

Abstract
Knottins are topologically complex peptides that are stabilised by a cystine knot and have exceptionally diverse functions, including protease inhibition. However, approaches for tuning their activity in situ are limited. Here, we demonstrate separate approaches for tuning the activity of knottin protease inhibitors using light or streptavidin. We show that the inhibitory activity and selectivity of an engineered knottin can be controlled with light by activating a second mode of action that switches the inhibitor ON against new targets. Guided by a knottin library screen, we also identify a position in the inhibitor's binding loop that permits insertion of a biotin tag without impairing activity. Using streptavidin, biotinylated knottins with nanomolar affinity can be switched OFF in activity assays, and the anticoagulant activity of a factor XIIa inhibitor can be rapidly switched OFF in human plasma. Our findings expand the scope of engineered knottins for precisely controlling protein function.
Keywords: Activity Switch, Enzymes, Inhibitors, Knottins, Photoactivation
Separate approaches for tuning the activity of cystine knot peptides are reported. Protease inhibitors were designed that had light‐controlled activity and could be switched ON against new targets, or had an affinity tag that allowed inhibitors to be switched OFF. These peptides enabled modulation of target activity with exquisite temporal control.
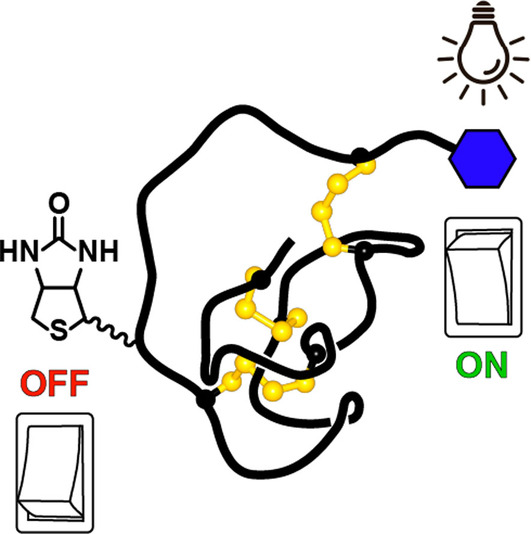
Introduction
The design of peptides and proteins with tuneable activity has paved the way for modulating chemical and biological processes with deft control. The new possibilities that can be realised with tuneable peptides and proteins have inspired several approaches for modulating their activity using light via photocaging[ 1 , 2 , 3 , 4 , 5 ] or photoswitching,[ 6 , 7 , 8 , 9 , 10 ] changes in pH,[ 11 , 12 ] or competing binding interactions.[ 13 , 14 , 15 ] These strategies have been successfully applied to diverse targets, including enzymes[ 1 , 3 , 7 ] and transmembrane proteins,[ 6 , 12 ] as well as peptides or proteins that modulate the activity of these targets, such as enzyme inhibitors.[ 16 , 17 , 18 ] For the latter, tuneable activity is particularly useful as it can shift the inhibitor's influence on its intended target or pathway.[ 16 , 17 , 18 ] However, efforts to engineer peptide‐based inhibitors with switchable activity have been largely confined to relatively short peptides with non‐complex topologies.
To generate topologically complex peptides with tuneable activity, we focused on peptides that feature a cystine knot—a key structural motif that serves as the cornerstone in a diverse array of bioactive peptides, including knottins and cyclotides (cyclic knottins).[ 19 , 20 , 21 , 22 ] The cystine knot comprises three disulfide bonds, where two disulfide bonds and their adjoining backbone segments form a macrocycle that is threaded by the third disulfide bond. [23] Segments between adjacent Cys residues form surface loops that confer biological function. Several knottins function as inhibitors of hydrolytic enzymes (proteases, amylase), with the most common enzyme targets for these knottins being serine proteases. [24]
Knottin protease inhibitors (PIs) bind reversibly to their targets with high affinity (K i values ≈10−10 to 10−12 M), which derives from their fast (substrate‐like) association rates and slow dissociation rates. [25] Knottins also have well‐defined structures, high stability in biological fluids, and multiple contact loops for optimisation. These properties have sparked considerable interest in knottin PIs for generating pharmaceutical leads or chemical probes.[ 26 , 27 , 28 , 29 , 30 , 31 ] For example, the 34‐amino acid cyclic knottin, Momordica cochinchinensis trypsin inhibitor‐II (MCoTI‐II), [32] has been increasingly used to design new inhibitors for protease therapeutic targets.[ 33 , 34 , 35 , 36 , 37 , 38 , 39 ] However, engineering efforts have relied solely on the inhibitor's ancient mode of action, and the design of knottin PIs with tuneable activity has yet to be demonstrated. These observations suggest that the potential of the knottin framework for inhibitor engineering has yet to be fully explored.
In this study, we demonstrate complementary approaches for modulating the activity of a synthetic knottin PI that can be used to install target selectivity, a light‐activated mechanism switch or an affinity tag‐based OFF switch (Scheme 1). We show that a potent but non‐selective knottin can be converted into a selective inhibitor with as few as two amino acid substitutions. We then modulate the activity and selectivity of this knottin with light by installing a photoreactive amino acid to switch the inhibitor's mode of action. By adding an affinity tag (biotin) at a position in the binding loop that we determined as not interfering with inhibitory activity, we also show that knottins can be rapidly switched OFF by adding an external effector (streptavidin). Using tuneable knottins, we modulate individual enzymes in activity assays and an enzyme cascade in human plasma, expanding the potential of knottins for precise temporal control of protein function.
Scheme 1.
