

Abstract
Exome sequencing (ES) in the clinical setting of inborn metabolic diseases (IMDs) has created tremendous improvement in achieving an accurate and timely molecular diagnosis for a greater number of patients, but it still leaves the majority of patients without a diagnosis. In parallel, (personalized) treatment strategies are increasingly available, but this requires the availability of a molecular diagnosis. IMDs comprise an expanding field with the ongoing identification of novel disease genes and the recognition of multiple inheritance patterns, mosaicism, variable penetrance, and expressivity for known disease genes. The analysis of trio ES is preferred over singleton ES as information on the allelic origin (paternal, maternal, “de novo”) reduces the number of variants that require interpretation. All ES data and interpretation strategies should be exploited including CNV and mitochondrial DNA analysis. The constant advancements in available techniques and knowledge necessitate the close exchange of clinicians and molecular geneticists about genotypes and phenotypes, as well as knowledge of the challenges and pitfalls of ES to initiate proper further diagnostic steps. Functional analyses (transcriptomics, proteomics, and metabolomics) can be applied to characterize and validate the impact of identified variants, or to guide the genomic search for a diagnosis in unsolved cases. Future diagnostic techniques (genome sequencing [GS], optical genome mapping, long‐read sequencing, and epigenetic profiling) will further enhance the diagnostic yield. We provide an overview of the challenges and limitations inherent to ES followed by an outline of solutions and a clinical checklist, focused on establishing a diagnosis to eventually achieve (personalized) treatment.
Keywords: diagnostic yield, exome‐negative, exome sequencing, genome sequencing, inborn metabolic disease, treatment
Synopsis.
We provide an overview of the challenges and limitations inherent to exome sequencing, followed by an outline of solutions and a clinical checklist, focused on establishing a diagnosis to eventually achieve (personalized) treatment in exome‐negative patients.
1. INTRODUCTION
Although each rare genetic disease (RGD) affects a small number of individuals, collectively they affect about 1% of all live births representing an important global disease burden. The total number of RGDs is still to be determined. Currently, OMIM (www.omim.org) lists 6281 disease‐gene associations, of which 1629 are inborn metabolic disease (IMD)s (www.iembase.org) (accession date 08.01.2022).
Over the past 10 years, achieving an accurate and timely molecular diagnosis in the clinical setting of RGD has seen tremendous improvement with a prominent role for whole‐exome sequencing (ES; Figure 1). This also influenced the definition of IMDs. The presence of an abnormal metabolite is no longer a prerequisite for a disease to be labeled as an IMD. Classification of a disorder as an IMD requires only that impairment of specific enzymes or biochemical pathways is intrinsic to the pathomechanism. 10
FIGURE 1.
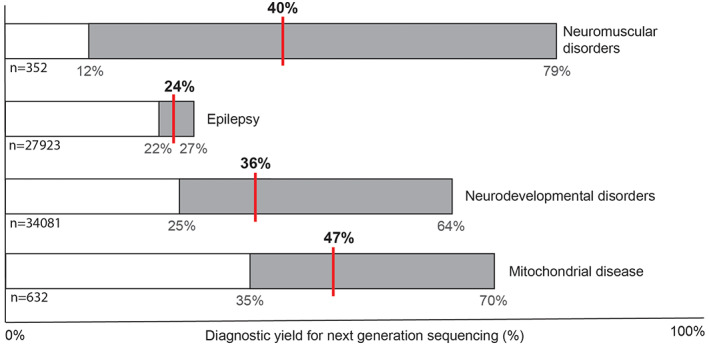
Median diagnostic yield of next‐generation sequencing for different groups of rare genetic neurological disease. The bars display data from multiple studies (“n” is the total number of individuals) showing the range in diagnostic yields in light gray and the mean with a red line. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9
In parallel, the development of personalized treatment options for RGD, and especially IMDs, has gained significant momentum. Both in vitro and in vivo studies increasingly identify treatment targets for a growing number of disorders, paving the way for (personalized) treatment opportunities either based on the pathomechanism (e.g., nutritional interventions) or aiming to directly target the genetic defect itself. 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 Assuming that treatment early in the course improves outcome, a timely diagnosis is key. Given the absence of known biomarkers in many recently molecularly defined RGD/IMDs, genetic (newborn) screening is the only way to the diagnosis. 19 Digital knowledgebases such as Treatabolome and Treatable ID (www.treatable-id.org) aim to shorten the path to therapy, by providing (digital) access to treatment information for specific genes, variants, or (groups of) disorders. 20 , 21 , 22 Treatment response has even been suggested as an addition to the American College of Medical Genetic (ACMG) criteria for variant interpretation. 23
Here, we provide a diagnostic strategic outline for those patients with (suspected) RGD/IMDs in which ES did not provide a diagnosis (ES‐negative) to optimize the diagnostic yield, and thus therapeutic potential. We will focus on clinical presentations as neurodevelopmental disorders (NDDs)/intellectual disability, epilepsy, and movement disorders.
2. ES SUCCESS: CURRENT CLINICAL PRACTICE
Whereas data is generated for all protein‐coding sequences, interpretation strategies are often divided into a targeted interpretation strategy first based on (rare) variants in genes with an established association to the disease (virtual panel) or when investigating all known gene‐disease associations listed in OMIM (clinical exome). Subsequently, an exome‐wide analysis allows the identification of variants in candidate disease‐genes without prior disease association. Accordingly, clinicians are no longer limited to sequencing data of only one gene at a time, but have access to data from the coding regions of virtually all genes allowing a comprehensive diagnostic strategy and making ES the first‐tier diagnostic tool for RGD/IMDs. 1 , 24 , 25 , 26
2.1. Analysis of copy number variation and mitochondrial DNA
Although the annotation of point mutations (SNVs) and small insertions or deletions (indels) from ES data is standard, the annotation of copy number variation (CNV) is not. CNVs are a prevalent source of genetic variation that has been implicated in many genomic disorders, 27 , 28 resulting in the widespread application of genomic microarrays as a first‐tier diagnostic tool for CNV detection. More recently, it has been shown that the majority (88%) of disease‐relevant CNVs (>3 exons) can be detected in short‐read ES data via read‐depth analysis. 29 , 30 , 31 , 32 Moreover, in a cohort unselected for cytogenetic abnormalities, CNV analysis from ES outperformed chromosomal micro‐arrays when applied as a second‐line test. 33 One has to bear in mind that genomic microarrays, single nucleotide polymorphism (SNP) chips, multiplex ligation‐dependent probe amplification (MLPA), and other technologies are superior for detecting certain types of CNVs.
The full potential of ES is often not exploited as the additional analysis of “off‐target reads” (alternatively beads‐based enrichment of mtDNA) deriving from the mtDNA can be used to analyze both, nuclear and mitochondrial, genomes. Additional analysis of the mtDNA in existing ES data has been shown to increase the diagnostic yield by nearly 2% in an IMD cohort. It showed a high concordance (96.2%) and excellent precision (99.5%) when compared to the gold standard of targeted mtDNA next‐generation sequencing and is thus of sufficient quality for clinical diagnostics. 34 One should, however, be aware of the tissue specificity of mtDNA alterations, and thus consider additional (targeted) next‐generation sequencing preferably in an affected tissue. Furthermore, mtDNA deletion(s) are usually not picked up by ES using DNA from blood since the proportion of deleted mtDNA is often too low. Again, in specific cases, ES cannot replace current techniques and this should be kept in mind.
3. CHALLENGES AND LIMITATIONS OF ES AND MITIGATION STRATEGIES
3.1. Data sharing and collaboration
The Matchmaker exchange network connects the data from six matchmaking initiatives (GeneMatcher, DECIPHER, PhenomeCentral, seqr, MyGene2, IRUD). 35 , 36 , 37 , 38 , 39 , 40 This creates a collective data set containing over 150 000 cases from more than 11 000 contributors for sharing of molecular and clinical data to establish gene‐disease associations for “newly” defined RGD. 41 It is difficult to quantify the success that can be attributed to these initiatives; however, the number of matches (e.g., in 2019, 56% of gene entries in GeneMatcher had at least one match) and publications (e.g., in 2019, GeneMatcher was cited by 267 publications of which 77% were novel disease gene discoveries) facilitated by these platforms show their importance in gene discovery.
3.2. The need for continuous data (re)analysis and (reverse) phenotyping
Data interpretation is a continuously evolving discipline; with increased access to sequencing, requiring less clinical preselection, milder presentations of diseases are uncovered giving rise to the realization that all diseases have a phenotypic spectrum rather than one “classic” phenotype. Variants should not be discarded just because single disease‐feature are absent or unusual features are present. A detailed family history and collection of medical data from other (similarly) affected family members can be of added value. Disease manifestations may emerge or disappear over time and may not be present when an individual is investigated at a single time point during the course of the disease. The term “reverse phenotyping” (re‐evaluating the clinical findings of the individual in light of a potential pathogenic genotype) illustrates the ongoing urgency of exchange between the diagnostic laboratory and the referring physician.
