

Summary
Sjögren’s syndrome (SjS) is an autoimmune disease characterized by the triad of sicca symptoms, fatigue and pain. This diagnosis is usually made in women at the average age of 60 years. Diagnosis is made when sicca symptoms persist for more than three months, after the exclusion of possible differential diagnoses, and using the ACR/EULAR 2016 classification criteria for SjS. Many organs can be affected in the course of this disease. Xerosis cutis and pruritus are the most common skin manifestations, followed by leukocytoclastic vasculitis and subacute cutaneous lupus erythematosus. In addition, SjS patients often have myoarthralgia and neuropsychiatric symptoms. In the long term, attention must be paid to the increased risk of cardiovascular disease and lymphoma. Due to the multiorgan involvement in SjS patients, interdisciplinary care is required.
Introduction
Sjögren’s syndrome (SjS) is a chronic autoimmune connective tissue disease. The clinical symptom triad includes sicca symptoms, fatigue, and pain [1]. Patients may also display additional organ manifestations.
History
In 1892, Mikulicz was the first scientist to report on a patient with sicca symptoms and bilateral swelling of the parotid glands [2]. Gougerot introduced the term “sicca syndrome” in 1925. Eight years later, Henrik Sjögren first reported on a systemic disease he called “keratoconjunctivitis sicca”. He described this as a combination of dry eyes and mouth associated with rheumatoid arthritis (RA).
Etiology and pathogenesis
SjS is often called “autoimmune epithelitis” [3] since epithelial cells play a key role in this disease – as both targets and initiators of the autoimmune process [4]. The epithelial cells produce pro‐inflammatory cytokines, which in turn leads to impaired function of the salivary glands [5].
Etiology and pathogenesis are not fully understood [6], but a multifactorial process is assumed to be the most likely explanation (Figure 1).
Figure 1.
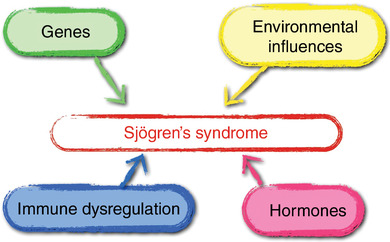
Pathogenesis of Sjögren’s syndrome (adapted from [133]).
Genetic predisposition is a prerequisite (HLA‐DRB1*03: 01, DQA1*05: 01, DQB1*02: 01 [7, 8], X chromosomes), but epigenetic modifications [6] are also required for the development of this autoimmune disease. An increased risk for connective tissue diseases (such as systemic lupus erythematosus [SLE], systemic scleroderma [SS]) or other autoimmune diseases has been demonstrated in families of SjS patients [9]. Other factors such as immune dysregulation, hormones, and environmental influences (among other factors, stress [10], infections [11], drugs [12], vaccines [13], or silicone breast implants [14] are currently being discussed) then lead to misdirected activation of the innate and adaptive immune system. This activates the type 1 and type 2 interferon signaling cascades that stimulate proliferation of B cells.
Viral infections, in particular [11], mainly the Epstein‐Barr virus [15] are suspected candidates for inducing SjS. Interestingly, an increased incidence of SjS as compared to earlier years was reported in Brazil during the COVID‐19 pandemic [16].
Epidemiology
SjS is one of the most common rheumatological diseases. Its prevalence is estimated to be about 0.5 % and its incidence about 4/1000 people per year [17]. It has an unusual age at diagnosis of around 60 years (between the 5th and the 7th decade of life) [18]. In cases where SjS is diagnosed in younger patients (less than 35 years of age), presentation is frequently associated with fever, lymphadenopathy, and high disease activity [19]. In clinical studies, disease activity is measured by the ESSDAI (European League against Rheuma Sjögren’s syndrom disease activity index).
SjS is diagnosed eight times more frequently in women than in men [18]. Some studies have investigated differences between male and female SjS patients [20], showing that male SjS patients have more severe courses and higher mortality rates [21, 22, 23].
There are rare cases of juvenile SjS, usually appearing around ten years of age. This is characterized by recurrent parotid gland inflammation (about 67 % of patients) and arthralgia. Laboratory findings (rheumatic factor, ANA, Ro/SS‐A antibodies) may be normal in patients with juvenile SjS [24, 25].
Sicca symptoms and systemic activity (ESSDAI) vary based on ethnicity. African Americans show higher disease activity scores, followed by European, Asian, and Hispanic patients [26]. In addition, SjS is frequently diagnosed at an earlier age in African Americans and Africans than in Europeans [27, 28]. Higher systemic activity has been reported for patients from more Southern latitudes (below the 50th parallel north in Europe, below the equator in America, and below the 30th parallel north in Asia) as compared with patients from more northern latitudes [26]. Possible causes are currently being discussed, including genetic factors, lifestyle (microbiome), and environmental influences such as metals in the earth, air pollution, or viral infection [26].
Diagnosis and classification
SjS is diagnosed based on the patient’s subjective sicca symptoms if these cannot be explained by other differential diagnoses, after excluding certain diseases and using the objective classification criteria.
Subjective criteria for sicca symptoms
Patients must have had sicca symptoms for at least three months before the SjS classification criteria can be applied. The following questions need to be clarified when talking with the patient:
-
–
Have you had a sensation of dry eyes every day for more than three months?
-
–
Do you often have the sensation of sand/grit in your eye?
-
–
Do you use artificial tears more than three times per day?
-
–
Have you had a sensation of a dry mouth every day for more than three months?
-
–
Do you frequently need to drink something to help you swallow dry food?
If one of these questions is answered in the affirmative, the patient has subjective sicca symptoms.
Differential diagnoses of xerostomia and xerophthalmia
Sicca symptoms should not be explainable by other possible causes. Differential diagnoses for xerostomia [29] and xerophthalmia [30] are listed in Table 1.
Table 00.
Differential diagnoses of xerostomia and xerophthalmia
| Xerostomia | Xerophthalmia | |
|---|---|---|
| General | Age, pollinosis, malignancy | |
| Metabolic causes | Diabetes mellitus | |
|
Vitamin deficiency (B1, B2, B6, B12), anemia, hypercalcemia Hyperthyreosis and hypothyreosis, dialysis, chronic renal failure |
Vitamin A deficiency, hypoandrogenemia | |
| Environmental influences | Radiotherapy, chemical and thermic burns, scarring, nicotine abuse/cigarette smoke | |
| Sialadenitis/sialolithiasis | Wind, low air humidity, computer work, contact lenses | |
| Hereditary causes | Hereditary gelsolin amyloidosis | Congenital alacrimy, familial dysautonomy |
| Neuropsychiatric causes | Anxiety disorder, depression, schizophrenia, Parkinson’s disease, Alzheimer’s disease | |
| Inflammatory diseases | Primary biliary cholangitis, amyloidosis, sarcoidosis, GvHD, mucosal pemphigoid, IgG4 associated disease | |
| Lichen planus | Rosacea, chronic blepharitis | |
| Infections | HIV, HCV | |
| Varicella, hand‐foot‐mouth disease, herpes stomatitis | Trachoma, post‐zoster neuropathy, adenoviruses | |
| Drugs | Antihistamines, anticholinergics, diuretics, tricyclic antidepressants | |
| Bronchodilators, psychotropic drugs (anxiolytics, neuroleptics), antihypertensives, opioids, interferon‐alpha, triptans, appetite suppressants | Retinoids, topical medications with preservatives | |
Exclusion diagnoses
The following diseases must be excluded before SjS can be diagnosed [31]: History of radiation therapy in the head and neck area, active hepatitis C infection (positive HCV‐PCR), AIDS, sarcoidosis, amyloidosis, graft‐versus‐host disease (GvHD), IgG4‐associated disease. Pre‐existing lymphoma is no longer considered an exclusion diagnosis. Patients with sicca symptoms who are taking an anticholinergic should discontinue this and be re‐evaluated after an appropriate interval (> 5 half‐lives).
Classification criteria 2016
There are different classification criteria for SjS [31, 32, 33]. The current classification criteria for primary SjS (pSjS) were published in 2016 by ACR/EULAR (American College of Rheumatology/European League against Rheumatism) [31]. Table 2 lists the classification criteria. The primary aim of the ACR/EULAR criteria is to define inclusion criteria for clinical studies. pSjS is diagnosed in patients with a total score of ≥ 4. Sensitivity (96 %) and specificity (95 %) are high, however a total score of ≥ 4 is sometimes not reached in early stages of the disease. For example: Xerophthalmia may have been present for five months and differential diagnoses excluded, but objective tests may only detect Ro/SS‐A antibodies (3 points). If there is still a justified suspicion of SjS, repeating the objective tests listed in the classification criteria after a few months may therefore make sense. Among the objective criteria, Ro/SS‐A antibodies and histological investigation of the salivary glands (focus score [FS] ≥ 1) are mandatory criteria. SjS can only be diagnosed if at least one of these criteria is fulfilled.
Table 2.
2016 ACR/EULAR classification criteria for primary Sjögren’s syndrome (adapted from [31]). After controlling the inclusion criteria (dry eyes and/or mouth for at least 3 months without other explanation) and exclusion criteria, the classification criteria can be used. The diagnosis of SjS is confirmed when ≥ 4 points of the classification criteria are reached

|
Eyes
Staining the cornea with lissamine green or fluoresceine shows degenerated and/or dead cells on the corneal surface. The Schirmer test measures the amount of lacrimal fluid with filter paper.
Mouth
The unstimulated whole saliva (UWS) flow rate is the amount of saliva produced within five to 15 minutes while the patient is sitting quietly without speaking or chewing [34].
