The identification of the pathways modulated by this fusion could help elucidate the pathogenesis of this disease and potentially develop new treatment options for mesenchymal chondrosarcoma.
We hypothesized that the HEY1–NCOA2 fusion protein would drive the expression of canonical HEY1 targets through the bHLH DNA‐binding and dimerization domains of HEY1, as well as the transactivation domains of NCOA2. This was confirmed by our ChIP‐seq data, which revealed extreme similarity in genome‐wide DNA‐binding patterns between HEY1–NCOA2 and wildtype HEY1. The intracellular localization and ChIP‐seq data indicate that the oncogenic activities of HEY1–NCOA2 likely reflect its impact on HEY1‐dependent pathways. Hence, in the subsequent differential gene expression analysis of RNA‐seq data, we focused on the relationship between HEY1–NCOA2 and HEY1 in searching for HEY1–NCOA2 key target genes.
The most intriguing key targets of HEY1–NCOA2 discovered in this study are PDGFB and PDGFRA. We demonstrated that both PDGFB and PDGFRA were directly targeted and transactivated by the HEY1–NCOA2 fusion protein. PDGFB is a signaling ligand, a member of the protein family comprised of both platelet‐derived growth factors (PDGF) and vascular endothelial growth factors (VEGF). PDGFs act via two receptors, PDGFRA and PDGFRB, and PDGF‐PDGFR interactions activate PDGF receptor tyrosine kinases. By RNA‐seq and RT‐qPCR, we demonstrated that PDGFB gene expression was extremely upregulated by HEY1–NCOA2 (~105‐fold by RNA‐seq and ~120‐fold by RT‐qPCR). In comparison to PDGFB, the upregulation of PDGFRA expression was moderate (~3.75‐fold).
PDGF–PDGFR signaling mediates a variety of downstream signaling effector pathways, including PI3K–AKT, among others [48]. Additionally, functional classification of HEY1‐NCOA2 upregulated genes highlighted PI3K–AKT signaling as one of top enriched pathways in the current study. We next evaluated the activation of PI3K–AKT. Our preliminary data demonstrated that with the expression of the HEY1–NCOA2 fusion protein, but not wildtype HEY1 or NCOA2, the level of phosphor‐AKT (Ser473) dramatically increased (supplementary material, Figure S8). Of note, using approaches based on immunohistochemistry (IHC), some previous studies of mesenchymal chondrosarcoma demonstrated increased expression of PKC‐α, PDGFR‐α, and phosphorylated‐mTOR in the malignant mesenchymal chondroblasts of mesenchymal chondrosarcoma tumor samples, indicating that these pathways' potential roles in the development or maintenance of mesenchymal chondrosarcoma [49, 50]. Hence, our data, to some extent, elucidated the biologic mechanism underlying the histopathologic features that had been identified in mesenchymal chondrosarcoma patients' tumor samples. Clearly, much more work is needed to better define the role(s) of PDGF signaling in mesenchymal chondrosarcoma tumorigenesis, but our study suggests that small‐molecule inhibitors targeting PDGF signaling should be evaluated in preclinical models of mesenchymal chondrosarcoma.
Another interesting finding in this study that may provide a fundamental rationale for exploring a new treatment approach for mesenchymal chondrosarcoma is the direct transactivation of BCL2 by the HEY1–NCOA2 fusion protein. BCL2 has been reported to exhibit stronger expression in malignant mesenchymal chondroblasts in comparison to more mature‐appearing chondrocytic counterparts in mesenchymal chondrosarcoma tumor samples, by IHC staining [49]. In addition, a recent study reported that the inhibition of Bcl‐2 family members restores the apoptotic machinery in mesenchymal chondrosarcoma, rendering the cells sensitive to conventional chemotherapy [51]. In the present report, our integrated genome‐wide ChIP‐seq and expression profiling (RNA‐seq) identified BCL2 as one of the 674 key‐direct targets of the HEY1–NCOA2 fusion protein. Further on, our RT‐qPCR and immunoblotting studies verified the RNA‐seq findings of BCL2 transactivation by the HEY1–NCOA2 fusion protein (Figure 5). Overall, our findings provide a biological rationale for exploring BCL2 inhibitors in treating mesenchymal chondrosarcoma.