Reanalysis of ES data 1–3 years after the initial analysis may increase the diagnostic yield by 3%–15% and should make use of both the updated “phenotypical” input, as well as, the “genotypic” knowledge to be most successful. 42 , 43 , 44 , 45 At initial analysis, there may have been insufficient evidence for candidate variants or gene causality (Box 1).
BOX 1. Online resources.
-
‐
The American College of Medical Genetics (ACMG) standards and guidelines for the interpretation of sequence variants': consists of five classes (benign, likely benign, variant of uncertain significance [VUS], likely pathogenic or pathogenic), and lists functional tests as an important factor in variant interpretation. 46 In line with the increasing pathomechanism‐based treatment options, we suggested to integrate “response to treatment” into the variant interpretation. 31 An online tool for variant interpretation with individual adjustment can be found at https://wintervar.wglab.org/). 47
-
‐
The ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) annually curates >10 000 disease variants.
-
‐
The Genome Aggregation Database (gnomAD, www.gnomad.org) contains the genomic data of more than 140 000 genomic data sets of “healthy/unaffected” individuals. 48 , 49
-
‐
GeneMatcher (www.genematcher.org) is a matchmaking initiative where researchers interested in the same gene can get in touch. Numerous examples of successful matchmaking for identifying rare genetic disease can be found on the website listing > 400 publications referencing to this initiative.
-
‐
GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1116/) comprehensively provide clinically relevant and medically actionable information (diagnosis, management, and genetic counseling) in a standardized format focused on the clinician (>800 chapters available).
-
‐
Genome Aggregation Database (gnomAD, www.gnomad.org): genomic data of more than 140 000 genomic datasets of healthy individuals. This database allows better interpretation of variants, especially when in‐house databases are small.
-
‐
The Leiden Open Variation Database (LOVD, www.lovd.nl) includes gene‐ and patient‐centered data containing >690 000 disease variants. This database allows better interpretation of variants, especially when in‐house databases are small.
-
‐
Online Mendelian Inheritance in Man (OMIM, www.omim.org) is the comprehensive, authoritative compendium of human genes and genetic phenotypes.
-
‐
Varsome (https://varsome.com/) is a variant knowledge community, data aggregator, and variant data discovery tool.
3.3. When reanalysis is not enough: when to consider a new ES?
The diagnostic yield is in part a result of the continuous development of bioinformatic tools to analyze and interpret ES data and constant updates of variant databases. At the same time, it is important to be aware of the data quality and completeness (coverage of the entire coding region of [clinically relevant] genes, as well as, sufficient read depth for the covered regions), especially of older ES data. For example, the Agilent V4 exome enrichment platform captured 89% of annotated genes on average while its follow‐up version V5 captured 95%. 50 As will be discussed later, genome sequencing (GS) data are generated enrichment‐free and this aspect can overcome the limitation of ES in sufficiently covering coding exons, especially GC‐rich regions, as well as in characterizing structural variants. Depending on local resources, both a new ES on an up‐to‐date exome enrichment platform or ideally GS should be considered in a ES‐negative case after reanalysis.
4. DISCOVERING VARIANTS FROM ES DATA (BOX 2)
4.1. Multiple inheritance models
The three most common patterns of Mendelian inheritance are: autosomal dominant (AD), autosomal recessive (AR), and X‐linked. The mode of inheritance for human diseases can, however, be more complex (Figure 2). An increasing number of genes are identified that can be associated with an AD, as well as, an AR disorder and the co‐occurrence of different modes of inheritance is not restricted to a specific type of protein or clinical phenotype (Table S1). Hence, it is within reason to expect that this phenomenon is to be uncovered for more disease genes. This is another factor complicating the variant interpretation and emphasizes the importance of reverse phenotyping and need to continue the search for confirmative biomarkers and functional (biochemical) assays, especially for potentially treatable conditions.
FIGURE 2.
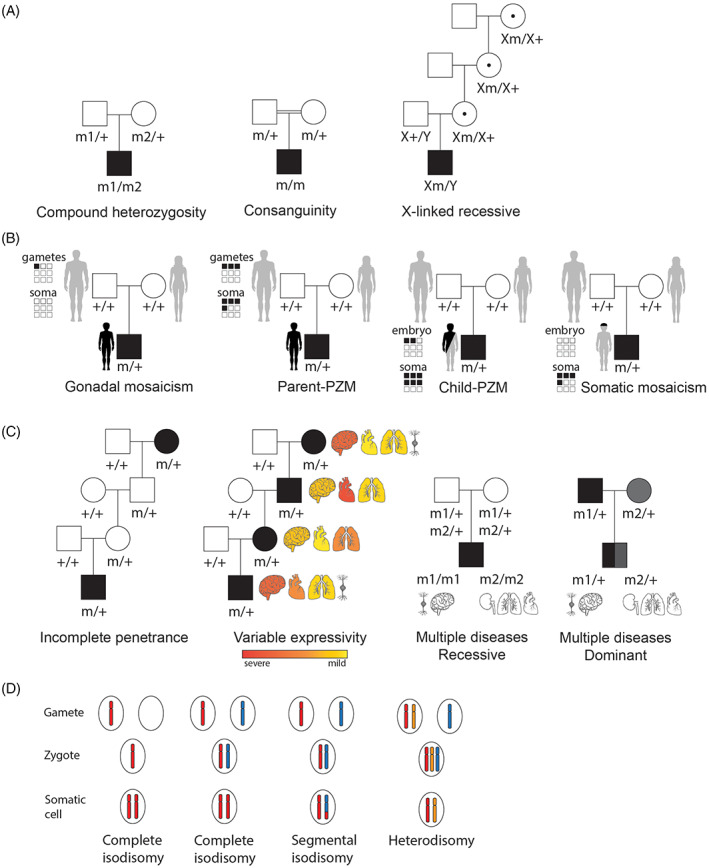
Example pedigrees explaining different inheritance patterns. (A) Recessive inheritance models. Compound heterozygosity: the parents each carry a different variant in the same gene. The child receives the variant carrying allele from each parent and is affected. Consanguinity: the parents are related and carry the same heterozygous variant. XL‐recessive inheritance: females are carriers of the pathogenic trait while males are affected. (B) Gonadal mosaicism: variant occurs in gonadal cells of a healthy parent and the pathogenic variant is transmitted to the child. Parent‐post‐zygotic variant (PZM): the PZM occurs during the embryonic stage of the parent resulting in both gametes and somatic cells (soma) to carry the pathogenic variant, which is transmitted to the affected child. Child‐PZM: the variant occurs during embryogenesis of the child and results in multiple mosaic tissues. Somatic mosaicism: the pathogenic variant occurs post‐zygotically at a later stage during development affecting a single or limited number of tissues. (C) Complex inheritance models. Incomplete penetrance: all individuals in the family carry the pathogenic variant, but not all individuals manifest the disease phenotype. Variable expressivity: all individuals in the family carry the pathogenic variant, but the expression of the phenotype is variable. Multiple diseases recessive: both parents are heterozygous carriers of two pathogenic variants that are associated with two distinct disease phenotypes. Both pathogenic variants are transmitted to the child who presents the two distinct disorders. Multiple diseases dominant: both parents manifest a dominant disease and transmit the variant allele to the child who presents two distinct disorders. Pathogenic variant (m). Pathogenic variant on chromosome X (Xm). (D) Uniparental disomy. Complete isodisomy occurs when both copies of the chromosome originate from one parent and none from the other parent. Segmental isodisomy is when only a segment of the chromosome originates from one parent and the rest of the chromosome has two origins, one from mother and one from father. Heterodisomy refers to the situation in which both homologs from one parent are inherited by the child.
The availability of ES data from multiple family members, parents (trio‐ES), or (affected) siblings, facilitates better interpretation of variants. For example, trio‐ES provides immediate data on the allelic origin of the variant and will allow for the detection of de novo variants (variants that are not inherited from either parent). 51 In addition, deep phenotyping data next to ES of multiple family members allow for accurate segregation analysis and aid in figuring out complex modes of inheritance. 52 Standardization of phenotypic terms (e.g., Human Phenotype Ontology [HPO]) is important to utilize phenotyping to its best, for example, when comparing patients from different institutes. 53 However, it needs to be reminded that when parents are only mildly affected and not recognized as affected individuals, inherited genetic variants will be filtered out in AD inherited disorders and therefore lost in analysis. Similarly, when a variant occurs as a mosaic in one of the parents, the analysis should be adapted accordingly (Box 2).
BOX 2. Illustrative examples of complex genomics in IMD.