Laboratory investigations
The only laboratory value valid for a SjS diagnosis according to the classification criteria is the SS‐A/Ro antibody. SS‐A/Ro antibodies are antinuclear antibodies (ANA) against Ro52 and/or Ro60. Detection of SS‐B/La antibodies in the absence of SS‐A/Ro antibodies is no longer relevant for diagnosing SjS according to the classification criteria.
Histology
Salivary gland biopsy remains a rarely used diagnostic instrument [35]. The specimen can be taken from the lower lip [36]. In salivary gland histology, the focus score is the gold standard for quantifying lymphocytic infiltration. (Grade 0: no lymphocytes/4 mm2, grade 1: minor lymphocyte infiltration, grade 2: moderate infiltration but less than one focus, grade 3: one focus, grade 4: more than one focus; a focus is an accumulation of ≥ 50 lymphocytes/4 mm2 salivary gland tissue) [37]. Specificity is high, but sensitivity is low [38]. Tissue samples can be particularly helpful in the early stages of the disease when other criteria are still negative and differential diagnoses must be excluded. Histology is only recommended if Ro/SS‐A antibodies cannot be detected [39, 40].
Diagnostic imaging
Diagnostic imaging methods (such as ultrasound, scintigraphy, sialography) are not currently recommended in the classification criteria, but in everyday clinical practice, ultrasound of the salivary gland is a simple, reliable, and helpful diagnostic procedure [39, 40].
Additional laboratory parameters
Additional laboratory parameters for diagnosing SjS or monitoring disease activity are currently being developed but are not yet validated [41]. In future diagnostics, it may be possible to detect some biomarkers in saliva (profilin and CA‐I or IL‐4, IL‐5 and mi‐RNA) or in tears (cathepsin S). Myxovirus resistance protein A (MxA) in correlation with interferon type‐1 activity may reflect disease activity [41].
pSjS and secondary SjS (sSjS)
The term sSjS is used when the SjS occurs together with other autoimmune diseases. However, the distinction between pSjS and sSjS varies in the scientific literature [42, 43]. Initially, SjS was called “secondary” when it occurred in conjunction with rheumatoid arthritis (RA) [44]. SjS is diagnosed in up to 30 % of RA patients [45]. Interestingly, patients with combined RA and sSjS have more severe, destructive arthritis compared to patients with RA alone [46, 47]. The term “sSjS” is mostly used for SjS in association with RA, systemic lupus erythematosus (SLE), or systemic sclerosis (SS). Associated SjS is diagnosed in up to 20 % of patients with SLE or SS [45]. There are also many similarities between LE and SjS (etiology, pathogenesis, epidemiology, treatment) [48]. The greatest difficulty concerning clinical presentation of SjS arises because of the frequent overlapping manifestations with SLE, SS, or overlap syndromes. Clear differentiation (SLE versus SjS versus SS) is often impossible.
If SjS and SS occur in association with primary biliary cholangitis, this is called Reynolds syndrome [49]. The term sSjS is sometimes also used if SjS occurs in association with autoimmune diseases that are not connective tissue diseases (such as primary biliary cholangitis, or Hashimoto thyreoiditis) [50]. Depending on how pSjS and sSjS are differentiated, the ratio of pSjS to sSjS varies between 1/3 [51] and 3/1 [50]. Note that the term “secondary” in this context does not signify any temporal sequence in the occurrence of the autoimmune diseases.
Clinical presentation
Sicca symptoms
Sicca symptoms impair quality of life in SjS patients [52]. Subjective symptoms frequently show little correlation with the objective measurements of gland functionality [53]. An association between the severity of xerophthalmia (in particular inflammatory keratolysis and scleritis) and mortality in SjS patients has been reported [54]. Quality of life is impaired by keratoconjunctivitis sicca since this is associated with foreign body sensation, burning, and increased sensitivity to light. In the long term, ocular involvement in SjS may lead to blindness [55]. SjS patients therefore need to be closely monitored by an ophthalmologist. Xerostomia is characterized by difficulty chewing and masticating, especially for dry foods, and also difficulty is speaking. Xerostomia is also associated with an increased risk of caries, early loss of teeth, and increased fungal infections with candida albicans.
The prevalence of vaginal dryness in SjS patients is comparable with that found in women of the same age in the general population [56, 57]. However, 61–68 % of SjS patients report dyspareunia [56, 58]. This is significantly more common than in studies on dyspareunia in women of the same age in the general population (8 %) [59]. One possible cause for the more frequent occurrence in SjS patient may be an altered composition of the vaginal secretions [57]. Sicca symptoms can also affect other mucous membranes, leading to symptoms like heartburn, sore throat, chronic cough (tracheobronchitis sicca), and recurrent urinary tract infections in SjS patients.
Swelling of the parotid gland
About one‐third of patients have recurrent parotitis [60, 61]. Parotitis is usually found in juvenile SjS. In adult patients, possible differential diagnoses such as non‐Hodgkin lymphoma need to be considered (Table 3).
Table 3.
Treatment of Sjögren’s syndrome (adapted from [1])
| Clinical Manifestation | Treatment | |
|---|---|---|
| Sicca symptoms | Xerophthalmia |
1) ED (≥ 2 x per day) and eye gels/ointments 2) Topical GC/NSAID 3) CyA AT 4) Serum tear drops 4) Pilocarpine or silicone plugs in the tear duct, scleral lenses |
| Xerostomia |
1) Topical fluorides/sugar‐free chewing gum/boiled sweets/artificial aliva (mouth sprays, gels, or rinsing solutions) 2) Pilocarpine |
|
| Xerosis cutis |
1) Moisturizers 2) Topical GC |
|
| Mucous membranes |
1) Rhinitis sicca: Nose oil 2) Tracheobronchitis sicca: Pilocarpine, bromhexine, inhalation with sodium chloride 3) Dyspareunia: estrogen‐containing suppositories |
|
| Immunosuppressants and rituximab are generally not recommended due to their potential side effects. | ||
| Parotitis | Acute |
NOTE: Exclude infection! 1) Symptomatic treatment 2) GC 3) RTX/BLM |
| Chronic |
NOTE: Exclude lymphoma or other causes! ± surgery |
|
| Joints | Arthralgia |
1) NSAID 2) HCQ |
| Arthritis |
1) NSAID + HCQ 2) HCQ + GC 3) SsI 4) RTX or ABA |
|
| Skin | Sunscreen! | |
| Cutaneous LE |
1) Topical GC or HCQ +/‐ GC 2) other antimalarials +/‐ GC 3) Retinoids, SsI |
|
| Cutaneous vasculitis |
1) GC 2) Oral SsI or RTX 3) CyC ± Pex |
|
| Fatigue |
1) Exercise, endurance training 2) HCQ |
|
| Raynaud symptoms |
Keeping warm, paraffin hand baths, Ca antagonists, AT‐II receptor blockers, phosphodiesterase inhibitors Statins ± aspirin |
|
| Kidney | Tubular |
1) symptomatic (Bicarbonate or electrolyte supplementation) 2) GC 3) SsI |
| Glomerulonephritis |
1) GC 2) RTX or CyC 3) Pex |
|
| Lung | Bronchitis | Inhalation treatment (ß2 mimetics, steroids) |
| ILD |
1) GC 2) SsI 3) CyC or RTX 4) Nintedanib |
|
| PNS | Mononeuritis multiplex |
1) GC 2) oral SsI or RTX 3) CyC ± Pex |
| Axonal PN |
1) Symptomatic 2) IVIG 3) Pulses MP 4) CyC |
|
| Ganglionopathy/CIDP |
1) IVIG 2) Pulses MP 3) CyC |
|
| CNS | CNS vasculitis/NMOSD |
1) GC 2) CyC 3) RTX ± Pex, eculimumab |
| Lymphocytic meningitis |
1) Symptomatic 2) GC 3) CyC 4) RTX ± Pex, eculimumab |
|
| Symptoms resembling multiple sclerosis | Treatment for multiple sclerosis | |
| Hematological Manifestation | Neutropenia < 500 |
1) consider G‐CSF 2) GC |
| Thrombocytopenia < 20,000 | GC | |
| Hemolytic anemia |
1) GC + IVIG 2) RTX 3) Pex or CyC |
|
| NOTE: if blood count values show persistent deviation from the normal range, lymphoma or other causes must be excluded. | ||
| B cell lymphoma | Depending on the type of lymphoma (surgery, radiotherapy, chemotherapy, RTX, R‐CHOP) | |
Abbr.: ABA, Abatacept; BLM, belimumab; CIDP, chronic‐inflammatory demyelinizing polyneuropathy; CNS, central nervous system; CyA, ciclosporin; CyC, cyclophosphamide pulsed therapy; ED, eye drops; GC, glucocorticoids; HCQ, hydroxychloroquine; ILD, interstitial lung disease; IVIG, intravenous immunoglobulins; LE, lupus erythematosus; NMOSD, Neuromyelitis optica spectrum diseases; NSAID, non‐steroidal anti‐inflammatory drugs; PEX, plasma exchange; PNS, peripheral nervous system; RTX, rituximab; SjS, Sjögren’s syndrome; SsI, steroid‐sparing immunosuppressants (MTX, methotrexate; AZA, azathioprine; MMF, mycophenolate mofetil).
Skin
Skin manifestations occur in 14–16 % [62, 63] of SjS patients; however “dry skin” was not included in these studies [62, 63].
Xerosis cutis and pruritus
Dry skin and pruritus are common cutaneous symptoms of SjS. Xerosis cutis occurs with a prevalence of up to 68.4 % in patients with SjS [64, 65]. For a long time, dysfunction of the sweat glands was assumed to be the underlying cause, but this has not been confirmed [66]. The current assumption is that a defective skin barrier causes this problem [66]. The prevalence of pruritus in SjS is estimated at about 53 % [65], compared to 25.5 % in the general population [67]. The pathophysiology of SjS‐associated pruritus in currently still unknown [65]. In SjS, pruritus does not correlate with the severity of xerosis cutis. Its intensity in SjS appears to be higher than in other connective tissue diseases [65].