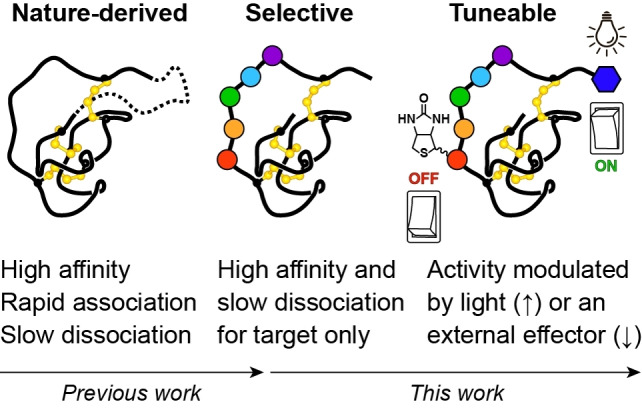
Nature‐derived knottin PIs are acyclic (solid line) or cyclic (dashed line) peptides that bind with high affinity to their targets and show fast association‐slow dissociation rates. In this work, we engineer selective knottin PIs with minimal modifications, then modulate their activity with light or an external effector.
Results and Discussion
Our approach for designing tuneable knottin PIs was initially guided by screening libraries of inhibitors with sequence diversity at one of four positions (P1′–P4′) in the binding loop. This approach is complementary to previous studies that identified selective knottins PIs by screening ribosomally‐synthesised libraries via mRNA display or yeast surface display.[ 33 , 36 ] The template for generating the synthetic libraries was based on MCoTI‐II, as we anticipated that this knottin could be used to design backbone cyclic or acyclic variants. To simplify access to the peptide libraries, we generated an acyclic MCoTI‐II variant (named MCoLib) that was linearised in loop 6 (between CysVI and CysI), and selected ten chemically diverse amino acids for substitution at the P1′–P4′ positions (Figure 1A). Further details on library design and synthesis are included in the Supporting Information (Supplementary Results 1).
Figure 1.

Synthetic knottin libraries for profiling protease specificity. A) Schematic representation of the knottin libraries, where each of the ten amino acids shown was substituted into the P1′ (blue), P2′ (green), P3′ (orange) or P4′ (red) positions. P1 (purple) was fixed as Arg (R). B) Heat maps illustrating the inhibitory activity (relative to enzyme activity in the absence of inhibitor) for knottin variants with selected amino acids (column titles) at a given position (row titles, centre). Inhibitory activity is shown as a gradient from low (blue) to high (green) as indicated in the key below the heat maps. Residues present in MCoTI‐II are highlighted with a white border (P1′ is Ile). Data are from three experiments (mean) and are shown as bar graphs (mean±SD) in Figure S10. Nle denotes norleucine.
The resulting 40 knottins were screened against a panel of serine proteases to identify amino acids compatible with activity at each position. Proteases screened were human cationic trypsin and three therapeutic targets inhibited by MCoTI‐II: factor XIIa (FXIIa),[ 33 , 34 ] matriptase,[ 35 , 36 ] and kallikrein‐related peptidase 4 (KLK4). [37] The knottin library was screened in competitive inhibition assays using a fixed concentration of inhibitor based on the activity of MCoLib, which had a low nanomolar (FXIIa: 3.3 nM) or sub‐nanomolar K i (trypsin: 0.4 nM, matriptase: 0.4 nM, KLK4: 0.07 nM) against each target.
Specificity data revealed that, although the native P1′–P4′ sequence (ILKK) in MCoTI‐II was broadly favoured, all positions were amenable to substitution (Figure 1B). Residues in close proximity to the scissile bond (P1′ and P2′) had greater influence on knottin activity and selectivity. At the P1′ position, Ile is highly conserved in knottin PIs[ 19 , 40 ] and hydrophobic residues were preferred by trypsin, led by norleucine (Nle) and Ile (Figure S10). However, a broader range of amino acids was favoured by other enzymes that included Phe (FXIIa) and hydrophobic or polar residues (matriptase and KLK4). At the P2′ position, the most common residue in nature‐derived knottin PIs is Leu,[ 19 , 40 ] which was well‐tolerated by all enzymes screened. Trypsin, FXIIa, and KLK4 favoured several additional amino acids, including Lys (trypsin and KLK4) or aromatic residues (FXIIa and KLK4). FXIIa appeared to be the only enzyme that tolerated Glu at the P2′ position. By contrast, the P2′ specificity of matriptase appeared to be relatively narrow, with only Nle or Leu generating potent inhibitors (Figure S10).
By comparison, amino acid substitutions at P3′ and P4′ had less influence on knottin activity. For trypsin, each substitution at P3′ produced little change in activity compared to Lys (present in MCoTI‐II). Similarly, most P3′ residues were well‐tolerated by FXIIa and KLK4, although variants with Phe, Asn or Glu showed less potent activity against FXIIa, and all substitutions except Glu led to slightly improved activity against KLK4 (compared to Lys). P3′ substitutions had larger effects on matriptase inhibition, with Lys, Ala, Trp, and Val being preferred, whereas Asn or Glu produced variants with weak activity. Substituting the P4′ residue in MCoLib did not lead to marked changes in activity against any of the proteases screened, although matriptase and KLK4 showed some degree of amino acid specificity.