3.1 Multiple inheritance models:
‐ disorders of organelle biogenesis, dynamics, and interactions (OPA1 54 and OPA3 55, 56 ); disorders of complex I subunits and assembly factors (DNAJC30 57 ); disorders of mtDNA replication and maintenance (TWNK 58, 59 ), other disorders of mitochondrial function (AFG3L2,60, 61, 62 CLPB, 63 , 64 and ATAD3A 65 ); disorders of organelle interplay (EMC1 66 ); disorders of mitochondrial and peroxisomal dynamics (GDAP1 67, 68 ); disorders of the synaptic vesicle cycle (PRRT2 69 and KIF1A 70, 71 ); disorders of non‐mitochondrial tRNA processing and aminoacyl‐tRNA synthetases (NSUN2 72 ); and disorders of carbohydrate transmembrane transport and absorption (GLUT1 73 ).
3.2 X‐linked IMDs:
‐ disorders of pyruvate metabolism (PDHA1 74 ); disorders of mitochondrial membrane biogenesis and remodeling (TAZ 75 ); disorders of peroxisomal fatty acid oxidation (ABCD1 76 ); and disorders of sphingolipid degradation (GLA 77 ), urea cycle disorders and inherited hyperammonemias (OTC 78 ).
3.3 Mosaicism:
‐ disorders of mitochondrial metabolite repair (IDH1, IDH2 79 ); disorders of sphingolipid degradation (GLA 80 ), urea cycle disorders, and inherited hyperammonemias (OTC 81 ); disorders of carbohydrate transmembrane transport and absorption (GLUT1 82 ); and disorders of niacin and NAD metabolism (NAXD 83 ).
3.4 Variable penetrance and expressivity (reviewed in ref. 84 ):
‐ general: copper‐transporting ATPase subunit beta deficiency (ATP7B), mtDNA‐related disorders (heteroplasmy, threshold), gender‐related penetrance: disorders of tetrahydrobiopterin metabolism (GCH1), age‐related penetrance: disorders of the synaptic vesicle cycle (LRRK2), ethnicity related penetrance: disorders of the synaptic vesicle cycle (LRRK2).
3.5 Multiple diseases in one individual or one family:
‐ disorders of phenylalanine and tyrosine metabolism (PAH) and disorders of glycogen metabolism (SLC37A4) (personal experience), disorders of glycolysis (PCK1), and disorders of glutamate (GRIN2B). 85
4.1 Intronic, regulatory and splice‐site variants (reviewed in ref. 86 ):
‐ exon skipping: disorders of mitochondrial fatty acid oxidation (ACADM), disorders of mitochondrial nucleotide pool maintenance (MPV17), gamma‐aminobutyric acid neurotransmitter disorders (SSADH), disorders of sphingolipid degradation (GLA), and disorders of mitochondrial membrane biogenesis and remodeling (SERAC1).
‐ poisonous exon: disorders of complex I subunits and assembly factors (TIMMDC1 87 ).
4.2 Complex DNA rearrangements:
‐ short tandem repeat expansion: disorders of glutamate/glutamine and aspartate/asparagine metabolism (GLS 88 ).
6. Epigenetics:
‐ compound epigenetic‐genetic heterozygosity: disorders of cobalamin metabolism (MMACHC/PRDX1, “epi‐CLBc”). 89
‐ uniparental disomy (UPD): disorders of glycerolipid metabolism (ABHD5 90 )*, disorders of amino acid transport (SLC7A7 91 ).*
(*in both cases, UPD led to homozygosity of a variant found in heterozygous state in parents, not to an epigenetic effect).
4.2. X‐linked disorders
X‐inactivation is a well‐established, physiological, dosage compensation mechanism ensuring that X‐chromosomal genes are expressed at comparable levels in males and females. 92 The phenotypic range in XL‐disorders is broad. Certain XL‐disorders are incompatible with life in males and therefore only affected females are seen, while others present with more severely affected males compared to females. In addition, the onset of disease in females can be (much) later in life and hence mothers of severely affected males can be without medical concerns at the time of ES. In some cases, skewed X‐inactivation can occur, meaning that the X‐inactivation of one X‐chromosome is favored over the other (here: the X chromosome with the mutation), and explain the phenomena seen.
4.3. Mosaicism
De novo pathogenic variants (DNMs) can occur during gametogenesis, as well as, the post‐zygotic phase resulting in mosaicism (Figure 2B). Post‐zygotic variants (PZMs) leading to disease often occur during early embryogenesis in the first 15 mitotic divisions. 93 Mosaic variants may be inherited from a parent (gonadal or somatic mosaic) or can arise in the child, which influences the recurrence risk and is therefore important to know for adequate counseling. 94 , 95 In an epilepsy disorder cohort 3.5% of the molecular diagnoses could be attributed to pathogenic mosaic variants and a study of 4293 patients with NDD showed that mosaicism was present in 40 cases, of which in 16 the variant was of parental origin and in 24 cases had occurred de novo in the child. 96 , 97 Upon detection or suspicion of a mosaic variant in the affected individual, it is important to perform sensitive testing in the parents to determine their genetic profile as this will affect counseling. The recurrence risk that parents have in a new pregnancy can roughly be divided into three groups. A moderate to high risk of up to 50% in case of parent‐PZM (~1%–2% of DNM) depending on the level of parental mosaicism. Low risk, 1%, when gonadal mosaicism is at play (~95% of DNM) and a negligible risk when the DNM occurs in the child (~3% of DNM). 97 Post‐zygotic de novo events in the child (child‐PZM) that result in mosaicism may contribute to the variable expressivity of single‐gene disorders through gene‐dose effect or by acting in addition with other (epi)genetic factors.
Somatic pathogenic variants, already known to be involved in cancer development, may also play a role in neurodevelopmental diseases, especially in patients with brain malformations. 98 , 99 Pathogenic somatic DNM affecting the brain may remain undetected in ES when the gene is not expressed in blood and require sequencing of other tissues, for example, fibroblasts or buccal swabs. Furthermore, the detection of mosaicism, in general, requires deep sequencing (preferably >100× coverage) which is not always achieved by ES. In addition, the analysis strategy must be adapted to enable the detection of low‐level variants.
4.4. Variable penetrance and expressivity
“Penetrance” of a monogenic disorder describes the conditional probability that an individual carrying a pathogenic variant manifests the disease phenotype (Figure 2C). If this probability does not equal 100% within a specific age range, the disorder displays incomplete penetrance. Hence, some AD disorders occasionally appear to skip a generation, meaning that individuals carrying a pathogenic variant do not express the disease phenotype (asymptomatic carriers) but can transmit the mutant allele to the offspring (reviewed in Mangrinelli et al. 100 ). It should be borne in mind that when using trio‐ES analysis, these variants can be missed. AR, XL, and mtDNA‐linked disorders can also exhibit incomplete penetrance. Among others, age, gender, and ethnicity are drivers of incomplete penetrance in certain diseases. Although it goes beyond the scope of this article, we would like to alert that for mtDNA‐related issues like heteroplasmy, threshold and the chosen tissue have to be taken into account as well.
“Expressivity” is the extent to which a given genotype is expressed at the phenotypic level (Figure 2C). When the same genetic variant is expressed, it can show quantitatively different effects among distinct individuals, even among members of the same family (intrafamilial variability). Variable expressivity is recognized in monogenic disorders with all patterns of inheritance and represents a major contributor to phenotypic heterogeneity. 101
Finally, genetic modifiers may play a role not only in the severity of the disease, but also in whether a carrier will manifest the disease. 102 These modifiers usually segregate independently from the disease‐causing variant, and are currently investigated for several inherited diseases to better understand the disease pathophysiology (and will need to be included in the diagnostics of specific genes in the future).
4.5. Multiple diseases in one individual or one family
It may not always be clear whether there is one or multiple (hereditary) diseases running in one family or, even more complicated, in one individual (Figure 2C). The occurrence of multiple genetic diagnoses underlying a rare disease phenotype in an individual is estimated about 5% and most often occurs in consanguineous families. 52 , 103 To maximize the chance of identifying multiple genetic diagnoses and their underlying molecular defects, ES should be extended to more family members beyond trio‐ES. One should not forget that family members may be hesitant to participate, fearing an unexpected genetic diagnosis especially in the absence of medical complaints. A diagnosis of multiple disorders obviously has implications for genetic counseling and patient management. Also, it is essential for our insights into rare diseases: blended phenotypes caused by multiple disorders should not be mistaken for an extended phenotype caused by a single disorder. In addition, patients may develop more common disorders during life, such as type II diabetes or cancer, that are not directly related to their genetic disorders.