Other targets of interest, such as HES1 and SOX4, have a well‐credentialled role in the transcriptional control of chondrocyte specification and differentiation [52]. Chondrocytes arise in development from mesenchymal stem cells/multipotent skeletogenic progenitor‐stem cells (SSCs). HES1 was reported to delay the differentiation of SSCs into chondrocytes [53], and SOX4, along with its closely related family members SOX11 and SOX12, is expressed strongly in SSCs and prechondrocytes, and weakly in chondrocytes [54]. In a previous study, Bhattaram et al demonstrated that Sox4/Sox11/Sox12 act redundantly to keep mesenchymal progenitor cells alive in a mouse model [55]. In our study, the promoters of both HES1 and SOX4 were bound directly by HEY1–NCOA2, and both genes were significantly upregulated by HEY1–NCOA2. Further study is needed to elucidate the interplay between HES1, SOX4, and HEY1–NCOA2 in the tumorigenesis of mesenchymal chondrosarcoma.
Our study represents the first comprehensive, genome‐wide analysis of HEY1–NCOA2 DNA binding sites and correlates these to HEY1–NCOA2‐associated gene expression profiles. Our results point to a pivotal role for the HEY1–NCOA2 fusion in mesenchymal chondrosarcoma tumorigenesis. While we do not yet know whether HEY1–NCOA2 alone is sufficient to induce malignant transformation in vivo, in our model system using iPSC‐MSC, HEY1–NCOA2 appeared to act on the key aspects of increasing cell cycle transition, a recognized facet of malignant transformation, and likewise induces genes that are implicated in chondrocyte differentiation. The findings of our present study, which were generated by using a forced gene expression system based on the iPSC‐MSC cell models, remains to be verified in isogenic cell lines with the HEY1–NCOA2 fusion. Additionally, the intriguing HEY1–NCOA2 downstream targets identified in this study will be evaluated in a larger cohort of mesenchymal chondrosarcoma tumor samples to establish more precisely the specificity of these dysregulated genes and pathways in mesenchymal chondrosarcoma development. Finally, further studies on the potential downstream target genes and pathways of HEY1–NCOA2 may help to develop new therapeutic approaches for this often‐lethal malignancy.
Author contributions statement
LW, WQ and WR conceived and designed the study. WQ, WR, ZY and HJ acquired data. WR, WQ, BX and LW analyzed and interpreted data. WR, BX, SW, YF and GW were responsible for bioinformatics. All the authors critically revised the article and read and approved the final version.
Supporting information
Supplementary materials and methods
Figure S1. The breakpoint DNA sequence of the inducible‐expression pINDUCER21‐HEY1‐NCOA2 construct
Figure S2. Genomic context of HEY1 and HEY1‐NCOA2 binding peaks and Venn diagram showing the number of HEY1–NCOA2 binding peaks with E‐box motif overlapping with HEY1 binding peaks with E‐box motif
Figure S3. HEY1–NCOA2 demonstrates distinct gene expression profile
Figure S4. HEY1 and HEY1–NCOA2 binding in promoters of target genes
Figure S5. Box plots visualizing the enrichment of the 674 HEY1–NCOA2 direct‐target‐and‐transactivating genes identified using the iPSC‐MSCs cell model
Figure S6. GSEA enrichment plots
Figure S7. HEY1‐NCOA2 enhances cell growth in 3D model
Figure S8. HEY1–NCOA2 fusion protein increased the level of phospho‐AKT (Ser473)
Table S1. Sequences of primers used for RT‐qPCR
Table S2. Differential gene expression (DGE) analysis
Table S3. HEY1–NCOA2 key‐direct‐target genes
Table S4. Samples accrued for the ssGSEA analysis
Table S5. The ssGSEA Normalized Enrichment Scores (NESs) of all 164 samples
Table S6. Functional pathways enriched in HEY1–NCOA2 upregulated genes (from supplementary material, Table S2)
Acknowledgements
We thank Dr. Jeffrey M. Klco for critical review of the article and for his valuable comments. We thank the staff at St. Jude Hartwell Center of Biotechnology for performing ChIP‐seq and RNA‐seq. The work was partially supported by ALSAC.