We next explored whether differences in P1′–P4′ specificity could be harnessed to design selective knottin PIs. The enzyme selected was FXIIa, an established target for developing next‐generation anticoagulants [41] that was the subject of a previous MCoTI‐II engineering study. [34] In that study, the P1′–P4′ segment received little attention, and the best variant showed modest potency (K i=490 nM) and limited selectivity (≈10‐fold) over trypsin and plasmin, despite sequence changes at five positions. [34]
In Figure 1, we identified the P1′ and P2′ residues as key activity determinants. Accordingly, we identified substitutions of interest at these positions, then set out to test chemically similar proteinogenic or non‐proteinogenic amino acids, before synthesising a focused peptide library to screen all possible combinations of the preferred amino acids at each position. Details on the design and activity of these variants are provided in the Supporting Information (Supplementary Results 2).
Initially, the new variants (1–7) were screened against FXIIa, trypsin, KLK4, and matriptase to identify potent and selective leads (Figure S14). We then determined K i values for 1, 3, and 7 against each enzyme (Figure 2A). The most selective variant (1) retained low nanomolar activity against FXIIa (K i=11 nM), which represents only three‐fold change in K i compared to MCoLib. However, changes in activity for off‐target enzymes were substantially higher: 7 350‐fold for trypsin, 7 075‐fold for matriptase, and at least another order of magnitude (>100 000‐fold) for KLK4. Inhibition data for a broader panel of human serine proteases (thrombin, FXa, FXIa, plasmin, plasma kallikrein, uPA, tPA) further verified the high selectivity of 1 (Table S7). We also explored the kinetic basis for the divergent activities of MCoLib and 1 against FXIIa and trypsin by comparing inhibitor dissociation rates, as described previously. [42] The parent molecule MCoLib is a potent inhibitor of both targets and progress curves at steady state yielded similar k off values of 4.8×10−3 s−1 and 2.2×10−3 s−1 for FXIIa and trypsin, respectively (Figure 2B). Calculating k on using experimentally determined values for K i (3.3 nM and 0.4 nM, Figure 2A) and k off indicated that k on was in the order of 106 M−1 s−1. For the selective inhibitor 1 and FXIIa, k off was similar to MCoLib (6.1×10−3 s−1), but for trypsin the progress curves were identical after adding 1 prior to, or simultaneously with, substrate which is consistent with fast on‐fast off kinetics. [43]
Figure 2.

Design of potent and selective FXIIa inhibitors via two substitutions. A) Sequence modifications in the binding loop for selected knottin variants (4FPhe denotes 4‐fluoro‐L‐Phe) and K i values (±standard error) against FXIIa, trypsin, matriptase, or KLK4. Data are from three experiments performed in triplicate, and >10 μM indicates less than 50 % inhibition at 10 μM. B) Reaction progress curves showing enzyme activity (y‐axis, mOD) after addition of substrate to FXIIa or trypsin pre‐incubated with inhibitor (orange) or simultaneously with inhibitor (purple). Enzyme activity in the absence of inhibitor (black) illustrates the control rate. Progress curves used to calculate k off for each enzyme‐inhibitor pair (except trypsin‐1) are shown in Figure S15. C) Inhibitory activity of compound (comp.) 1 in coagulation assays using human plasma. Activated partial thromboplastin time (aPTT) assays measure the FXIIa‐initiated intrinsic pathway, and prothrombin time (PT) assays measure the FVIIa/tissue factor‐initiated extrinsic pathway. Data show mean±SD from n=3, and control indicates clotting time in the absence of inhibitor.
We also examined whether the P1′ and P2′ substitutions from the backbone acyclic variant 1 could be used to generate a backbone cyclic inhibitor by restoring loop 6 from MCoTI‐II. This peptide was produced by enzymatic cyclisation of a synthetic precursor[ 44 , 45 ] using the native cyclase from Momordica cochinchinensis MCoAEP2. [46] Although 1 remained a more potent FXIIa inhibitor than the resulting cyclic knottin (K i=99±4 nM), this level of activity represents a slight improvement on cyclic MCoTI‐II (K i=129±9 nM). [33]
Having identified 1 as a potent and selective FXIIa inhibitor, in vitro coagulation assays were performed to assess its anticoagulant activity. Activation of the coagulation system is mediated by two converging protease cascades: the intrinsic/contact pathway and the extrinsic/tissue factor pathway. [47] FXIIa is the lead enzyme in the intrinsic pathway, and its activation is measured via activated partial thromboplastin time (aPTT) assays. Inhibitory activity is observed as an increase in clotting time compared to the control without inhibitor. For 1, a concentration‐dependent effect on plasma clotting was observed (Figure 2C), with the inhibitor concentration required to double the clotting time (EC2×) calculated as 2.9 μM. To verify the selectivity of 1 for FXIIa and the intrinsic pathway, prothrombin time (PT) assays were performed to measure clotting via the extrinsic pathway. No effect on clotting time was observed at 10 μM inhibitor.
In converting the non‐selective inhibitor MCoLib into a selective FXIIa inhibitor 1, we showed that substitutions in the binding loop led to marked changes in the dissociation rate for trypsin, but not FXIIa (Figure 2B). By extension, these findings also suggested that an approach capable of modulating the dissociation rate could enable tuneable activity and selectivity—we anticipated that this effect could be achieved by covalently linking the inhibitor and protease. If the effect could be initiated using an external stimulus rather than an enzyme‐dependent reaction, it would potentially allow both the activity and selectivity of an inhibitor to be modulated under specific conditions. To test this concept, we focused on 1 and evaluated two sites for introducing a photoreactive amino acid (4‐azido‐L‐phenylalanine [AzF], Figure 3A): either the P4′ position (1X1, X1 denotes crosslinking position 1), as our library screen indicated that substitutions at this position were well‐tolerated by several enzymes, or at the N‐terminus (1X2), as several nature‐derived knottin PIs have sequence extensions at this position. [40] Both sites are located more than three residues from the scissile bond.