4.6. Digenic disease/synergistic heterozygosity, polygenic risk scores
Different inheritance models, such as digenic disease and synergistic heterozygosity, are being explored in which not a single gene, but variants in multiple genes are causative for a genetic disorder in an individual. 104 , 105 Furthermore, the genetic burden (amount of genetic variants in disease‐associated genes) that individuals carry can be expressed in a so‐called polygenic risk score. 106 The exact role that these models play in the origin of RGDs/IMDs is still under investigation and not yet ready for diagnostic care.
5. TECHNICAL LIMITATIONS OF ES
5.1. Intronic, regulatory, and splice site variants
Roughly 1%–2% of our DNA is protein‐coding and referred to as the exome. The majority of disease‐causing variants that we currently know are located in this “small” coding section of our DNA. The remainder is called non‐coding DNA which consists of intergenic (between genes) and intronic (between the exons of a gene) DNA. The exact function of most of the non‐coding DNA remains to be elucidated, however, for certain evolutionary conserved regions, that is, promotor sites, (non) canonical splice sites, untranslated regions (UTRs), and long non‐coding RNAs (lncRNA), the importance in causing disease has been shown. 84 , 107 , 108
The essential role of UTRs and introns in gene regulation, and thereby disease association, is becoming more apparent. Variants in the splice site regions (−3 bases and +8 bases of each exon) are known to often cause aberrant transcripts. Moreover, in recent years the essential role of UTRs and deep‐intronic sequences in gene regulation has become evident. 109 Deep‐intronic variants, as well as, variants in the UTR can influence gene regulation by affecting the binding site for regulatory proteins or by altering the gene transcript. Such variants can, for example, introduce a new start codon in the 5′ UTR or cause the inclusion of a pseudoexon that can in turn lead to an out‐of‐frame transcript that may be dysfunctional (Figure S1). 84 A recent publication showed the significant contribution of pathogenic variants in the 5′ UTR region of MEF2C, emphasizing the importance of screening the 5′ UTR of disease‐relevant genes. 108 Another element that should be considered during genome analysis are variants surrounding the naturally occurring poison exons, which are exons that contain a termination codon and cause degradation of the transcript via nonsense‐mediated decay when included. Several publications have now shown the impact of poison exons in NDD. 110 Furthermore, intergenic DNA contains non‐protein‐coding DNA but sequences important for transcriptional and translational regulation of protein‐coding sequences (e.g., enhancers) and can encode for RNA genes. 111 These RNA genes in turn play a role in transcriptional and translational processes (Figure S1). More and more examples arise showing the pathogenic effect of variants in non‐protein‐coding DNA on human biology. 112 , 113 During embryogenesis, precise gene transcription in space and time requires that distal enhancers and promoters communicate by physical proximity within gene regulatory landscapes. To achieve this, regulatory landscapes fold in the nuclear space, creating complex 3D structures that influence enhancer‐promoter communication and gene expression and that, when disrupted, can cause disease. 114 RNA‐sequencing aids in estimating the effect of a variant on transcription, but each non‐coding DNA variant requires adequate functional testing before a final molecular diagnosis can be made.
5.2. Complex DNA rearrangements
Disease‐causing variants are not limited to SNVs or CNVs, but include structural variants (SVs) that can result in complex rearrangements of our DNA. SVs are large genomic alterations that include DNA duplications, repeats, deletions, insertions, inversions, and translocations. CNVs are a subtype of SVs that are not addressed in this paragraph. Standard ES analysis is based on short sequence reads that are aligned to the best matching position in the reference genome. In some cases, for example, in case of a translocation or a transposable element, the mapped position does not resemble the true genomic position in the patient. Mobile or transposable elements are DNA sequences that move around or change the number of copies in the genome. Mobile elements can be inserted within a gene, causing a frameshift, or nearby affecting splicing or gene regulation. 18 A switch from short‐read sequencing (SRS, 150–300 bp reads) to long‐read sequencing (LRS, >10 kb reads) allows for better sequence mapping and therefore better SV detection, including complex rearrangements and repetitive sequences. 115 A recent publication comparing SRS to LRS showed that 80% of LRS SV calls were not identified by SRS. 116 Short‐tandem repeats (STRs) are another type of SV that are missed in ES analysis. STR variants and mobile elements can also be detected by ES when specific bio‐informatic tools are used, which are not yet standard in diagnostic settings. 117 , 118 STR expansions are often associated with neurologic disease, for example, Fragile X syndrome, Huntington's disease and several hereditary ataxias, and will likely be the underlying genetic pathomechanism in subgroups of unsolved patients (e.g., glutaminase deficiency 119 ). Another suitable new technique is optical genome mapping based on high‐resolution genome imaging instead of sequencing. In short, long linear single DNA molecules are labeled at specific sites creating a whole‐genome image of which patient patterns can be compared to the expected labeling pattern of the reference genome. This technique proves to be especially suitable for large and complex rearrangements (>500 bp), and also shows high sensitivity to somatic SVs, but can also detect STR expansions. 120 , 121 , 122 This technique is complementary to sequencing, and rather a potential replacement of all cytogenetic technologies, such as karyotyping, FISH and CNV‐microarrays. Altogether, the detection of SVs can be improved by moving from ES to GS since inclusion of the non‐coding parts will improve sequence read mapping. However, important to note is that GS files are considerably larger and contain more variants that make the interpretation of the data more challenging. Currently, the analysis pipeline for GS data is ready for clinical interpretation when it comes to the coding region of our DNA, but it will take time, research, and experience to be able to clinically interpret the non‐coding region.
6. THE ADDED VALUE OF FUNCTIONAL DATA
6.1. Transcriptomics (RNA‐seq) and proteomics
For the accurate interpretation of variants, DNA sequence data alone may not always suffice. In some cases, transcriptomics is performed before or simultaneous with ES to highlight those genes that show an aberrant transcription profile. 123 It is important to note that the choice of tissue used for transcriptomic studies could impact the results as not all genes are equally relevant and expressed in every tissue. 123 , 124
RNA analysis may also be used to study up‐ or downregulated genes or pathways just as proteomics can provide insights into protein interaction networks. The effect of missense variants is not always straightforward as they may lead to aberrant splicing, incorrect folding of the protein, or abnormal expression levels. The latter could be a result of allelic expression imbalance (AEI), where the mutant allele is higher expressed than the wildtype allele causing a seemingly dominant presentation (heterozygous variant) of a recessive disorder. 88 ES (or preferably GS) to identify intronic and regulatory variants, in combination with quantitative RNA studies are required to detect AEI. Again other techniques can aid in confirming pathogenicity. It has been shown that additional RNA‐seq can enhance the diagnostic yield by 10%–16% compared to ES alone and can be integrated at reasonable costs and turnaround times. 124 , 125 The diagnostic value of integrating proteomic data with genomic, transcriptomic, and phenotypic data from 145 individuals has been shown in a study on individuals suspected of having a mitochondrial disease and achieved genetic resolution of 21% in ES‐negative cases. 126 Although RNA‐seq still focuses on the genetic level, additional functional analyses can help in variant interpretation, especially for IMD. As for genetic analyses, these ‐omics techniques rely on finding “outliers” and need comparison with a control cohort of sufficient size.
6.2. Metabolomics
Targeted metabolic screenings, covering a specific class of metabolites such as amino acids, organic acids, or acylcarnitines, have already been used for decades in diagnostics for IMD, providing a variant‐transcending read‐out of biochemical function of a metabolic pathway. Currently, untargeted metabolomics is replacing targeted metabolic screening, moving from multiple targeted analyses performed in parallel, to a “one‐size‐fits most” ‐omics approach (Next Generation Metabolic Screening [NGMS]; validated for use in clinical diagnostics under ISO:15189 accreditation). 127 , 128 Furthermore, NGMS offers the possibility to find novel biomarkers in (neuro) IMDs that in some cases have been found to play an essential role in the pathophysiology of the disease. 87 , 129 , 130 Untargeted metabolomics holds the potential to serve as a functional counterpart to ES data, as it provides unbiased functional data that may not otherwise be available from targeted biochemical assays. 131 , 132 , 133 Besides the additional functional evidence to determine the pathogenicity of variant of unknown significance (VUS) in known IMD genes, untargeted metabolomics may detect aberrant metabolite profiles that highlight certain pathways and may lead to the discovery of novel disease‐gene associations (Figure 3). This approach specifically helps to elucidate the pathogenicity of variants in genes that encode for enzymes or transporters. A recent study with 170 individuals presenting predominantly with neurological symptoms showed that metabolomics data contributed to the variant interpretation in 73 different IMD genes in 43% of investigated cases. 134 Importantly, untargeted metabolomics can increase our mechanistic understanding of the metabolic pathways involved, as well as identify novel biomarkers and treatment targets (Box 3). Until now, untargeted metabolomics has mostly been applied to body fluid analysis and as such will only evaluate metabolites that will be able to cross the cell outer membrane. The technique surely will be able to gain momentum when studies will start targeting the intracellular metabolome of various cell types.