No conflicts of interest were declared.
Data availability statement
The ChIP‐seq and RNA‐seq data in this study are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196003.
References
References 56–75 are cited only in the supplementary material.
- 1. Mertens F, Antonescu CR, Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 2016; 55: 291–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oda Y, Yamamoto H, Kohashi K, et al. Soft tissue sarcomas: from a morphological to a molecular biological approach. Pathol Int 2017; 67: 435–446. [DOI] [PubMed] [Google Scholar]
- 3. Mertens F, Johansson B, Fioretos T, et al. The emerging complexity of gene fusions in cancer. Nat Rev Cancer 2015; 15: 371–381. [DOI] [PubMed] [Google Scholar]
- 4. WHO . Classification of Tumours of Soft Tissue and Bone (5th edn). IARC Press: Lyon, 2020. [Google Scholar]
- 5. Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1‐NCOA2 fusion in mesenchymal chondrosarcoma based on a genome‐wide screen of exon‐level expression data. Genes Chromosomes Cancer 2012; 51: 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakayama R, Miura Y, Ogino J, et al. Detection of HEY1‐NCOA2 fusion by fluorescence in‐situ hybridization in formalin‐fixed paraffin‐embedded tissues as a possible diagnostic tool for mesenchymal chondrosarcoma. Pathol Int 2012; 62: 823–826. [DOI] [PubMed] [Google Scholar]
- 7. Fritchie KJ, Jin L, Ruano A, et al. Are meningeal hemangiopericytoma and mesenchymal chondrosarcoma the same?: a study of HEY1‐NCOA2 fusion. Am J Clin Pathol 2013; 140: 670–674. [DOI] [PubMed] [Google Scholar]
- 8. Panagopoulos I, Gorunova L, Bjerkehagen B, et al. Chromosome aberrations and HEY1‐NCOA2 fusion gene in a mesenchymal chondrosarcoma. Oncol Rep 2014; 32: 40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andersson C, Osterlundh G, Enlund F, et al. Primary spinal intradural mesenchymal chondrosarcoma with detection of fusion gene HEY1‐NCOA2: a paediatric case report and review of the literature. Oncol Lett 2014; 8: 1608–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen JN, Solomon DA, Horvai AE, et al. Pancreatic involvement by mesenchymal chondrosarcoma harboring the HEY1‐NCOA2 gene fusion. Hum Pathol 2016; 58: 35–40. [DOI] [PubMed] [Google Scholar]
- 11. Toki S, Motoi T, Miyake M, et al. Minute mesenchymal chondrosarcoma within osteochondroma: an unexpected diagnosis confirmed by HEY1‐NCOA2 fusion. Hum Pathol 2018; 81: 255–260. [DOI] [PubMed] [Google Scholar]
- 12. Bishop MW, Somerville JM, Bahrami A, et al. Mesenchymal chondrosarcoma in children and young adults: a single institution retrospective review. Sarcoma 2015; 2015: 608279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mantilla JG, Ricciotti RW, Chen E, et al. Detecting disease‐defining gene fusions in unclassified round cell sarcomas using anchored multiplex PCR/targeted RNA next‐generation sequencing‐molecular and clinicopathological characterization of 16 cases. Genes Chromosomes Cancer 2019; 58: 713–722. [DOI] [PubMed] [Google Scholar]
- 14. de Andrea CE, San‐Julian M, Bovee J. Integrating morphology and genetics in the diagnosis of cartilage tumors. Surg Pathol Clin 2017; 10: 537–552. [DOI] [PubMed] [Google Scholar]
- 15. Sajjad EA, Sikora K, Paciejewski T, et al. Intraparenchymal mesenchymal chondrosarcoma of the frontal lobe‐‐a case report and molecular detection of specific gene fusions from archival FFPE sample. Clin Neuropathol 2015; 34: 288–293. [DOI] [PubMed] [Google Scholar]