Vasculitis
Vasculitis is the second most common skin manifestation. Depending on the study, vasculitis has been reported in up to 30 % of SjS patients [63, 64, 68]. Patients present with palpable purpura, but in some cases also non‐palpable purpura (Figure 2a) as well as urticarial eruptions (Figure 2b) on the lower legs. Histology will usually confirm leukocytoclastic vasculitis. Medium‐sized vessels are only rarely affected [63, 69]. Vasculitis in SjS patients appears to be associated with severe disease [63] and an increased risk of lymphoma [70].
Figure 2.
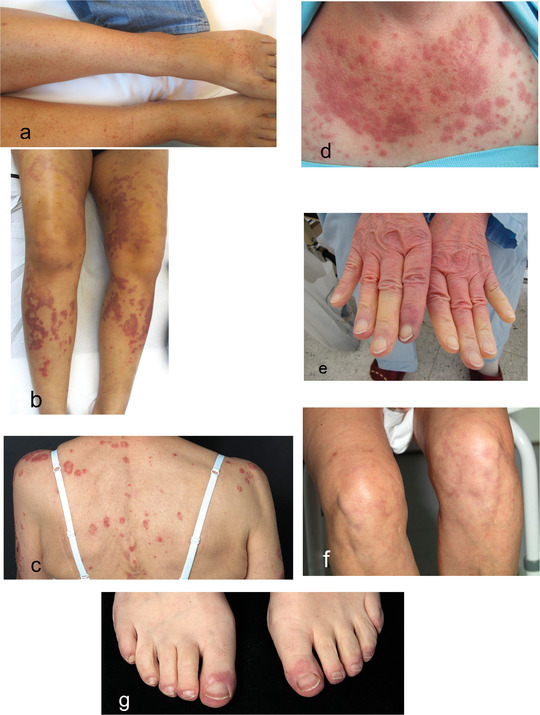
Dermatological clinical pictures in the context of Sjögren’s syndrome. purpura as a clinical manifestation of leukocytoclastic vasculitis (a). Urticarial vasculitis (b). Subacute cutaneous lupus erythematosus (c). Lupus erythematosus tumidus (d). Raynaud phenomenon (e). Livedo racemosa (f). Chilblain lupus erythematosus (g).
Lupus erythematosus
Subacute cutaneous LE (Figure 2c) is the most common LE‐specific skin manifestation in SjS. Out of 185 SjS patients treated at our hospital between January 2000 and December 2016, about 10 % had subacute cutaneous LE (unpublished data on our files). Conversely, SjS was found in 14 % of patients with subacute cutaneous LE [71]. According to a Japanese study, in Asian patients annular erythema is the equivalent of subacute cutaneous LE in patients of European descent [72]. Annular erythema of SjS is reported almost exclusively in Asian patients [73]. Only one study [74] reported this annular erythema in 9 % of non‐Asian SjS patients. Its clinical presentation with urticarial plaques, however, is comparable with LE tumidus (Figure 2d). Histology shows perivascular and periadnexial lymphocytic infiltration with dermal mucin deposits. Healing is achieved with hydroxychloroquine. In summary, it is still unclear if annular erythema of SjS is truly a separate entity or if it represents a manifestation of tumid LE/subacute cutaneous LE in the context of SjS [72, 75].
Other
Raynaud’s phenomenon (Figure 2e) occurs in 16–35 % [19, 64] of SjS patients. The clinical symptoms of Raynaud’s phenomenon in SjS are usually milder than in other connective tissue diseases. In severe cases of Raynaud’s syndrome in SjS, clinicians need to consider concomitant or developing SS. Other skin conditions associated with SjS include chronic urticaria [76], frontal fibrosing alopecia [77], vitiligo [78], and lichen planus [79].
In retrospective studies and case series, the following additional skin manifestations in SjS have been published [80, 81, 82, 83]: Erythema‐multiforme‐like eruptions, Sweet syndrome, erythema nodosum, cutaneous nodular amyloidosis, lymphomatoid papulosis, and cutaneous T‐cell lymphoma. Livedo patterns [81] have been reported in some cases, yet without coagulation diagnostics. There is a general association between antiphospholipid syndrome and connective tissue disease [84, 85, 86], thus coagulation diagnostics (antiphospholipid antibodies and lupus anticoagulant) need to be initiated in SjS patients with corresponding skin manifestations (livedo racemosa, Raynaud’s symptoms, purpura) (Figure 2f). Chilblains have also been observed in SjS patients [83], and it is debatable whether some of these patients may have had Chilblain LE (Figure 2g), and if patients with erythema multiforme‐like eruptions may have had subacute cutaneous LE as also named Rowell syndrome. The abovementioned dermatological diagnoses in the literature were usually made by clinical means and were not confirmed by tissue sampling.
Other organs
Serositis
In SjS, as compared with SLE [87], serositis is very rare. Asymptomatic pericarditis is common (33 %), but symptomatic pericarditis is only observed in 1 % of SjS patients [88]. Pleuritis has only been described in individual SjS patients [89].
Liver
In a retrospective study, increased levels of liver enzymes were detected in one‐third of SjS patients [90]. When liver enzymes are elevated in SjS patients, infectious causes need to be excluded first [91]. The prevalence of autoimmune hepatitis in SjS is estimated at 1–4 %, and that of primary biliary cholangitis at 4–9 % [91].
Lungs
The lungs are affected in up to 20 % of SjS, mostly presenting as a dry cough [92]. Recurrent respiratory infections are observed in 10–35 % of SjS patients. Interstitial lung disease and its sequelae are frequent causes of death in SjS patients; therefore, early diagnosis and treatment are essential. Conventional X‐ray investigations are insufficient for early diagnosis. Initially, and also later for monitoring purposes, high‐resolution computed tomography (HR‐CT) as well as pulmonary function testing including measuring of diffusion capacity are recommended. Interstitial lung disease frequently presents as lymphocytic interstitial lung disease, characterized by cystic changes of the lung parenchyma and ground‐glass opacities [93].
Kidneys
Affection of the kidneys in SjS is rare (≤ 10 %) [94]. The most common changes are tubulo‐interstitial nephritis or membranoproliferative glomerulonephritis. In the early stages, tubulointerstitial nephritis presents with renal‐tubular, metabolic acidosis. Investigation of the pH status is important for early diagnostics. Membranoproliferative glomerulonephritis is frequently caused by cryoglobulinemia. Prognosis is generally favorable. Since affection of the kidneys is often asymptomatic, regular monitoring is indicated (renal function parameters, protein‐creatinine ratio, pH status [venous blood gases]) [94].
Laboratory investigations
Typical laboratory findings in SjS include ANA (79 %), Ro/SS‐A antibodies (73 %), La/SS‐B antibodies (45 %), rheumatoid factor RF (48 %), cryoglobulinemia (7 %), hypergammaglobulinemia, complement consumption (C3 and/or C4) [95, 96], and increased erythrocyte sedimentation rate (ESR) [50]. In some cases, patients may also display cytopenia [61]. A recent study with 10,500 patients showed that cryoglobulinemia, complement consumption, and La/SS‐B antibodies were most highly correlated with systemic activity as measured by the ESSDAI [95].
Joints and muscles
Arthritis is one of the most common manifestations of SjS (15–90 %) [97]. It is characterized by intermittent, symmetric, non‐destructive polyarthritis. Axial involvement has been reported less frequently. If a patient presents with erosive arthritis, RA should be considered [97]. SjS patients with erosive arthritis have clinical, serological, and radiological characteristics of RA. Nevertheless, they differ from patients with “simple RA” in terms of additional organ involvement and genetic background [98].
Myalgia is common in SjS. Active myositis is rare, at 2 % [96], and is diagnosed within the context of inflammatory idiopathic myopathy or overlap syndrome.
Neuropsychiatric manifestation
Depending on the publication, neurological symptoms occur in 18–45 % of SjS cases [99], frequently even before the diagnosis of SjS is confirmed [100]. We can differentiate between affection of the peripheral nervous system (PNS, 5.3–21 %, such as mono/polyneuropathy), the central nervous system (CNS, 2.5–60 %, such as ischemic stroke, neuromyelitis‐optica spectrum diseases [NMOSD], symptoms resembling multiple sclerosis or amyotrophic lateral sclerosis) and autonomous nervous system (2–50 %, such as orthostatic hypotension, functional disorders of the bladder) [99]. Aquaporin‐4 antibodies are detected in more than 80 % of SjS patients presenting with acute CNS manifestations [101]. These antibodies are typical for NMOSD. Coexistence of both diseases (SjS and NMOSD) in the same patient appears to be associated with severe neurological progression [101]. Three‐fourths of SjS patients show cognitive impairment [102], and many also have psychiatric disease (such as depression or anxiety disorders [103]). After lymphoma, neuropsychiatric manifestations are the most common severe systemic manifestations of SjS [96].
Cardiovascular events
pSjS is associated with an increased risk for cerebrovascular events and myocardial infarction [68, 104]. Cardiovascular risk factors such as arterial hypertension, hypertriglyceridemia, dyslipidemia, and obesity are more common in pSjS, whereas SjS patients smoke less frequently compared to the general population [105]. Future studies are needed to determine if the risk of atherosclerosis is linked to the activity of SjS. SjS patients who have both Ro/SS‐A antibodies and La/SS‐B antibodies have a much higher risk of cerebrovascular events compared with the general population [104]. Antiphospholipid antibodies are also found more frequently in SjS patients than in the general population [86]; however, only about one‐third of these SjS patients will develop obvious antiphospholipid syndrome [85].