FIGURE 3.
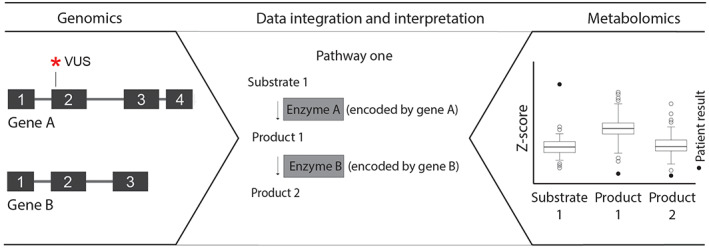
Added value of genomics and metabolomics data integration. Exome or genome sequencing (genomics) provides data on variants found in the DNA of a patient, whereas targeted or untargeted metabolomics provides data on aberrant metabolites. The integration of both data sets may aid in the interpretation of variants. For example, the metabolic profile of a patient showed an elevated level of substrate 1 and a decrease of products 1 and 2, implicating that there is a defect in pathway one that in turn points to gene A with a VUS. VUS, variant of unknown significance.
BOX 3. IMDs characterized with support of ‐omics techniques.
Transcriptomics: disorders of complex I subunits and assembly factors (TIMMDC1 87 ).
Metabolomics: disorders of sialic acid metabolism (NANS 133 ), disorders of pyridoxine metabolism (ALDH7A1 135, 136).
Glycomics: disorders of N‐linked protein glycosylation (GFUS 137 ).
Lipidomics: disorders of mitochondrial membrane biogenesis and remodeling (SERAC1 138 ); disorders of phosphatidylcholine, phosphatidylserine, and phosphatidylethanolamine metabolism (PCYT2 139 ); disorders of fatty acyl synthesis, elongation, and recycling (ALDH3A2 140 ); and disorders of peroxisomal fatty acid oxidation (reviewed in ref. 141 ).
Complexomics: other disorders of mitochondrial function (CLPB 63 ).
6.3. Other ‐omics
Although proteomics and metabolomics are quite universal approaches, the utility and yield of other ‐omics techniques (e.g., lipidomics and glycomics) 135 , 142 , 143 require further evaluation. Fluxomics using stable isotope labeling, to study the flux of metabolites through a metabolic pathways, is a relatively new technique. 144 It has the capacity to analyze entire metabolic pathways in vitro and in vivo in a dynamic fashion, useful especially for IMDs to delineate the reprogrammed metabolism due to a specific enzymatic or other biochemical defect. 145
In summary, (targeted) metabolomics, RNA‐seq, and proteomics are the most established ‐omics technologies alongside ES/GS and “other ‐omics” have mainly been used to validate or reject single candidate variants or to study novel gene‐disease phenotypes (Box 3). Evidence for their utility in terms of pathophysiologic delineation, prognostication, and therapeutic effect monitoring is clearly emerging. 146 , 147
7. EPIGENETICS
Besides changes in the DNA sequence itself, epigenetic changes can cause disease. Modifications of DNA or histones regulate gene expression by remodeling the chromatin structure, making DNA accessible or inaccessible for the transcriptional machinery. One of these so‐called epigenetic modifications is methylation of cytosines at CpG dinucleotides in the DNA molecule. Methylation of the promoter region of a gene is generally associated with gene silencing. Epigenetic changes are thought to be caused by stochastic errors in the establishment or maintenance of the epigenome or induced by underlying variations in DNA sequence. Changes in methylation status of the DNA are not detectable by ES.
7.1. Targeted or genome‐wide analysis of promoter methylation
The best known example of promoter hypermethylation associated with gene silencing is Fragile X syndrome. A repeat expansion in the promoter region of FRM1 causes hypermethylation and loss of expression of that allele. 139 A small number of other examples exist where methylation analysis revealed the disease‐causing mechanism while the underlying genetic defects (repeat expansions, variants in non‐coding regions, or in neighboring genes) are usually not detected by ES. Methylation analysis, either targeted or genome‐wide, will detect these epigenetic defects regardless of the underlying genetic defect. Only few examples are known today. A study of 489 patients with NDD and congenital anomalies without an identified cause showed an increase in de novo epigenetic aberrations in patients compared to controls. Causality needs to be established, but this indicates that epigenetic variations may play a substantial role in the etiology of NDD. 148 Recently “epi‐cblC” has been described as a first example of an IMD with compound epigenetic‐genetic heterozygosity. Affected individuals are compound heterozygous for a genetic variant and an epimutation at the MMACHC locus, which is secondary to a splicing variant at the adjacent PRDX1 gene. Both these variants cause aberrant antisense transcription and cis‐hypermethylation of the MMACHC gene promotor with subsequent silencing. 149
7.2. Methylation status of imprinted genes
Genomic imprinting describes the phenomenon that the expression of a gene is dependent on the parental origin of the gene. In humans, already 100 genes are known to be subject to imprinting and are expressed only from the paternal or the maternal allele. Imprinted genes tend to be organized in clusters under the control of imprinting centers. These imprinting centers are differentially methylated, that is, methylation is present only on one of the two alleles. The most well‐known clusters are those on 11p15, involved in Beckwith–Wiedemann/Silver–Russel syndrome, and on 15q13, involved in Prader–Willi/Angelman syndrome. Disturbances of imprinting result in overexpression or loss of expression of imprinted genes. This can be due to loss of one allele, uniparental disomy (UPD), or imprinting defects (often primary epigenetic defects). UPD occurs when both copies of a chromosome originate from one parent (Figure 2D). This can involve the entire chromosome (complete isodisomy or heterodisomy) or only a small segment (segmental disomy). As a consequence, UPD may result in aberrant dosage of genes regulated by genomic imprinting or homozygosity of a recessive mutation (reviewed in 150 ). Targeted methylation analysis of imprinted regions to detect imprinting disorders is usually done by methylation‐specific multiplex ligation‐dependent probe amplification (MLPA) assays, but can also be detected with genome‐wide methylation analysis. 151 , 152
7.3. Methylation signatures
In addition to methylation defects at specific loci, variants in genes encoding proteins involved in epigenetic regulation may cause a broader, genome‐wide aberrant methylation profile. Patients with variants in such genes exhibit DNA methylation “episignatures” that are detectable in peripheral blood and have shown to be highly sensitive and specific for specific disorder. 151 , 153 Currently, a characteristic methylation signature has been recognized for over 40 syndromes, in association with more than 60 genes. These signatures can be used as biomarkers for ES‐negative patients or for variant interpretation. Epigenetic profiling is available for diagnostic care. 152 From a cohort of 207 affected individuals, ~28% were positive for an episignature that supported the diagnosis of the associated syndrome. The number of identified disorder‐ or gene‐specific epigenetic signatures is growing rapidly, increasing the diagnostic power and yield of this type of test.
In addition to genetic variations, environment may impact the epigenome. Prenatal exposure to, for instance, maternal smoking, alcohol consumption, or famine affects the health of the child and is associated with changes in the genome‐wide methylation profile. 89 , 154 , 155 Especially fetal alcohol spectrum disorder, a non‐inherited mimicker of genetic NDD, seems to be associated with specific epigenetic profiles, which is promising for future diagnostic use to discriminate between the two disorders. 156
7.4. The genomic checklist
“The modern world has given us stupendous know‐how. Yet avoidable failures continue to plague us in health care […]—in almost every realm of organized activity. And the reason is simple: the volume and complexity of knowledge today has exceeded our ability as individuals to properly deliver it to people—consistently, correctly, safely. We train longer, specialize more, use ever‐advancing technologies, and still we fail.” 157 Atul Gawande makes this compelling argument that we can do better, using the simplest of methods: the checklist. For surgical safety and on intensive care units, World Health Organization checklists have been adopted worldwide as a standard for care, decreasing errors and improving success. 158 , 159 , 160 Analogous to these, we propose the genomic checklist (Figure 4), with the goal of standardizing analysis and interpretation methods, to enhance diagnostic success as well as minimize delay and costs.
FIGURE 4.