- 16. Nyquist KB, Panagopoulos I, Thorsen J, et al. Whole‐transcriptome sequencing identifies novel IRF2BP2‐CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS One 2012; 7: e49705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uneda A, Kurozumi K, Fujimura A, et al. Intracranial mesenchymal chondrosarcoma lacking the typical histopathological features diagnosed by HEY1‐NCOA2 gene fusion. NMC Case Rep J 2020; 7: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen C‐W, Chen I‐H, Hu M‐H, et al. Primary intradural extramedullary spinal mesenchymal chondrosarcoma: case report and literature review. BMC Musculoskelet Disord 2019; 20: 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moriya K, Katayama S, Onuma M, et al. Mesenchymal chondrosarcoma diagnosed on FISH for HEY1‐NCOA2 fusion gene. Pediatr Int 2014; 56: e55–e57. [DOI] [PubMed] [Google Scholar]
- 20. Low SYY, Kuick CH, Seow WY, et al. Primary paediatric epidural sarcomas: molecular exploration of three cases. BMC Cancer 2019; 19: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Folpe AL, Graham RP, Martinez A, et al. Mesenchymal chondrosarcomas showing immunohistochemical evidence of rhabdomyoblastic differentiation: a potential diagnostic pitfall. Hum Pathol 2018; 77: 28–34. [DOI] [PubMed] [Google Scholar]
- 22. Chang KTE, Goytain A, Tucker T, et al. Development and evaluation of a pan‐sarcoma fusion gene detection assay using the NanoString nCounter Platform. J Mol Diagn 2018; 20: 63–77. [DOI] [PubMed] [Google Scholar]
- 23. Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 2003; 194: 237–255. [DOI] [PubMed] [Google Scholar]
- 24. Weber D, Wiese C, Gessler M. Hey bHLH transcription factors. Curr Top Dev Biol 2014; 110: 285–315. [DOI] [PubMed] [Google Scholar]
- 25. Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol 2003; 17: 1681–1692. [DOI] [PubMed] [Google Scholar]
- 26. Strehl S, Nebral K, König M, et al. ETV6‐NCOA2: a novel fusion gene in acute leukemia associated with coexpression of T‐lymphoid and myeloid markers and frequent NOTCH1 mutations. Clin Cancer Res 2008; 14: 977–983. [DOI] [PubMed] [Google Scholar]
- 27. Jin Y, Möller E, Nord KH, et al. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosomes Cancer 2012; 51: 510–520. [DOI] [PubMed] [Google Scholar]
- 28. Bekers EM, Groenen PJTA, Verdijk MAJ, et al. Soft tissue angiofibroma: Clinicopathologic, immunohistochemical and molecular analysis of 14 cases. Genes Chromosomes Cancer 2017; 56: 750–757. [DOI] [PubMed] [Google Scholar]
- 29. Panagopoulos I, Gorunova L, Viset T, et al. Gene fusions AHRR‐NCOA2, NCOA2‐ETV4, ETV4‐AHRR, P4HA2‐TBCK, and TBCK‐P4HA2 resulting from the translocations t(5;8;17)(p15;q13;q21) and t(4;5)(q24;q31) in a soft tissue angiofibroma. Oncol Rep 2016; 36: 2455–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhuravleva J, Paggetti J, Martin L, et al. MOZ/TIF2‐induced acute myeloid leukaemia in transgenic fish. Br J Haematol 2008; 143: 378–382. [DOI] [PubMed] [Google Scholar]
- 31. Carapeti M, Aguiar RC, Goldman JM, et al. A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 1998; 91: 3127–3133. [PubMed] [Google Scholar]
- 32. Sumegi J, Streblow R, Frayer RW, et al. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX‐FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer 2010; 49: 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshida H, Miyachi M, Sakamoto K, et al. PAX3‐NCOA2 fusion gene has a dual role in promoting the proliferation and inhibiting the myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014; 33: 5601–5608. [DOI] [PubMed] [Google Scholar]
- 34. Brunetti M, Panagopoulos I, Gorunova L, et al. RNA‐sequencing identifies novel GREB1‐NCOA2 fusion gene in a uterine sarcoma with the chromosomal translocation t(2;8)(p25;q13). Genes Chromosomes Cancer 2018; 57: 176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mosquera JM, Sboner A, Zhang L, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer 2013; 52: 538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whittle SB, Hicks MJ, Roy A, et al. Congenital spindle cell rhabdomyosarcoma. Pediatr Blood Cancer 2019; 66: e27935. [DOI] [PubMed] [Google Scholar]