Figure 3.

A photoreactive knottin with light‐controlled activity. A) Location of the AzF residue in 1X1 or 1X2, and reaction scheme for photoactivation of AzF via formation of a reactive nitrene that can directly insert into C−H or N−H bonds, or undergo ring expansion to form a dehydroazepine intermediate. B) Crosslinking of 1X1 or 1X2 to FXIIa (5 min UV exposure) analysed by SDS‐PAGE (C=complex, P=protease, K=knottin). Control is a 1 variant that lacks a photoreactive amino acid. C) Data from competitive inhibition assays comparing the activity of 1X1 or 1X2 against FXIIa before (grey) or after (blue) UV crosslinking. Data (mean±SD) for each condition are normalised to controls (FXIIa without inhibitor) with or without UV exposure. D) Apparent IC50 values for 1X1 or 1X2 against FXIIa before or after UV crosslinking.
The two sites for introducing AzF were evaluated by comparing the protein crosslinking efficiency and inhibitory activity for 1X1 and 1X2. Crosslinking assays were performed by incubating FXIIa with inhibitor (1 : 2 ratio) for 10 min, followed by exposure to UV light (5 min) and analysis of the reaction products by SDS‐PAGE (Figure 3B). For both inhibitors, appearance of a higher molecular weight band was observed, consistent with formation of a covalent FXIIa‐knottin complex. Band intensities indicated higher crosslinking efficiency for 1X2, with the complex‐to‐protease ratio similar to a previous study on EETI‐II bearing a photo‐methionine residue at the P2′ position. [48] This approach was also used in a recent study to generate photoactive knottins that target a voltage‐gated sodium channel. [49] We also assessed changes in FXIIa inhibition before and after UV crosslinking (Figure 3C, D). Competitive inhibition assays before crosslinking revealed that both inhibitors maintained activity against FXIIa (IC50=84 nM and 54 nM for 1X1 and 1X2, respectively). After crosslinking 1X2 and FXIIa, we observed a 7.7‐fold improvement in apparent IC50, whereas no gain in activity was observed for 1X1 under the same conditions.
Since 1X2 emerged as the best variant, we performed further experiments to verify formation of the crosslinked complex and that the improved activity was due to crosslinking within the active site. MS analysis of the FXIIa‐1X2 complex revealed a mass shift of +3 421 Da compared to non‐crosslinked FXIIa, corresponding to attachment of a single 1X2 molecule to FXIIa (Figure S17). Additionally, control assays where 1X2 was exposed to UV light prior to adding FXIIa revealed only a small shift in IC50 (Figure S18A), confirming that improved activity required exposure of both 1X2 and FXIIa to UV light. We also examined crosslinking in the presence of a high‐affinity competing ligand (7, K i=2.5 nM) to demonstrate that crosslinking occurred within the active site. Pre‐incubating FXIIa with 7 led to a marked decrease in levels of the covalent FXIIa‐knottin complex (Figure S17). Additionally, no complex was observed when FXIIa and 1X2 were mixed, but not exposed to UV light (Figure S17).
We next examined whether 1X2 could be crosslinked to trypsin and, thus, overcome the rapid dissociation of the parent molecule 1, which is a weak trypsin inhibitor. These assays were performed using a range of UV exposure times (30 s to 5 min) to track the shift in apparent IC50. For FXIIa, improved inhibition was observed with increasing UV exposure times, with maximum effects observed after 2–5 min exposure (Figure 4). For trypsin, 1X2 was a weak inhibitor without UV exposure (19 % inhibition at 10 μM). However, exposure to UV light for 30 s was sufficient to substantially shift the inhibitor's activity against trypsin (apparent IC50=510 nM), and 5 min UV exposure lowered the IC50 further to 220 nM. To explore whether this effect extended to other proteases, we performed similar assays with matriptase and KLK4 where the IC50 for 1X2 also exceeds 10 μM. Only slight changes in activity were observed for matriptase (Figure S18), which may be attributable to inefficient crosslinking. However, for KLK4, 30 s exposure to UV light produced a marked shift in activity (apparent IC50=2.5 μM), with further improvement observed after 5 min UV exposure where the IC50 reached 920 nM. These findings demonstrate that knottin PIs with dual modes of action can be generated by installing a photoreactive amino acid, which enables the inhibitor to be switched ON against an expanded set of targets using light.
Figure 4.

A photoreactive knottin with light‐controlled selectivity. Graphs show data (mean±SD, three experiments in duplicate) from competitive inhibition assays comparing the activity of 1X2 against FXIIa, trypsin or KLK4 with or without exposure to UV light. Exposure times are indicated in the key below the graphs. Data for each exposure time are normalised to controls (protease without inhibitor) exposed to UV light for the same time. IC50 values calculated for each exposure time are shown below the key. For trypsin and KLK4, exposure times of 1–3 min were not performed (N.D.=not determined).