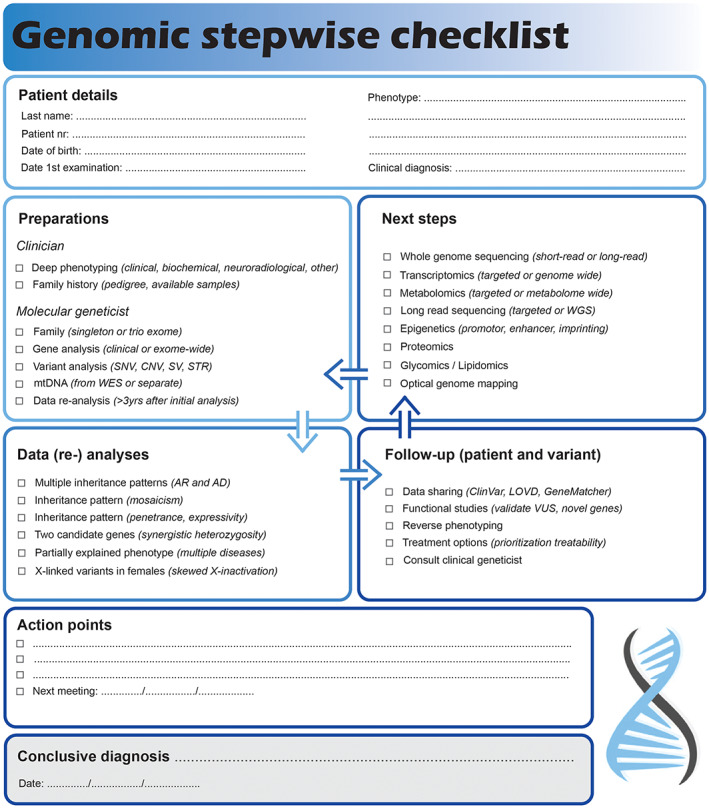
Genomic stepwise checklist to solve exome negative neurological cases. This checklist can be used to standardize analysis and interpretation methods to improve diagnostic care.
8. CONCLUSIONS
We have shown that a “negative” ES should not be considered the end of the road in the quest for a diagnosis in patients suspected of having an RGD. Rather the contrary, there is more to explore still.
AUTHOR CONTRIBUTIONS
Saskia B. Wortmann, Machteld M. Oud, Lisenka E. L. M. Vissers, and Clara D. M. van Karnebeek conceptualized the study; Saskia B. Wortmann and Machteld M. Oud drafted the manuscript; all other authors revised it critically for important intellectual content.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ETHICS STATEMENT
Not applicable.
Supporting information
Appendix S1 Supporting information
ACKNOWLEDGMENTS
The authors thank Han G. Brunner for his input during fruitful discussions, the TIDEX team for our multiomics collaborations, and Stichting Metakids for their support of United for Metabolic Diseases. NI Wolf, BP van de Warrenburg, and MA Willemsen are members of the European Reference Network “Rare Neurological Disorders” (ERN‐RND), project ID 739510. SB Wortmann, RG Feichtinger, JA Mayr, and CD van Karnebeek are (affiliated) members of the European Reference Network “MetabERN‐ European Reference Network for Hereditary Metabolic Disorders.” LELM Vissers is a member of the European Reference Network on “Rare Congenital Malformations and Rare Intellectual Disability ERN‐ITHACA” [EU Framework Partnership Agreement ID: 3HP‐HP‐FPA ERN‐01‐2016/739516]. SBW receives funding from PMU‐FFF A‐20/01/040‐WOS, ERAPERMED2019‐310, FWF‐18023. LELMV receives funding for the SOLVE‐RD project from the European Union's Horizon 2020 research and innovation program under grant agreement no. 779257. HP was supported by the German Federal Ministry of Education and Research (BMBF, Bonn, Germany) and Horizon2020 through the European Joint Program on Rare Diseases project GENOMIT (01GM1920A) and the ERA PerMed project PerMiM (01KU2016A). BvdW receives grants from Hersenstichting, Radboudumc, ZonMW, Gossweiler Foundation, and Michael J Fox Foundation. CvK receives funding from the European Union's Horizon 2020 research and innovation program under the EJP RD COFUND‐EJP no. 825575, and Stichting Metakids NL.
Wortmann SB, Oud MM, Alders M, et al. How to proceed after “negative” exome: A review on genetic diagnostics, limitations, challenges, and emerging new multiomics techniques. J Inherit Metab Dis. 2022;45(4):663‐681. doi: 10.1002/jimd.12507
Saskia B. Wortmann, Machteld M. Oud, Lisenka E. L. M. Vissers, Clara D. M. van Karnebeek contributed equally to this study.
Funding information Bundesministerium für Bildung und Forschung, Grant/Award Number: GENOMIT (01GM1920A); ERA‐Permed, Grant/Award Number: PerMiM (01KU2016A); ERAPERMED, Grant/Award Number: ERAPERMED2019‐310; FWF, Grant/Award Number: FWF‐18023; Hersenstichting; Horizon 2020, Grant/Award Numbers: No 779257, EJP RD COFUND‐EJP N° 825575; Michael J. Fox Foundation for Parkinson's Research; PMU‐FFF, Grant/Award Number: PMU‐FFF A‐20/01/040‐WOS; Radboud Universitair Medisch Centrum; Stichting metakids; ZonMW
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Srivastava S, Love‐Nichols JA, Dies KA, et al. Meta‐analysis and multidisciplinary consensus statement: exome sequencing is a first‐tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413‐2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stefanski A, Calle‐Lopez Y, Leu C, et al. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: a systematic review and meta‐analysis. Epilepsia. 2021;62:143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stenton SL, Prokisch H. Genetics of mitochondrial diseases: identifying mutations to help diagnosis. EBioMedicine. 2020;56:102784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schofield D, Alam K, Douglas L, et al. Cost‐effectiveness of massively parallel sequencing for diagnosis of paediatric muscle diseases. NPJ Genom Med. 2017;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herman I, Lopez MA, Marafi D, et al. Clinical exome sequencing in the diagnosis of pediatric neuromuscular disease. Muscle Nerve. 2021;63:304‐310. [DOI] [PubMed] [Google Scholar]
- 6. Tsang MHY, Chiu ATG, Kwong BMH, et al. Diagnostic value of whole‐exome sequencing in Chinese pediatric‐onset neuromuscular patients. Mol Genet Genomic Med. 2020;8:e1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krenn M, Tomschik M, Rath J, et al. Genotype‐guided diagnostic reassessment after exome sequencing in neuromuscular disorders: experiences with a two‐step approach. Eur J Neurol. 2020;27:51‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haskell GT, Adams MC, Fan Z, et al. Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol Genet. 2018;4:e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaplanis J, Samocha KE, Wiel L, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morava E, Rahman S, Peters V, Baumgartner MR, Patterson M, Zschocke J. Quo vadis: the re‐definition of "inborn metabolic diseases". J Inherit Metab Dis. 2015;38:1003‐1006. [DOI] [PubMed] [Google Scholar]
- 11. Hoytema van Konijnenburg EMM, Wortmann SB, Koelewijn MJ, et al. Treatable inherited metabolic disorders causing intellectual disability: 2021 review and digital app. Orphanet J Rare Dis. 2021;16(16):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wortmann SB, Van Hove JLK, Derks TGJ, et al. Treating neutropenia and neutrophil dysfunction in glycogen storage disease type Ib with an SGLT2 inhibitor. Blood. 2020;136:1033‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rymen D, Lindhout M, Spanou M, et al. Expanding the clinical and genetic spectrum of CAD deficiency: an epileptic encephalopathy treatable with uridine supplementation. Genet Med. 2020;22:1589‐1597. [DOI] [PubMed] [Google Scholar]
- 14. Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile‐onset spinal muscular atrophy with nusinersen: final report of a phase 2, open‐label, multicentre, dose‐escalation study. Lancet Child Adolesc Health. 2021;5:491‐500. [DOI] [PubMed] [Google Scholar]
- 15. Mendell JR, Al‐Zaidy SA, Lehman KJ, et al. Five‐year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021;78:834‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Day JW, Finkel RS, Chiriboga CA, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile‐onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open‐label, single‐arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:284‐293. [DOI] [PubMed] [Google Scholar]
- 17. Dhillon S. Risdiplam: first approval. Drugs. 2020;80:1853‐1858. [DOI] [PubMed] [Google Scholar]
- 18. Kim J, Hu C, Moufawad El Achkar C, et al. Patient‐customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381:1644‐1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vill K, Kolbel H, Schwartz O, et al. One year of newborn screening for SMA – results of a German pilot project. J Neuromuscul Dis. 2019;6:503‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonne G. The Treatabolome, an emerging concept. J Neuromuscul Dis. 2021;8:337‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Atalaia A, Thompson R, Corvo A, et al. A guide to writing systematic reviews of rare disease treatments to generate FAIR‐compliant datasets: building a Treatabolome. Orphanet J Rare Dis. 2020;15:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Karnebeek CD, Houben RF, Lafek M, Giannasi W, Stockler S. The treatable intellectual disability APP www.treatable‐id.org: a digital tool to enhance diagnosis & care for rare diseases. Orphanet J Rare Dis. 2012;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen JJ, Wortmann SB, de Boer L, et al. The role of clinical response to treatment in determining pathogenicity of genomic variants. Genet Med. 2021;23:581‐585. [DOI] [PubMed] [Google Scholar]
- 24. Wortmann SB, Mayr JA, Nuoffer JM, Prokisch H, Sperl W. A guideline for the diagnosis of pediatric mitochondrial disease: the value of muscle and skin biopsies in the genetics era. Neuropediatrics. 2017;48:309‐314. [DOI] [PubMed] [Google Scholar]
- 25. Thomas Q, Vitobello A, Tran Mau‐Them F, et al. High efficiency and clinical relevance of exome sequencing in the daily practice of neurogenetics. J Med Genet. 2021;59:445‐452. [DOI] [PubMed] [Google Scholar]
- 26. Westra D, Schouten MI, Stunnenberg BC, et al. Panel‐based exome sequencing for neuromuscular disorders as a diagnostic service. J Neuromuscul Dis. 2019;6:241‐258. [DOI] [PubMed] [Google Scholar]
- 27. Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437‐455. [DOI] [PubMed] [Google Scholar]