- 37. Alaggio R, Zhang L, Sung Y‐S, et al. A molecular study of pediatric spindle and Sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2‐related fusions in infantile cases. Am J Surg Pathol 2016; 40: 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Argani P, Reuter VE, Kapur P, et al. Novel MEIS1‐NCOA2 gene fusions define a distinct primitive spindle cell sarcoma of the kidney. Am J Surg Pathol 2018; 42: 1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agaram NP, Zhang L, Sung Y‐S, et al. Expanding the Spectrum of intraosseous rhabdomyosarcoma: correlation between 2 distinct gene fusions and phenotype. Am J Surg Pathol 2019; 43: 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raha D, Hong M, Snyder M. ChIP‐Seq: a method for global identification of regulatory elements in the genome. Curr Protoc Mol Biol 2010; Chapter 21: Unit 21: 19.1‐14. [DOI] [PubMed] [Google Scholar]
- 41. Szwarc MM, Kommagani R, Lessey BA, et al. The p160/steroid receptor coactivator family: potent arbiters of uterine physiology and dysfunction. Biol Reprod 2014; 91: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heisig J, Weber D, Englberger E, et al. Target gene analysis by microarrays and chromatin immunoprecipitation identifies HEY proteins as highly redundant bHLH repressors. PLoS Genet 2012; 8: e1002728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aigner T, Loos S, Müller S, et al. Cell differentiation and matrix gene expression in mesenchymal chondrosarcomas. Am J Pathol 2000; 156: 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Almalki SG, Agrawal DK. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 2016; 92: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ryu N‐E, Lee S‐H, Park H. Spheroid culture system methods and applications for mesenchymal stem cells. Cells 2019; 8: 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frezza AM, Cesari M, Baumhoer D, et al. Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. A European musculoskeletal oncology society study. Eur J Cancer 2015; 51: 374–381. [DOI] [PubMed] [Google Scholar]
- 47. Schneiderman BA, Kliethermes SA, Nystrom LM. Survival in mesenchymal chondrosarcoma varies based on age and tumor location: a survival analysis of the SEER database. Clin Orthop Relat Res 2017; 475: 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andrae J, Gallini R, Betsholtz C. Role of platelet‐derived growth factors in physiology and medicine. Genes Dev 2008; 22: 1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brown RE, Boyle JL. Mesenchymal chondrosarcoma: molecular characterization by a proteomic approach, with morphogenic and therapeutic implications. Ann Clin Lab Sci 2003; 33: 131–141. [PubMed] [Google Scholar]
- 50. Brown RE. Morphoproteomic portrait of the mTOR pathway in mesenchymal chondrosarcoma. Ann Clin Lab Sci 2004; 34: 397–399. [PubMed] [Google Scholar]
- 51. de Jong Y, van Maldegem AM, Marino‐Enriquez A, et al. Inhibition of Bcl‐2 family members sensitizes mesenchymal chondrosarcoma to conventional chemotherapy: report on a novel mesenchymal chondrosarcoma cell line. Lab Invest 2016; 96: 1128–1137. [DOI] [PubMed] [Google Scholar]