Although the P4′ position was not amenable to crosslinking (1X1), we speculated that this site might be useful for peptide functionalisation where interaction with the target protease is not essential but benefits from proximity to the interacting residues. To explore this concept, we labelled 1 by incorporating a biotin tag on the side chain of a Lys residue at P4′. Since all modifications in the new inhibitor (1B) are N‐terminal to CysII, we examined whether modified knottins could be assembled by ligating a labelled peptide thioester to the knottin core using native chemical ligation, [50] followed by oxidative folding in a one pot reaction. This approach has been successfully demonstrated using Boc SPPS,[ 48 , 51 ] and we explored whether it could be extended to Fmoc SPPS by synthesising the nine‐residue N‐terminal fragment as a peptide hydrazide [52] separate to the C‐terminal knottin core. The purified peptide hydrazide was converted to a peptide thioester via an acyl pyrazole intermediate, [53] followed by overnight ligation after adding the 20‐residue C‐terminal fragment and adjusting to pH 7. The crude ligation products were diluted 1 : 50 in 0.1 M ammonium bicarbonate pH 8.3 containing 3 mM GSSG to promote formation of the cystine knot, and the desired knottin 1B was isolated after a single HPLC purification (yield=42 %). This type of approach may be beneficial for rapidly generating knottin variants, or for sequences and labelling applications that would otherwise present synthetic challenges.
Having generated the labelled knottin 1B, we assessed its activity against FXIIa in competitive inhibition assays. 1B remained a potent FXIIa inhibitor (K i=12 nM) with equipotent activity to its non‐labelled counterpart 1 (K i=11 nM), indicating that the biotin tag at P4′ does not have a detrimental effect on activity. We then expanded our labelling study to MCoLib. This knottin is a potent inhibitor of four protease targets (Figure 2A), enabling a broader analysis of whether P4′ labelling diminishes inhibitory activity. The labelled analogue LibB remained a potent inhibitor of all four targets, with K i values in the low to sub‐nanomolar range (Figure 5A). These findings verify that, even with high affinity inhibitors, biotin labelling at P4′ is compatible with potent inhibition.
Figure 5.

Switching OFF biotin‐labelled knottins using an external effector. A) K i values (±standard error) for the biotin‐labelled inhibitor LibB compared with its non‐labelled counterpart MCoLib. Data are from three experiments performed in triplicate. B) Schematic illustrating the concept of an affinity tag OFF switch based on knottin labelling at P4′ that introduces overlapping binding sites for streptavidin and the target protease. C) Recovery of enzyme activity (y‐axis) after adding 0.25–1 equiv streptavidin (x‐axis) to FXIIa incubated with 1B (≈1 : 20 ratio FXIIa:1B). Activity data are expressed as a % relative to controls with FXIIa and substrate only (mean±SD from three experiments). No recovery of activity was observed for the unlabelled inhibitor 1. D) Recovery of enzyme activity after adding streptavidin to FXIIa (purple), trypsin (blue), matriptase (green) or KLK4 (orange) incubated with LibB. E) No recovery of activity was observed for the non‐labelled inhibitor MCoLib.
Successfully installing an affinity tag within the protease binding loop suggested that it might be possible to switch OFF an inhibitor by adding an external tag‐binding effector. We anticipated that a biotin‐labelled knottin could bind to either a protease or streptavidin, but not both simultaneously as interaction with streptavidin would obstruct the protease binding loop (Figure 5B). For reversible inhibitors, sequestering free inhibitor using streptavidin would shift the equilibrium between free and protease‐bound states, ultimately recovering enzyme activity.
We tested this concept by incubating FXIIa with 1B then adding varying concentrations of streptavidin (Figure 5C). Initially, we incubated FXIIa with 1B for 10 min, then added streptavidin (0.25–1 equiv relative to 1B) and incubated for 10 min prior to adding substrate and monitoring activity. Adding streptavidin (four biotin binding sites per molecule) led to a concentration‐dependent recovery in FXIIa activity, with 95 % activity recovered after adding 0.5 or 1 equiv streptavidin (Figure 5C). By contrast, adding streptavidin to FXIIa incubated with the non‐labelled inhibitor 1 did not recover FXIIa activity. We then explored whether streptavidin could switch OFF the higher affinity inhibitor LibB against an expanded set of protease targets (Figure 5D). As with 1B, we observed a concentration‐dependent recovery in enzyme activity that reached 80–95 % for matriptase, trypsin, and FXIIa after adding 1 equiv streptavidin 10 min prior to adding substate. Recovery of KLK4 activity was less pronounced, which may reflect the higher affinity of LibB for KLK4 (K i=50 pM). No recovery of activity was observed for the non‐labelled inhibitor MCoLib (Figure 5E). We also explored whether a biotin tag could be added to 1X2 to generate a labelled knottin (1X2B) that has light‐controlled activity. Exposure to UV light led to improvements in 1X2B activity against FXIIa and trypsin that were similar to 1X2, and forming crosslinked complexes diminished the effect of streptavidin on recovering activity (Supplementary Results 3).
The binding kinetics of knottin PIs and substantial recovery of activity for FXIIa, trypsin, and matriptase after adding streptavidin suggested that this system could enable rapid, fine‐tuned control over an enzymatic reaction in progress. To demonstrate this concept, we started reactions by mixing FXIIa with a chromogenic substrate, then added 1B to stop the reaction after 5 min, followed by 1 equiv streptavidin to restart FXIIa activity after a further 5 min. Progress curves (Figure 6A) show that adding 500 nM 1B (I) was sufficient to rapidly stop the reaction. Subsequently adding 500 nM streptavidin (II) to switch OFF the inhibitor enabled ≈95 % recovery of FXIIa activity, whereas near‐complete inhibition was maintained in reactions without streptavidin (Figure 6A). These findings demonstrate that, with 1B and streptavidin, we could start, stop, then restart an enzymatic reaction in the space of 15 min. To explore whether additional stop‐start cycles are possible, we repeated the experiment shown in Figure 6A, then added 2 μM 1B at 17 min, followed by 2 μM streptavidin at 22 min. Progress curves (Figure S21) revealed that we could successfully re‐stop the reaction by saturating the streptavidin present with additional 1B, then restart activity by adding streptavidin to counterbalance the excess 1B.