Malignancy
SjS patients have a significantly increased (10–44‐fold) risk for lymphoma [106]). Mucous‐membrane‐associated non‐Hodgkin lymphoma [95] and other non‐Hodgkin lymphomas were the most frequently observed types of lymphoma. Apart from Raynaud’s symptoms, palpable purpura, and swelling of the parotid glands, the following parameters are being discussed as independent risk factors for the development of lymphoma in SjS patients: cryoglobulinemia, hypergammaglobulinemia, complement consumption in C4, FS ≥ 1 in tissue samples of the labial salivary glands, and lastly, ESSDAI at the time of diagnosis [70, 107, 108]. The higher the FS at the time of diagnosis in SjS patients, the earlier the occurrence of lymphoma is expected [107]. This appears to be caused by chronic stimulation of B cells in the exocrine glands and mucous membranes. An increased risk of malignancy (melanoma, non‐Hodgkin lymphoma, breast cancer) has been reported in Ro/SS‐A antibody‐positive SjS patients [109]. SjS patients also have an increased risk for other malignancies apart from non‐Hodgkin lymphoma [110, 111]. Other factors (sex, ethnicity, dysfunctional exocrine glands, immune dysregulation) appear to play a role in the development of malignancy. Thyroid cancer was more frequently found in women with SjS, and lung cancer more frequently in men with SjS [110].
ESSDAI/ESSPRI
Originally, scores for measuring SjS activity were focused solely on sicca symptoms [53]. Additional scores have been developed in recent years to quantify systemic activity of SjS. These are used primarily for clinical studies [112]. The EULAR Sjögren’s Syndrome Disease Activity Index (ESSDAI) [113] is used to assess disease activity. It comprises twelve areas: General health (fever, night sweats, weight loss), lymph nodes, involvement of glands (swelling of the salivary and/or tear glands), joints, skin, lungs, kidneys, or nerves, also hematological changes and other laboratory investigations (complement, cryoglobulins, hypergammaglobulinemia). Depending on activity, every area is allotted a score of 0–4; the scores are added and thereby provide a numerical activity score.
The EULAR Sjögren’s Syndrome‐Patient Reported Index (ESSPRI) [114] is used for quantifying symptoms reported by the patient and thus reflect her point of view. This score comprises the patient’s subjective assessment of sicca symptoms, fatigue, and pain. ESSDAI and ESSPRI complement each other but do not necessarily correlate [115].
Treatment
Since SjS can affect multiple organ systems, an interdisciplinary approach is required for individual treatment. Basically, we must differentiate between symptomatic treatment of sicca symptoms, and immunosuppressant treatment of organ involvement. Updated EULAR recommendations for treating SjS were published in 2020 [1]. These are presented in the next section (Table 3), but have also been modified according to the authors’ experience and the current literature.
Sicca symptoms
Xerostomia
Reduced secretion of saliva results in higher incidences of caries in SjS patients. Topical fluorides, meticulous oral hygiene (dentist and patient), and refraining from smoking are recommended for prevention of caries. If the mouth is only slightly dry, non‐pharmacological stimulation (such as sugar‐free chewing gums or hard candies) may be prescribed. Moderate to severe xerostomia requires artificial saliva (mouth sprays, gels, or rinses) and/or pharmacological stimulation. This is achieved by muscarinic agonists (pilocarpine). Possible and also common side effects of pilocarpine include hyperhidrosis, hot flushes, headache, and nausea.
Xerophthalmia
Dry eyes are primarily treated with eye drops (at least twice a day) as well as eye gels or ointments. If eye drops are used four times a day or even more frequently, preservative‐free preparations are recommended. Placing silicone plugs into the tear duct results in short‐term, reversible obstruction of the tear duct and prevents rapid draining of the tear fluid. Ophthalmologists can also prescribe scleral lenses. Topical corticosteroids or topical non‐steroidal anti‐inflammatory drugs (NSAID) may be used for short‐term treatment of severe keratoconjunctivitis sicca. If treatment is required for longer periods, ciclosporin eye drops are recommended. If patients do not respond to ciclosporin eye drops or cannot tolerate them, autologous serum eye drops may be used. Oral muscarinic agonists are considered a treatment of last resort for dry eyes.
Hydroxychloroquine is not recommended for treating xerophthalmia since studies have not shown a significant therapeutic effect as compared to placebo [116]. Steroid‐sparing immunosuppressants (SsI) such as methotrexate, azathioprine, and mycophenolate mofetil are not recommended due to their potential side effects. Rituximab (anti‐CD20 antibody) for treating sicca symptoms is still the subject of intense discussion since various randomized controlled trials have not consistently shown superiority to placebo. One study did observe a positive effect of rituximab on stimulated salivary secretion [117], but two others did not [118, 119]. In summary: Rituximab remains the most thoroughly studied and currently best therapeutic option for treating severe treatment‐refractory sicca symptoms in SjS, particularly in view of severe long‐term complications of sicca symptoms such as blindness. Any use of rituximab in SjS patients currently requires a strict indication. A recently published retrospective study found an increased risk of severe COVID‐19 infection in rheumatological patients, especially those pre‐treated with rituximab [120].
Systemic symptoms
Glucocorticoids should only be administered at the minimum dose and duration required for controlling disease activity. Steroid‐sparing immunosuppressants are to be preferred to save on glucocorticoid use. No specific substance from the group of SsI has proven superior to the others. Regarding the role of B cells in the pathogenesis of SjS, rituximab and belimumab (anti‐BAFF antibody) represent promising therapeutic options which have proven their efficacy in treating SjS [121, 122, 123]. At this point in time, however, they are only recommended for patients with severe, refractory systemic disease. The indication for rituximab in SjS is usually in the context of vasculitis [124]. Regarding fatigue, rituximab usage remains controversial [117–119, 123, 125]. Fatigue in general is a huge diagnostic and therapeutic challenge in connective tissue disease including SjS. It may on the one hand be a symptom of related conditions (such as fibromyalgia), but on the other hand it may also be interpreted as a sign of SjS disease activity or even indicate malignancy, mostly non‐Hodgkin’s lymphoma. Regular physical exercise (endurance training) may prove beneficial. Hydroxychloroquine has been mentioned in reviews as a possible treatment option for fatigue, but there is currently no evidence of statistically significant improvement [126].
Future therapeutic options
The efficacy of various medications is currently being investigated in clinical studies [127]. It should be stressed that future therapeutic approaches are not limited to B cells but also target other areas of the immune response (janus kinase inhibitors, BDCA‐2 antibodies, Il‐12/23 antibodies). In SjS, as in other diseases, the long‐term goal is to offer every patient an individualized treatment concept.
SjS and pregnancy
If maternal Ro/SS‐A antibodies are passed through the placenta into the fetal circulation, autoimmune congenital heart block (CHB) in the fetus may occur as early as the 11th week of pregnancy [128]. CHB is found in 2–5 % of fetuses from Ro/SS‐A antibody‐positive mothers [128]. Fetuses from mothers with SjS have an increased risk of CHB for several reasons. Most pregnancies in women with Ro/SS‐A antibodies occur before full clinical manifestation of SjS, so SjS is usually neither suspected nor diagnosed in these young women before they become pregnant. These patients are therefore not treated with the appropriate medication [129]. SjS patients have high Ro/SS‐A antibody titers, which is associated with an increased risk of fetal CHB [130]. In a subsequent pregnancy, the risk of fetal CHB may increase up to 18 % in Ro/SS‐A antibody‐positive women who have already borne an infant with CHB [131]. In Ro/SS‐A antibody‐positive women, weekly sonography of the fetal heart is recommended between the 16th and the 31st week of pregnancy to assess cardiac rhythm in the fetus. Such women were previously treated with glucocorticosteroids or intravenous immunoglobulins during pregnancy to prevent fetal CHB. However, this recommendation was not evidence‐based [132]. A recent multicenter clinical study with hydroxychloroquine showed a reduction of CHB recurrence by more than half (from 18 % to 7.4 %) [131]. Use of hydroxychloroquine by Ro/SS‐A antibody‐positive women to prevent fetal CHB may therefore be considered. Neonatal LE is rare and may present with subacute cutaneous LE, blood count anomalies (leukopenia, anemia, thrombocytopenia) and involvement of the liver [132].
Interdisciplinary patient management
Patients should visit an ophthalmologist and a pulmonologist once a year, and a dentist twice a year including professional teeth cleaning. Diagnostic imaging such as echocardiography, abdominal, lymph node, and parotid sonography once a year is useful. This also applies to laboratory investigations (blood count, differential blood count, renal function parameters, liver enzymes, C‐reactive protein, erythrocyte sedimentation rate [ESR], complement, electrophoresis with quantitative analysis of immunoglobulins [IgG, IgA, IgM], venous blood gases [pH status], urinalysis, urine sedimentation with acanthocytes, protein/creatinine ratio in spontaneous urine).
Barbara C. Böckle Finanzielle Interessen: Nein Erklärung zu nicht‐finanziellen Interessen: ÖGDV, EADV André Fiona Finanzielle Interessen: Ja, von einer anderen Institution Erklärung zu nicht‐finanziellen Interessen: ÖGDV, ESDR
CME Questions/Lernerfolgskontrolle
-
Welche Faktoren spielen vermutlich eine Rolle bei der Pathogenese des SjS?
Infektionen
Rauchen
Medikamente
Hormone
Alle Antworten sind richtig.
-
Hinsichtlich der Epidemiologie des SjS – Welche Antwort ist richtig?
Das durchschnittliche Alter, in dem die Diagnose gestellt wird, liegt meist bei 20 Jahren.
Charakteristisch für das juvenile SjS sind rezidivierende Pleuritis und Myositis.
Das SjS ist häufiger bei Männern als bei Frauen.
Männer haben einen schwereren Verlauf der Erkrankung als Frauen.
Das SjS ist eine seltene rheumatologische Erkrankung.