- 52. Liu C‐F, Samsa WE, Zhou G, et al. Transcriptional control of chondrocyte specification and differentiation. Semin Cell Dev Biol 2017; 62: 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rutkowski TP, Kohn A, Sharma D, et al. HES factors regulate specific aspects of chondrogenesis and chondrocyte hypertrophy during cartilage development. J Cell Sci 2016; 129: 2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bhattaram P, Penzo‐Méndez A, Kato K, et al. SOXC proteins amplify canonical WNT signaling to secure nonchondrocytic fates in skeletogenesis. J Cell Biol 2014; 207: 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bhattaram P, Penzo‐Méndez A, Sock E, et al. Organogenesis relies on SoxC transcription factors for the survival of neural and mesenchymal progenitors. Nat Commun 2010; 1: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnetjournal 2011; 17: 10–12. [Google Scholar]
- 57. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics 2010; 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tischler G, Leonard S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol Med 2014; 9: 13. [Google Scholar]
- 60. Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP‐seq experiments for DNA‐binding proteins. Nat Biotechnol 2008; 26: 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform 2013; 14: 144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang Y, Liu T, Meyer CA, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 2008; 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Frankish A, Diekhans M, Ferreira AM, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019; 47: D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jolma A, Yan J, Whitington T, et al. DNA‐binding specificities of human transcription factors. Cell 2013; 152: 327–339. [DOI] [PubMed] [Google Scholar]
- 66. Shen W, Le S, Li Y, et al. SeqKit: a cross‐platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 2016; 11: e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 2011; 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015; 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Law CW, Chen Y, Shi W, et al. voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol 2014; 15: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016; 44: W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rusch M, Nakitandwe J, Shurtleff S, et al. Clinical cancer genomic profiling by three‐platform sequencing of whole genome, whole exome and transcriptome. Nat Commun 2018; 9: 3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stewart E, Federico S, Karlstrom A, et al. The Childhood Solid Tumor Network: a new resource for the developmental biology and oncology research communities. Dev Biol 2016; 411: 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Downing JR, Wilson RK, Zhang J, et al. The pediatric cancer genome project. Nat Genet 2012; 44: 619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. The breakpoint DNA sequence of the inducible‐expression pINDUCER21‐HEY1‐NCOA2 construct
Figure S2. Genomic context of HEY1 and HEY1‐NCOA2 binding peaks and Venn diagram showing the number of HEY1–NCOA2 binding peaks with E‐box motif overlapping with HEY1 binding peaks with E‐box motif
Figure S3. HEY1–NCOA2 demonstrates distinct gene expression profile
Figure S4. HEY1 and HEY1–NCOA2 binding in promoters of target genes
Figure S5. Box plots visualizing the enrichment of the 674 HEY1–NCOA2 direct‐target‐and‐transactivating genes identified using the iPSC‐MSCs cell model
Figure S6. GSEA enrichment plots
Figure S7. HEY1‐NCOA2 enhances cell growth in 3D model
Figure S8. HEY1–NCOA2 fusion protein increased the level of phospho‐AKT (Ser473)
Table S1. Sequences of primers used for RT‐qPCR
Table S2. Differential gene expression (DGE) analysis
Table S3. HEY1–NCOA2 key‐direct‐target genes
Table S4. Samples accrued for the ssGSEA analysis
Table S5. The ssGSEA Normalized Enrichment Scores (NESs) of all 164 samples
Table S6. Functional pathways enriched in HEY1–NCOA2 upregulated genes (from supplementary material, Table S2)
Data Availability Statement
The ChIP‐seq and RNA‐seq data in this study are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196003.