Figure 6.

Streptavidin rapidly switches OFF a biotin‐labelled knottin in kinetic assays and in human plasma. A) Progress curves showing substrate cleavage by FXIIa (y‐axis, OD). Reactions were initiated by adding FXIIa (5 nM) to substrate (150 μM). At 5 min (I), 1B (500 nM) was added to stop the indicated reactions (blue circles or open circles). At 10 min (II), streptavidin (500 nM, blue circles) or an equivalent volume of buffer (open circles) was added. Recovery of FXIIa activity after adding streptavidin was ≈95 % relative to controls without 1B (black circles). Data show representative curves from one of three experiments. B) Modified aPTT assay for switching OFF 1B in human plasma. Adding phospholipid (PL) and kaolin (1) to citrated plasma initiates activation of factor XII (FXII), which subsequently activates plasma kallikrein (PK) and factor XI (FXI). CaCl2 is required for activation of downstream enzymes: factor IX (FIX), factor X (FX), and thrombin (Th). Adding 1B during the first incubation (1) allows inhibition of FXIIa, which can be switched OFF in the second incubation (2) by adding streptavidin. Clotting times are shown in C). For controls (black bars), buffer was added in the first incubation and buffer or streptavidin in the second. 10 μM 1B was added in the first incubation for the remaining conditions, followed by buffer (open bar) or 0.5–1 equiv streptavidin (blue bars) in the second. Data show the mean±SD from n=4.
Finally, we explored whether streptavidin could switch OFF a labelled knottin in human plasma. In standard aPTT clotting assays, incubating citrated plasma with phospholipid and kaolin facilitates activation of FXII to FXIIa, which initiates a positive feedback loop with plasma kallikrein and enables activation of FXI to FXIa (Figure 6B). Activation of downstream coagulation enzymes requires plasma recalcification. Therefore, we designed an assay where an initial incubation (1) was performed after adding phospholipid and kaolin to plasma with 10 μM 1B to allow inhibition of FXIIa‐mediated activation events, then repeated this incubation (2) after adding streptavidin (to enable recovery of FXIIa activity) or buffer, before adding CaCl2 and measuring the clotting time. Compared to typical aPTT assays, faster clotting times are observed due to the incubation time prior to adding CaCl2 being extended. In this modified clotting assay, adding 1B without streptavidin increased the clotting time by more than two‐fold, whereas adding streptavidin without 1B had no effect (Figure 6C). Strikingly, adding 1B to inhibit FXIIa, then adding 0.5 or 1 equiv streptavidin to switch OFF 1B enabled recovery of FXIIa activity, such that the clotting time (38 s) approached the control (33 s) where the inhibitor is absent. These findings demonstrate that applications of the affinity tag‐based OFF switch extend from a simple kinetic assay with purified components to a complex biological setting that involves modulation of an enzyme cascade in human plasma.
Conclusion
Tuneable peptides and proteins have broad applications for modulating chemical or biological processes. Our study has revealed separate approaches for tuning the activity of knottin PIs, a class of cystine knot peptides that have evolved as reversible serine protease inhibitors. Using a photoreactive knottin, we demonstrate that the activity and selectivity of a cystine knot peptide can be modulated with light, which allows the inhibitor to be switched ON against an expanded set of targets. This approach pairs the generally rapid association rates of knottin PIs with a mechanism switch that facilitates crosslinking of the inhibitor to the protease. We also demonstrate that knottin PIs can be switched OFF when a biotin affinity tag is installed to introduce overlapping binding sites for the protease and streptavidin. This approach relied on identifying a site for labelling that does not interfere with inhibitory activity, which we showed by labelling P4′ Lys with biotin. Adding streptavidin allowed us to tune the influence of a labelled knottin on an enzymatic reaction, such that we could rapidly switch OFF an inhibitor in kinetic assays and in human plasma. These approaches were guided by specificity data from a knottin library screen, which identified model peptides and potential sites for functionalisation, and provide new insights into sequence tolerance at key binding determinants.
Knottin PIs, and cystine knot peptides more broadly, are becoming increasingly recognised as valuable scaffolds for designing pharmaceutical leads or molecular probes. We anticipate that the approaches developed in this study will be applicable to other cystine knot peptides, including those with functions beyond enzyme inhibition. The degree to which individual peptides can be switched ON or OFF will rely on the specific interactions that confer activity, as well as the association and dissociation kinetics that underpin binding. Our findings reveal that these topologically complex peptides can be engineered to allow fine‐tuned control over target activity, which dramatically expands their scope for modulating protein function.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was funded by grants from the National Health and Medical Research Council (NHMRC, 1164412 to D.J.C. and S.J.D., and 1120066 to S.J.D.) and a NHMRC Investigator Grant to D.J.C. (2009564). The study was supported by access to the facilities of the ARC Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012). Open access publishing facilitated by The University of Queensland, as part of the Wiley ‐ The University of Queensland agreement via the Council of Australian University Librarians.
C. Y. Li, F. B. H. Rehm, K. Yap, C. N. Zdenek, M. D. Harding, B. G. Fry, T. Durek, D. J. Craik, S. J. de Veer, Angew. Chem. Int. Ed. 2022, 61, e202200951; Angew. Chem. 2022, 134, e202200951.
Contributor Information
David J. Craik, Email: d.craik@imb.uq.edu.au.