- 28. Vissers LE, de Vries BB, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet. 2010;47:289‐297. [DOI] [PubMed] [Google Scholar]
- 29. Plagnol V, Curtis J, Epstein M, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747‐2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525‐1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pfundt R, Del Rosario M, Vissers L, et al. Detection of clinically relevant copy‐number variants by exome sequencing in a large cohort of genetic disorders. Genet Med. 2017;19:667‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Ligt J, Boone PM, Pfundt R, et al. Detection of clinically relevant copy number variants with whole‐exome sequencing. Hum Mutat. 2013;34:1439‐1448. [DOI] [PubMed] [Google Scholar]
- 33. Marchuk DS, Crooks K, Strande N, et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS One. 2018;13:e0209185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wagner M, Berutti R, Lorenz‐Depiereux B, et al. Mitochondrial DNA mutation analysis from exome sequencing‐a more holistic approach in diagnostics of suspected mitochondrial disease. J Inherit Metab Dis. 2019;42:909‐917. [DOI] [PubMed] [Google Scholar]
- 35. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. 2009;84:524‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buske OJ, Girdea M, Dumitriu S, et al. PhenomeCentral: a portal for phenotypic and genotypic matchmaking of patients with rare genetic diseases. Hum Mutat. 2015;36:931‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arachchi H, Wojcik MH, Weisburd B, et al. Matchbox: an open‐source tool for patient matching via the matchmaker exchange. Hum Mutat. 2018;39:1827‐1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levenson D. Website aims to accelerate gene discovery, diagnosis, treatment: MyGene2.org fosters open sharing among families, researchers, and clinicians. Am J Med Genet A. 2016;170:1388‐1389. [DOI] [PubMed] [Google Scholar]
- 40. Adachi T, Kawamura K, Furusawa Y, et al. Japan's initiative on rare and undiagnosed diseases (IRUD): towards an end to the diagnostic odyssey. Eur J Hum Genet. 2017;25:1025‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Philippakis AA, Azzariti DR, Beltran S, et al. The matchmaker exchange: a platform for rare disease gene discovery. Hum Mutat. 2015;36:915‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matalonga L, Hernandez‐Ferrer C, Piscia D, et al. Solving patients with rare diseases through programmatic reanalysis of genome‐phenome data. Eur J Hum Genet. 2021;29:1337‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209‐214. [DOI] [PubMed] [Google Scholar]
- 44. Tan NB, Stapleton R, Stark Z, et al. Evaluating systematic reanalysis of clinical genomic data in rare disease from single center experience and literature review. Mol Genet Genomic Med. 2020;8:e1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Basel‐Salmon L, Orenstein N, Markus‐Bustani K, et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med. 2019;21:1443‐1451. [DOI] [PubMed] [Google Scholar]
- 46. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG‐AMP guidelines. Am J Hum Genet. 2017;100:267‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meienberg J, Zerjavic K, Keller I, et al. New insights into the performance of human whole‐exome capture platforms. Nucleic Acids Res. 2015;43:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109‐1112. [DOI] [PubMed] [Google Scholar]
- 52. Retterer K, Juusola J, Cho MT, et al. Clinical application of whole‐exome sequencing across clinical indications. Genet Med. 2016;18:696‐704. [DOI] [PubMed] [Google Scholar]
- 53. Kohler S, Gargano M, Matentzoglu N, et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021;49:D1207‐D1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nasca A, Rizza T, Doimo M, et al. Not only dominant, not only optic atrophy: expanding the clinical spectrum associated with OPA1 mutations. Orphanet J Rare Dis. 2017;12:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reynier P, Amati‐Bonneau P, Verny C, et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet. 2004;41:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Anikster Y, Kleta R, Shaag A, Gahl WA, Elpeleg O. Type III 3‐methylglutaconic aciduria (optic atrophy plus syndrome, or Costeff optic atrophy syndrome): identification of the OPA3 gene and its founder mutation in Iraqi Jews. Am J Hum Genet. 2001;69:1218‐1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stenton SL, Sheremet NL, Catarino CB, et al. Impaired complex I repair causes recessive Leber's hereditary optic neuropathy. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morino H, Pierce SB, Matsuda Y, et al. Mutations in Twinkle primase‐helicase cause Perrault syndrome with neurologic features. Neurology. 2014;83:2054‐2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baloh RH, Salavaggione E, Milbrandt J, Pestronk A. Familial parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch Neurol. 2007;64:998‐1000. [DOI] [PubMed] [Google Scholar]
- 60. Pierson TM, Adams D, Bonn F, et al. Whole‐exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia‐neuropathy syndrome linked to mitochondrial m‐AAA proteases. PLoS Genet. 2011;7:e1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cagnoli C, Stevanin G, Brussino A, et al. Missense mutations in the AFG3L2 proteolytic domain account for approximately 1.5% of European autosomal dominant cerebellar ataxias. Hum Mutat. 2010;31:1117‐1124. [DOI] [PubMed] [Google Scholar]
- 62. Di Bella D, Lazzaro F, Brusco A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313‐321. [DOI] [PubMed] [Google Scholar]
- 63. Wortmann SB, Zietkiewicz S, Guerrero‐Castillo S, et al. Neutropenia and intellectual disability are hallmarks of biallelic and de novo CLPB deficiency. Genet Med. 2021;23:1705‐1714. [DOI] [PubMed] [Google Scholar]
- 64. Wortmann SB, Zietkiewicz S, Kousi M, et al. CLPB mutations cause 3‐methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet. 2015;96:245‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Harel T, Yoon WH, Garone C, et al. Recurrent de novo and biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am J Hum Genet. 2016;99:831‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Harel T, Yesil G, Bayram Y, et al. Monoallelic and biallelic variants in EMC1 identified in individuals with global developmental delay, hypotonia, scoliosis, and cerebellar atrophy. Am J Hum Genet. 2016;98:562‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Birouk N, Azzedine H, Dubourg O, et al. Phenotypical features of a Moroccan family with autosomal recessive Charcot‐Marie‐Tooth disease associated with the S194X mutation in the GDAP1 gene. Arch Neurol. 2003;60:598‐604. [DOI] [PubMed] [Google Scholar]
- 68. Chung KW, Kim SM, Sunwoo IN, et al. A novel GDAP1 Q218E mutation in autosomal dominant Charcot‐Marie‐Tooth disease. J Hum Genet. 2008;53:360‐364. [DOI] [PubMed] [Google Scholar]
- 69. Ebrahimi‐Fakhari D, Moufawad El Achkar C, Klein C. PRRT2‐associated paroxysmal movement disorders. In: Adam MP et al., eds. GeneReviews((R)); 1993. [Google Scholar]
- 70. Lee JR, Srour M, Kim D, et al. De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum Mutat. 2015;36:69‐78. [DOI] [PubMed] [Google Scholar]
- 71. Riviere JB, Ramalingam S, Lavastre V, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet. 2011;89:219‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Innes AM, McInnes BL, Dyment DA. Clinical and genetic heterogeneity in Dubowitz syndrome: implications for diagnosis, management and further research. Am J Med Genet C Semin Med Genet. 2018;178:387‐397. [DOI] [PubMed] [Google Scholar]
- 73. Rotstein M, Engelstad K, Yang H, et al. Glut1 deficiency: inheritance pattern determined by haploinsufficiency. Ann Neurol. 2010;68:955‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ganetzky R, McCormick EM, Falk MJ. Primary pyruvate dehydrogenase complex deficiency overview. In: Adam MP et al., eds. GeneReviews((R)). Seattle, WA; 1993. [Google Scholar]
- 75. Ferreira C, Pierre G, Thompson R, Vernon H. Barth syndrome. In: Adam MP et al., eds. GeneReviews((R)). Seattle, WA; 1993. [Google Scholar]
- 76. Raymond GV, Moser AB, Fatemi A. X‐linked adrenoleukodystrophy. In: Adam MP et al., eds. GeneReviews((R)). Seattle, WA; 1993. [PubMed] [Google Scholar]
- 77. Mehta A, Hughes DA. Fabry disease. In: Adam MP et al., eds. GeneReviews((R)). Seattle, WA; 1993. [Google Scholar]
- 78. Lichter‐Konecki U, Caldovic L, Morizono H, et al. Ornithine transcarbamylase deficiency. In: Adam MP et al., eds. GeneReviews((R)). Seattle, WA; 1993. [Google Scholar]