-
Welches Kriterium wird nicht zu den ACR/EULAR‐Klassifikationskriterien 2016 gezählt?
positive Ro/SS‐A‐Antikörper
positive La/SS‐B‐Antikörper
ocular staining score ≥ 5 in wenigstens einem Auge
Schirmer‐Test ≤ 5mm/5min in wenigstens einem Auge
unstimulated whole saliva flow rate UWS ≤ 0,1 ml/min
-
Welche der unten angeführten Diagnosen muss ausgeschlossen werden, bevor die ACR/EULAR‐Klassifikationskriterien von 2016 für das SjS angewendet werden können?
Psoriasis vulgaris
Basaliom
aktive Hepatitis‐C‐Infektion
aktinische Keratosen
Hidradenitis suppurativa
-
Folgende Hautmanifestation wird bei Patienten mit SjS häufig beobachtet?
leukozytoklastische Vaskulitis
Psoriasis vulgaris
Periorale Dermatitis
Herpes zoster
Akne vulgaris
-
Welche Therapie wird bei Xerophthalmie bei SjS nicht empfohlen?
befeuchtende Augentropfen
topische Kortikosteroide
TNF‐alpha‐Blocker
Ciclosporin A‐Augentropfen
Pilocarpin‐Augentropfen
-
Was trifft zu?
Trockene Haut und Juckreiz sind häufige kutane Symptome des SjS.
Migräne geht mit erhöhtem Risiko für ein Lymphom einher.
Bei schwerer Raynaud‐Symptomatik im Rahmen eines SjS muss eine begleitende Psoriasis vulgaris in Betracht gezogen werden.
Fumarsäure kann als Zweitlinientherapie bei kutaner Vaskulitis eingesetzt werden.
Die Prävalenz des Juckreizes bei Patienten mit SjS ist niedriger im Vergleich zu der allgemeinen Bevölkerung.
-
Bezüglich klinischer Manifestationen beim SjS trifft zu?
Der ESSDAI ist ein Score, um die Krankheitsaktivität bei SjS zu messen.
Patienten mit SjS zeigen kaum neuropsychiatrische Manifestationen.
Eine Nierenbeteiligung beim SjS ist sehr häufig.
SjS‐Patienten haben oft positive antimitochondriale Antikörper.
Die subjektiven und objektiven Symptome der Sicca‐Symptomatik korrelieren miteinander.
-
Bezüglich Organmanifestationen bei SjS trifft zu?
Neurologische Manifestationen beim SjS sind extrem selten.
Das Raynaud‐Phänomen tritt bei weniger als 5 % der SjS Patienten
Eine Autoimmunhepatitis wird bei mehr als 10 % der SjS‐Patienten festgestellt.
Die Nierenbeteiligung beim SjS tritt sehr häufig auf.
SjS‐Patienten haben oft Arthralgien und Myalgien.
-
Patienten mit SjS haben eine gute Prognose, …
jedoch besteht ein erhöhtes Risiko für die Entwicklung eines Lymphoms.
eine Optimierung kardiovaskulärer Risikofaktoren ist nicht notwendig.
aber sie haben oft eine symptomatische Serositis.
allerdings besteht kein erhöhtes Risiko für einen fetalen AV‐Block während der Schwangerschaft.
aber es besteht kein erhöhtes Risiko für die Entwicklung von Schilddrüsenkarzinomen.
Liebe Leserinnen und Leser, der Einsendeschluss an die DDA für diese Ausgabe ist der 30. September 2022. Die richtige Lösung zum Thema „Das Spektrum melanozytärer Nävi und deren klinische Bedeutung“ in Heft 4 (April 2022) ist: 1c, 2d, 3c, 4e, 5e, 6a, 7d, 8c, 9a, 10c
Bitte verwenden Sie für Ihre Einsendung das aktuelle Formblatt auf der folgenden Seite oder aber geben Sie Ihre Lösung online unter http://jddg.akademie‐dda.de ein.
Section Editor
Prof. Dr. Trautinger, St. Pölten
Sjögren’s syndrome (SjS) is a connective tissue disease. It is defined by the clinical triad of sicca symptoms, fatigue, and pain.
Etiology and pathogenesis are not fully understood, but it is assumed that various factors such as genetics, environmental factors, hormones, and immune dysregulation play a role in the pathogenesis of this disease.
SjS is one of the most common rheumatological diseases.
SjS is most commonly diagnosed in women between the 5th and 7th decade of life.
SjS is diagnosed based on the patient’s subjective sicca symptoms if these cannot be explained by other differential diagnoses, after excluding certain diseases and using the objective classification criteria.
Patients must have had sicca symptoms for at least three months before the SjS classification criteria can be applied.
The following diseases must be excluded before SjS can be diagnosed [31]: History of radiation therapy in the head and neck area, active hepatitis C infection (positive HCV‐PCR), AIDS, sarcoidosis, amyloidosis, graft‐versus‐host disease (GvHD), IgG4‐associated disease.
The current classification criteria for primary SjS were published in 2016 by ACR/EULAR.
Among the objective criteria, Ro/SS‐A antibodies and histological investigation of the salivary glands (focus score [FS] ≥ 1) are mandatory criteria.
The term secondary SjS (sSjS) is used when SjS occurs in association with other autoimmune diseases. However, the distinction between pSjS and sSjS varies in the scientific literature.
Sicca symptoms impair quality of life in SjS patients.
Skin manifestations occur in 14–16 % of SjS patients.
Dry skin and pruritus are common cutaneous symptoms of SjS.
Vasculitis is the second most common skin manifestation.
Subacute cutaneous LE is the most common LE‐specific skin manifestation in the context of SjS.
Raynaud’s phenomenon occurs in 16–35 % of SjS patients.
Increased levels of liver enzymes are found in about one‐third of SjS patients. Infectious causes need to be excluded first.
Interstitial lung disease and its sequelae are frequent causes of death in SjS patients; therefore, early diagnosis and treatment are essential.
Affection of the kidneys in SjS is rare (≤ 10 %).
Arthritis is one of the most common manifestations of SjS (15–90 %).
Three‐fourths of SjS patients show cognitive impairment, and many also have psychiatric disease (depression, anxiety disorders).
SjS is associated with an increased cardiovascular risk.
SjS patients have a significantly increased (10–44‐fold) risk for lymphoma.
Since SjS can affect multiple organ systems, an interdisciplinary approach is required for individual treatment. Basically, we must differentiate between symptomatic treatment of sicca symptoms, and immunosuppressant treatment of organ involvement.
If maternal Ro/SS‐A antibodies are passed through the placenta into the fetal circulation, autoimmune congenital heart block (CHB) in the fetus may occur as early as the 11th week of pregnancy.
References
- 1. Ramos‐Casals M, Brito‐Zeron P, Bombardieri S et al. EULAR recommendations for the management of Sjogren’s syndrome with topical and systemic therapies. Ann Rheum Dis 2020; 79(1): 3–18. [DOI] [PubMed] [Google Scholar]
- 2. von Mikulicz JH. Über eine eigenartige symmetrische Erkrankung der Tränen‐ und Mundspeicheldrüsen. Billroth GT Beitr Chir Fortschr Stuttgart 1892: 610–30. [Google Scholar]
- 3. Skopouli FN, Moutsopoulos HM. Autoimmune epitheliitis: Sjogren’s syndrome. Clin Exp Rheumatol 1994; 12 (Suppl 11): S9–11. [PubMed] [Google Scholar]
- 4. Manoussakis MN, Kapsogeorgou EK. The role of intrinsic epithelial activation in the pathogenesis of Sjogren’s syndrome. J Autoimmun 2010; 35(3): 219–24. [DOI] [PubMed] [Google Scholar]
- 5. Verstappen GM, Pringle S, Bootsma H, Kroese FGM. Epithelial‐immune cell interplay in primary Sjogren syndrome salivary gland pathogenesis. Nat Rev Rheumatol 2021; 17(6): 333–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bombardieri M, Argyropoulou OD, Ferro F et al. One year in review 2020: pathogenesis of primary Sjogren’s syndrome. Clin Exp Rheumatol 2020; 38 (Suppl 126(4)): 3–9. [PubMed] [Google Scholar]
- 7. Lessard CJ, Li H, Adrianto I et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren’s syndrome. Nat Genet 2013; 45(11): 1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gottenberg JE, Busson M, Loiseau P et al. In primary Sjogren’s syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum 2003; 48(8): 2240–5. [DOI] [PubMed] [Google Scholar]
- 9. Kuo CF, Grainge MJ, Valdes AM et al. Familial risk of Sjogren’s syndrome and co‐aggregation of autoimmune diseases in affected families: a nationwide population study. Arthritis Rheumatol 2015; 67(7): 1904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skopouli FN, Katsiougiannis S. How stress contributes to autoimmunity‐lessons from Sjogren’s syndrome. FEBS Lett 2018; 592(1): 5–14. [DOI] [PubMed] [Google Scholar]
- 11. Utomo SW, Putri JF. Infections as risk factor of Sjogren’s syndrome. Open Access Rheumatol 2020; 12: 257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramos‐Casals M, Maria A, Suarez‐Almazor ME et al. Sicca/Sjogren’s syndrome triggered by PD‐1/PD‐L1 checkpoint inhibitors. Data from the International ImmunoCancer Registry (ICIR). Clin Exp Rheumatol 2019; 37 (Suppl 118(3)): 114–22. [PubMed] [Google Scholar]