Simon J. de Veer, Email: s.deveer@imb.uq.edu.au.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Wang J., Liu Y., Liu Y., Zheng S., Wang X., Zhao J., Yang F., Zhang G., Wang C., Chen P. R., Nature 2019, 569, 509–513. [DOI] [PubMed] [Google Scholar]
- 2. Bridge T., Shaikh S. A., Thomas P., Botta J., McCormick P. J., Sachdeva A., Angew. Chem. Int. Ed. 2019, 58, 17986–17993; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18154–18161. [Google Scholar]
- 3. Nguyen D. P., Mahesh M., Elsasser S. J., Hancock S. M., Uttamapinant C., Chin J. W., J. Am. Chem. Soc. 2014, 136, 2240–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Petersen S., Alonso J. M., Specht A., Duodu P., Goeldner M., del Campo A., Angew. Chem. Int. Ed. 2008, 47, 3192–3195; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3236–3239. [Google Scholar]
- 5. Salveson P. J., Haerianardakani S., Thuy-Boun A., Kreutzer A. G., Nowick J. S., J. Am. Chem. Soc. 2018, 140, 5842–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koçer A., Walko M., Meijberg W., Feringa B. L., Science 2005, 309, 755–758. [DOI] [PubMed] [Google Scholar]
- 7. Schierling B., Noel A. J., Wende W., Hien le T., Volkov E., Kubareva E., Oretskaya T., Kokkinidis M., Rompp A., Spengler B., Pingoud A., Proc. Natl. Acad. Sci. USA 2010, 107, 1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Poli M., Zawodny W., Quinonero O., Lorch M., Webb S. J., Clayden J., Science 2016, 352, 575–580. [DOI] [PubMed] [Google Scholar]
- 9. Babii O., Afonin S., Berditsch M., Reibetaer S., Mykhailiuk P. K., Kubyshkin V. S., Steinbrecher T., Ulrich A. S., Komarov I. V., Angew. Chem. Int. Ed. 2014, 53, 3392–3395; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3460–3463. [Google Scholar]
- 10. Beharry A. A., Wong L., Tropepe V., Woolley G. A., Angew. Chem. Int. Ed. 2011, 50, 1325–1327; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1361–1363. [Google Scholar]
- 11. Boyken S. E., Benhaim M. A., Busch F., Jia M., Bick M. J., Choi H., Klima J. C., Chen Z., Walkey C., Mileant A., Sahasrabuddhe A., Wei K. Y., Hodge E. A., Byron S., Quijano-Rubio A., Sankaran B., King N. P., Lippincott-Schwartz J., Wysocki V. H., Lee K. K., Baker D., Science 2019, 364, 658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koçer A., Walko M., Bulten E., Halza E., Feringa B. L., Meijberg W., Angew. Chem. Int. Ed. 2006, 45, 3126–3130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 3198–3202. [Google Scholar]
- 13. Langan R. A., Boyken S. E., Ng A. H., Samson J. A., Dods G., Westbrook A. M., Nguyen T. H., Lajoie M. J., Chen Z., Berger S., Mulligan V. K., Dueber J. E., Novak W. R. P., El-Samad H., Baker D., Nature 2019, 572, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lebar T., Lainscek D., Merljak E., Aupic J., Jerala R., Nat. Chem. Biol. 2020, 16, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schena A., Griss R., Johnsson K., Nat. Commun. 2015, 6, 7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Babii O., Afonin S., Diel C., Huhn M., Dommermuth J., Schober T., Koniev S., Hrebonkin A., Nesterov-Mueller A., Komarov I. V., Ulrich A. S., Angew. Chem. Int. Ed. 2021, 60, 21789–21794; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 21958–21964. [Google Scholar]
- 17. Hansen M. J., Velema W. A., de Bruin G., Overkleeft H. S., Szymanski W., Feringa B. L., ChemBioChem 2014, 15, 2053–2057. [DOI] [PubMed] [Google Scholar]
- 18. Toupin N. P., Arora K., Shrestha P., Peterson J. A., Fischer L. J., Rajagurubandara E., Podgorski I., Winter A. H., Kodanko J. J., ACS Chem. Biol. 2019, 14, 2833–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Postic G., Gracy J., Perin C., Chiche L., Gelly J. C., Nucleic Acids Res. 2018, 46, D454–D458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le Nguyen D., Heitz A., Chiche L., Castro B., Boigegrain R. A., Favel A., Coletti-Previero M. A., Biochimie 1990, 72, 431–435. [DOI] [PubMed] [Google Scholar]
- 21. Craik D. J., Daly N. L., Bond T., Waine C., J. Mol. Biol. 1999, 294, 1327–1336. [DOI] [PubMed] [Google Scholar]
- 22. de Veer S. J., Kan M. W., Craik D. J., Chem. Rev. 2019, 119, 12375–12421. [DOI] [PubMed] [Google Scholar]
- 23. Pallaghy P. K., Nielsen K. J., Craik D. J., Norton R. S., Protein Sci. 1994, 3, 1833–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiche L., Heitz A., Gelly J. C., Gracy J., Chau P. T., Ha P. T., Hernandez J. F., Le-Nguyen D., Curr. Protein Pept. Sci. 2004, 5, 341–349. [DOI] [PubMed] [Google Scholar]
- 25. Otlewski J., Zbyryt T., Biochemistry 1994, 33, 200–207. [DOI] [PubMed] [Google Scholar]