- 13. Colafrancesco S, Perricone C, Priori R et al. Sjogren’s syndrome: another facet of the autoimmune/inflammatory syndrome induced by adjuvants (ASIA). J Autoimmun 2014; 51: 10–6. [DOI] [PubMed] [Google Scholar]
- 14. Coroneos CJ, Selber JC, Offodile AC 2nd, Butler CE, Clemens MW. US FDA Breast Implant Postapproval Studies: Long‐term outcomes in 99,993 patients. Ann Surg 2019; 269(1): 30–6. [DOI] [PubMed] [Google Scholar]
- 15. Croia C, Astorri E, Murray‐Brown W et al. Implication of Epstein‐Barr virus infection in disease‐specific autoreactive B cell activation in ectopic lymphoid structures of Sjogren’s syndrome. Arthritis Rheumatol 2014; 66(9): 2545–57. [DOI] [PubMed] [Google Scholar]
- 16. Martelli Junior H, Gueiros LA, de Lucena EG, Coletta RD. Increase in the number of Sjogren’s syndrome cases in Brazil in the COVID‐19 Era. Oral Dis 2021. May 27. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel R, Shahane A. The epidemiology of Sjogren’s syndrome. Clin Epidemiol 2014; 6: 247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ji J, Sundquist J, Sundquist K. Gender‐specific incidence of autoimmune diseases from national registers. J Autoimmun 2016; 69: 102–6. [DOI] [PubMed] [Google Scholar]
- 19. Garcia‐Carrasco M, Ramos‐Casals M, Rosas J et al. Primary Sjogren syndrome: clinical and immunologic disease patterns in a cohort of 400 patients. Medicine (Baltimore) 2002; 81(4): 270–80. [DOI] [PubMed] [Google Scholar]
- 20. Brandt JE, Priori R, Valesini G, Fairweather D. Sex differences in Sjogren’s syndrome: a comprehensive review of immune mechanisms. Biol Sex Differ 2015; 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yazisiz V, Gocer M, Erbasan F et al. Survival analysis of patients with Sjogren’s syndrome in Turkey: a tertiary hospital‐based study. Clin Rheumatol 2020; 39(1): 233–41. [DOI] [PubMed] [Google Scholar]
- 22. Weng MY, Huang YT, Liu MF, Lu TH. Incidence and mortality of treated primary Sjogren’s syndrome in Taiwan: a population‐based study. J Rheumatol 2011; 38(4): 706–8. [DOI] [PubMed] [Google Scholar]
- 23. Anaya JM, Liu GT, D’Souza E et al. Primary Sjogren’s syndrome in men. Ann Rheum Dis 1995; 54(9): 748–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Souza TR, Silva IH, Carvalho AT et al. Juvenile Sjogren syndrome: distinctive age, unique findings. Pediatr Dent. 2012; 34(5): 427–30. [PubMed] [Google Scholar]
- 25. Marino A, Romano M, Giani T et al. Childhood Sjogren’s syndrome: An Italian case series and a literature review‐based cohort. Semin Arthritis Rheum 2021; 51(4): 903–10. [DOI] [PubMed] [Google Scholar]
- 26. Brito‐Zeron P, Acar‐Denizli N, Ng WF et al. Epidemiological profile and north‐south gradient driving baseline systemic involvement of primary Sjogren’s syndrome. Rheumatology (Oxford) 2020; 59(9): 2350–9. [DOI] [PubMed] [Google Scholar]
- 27. Brito‐Zeron P, Acar‐Denizli N, Zeher M et al. Influence of geolocation and ethnicity on the phenotypic expression of primary Sjogren’s syndrome at diagnosis in 8310 patients: a cross‐sectional study from the Big Data Sjogren Project Consortium. Ann Rheum Dis 2017; 76(6): 1042–50. [DOI] [PubMed] [Google Scholar]
- 28. Maldini C, Seror R, Fain O et al. Epidemiology of primary Sjogren’s syndrome in a French multiracial/multiethnic area. Arthritis Care Res (Hoboken) 2014; 66(3): 454–63. [DOI] [PubMed] [Google Scholar]
- 29. Tanasiewicz M, Hildebrandt T, Obersztyn I. Xerostomia of various etiologies: a review of the literature. Adv Clin Exp Med 2016; 25(1): 199–206. [DOI] [PubMed] [Google Scholar]
- 30. The definition and classification of dry eye disease: report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf 2007; 5(2): 75–92. [DOI] [PubMed] [Google Scholar]
- 31. Shiboski CH, Shiboski SC, Seror R et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjogren’s syndrome: A consensus and data‐driven methodology involving three international patient cohorts. Ann Rheum Dis 2017; 76(1): 9–16. [DOI] [PubMed] [Google Scholar]
- 32. Vitali C, Bombardieri S, Jonsson R et al. Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American‐European Consensus Group. Ann Rheum Dis 2002; 61(6): 554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shiboski SC, Shiboski CH, Criswell L et al. American College of Rheumatology classification criteria for Sjogren’s syndrome: a data‐driven, expert consensus approach in the Sjogren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken) 2012; 64(4): 475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Navazesh M, Kumar SK, University of Southern California School of D. Measuring salivary flow: challenges and opportunities. J Am Dent Assoc 2008; 139 (Suppl): 35S–40S. [DOI] [PubMed] [Google Scholar]
- 35. Fisher BA, Jonsson R, Daniels T et al. Standardisation of labial salivary gland histopathology in clinical trials in primary Sjogren’s syndrome. Ann Rheum Dis 2017; 76(7): 1161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Daniels TE, Cox D, Shiboski CH et al. Associations between salivary gland histopathologic diagnoses and phenotypic features of Sjogren’s syndrome among 1,726 registry participants. Arthritis Rheum 2011; 63(7): 2021–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chisholm DM, Mason DK. Labial salivary gland biopsy in Sjogren’s disease. J Clin Pathol 1968; 21(5): 656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guellec D, Cornec D, Jousse‐Joulin S et al. Diagnostic value of labial minor salivary gland biopsy for Sjogren’s syndrome: a systematic review. Autoimmun Rev 2013; 12(3): 416–20. [DOI] [PubMed] [Google Scholar]
- 39. Santiago ML, Seisdedos MR, Garcia Salinas RN et al. Usefulness of antibodies and minor salivary gland biopsy in the study of sicca syndrome in daily clinical practice. Reumatol Clin 2015; 11(3): 156–60. [DOI] [PubMed] [Google Scholar]
- 40. Kessel A, Toubi E, Rozenbaum M et al. Sjogren’s syndrome in the community: can serology replace salivary gland biopsy? Rheumatol Int 2006; 26(4): 337–9. [DOI] [PubMed] [Google Scholar]
- 41. Chen W, Cao H, Lin J et al. Biomarkers for primary Sjogren’s syndrome. Genomics Proteomics Bioinformatics 2015; 13(4): 219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mavragani CP, Moutsopoulos HM. Primary versus secondary Sjogren Syndrome: Is it time to reconsider these terms? J Rheumatol 2019; 46(7): 665–6. [DOI] [PubMed] [Google Scholar]
- 43. Kollert F, Fisher BA. Equal rights in autoimmunity: is Sjogren’s syndrome ever “secondary”? Rheumatology (Oxford) 2020; 59(6): 1218–25. [DOI] [PubMed] [Google Scholar]
- 44. Moutsopoulos HM, Chused TM, Mann DL et al. Sjogren’s syndrome (Sicca syndrome): current issues. Ann Intern Med 1980; 92(2 Pt 1): 212–26. [DOI] [PubMed] [Google Scholar]
- 45. Ramos‐Casals M, Brito‐Zeron P, Font J. The overlap of Sjogren’s syndrome with other systemic autoimmune diseases. Semin Arthritis Rheum 2007; 36(4): 246–55. [DOI] [PubMed] [Google Scholar]
- 46. He J, Ding Y, Feng M et al. Characteristics of Sjogren’s syndrome in rheumatoid arthritis. Rheumatology (Oxford) 2013; 52(6): 1084–9. [DOI] [PubMed] [Google Scholar]
- 47. Brown LE, Frits ML, Iannaccone CK et al. Clinical characteristics of RA patients with secondary SS and association with joint damage. Rheumatology (Oxford) 2015; 54(5): 816–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pasoto SG, Adriano de Oliveira Martins V, Bonfa E. Sjogren’s syndrome and systemic lupus erythematosus: links and risks. Open Access Rheumatol 2019; 11: 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bellelli A, Tumiati B, Rossi F et al. [The Reynolds syndrome. Clinical case and review of the literature. Analogy with graft‐versus‐host disease]. G Clin Med 1981; 62(9): 656–64. [PubMed] [Google Scholar]
- 50. Ye W, Chen S, Huang X et al. Clinical features and risk factors of neurological involvement in Sjogren’s syndrome. BMC Neurosci 2018; 19(1): 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lazarus MN, Isenberg DA. Development of additional autoimmune diseases in a population of patients with primary Sjogren’s syndrome. Ann Rheum Dis 2005; 64(7): 1062–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cornec D, Devauchelle‐Pensec V, Mariette X et al. Severe health‐related quality of life impairment in active primary Sjogren’s syndrome and patient‐reported outcomes: data from a large therapeutic trial. Arthritis Care Res (Hoboken) 2017; 69(4): 528–35. [DOI] [PubMed] [Google Scholar]
- 53. Alunno A, Bartoloni E, Valentini V et al. Discrepancy between subjective symptoms, objective measures and disease activity indexes: the lesson of primary Sjogren’s syndrome. Clin Exp Rheumatol 2018; 36 (Suppl 112(3)): 210–4. [PubMed] [Google Scholar]
- 54. Mathews PM, Robinson SA, Gire A et al. Extraglandular ocular involvement and morbidity and mortality in primary Sjogren’s Syndrome. PLoS One 2020; 15(9): e0239769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jacobi C, Cursiefen C. [Ophthalmological complications in Sjogren’s syndrome]. Z Rheumatol 2010; 69(1): 32–40. [DOI] [PubMed] [Google Scholar]
- 56. Marchesoni D, Mozzanega B, De Sandre P et al. Gynaecological aspects of primary Sjogren’s syndrome. Eur J Obstet Gynecol Reprod Biol 1995; 63(1): 49–53. [DOI] [PubMed] [Google Scholar]
- 57. Sellier S, Courville P, Joly P. [Dyspareunia and Sjogren’s syndrome]. Ann Dermatol Venereol 2006; 133(1): 17–20. [DOI] [PubMed] [Google Scholar]
- 58. Capriello P, Barale E, Cappelli N et al. Sjogren’s syndrome: clinical, cytological, histological and colposcopic aspects in women. Clin Exp Obstet Gynecol 1988; 15(1–2): 9–12. [PubMed] [Google Scholar]
- 59. Osborn M, Hawton K, Gath D. Sexual dysfunction among middle aged women in the community. Br Med J (Clin Res Ed) 1988; 296(6627): 959–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brito‐Zeron P, Theander E, Baldini C et al. Early diagnosis of primary Sjogren’s syndrome: EULAR‐SS task force clinical recommendations. Expert Rev Clin Immunol 2016; 12(2): 137–56. [DOI] [PubMed] [Google Scholar]
- 61. Ramos‐Casals M, Brito‐Zeron P, Solans R et al. Systemic involvement in primary Sjogren’s syndrome evaluated by the EULAR‐SS disease activity index: analysis of 921 Spanish patients (GEAS‐SS Registry). Rheumatology (Oxford) 2014; 53(2): 321–31. [DOI] [PubMed] [Google Scholar]
- 62. Durigan VSA, Duarte V, Troitiño C et al. Manifestaciones cutáneas extraglandulares en pacientes con síndrome de Sjögren primario. Revista Colombiana de Reumatología 2018; 25(2): 79–84. [Google Scholar]
- 63. Ramos‐Casals M, Anaya JM, Garcia‐Carrasco M et al. Cutaneous vasculitis in primary Sjogren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore) 2004; 83(2): 96–106. [DOI] [PubMed] [Google Scholar]
- 64. Bernacchi E, Amato L, Parodi A et al. Sjogren’s syndrome: a retrospective review of the cutaneous features of 93 patients by the Italian Group of Immunodermatology. Clin Exp Rheumatol 2004; 22(1): 55–62. [PubMed] [Google Scholar]
- 65. Valdes‐Rodriguez R, Rowe B, Lee HG et al. Chronic pruritus in primary Sjogren’s syndrome: characteristics and effect on quality of life. Acta Derm Venereol 2017; 97(3): 385–6. [DOI] [PubMed] [Google Scholar]
- 66. Bernacchi E, Bianchi B, Amato L et al. Xerosis in primary Sjogren syndrome: immunohistochemical and functional investigations. J Dermatol Sci 2005; 39(1): 53–5. [DOI] [PubMed] [Google Scholar]
- 67. Matterne U, Apfelbacher CJ, Vogelgsang L et al. Incidence and determinants of chronic pruritus: a population‐based cohort study. Acta Derm Venereol 2013; 93(5): 532–7. [DOI] [PubMed] [Google Scholar]
- 68. Bartoloni E, Baldini C, Schillaci G et al. Cardiovascular disease risk burden in primary Sjogren’s syndrome: results of a population‐based multicentre cohort study. J Intern Med 2015; 278(2): 185–92. [DOI] [PubMed] [Google Scholar]
- 69. Bockle BC, Jara D, Aichhorn K et al. Cerebral large vessel vasculitis in systemic lupus erythematosus. Lupus 2014; 23(13): 1417–21. [DOI] [PubMed] [Google Scholar]
- 70. Kapsogeorgou EK, Voulgarelis M, Tzioufas AG. Predictive markers of lymphomagenesis in Sjogren’s syndrome: From clinical data to molecular stratification. J Autoimmun 2019; 104: 102316. [DOI] [PubMed] [Google Scholar]
- 71. Biazar C, Sigges J, Patsinakidis N et al. Cutaneous lupus erythematosus: first multicenter database analysis of 1002 patients from the European Society of Cutaneous Lupus Erythematosus (EUSCLE). Autoimmun Rev 2013; 12(3): 444–54. [DOI] [PubMed] [Google Scholar]
- 72. Watanabe T, Tsuchida T, Ito Y et al. Annular erythema associated with lupus erythematosus/Sjogren’s syndrome. J Am Acad Dermatol 1997; 36(2 Pt 1): 214–8. [DOI] [PubMed] [Google Scholar]
- 73. Teramoto N, Katayama I, Arai H et al. Annular erythema: a possible association with primary Sjogren’s syndrome. J Am Acad Dermatol 1989; 20(4): 596–601. [DOI] [PubMed] [Google Scholar]
- 74. Brito‐Zeron P, Retamozo S, Akasbi M et al. Annular erythema in primary Sjogren’s syndrome: description of 43 non‐Asian cases. Lupus 2014; 23(2): 166–75. [DOI] [PubMed] [Google Scholar]
- 75. Kuhn A, Richter‐Hintz D, Schuppe HC et al. [Annular erythema in Sjogren syndrome. A variant of cutaneous lupus erythematosus?]. Hautarzt 2000; 51(4): 270–5. [DOI] [PubMed] [Google Scholar]
- 76. Chiu HY, Muo CH, Sung FC. Associations of chronic urticaria with atopic and autoimmune comorbidities: a nationwide population‐based study. Int J Dermatol 2018; 57(7): 822–9. [DOI] [PubMed] [Google Scholar]
- 77. Aragon CC, Ruiz‐Ordonez I Nieto‐Aristizabal I et al. Letter to the editor: Frontal fibrosing alopecia: An autoimmune manifestation? Autoimmun Rev 2021; 20(2): 102728. [DOI] [PubMed] [Google Scholar]
- 78. Chen YT, Chen YJ, Hwang CY et al. Comorbidity profiles in association with vitiligo: a nationwide population‐based study in Taiwan. J Eur Acad Dermatol Venereol 2015; 29(7): 1362–9. [DOI] [PubMed] [Google Scholar]
- 79. Collet E, Dalac S, Brichon P et al. [The association of lichen planus and primary Gougerot‐Sjogren syndrome]. Ann Dermatol Venereol 1989; 116(6–7): 483–6. [PubMed] [Google Scholar]
- 80. Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjogren syndrome. J Cutan Med Surg 2011; 15(1): 8–14. [DOI] [PubMed] [Google Scholar]
- 81. Ueki H, Inagaki Y, Hamasaki Y, Ono M. [Dermatological manifestations of Sjogren’s syndrome]. Hautarzt 1991; 42(12): 741–7. [PubMed] [Google Scholar]
- 82. Alexander EL, Provost TT. Cutaneous manifestations of primary Sjogren’s syndrome: a reflection of vasculitis and association with anti‐Ro(SSA) antibodies. J Invest Dermatol 1983; 80(5): 386–91. [DOI] [PubMed] [Google Scholar]
- 83. Soy M, Piskin S. Cutaneous findings in patients with primary Sjogren’s syndrome. Clin Rheumatol 2007; 26(8): 1350–2. [DOI] [PubMed] [Google Scholar]
- 84. Cervera R, Piette JC, Font J et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46(4): 1019–27. [DOI] [PubMed] [Google Scholar]
- 85. Fauchais AL, Lambert M, Launay D et al. Antiphospholipid antibodies in primary Sjogren’s syndrome: prevalence and clinical significance in a series of 74 patients. Lupus 2004; 13(4): 245–8. [DOI] [PubMed] [Google Scholar]
- 86. Pasoto SG, Chakkour HP, Natalino RR et al. Lupus anticoagulant: a marker for stroke and venous thrombosis in primary Sjogren’s syndrome. Clin Rheumatol 2012; 31(9): 1331–8. [DOI] [PubMed] [Google Scholar]
- 87. Liang Y, Leng RX, Pan HF, Ye DQ. The prevalence and risk factors for serositis in patients with systemic lupus erythematosus: a cross‐sectional study. Rheumatol Int 2017; 37(2): 305–11. [DOI] [PubMed] [Google Scholar]
- 88. Gyongyosi M, Pokorny G, Jambrik Z et al. Cardiac manifestations in primary Sjogren’s syndrome. Ann Rheum Dis 1996; 55(7): 450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hosoda C, Hosaka Y, Ryu K et al. Pleuritis associated with primary Sjogren syndrome. Respirol Case Rep 2018; 6(2): e00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Karp JK, Akpek EK, Anders RA. Autoimmune hepatitis in patients with primary Sjogren’s syndrome: a series of two‐hundred and two patients. Int J Clin Exp Pathol 2010; 3(6): 582–6. [PMC free article] [PubMed] [Google Scholar]
- 91. Zeron PB, Retamozo S, Bove A et al. Diagnosis of Liver Involvement in primary Sjogren syndrome. J Clin Transl Hepatol 2013; 1(2): 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Papiris SA, Maniati M, Constantopoulos SH et al. Lung involvement in primary Sjogren’s syndrome is mainly related to the small airway disease. Ann Rheum Dis 1999; 58(1): 61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yoo H, Hino T, Hwang J et al. Connective tissue disease‐related interstitial lung disease (CTD‐ILD) and interstitial lung abnormality (ILA): Evolving concept of CT findings, pathology and management. Eur J Radiol Open 2022; 9: 100419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Francois H, Mariette X. [Renal involvement in Sjogren’s syndrome]. Nephrol Ther 2020; 16(7): 440–52. [DOI] [PubMed] [Google Scholar]
- 95. Brito‐Zeron P, Acar‐Denizli N, Ng WF et al. How immunological profile drives clinical phenotype of primary Sjogren’s syndrome at diagnosis: analysis of 10,500 patients (Sjogren Big Data Project). Clin Exp Rheumatol 2018; 36 (Suppl 112(3)): 102–12. [PubMed] [Google Scholar]