- 26. Kintzing J. R., Cochran J. R., Curr. Opin. Chem. Biol. 2016, 34, 143–150. [DOI] [PubMed] [Google Scholar]
- 27. Kolmar H., Curr. Opin. Pharmacol. 2009, 9, 608–614. [DOI] [PubMed] [Google Scholar]
- 28. de Veer S. J., Weidmann J., Craik D. J., Acc. Chem. Res. 2017, 50, 1557–1565. [DOI] [PubMed] [Google Scholar]
- 29. Cox N., Kintzing J. R., Smith M., Grant G. A., Cochran J. R., Angew. Chem. Int. Ed. 2016, 55, 9894–9897; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10048–10051. [Google Scholar]
- 30. Miller C. L., Sagiv-Barfi I., Neuhofer P., Czerwinski D. K., Artandi S. E., Bertozzi C. R., Levy R., Cochran J. R., Cell Chem. Biol. 2021, 10.1016/j.chembiol.2021.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lesniak W. G., Aboye T., Chatterjee S., Camarero J. A., Nimmagadda S., Chem. Eur. J. 2017, 23, 14469–14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hernandez J. F., Gagnon J., Chiche L., Nguyen T. M., Andrieu J. P., Heitz A., Trinh Hong T., Pham T. T., Le Nguyen D., Biochemistry 2000, 39, 5722–5730. [DOI] [PubMed] [Google Scholar]
- 33. Liu W., de Veer S. J., Huang Y. H., Sengoku T., Okada C., Ogata K., Zdenek C. N., Fry B. G., Swedberg J. E., Passioura T., Craik D. J., Suga H., J. Am. Chem. Soc. 2021, 143, 18481–18489. [DOI] [PubMed] [Google Scholar]
- 34. Swedberg J. E., Mahatmanto T., Abdul Ghani H., de Veer S. J., Schroeder C. I., Harris J. M., Craik D. J., J. Med. Chem. 2016, 59, 7287–7292. [DOI] [PubMed] [Google Scholar]
- 35. Quimbar P., Malik U., Sommerhoff C. P., Kaas Q., Chan L. Y., Huang Y. H., Grundhuber M., Dunse K., Craik D. J., Anderson M. A., Daly N. L., J. Biol. Chem. 2013, 288, 13885–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Glotzbach B., Reinwarth M., Weber N., Fabritz S., Tomaszowski M., Fittler H., Christmann A., Avrutina O., Kolmar H., PLoS One 2013, 8, e76956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Swedberg J. E., Ghani H. A., Harris J. M., de Veer S. J., Craik D. J., ACS Med. Chem. Lett. 2018, 9, 1258–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sommerhoff C. P., Avrutina O., Schmoldt H. U., Gabrijelcic-Geiger D., Diederichsen U., Kolmar H., J. Mol. Biol. 2010, 395, 167–175. [DOI] [PubMed] [Google Scholar]
- 39. Thongyoo P., Bonomelli C., Leatherbarrow R. J., Tate E. W., J. Med. Chem. 2009, 52, 6197–6200. [DOI] [PubMed] [Google Scholar]
- 40. Kowalska J., Zablocka A., Wilusz T., Biochim. Biophys. Acta Gen. Subj. 2006, 1760, 1054–1063. [DOI] [PubMed] [Google Scholar]
- 41. Maas C., Renne T., Blood 2018, 131, 1903–1909. [DOI] [PubMed] [Google Scholar]
- 42. Baici A., Gyger-Marazzi M., Eur. J. Biochem. 1982, 129, 33–41. [DOI] [PubMed] [Google Scholar]
- 43. de Veer S. J., Swedberg J. E., Akcan M., Rosengren K. J., Brattsand M., Craik D. J., Harris J. M., Biochem. J. 2015, 469, 243–253. [DOI] [PubMed] [Google Scholar]
- 44. Yap K., Du J., Looi F. Y., Tang S. R., de Veer S. J., Bony A. R., Rehm F. B. H., Xie J., Chan L. Y., Wang C. K., Adams D. J., Lua L. H. L., Durek T., Craik D. J., Green Chem. 2020, 22, 5002–5016. [Google Scholar]
- 45. Yap K., Du J., Rehm F. B. H., Tang S. R., Zhou Y., Xie J., Wang C. K., de Veer S. J., Lua L. H. L., Durek T., Craik D. J., Nat. Protoc. 2021, 16, 1740–1760. [DOI] [PubMed] [Google Scholar]
- 46. Du J., Yap K., Chan L. Y., Rehm F. B. H., Looi F. Y., Poth A. G., Gilding E. K., Kaas Q., Durek T., Craik D. J., Nat. Commun. 2020, 11, 1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Furie B., Furie B. C., Cell 1988, 53, 505–518. [DOI] [PubMed] [Google Scholar]
- 48. Durek T., Zhang J., He C., Kent S. B. H., Org. Lett. 2007, 9, 5497–5500. [DOI] [PubMed] [Google Scholar]
- 49. Tzakoniati F., Xu H., Li T., Garcia N., Kugel C., Payandeh J., Koth C. M., Tate E. W., Cell Chem. Biol. 2020, 27, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B. H., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 51. Bang D., Kent S. B. H., Angew. Chem. Int. Ed. 2004, 43, 2534–2538; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2588–2592. [Google Scholar]
- 52. Fang G. M., Li Y. M., Shen F., Huang Y. C., Li J. B., Lin Y., Cui H. K., Liu L., Angew. Chem. Int. Ed. 2011, 50, 7645–7649; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7787–7791. [Google Scholar]
- 53. Flood D. T., Hintzen J. C. J., Bird M. J., Cistrone P. A., Chen J. S., Dawson P. E., Angew. Chem. Int. Ed. 2018, 57, 11634–11639; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11808–11813. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.