- 96. Flores‐Chavez A, Kostov B, Solans R et al. Severe, life‐threatening phenotype of primary Sjogren’s syndrome: clinical characterisation and outcomes in 1580 patients (GEAS‐SS Registry). Clin Exp Rheumatol 2018; 36 (Suppl 112(3)): 121–9. [PubMed] [Google Scholar]
- 97. Fauchais AL, Ouattara B, Gondran G et al. Articular manifestations in primary Sjogren’s syndrome: clinical significance and prognosis of 188 patients. Rheumatology (Oxford) 2010; 49(6): 1164–72. [DOI] [PubMed] [Google Scholar]
- 98. Mohammed K, Pope J, Le Riche N et al. Association of severe inflammatory polyarthritis in primary Sjogren’s syndrome: clinical, serologic, and HLA analysis. J Rheumatol 2009; 36(9): 1937–42. [DOI] [PubMed] [Google Scholar]
- 99. Alunno A, Carubbi F, Bartoloni E et al. The kaleidoscope of neurological manifestations in primary Sjogren’s syndrome. Clin Exp Rheumatol 2019; 37 (Suppl 118(3)): 192–8. [PubMed] [Google Scholar]
- 100. Tobon GJ, Pers JO, Devauchelle‐Pensec V, Youinou P. Neurological disorders in primary Sjogren’s syndrome. Autoimmune Dis 2012; 2012: 645967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Akaishi T, Takahashi T, Fujihara K et al. Impact of comorbid Sjogren syndrome in anti‐aquaporin‐4 antibody‐positive neuromyelitis optica spectrum disorders. J Neurol 2021; 268(5): 1938–44. [DOI] [PubMed] [Google Scholar]
- 102. Tezcan ME, Kocer EB, Haznedaroglu S et al. Primary Sjogren’s syndrome is associated with significant cognitive dysfunction. Int J Rheum Dis 2016; 19(10): 981–8. [DOI] [PubMed] [Google Scholar]
- 103. Wong JK, Nortley R, Andrews T, D’Cruz D. Psychiatric manifestations of primary Sjogren’s syndrome: a case report and literature review. BMJ Case Rep 2014; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mofors J, Holmqvist M, Westermark L et al. Concomitant Ro/SSA and La/SSB antibodies are biomarkers for the risk of venous thromboembolism and cerebral infarction in primary Sjogren’s syndrome. J Intern Med 2019; 286(4): 458–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Valim V, Gerdts E, Jonsson R et al. Atherosclerosis in Sjogren’s syndrome: evidence, possible mechanisms and knowledge gaps. Clin Exp Rheumatol 2016; 34(1): 133–42. [PubMed] [Google Scholar]
- 106. Retamozo S, Brito‐Zeron P, Ramos‐Casals M. Prognostic markers of lymphoma development in primary Sjogren syndrome. Lupus 2019; 28(8): 923–36. [DOI] [PubMed] [Google Scholar]
- 107. Chatzis L, Goules AV, Pezoulas V et al. A biomarker for lymphoma development in Sjogren’s syndrome: Salivary gland focus score. J Autoimmun 2021; 121: 102648. [DOI] [PubMed] [Google Scholar]
- 108. Chatzis LG, Stergiou IE, Goules AV et al. Clinical picture, outcome, and predictive factors of lymphoma in primary Sjogren’s syndrome. Results from a harmonized dataset (1981–2021). Rheumatology (Oxford) 2021. Dec 23. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 109. Bockle BC, Stanarevic G, Ratzinger G, Sepp NT. Analysis of 303 Ro/SS‐A antibody‐positive patients: is this antibody a possible marker for malignancy? Br J Dermatol 2012; 167(5): 1067–75. [DOI] [PubMed] [Google Scholar]
- 110. Kang J, Kim H, Kim J et al. Risk of malignancy in Korean patients with primary Sjogren’s syndrome. Int J Rheum Dis 2020; 23(9): 1240–7. [DOI] [PubMed] [Google Scholar]
- 111. Brito‐Zeron P, Kostov B, Fraile G et al. Characterization and risk estimate of cancer in patients with primary Sjogren syndrome. J Hematol Oncol 2017; 10(1): 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Seror R, Theander E, Bootsma H et al. Outcome measures for primary Sjogren’s syndrome: a comprehensive review. J Autoimmun 2014; 51: 51–6. [DOI] [PubMed] [Google Scholar]
- 113. Seror R, Ravaud P, Bowman SJ et al. EULAR Sjogren’s syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjogren’s syndrome. Ann Rheum Dis 2010; 69(6): 1103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Seror R, Ravaud P, Mariette X et al. EULAR Sjogren’s syndrome patient reported index (ESSPRI): development of a consensus patient index for primary Sjogren’s syndrome. Ann Rheum Dis 2011; 70(6): 968–72. [DOI] [PubMed] [Google Scholar]
- 115. Seror R, Gottenberg JE, Devauchelle‐Pensec V et al. European League Against Rheumatism Sjogren’s Syndrome Disease Activity Index and European League Against Rheumatism Sjogren’s Syndrome Patient‐Reported Index: a complete picture of primary Sjogren’s syndrome patients. Arthritis Care Res (Hoboken) 2013; 65(8): 1358–64. [DOI] [PubMed] [Google Scholar]
- 116. Yoon CH, Lee HJ, Lee EY et al. Effect of hydroxychloroquine treatment on dry eyes in subjects with primary Sjogren’s syndrome: a double‐blind randomized control study. J Korean Med Sci 2016; 31(7): 1127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Meijer JM, Meiners PM, Vissink A et al. Effectiveness of rituximab treatment in primary Sjogren’s syndrome: a randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum 2010; 62(4): 960–8. [DOI] [PubMed] [Google Scholar]
- 118. Devauchelle‐Pensec V, Mariette X, Jousse‐Joulin S et al. Treatment of primary Sjogren syndrome with rituximab: a randomized trial. Ann Intern Med 2014; 160(4): 233–42. [DOI] [PubMed] [Google Scholar]
- 119. Bowman SJ, Everett CC, O’Dwyer JL et al. Randomized controlled trial of rituximab and cost‐effectiveness analysis in treating fatigue and oral dryness in primary Sjogren’s syndrome. Arthritis Rheumatol 2017; 69(7): 1440–50. [DOI] [PubMed] [Google Scholar]
- 120. Bachiller‐Corral J, Boteanu A, Garcia‐Villanueva MJ et al. Risk of severe COVID‐19 infection in patients with inflammatory rheumatic diseases. J Rheumatol 2021; 48(7): 1098–102. [DOI] [PubMed] [Google Scholar]
- 121. De Vita S, Quartuccio L, Seror R et al. Efficacy and safety of belimumab given for 12 months in primary Sjogren’s syndrome: the BELISS open‐label phase II study. Rheumatology (Oxford) 2015; 54(12): 2249–56. [DOI] [PubMed] [Google Scholar]
- 122. Mariette X, Seror R, Quartuccio L et al. Efficacy and safety of belimumab in primary Sjogren’s syndrome: results of the BELISS open‐label phase II study. Ann Rheum Dis 2015; 74(3): 526–31. [DOI] [PubMed] [Google Scholar]
- 123. Fasano S, Isenberg DA. Present and novel biologic drugs in primary Sjogren’s syndrome. Clin Exp Rheumatol 2019; 37 (Suppl 118(3)): 167–74. [PubMed] [Google Scholar]
- 124. Roccatello D, Saadoun D, Ramos‐Casals M et al. Cryoglobulinaemia. Nat Rev Dis Primers 2018; 4(1): 11. [DOI] [PubMed] [Google Scholar]
- 125. Dass S, Bowman SJ, Vital EM et al. Reduction of fatigue in Sjogren syndrome with rituximab: results of a randomised, double‐blind, placebo‐controlled pilot study. Ann Rheum Dis 2008; 67(11): 1541–4. [DOI] [PubMed] [Google Scholar]
- 126. Wang X, Zhang T, Guo Z et al. The efficiency of hydroxychloroquine for the treatment of primary Sjogren’s syndrome: a systematic review and meta‐analysis. Front Pharmacol 2021; 12: 693796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Seror R, Nocturne G, Mariette X. Current and future therapies for primary Sjogren syndrome. Nat Rev Rheumatol 2021; 17(8): 475–86. [DOI] [PubMed] [Google Scholar]
- 128. Pruetz JD, Miller JC, Loeb GE et al. Prenatal diagnosis and management of congenital complete heart block. Birth Defects Res 2019; 111(8): 380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Julkunen H, Kaaja R, Kurki P et al. Fetal outcome in women with primary Sjogren’s syndrome. A retrospective case‐control study. Clin Exp Rheumatol 1995; 13(1): 65–71. [PubMed] [Google Scholar]
- 130. Brucato A, Doria A, Frassi M et al. Pregnancy outcome in 100 women with autoimmune diseases and anti‐Ro/SSA antibodies: a prospective controlled study. Lupus 2002; 11(11): 716–21. [DOI] [PubMed] [Google Scholar]
- 131. Izmirly P, Kim M, Friedman DM et al. Hydroxychloroquine to prevent recurrent congenital heart block in fetuses of Anti‐SSA/Ro‐positive mothers. J Am Coll Cardiol 2020; 76(3): 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Brucato A, Cimaz R, Caporali R et al. Pregnancy outcomes in patients with autoimmune diseases and anti‐Ro/SSA antibodies. Clin Rev Allergy Immunol 2011; 40(1): 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Agmon‐Levin N, Lian Z, Shoenfeld Y. Explosion of autoimmune diseases and the mosaic of old and novel factors. Cell Mol Immunol 2011; 8(3): 189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]