- 79. Nota B, Hamilton EM, Sie D, et al. Novel cases of D‐2‐hydroxyglutaric aciduria with IDH1 or IDH2 mosaic mutations identified by amplicon deep sequencing. J Med Genet. 2013;50:754‐759. [DOI] [PubMed] [Google Scholar]
- 80. Bae EH, Choi JM, Ki CS, et al. A late‐onset male Fabry disease patient with somatic mosaicism of a classical GLA mutation: a case report. Ann Palliat Med. 2021;10:4926‐4931. [DOI] [PubMed] [Google Scholar]
- 81. Bowling F, McGown I, McGill J, Cowley D, Tuchman M. Maternal gonadal mosaicism causing ornithine transcarbamylase deficiency. Am J Med Genet. 1999;85:452‐454. [DOI] [PubMed] [Google Scholar]
- 82. Takahashi S, Matsufuji M, Yonee C, et al. Somatic mosaicism for a SLC2A1 mutation: implications for genetic counseling for GLUT1 deficiency syndrome. Clin Genet. 2017;91:932‐933. [DOI] [PubMed] [Google Scholar]
- 83. Van Bergen NJ, Guo Y, Rankin J, et al. NAD(P)HX dehydratase (NAXD) deficiency: a novel neurodegenerative disorder exacerbated by febrile illnesses. Brain. 2019;142:50‐58. [DOI] [PubMed] [Google Scholar]
- 84. Willemsen MA, Vissers LE, Verbeek MM, et al. Upstream SLC2A1 translation initiation causes GLUT1 deficiency syndrome. Eur J Hum Genet. 2017;25:771‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Adams DR, Yuan H, Holyoak T, et al. Three rare diseases in one Sib pair: RAI1, PCK1, GRIN2B mutations associated with Smith‐Magenis Syndrome, cytosolic PEPCK deficiency and NMDA receptor glutamate insensitivity. Mol Genet Metab. 2014;113:161‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kremer LS, Wortmann SB, Prokisch H. "Transcriptomics": molecular diagnosis of inborn errors of metabolism via RNA‐sequencing. J Inherit Metab Dis. 2018;41:525‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Engelke UF, van Outersterp RE, Merx J, et al. Untargeted metabolomics and infrared ion spectroscopy identify biomarkers for pyridoxine‐dependent epilepsy. J Clin Invest. 2021;131:e148272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Falkenberg KD, Braverman NE, Moser AB, et al. Allelic expression imbalance promoting a mutant PEX6 allele causes Zellweger Spectrum disorder. Am J Hum Genet. 2017;101:965‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tobi EW, Goeman JJ, Monajemi R, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kopp J, Has C, Hotz A, Grunert SC, Fischer J. Maternal isodisomy of chromosome 3 combined with a de novo mutation in the ABHD5 gene causes autosomal recessive Chanarin‐Dorfman syndrome. Genes. 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kang E, Kim T, Oh A, et al. Lysinuric protein intolerance with homozygous SLC7A7 mutation caused by maternal uniparental isodisomy of chromosome 14. J Hum Genet. 2019;64:1137‐1140. [DOI] [PubMed] [Google Scholar]
- 92. Deng X, Berletch JB, Nguyen DK, Disteche CM. X chromosome regulation: diverse patterns in development, tissues and disease. Nat Rev Genet. 2014;15:367‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rahbari R, Wuster A, Lindsay SJ, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Acuna‐Hidalgo R, Bo T, Kwint MP, et al. Post‐zygotic point mutations are an Underrecognized source of de novo genomic variation. Am J Hum Genet. 2015;97:67‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Campbell IM, Yuan B, Robberecht C, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014;95:173‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Stosser MB, Lindy AS, Butler E, et al. High frequency of mosaic pathogenic variants in genes causing epilepsy‐related neurodevelopmental disorders. Genet Med. 2018;20:403‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wright CF, Prigmore E, Rajan D, et al. Clinically‐relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data. Nat Commun. 2019;10:2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jamuar SS, Lam AT, Kircher M, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Renkema GH, Wortmann SB, Smeets RJ, et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur J Hum Genet. 2015;23:202‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Magrinelli F, Balint B, Bhatia KP. Challenges in Clinicogenetic correlations: one gene – many phenotypes. Mov Disord Clin Pract. 2021;8:299‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cooper DN, Krawczak M, Polychronakos C, Tyler‐Smith C, Kehrer‐Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132:1077‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kearney JA. Genetic modifiers of neurological disease. Curr Opin Genet Dev. 2011;21:349‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Vockley J, Rinaldo P, Bennett MJ, Matern D, Vladutiu GD. Synergistic heterozygosity: disease resulting from multiple partial defects in one or more metabolic pathways. Mol Genet Metab. 2000;71:10‐18. [DOI] [PubMed] [Google Scholar]
- 105. Lowik M, Levtchenko E, Westra D, et al. Bigenic heterozygosity and the development of steroid‐resistant focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2008;23:3146‐3151. [DOI] [PubMed] [Google Scholar]
- 106. Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 2020;12:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Allou L, Balzano S, Magg A, et al. Non‐coding deletions identify Maenli lncRNA as a limb‐specific En1 regulator. Nature. 2021;592:93‐98. [DOI] [PubMed] [Google Scholar]
- 108. Wright CF, Quaife NM, Ramos‐Hernandez L, et al. Non‐coding region variants upstream of MEF2C cause severe developmental disorder through three distinct loss‐of‐function mechanisms. Am J Hum Genet. 2021;108:1083‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Vaz‐Drago R, Custodio N, Carmo‐Fonseca M. Deep intronic mutations and human disease. Hum Genet. 2017;136:1093‐1111. [DOI] [PubMed] [Google Scholar]
- 110. Carvill GL, Mefford HC. Poison exons in neurodevelopment and disease. Curr Opin Genet Dev. 2020;65:98‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Biswas A, Narlikar L. A universal framework for detecting cis‐regulatory diversity in DNA regions. Genome Res. 2021;31:1646‐1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kumar V, Westra HJ, Karjalainen J, et al. Human disease‐associated genetic variation impacts large intergenic non‐coding RNA expression. PLoS Genet. 2013;9:e1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bartonicek N, Clark MB, Quek XC, et al. Intergenic disease‐associated regions are abundant in novel transcripts. Genome Biol. 2017;18:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Robson MI, Ringel AR, Mundlos S. Regulatory landscaping: how enhancer‐promoter communication is sculpted in 3D. Mol Cell. 2019;74:1110‐1122. [DOI] [PubMed] [Google Scholar]
- 115. Mantere T, Kersten S, Hoischen A. Long‐read sequencing emerging in medical genetics. Front Genet. 2019;10:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pauper M, Kucuk E, Wenger AM, et al. Long‐read trio sequencing of individuals with unsolved intellectual disability. Eur J Hum Genet. 2021;29:637‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. van der Sanden B, Corominas J, de Groot M, et al. Systematic analysis of short tandem repeats in 38,095 exomes provides an additional diagnostic yield. Genet Med. 2021;23:1569‐1573. [DOI] [PubMed] [Google Scholar]
- 118. Torene RI, Galens K, Liu S, et al. Mobile element insertion detection in 89,874 clinical exomes. Genet Med. 2020;22:974‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. van Kuilenburg ABP, Tarailo‐Graovac M, Richmond PA, et al. Glutaminase deficiency caused by short tandem repeat expansion in GLS. N Engl J Med. 2019;380:1433‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mantere T, Neveling K, Pebrel‐Richard C, et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am J Hum Genet. 2021;108:1409‐1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Neveling K, Mantere T, Vermeulen S, et al. Next‐generation cytogenetics: comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am J Hum Genet. 2021;108:1423‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhang Q, Xu X, Ding L, et al. Clinical application of single‐molecule optical mapping to a multigeneration FSHD1 pedigree. Mol Genet Genomic Med. 2019;7:e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yepez VA, Mertes C, Muller MF, et al. Detection of aberrant gene expression events in RNA sequencing data. Nat Protoc. 2021;16:1276‐1296. [DOI] [PubMed] [Google Scholar]
- 124. Yépez VA, Gusic M, Kopajtich R, et al. Blessing H clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022;14:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kremer LS, Bader DM, Mertes C, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kopajtich R, Smirnov D, Stenton SL, et al. Integration of proteomics with genomics and transcriptomics increases the diagnostic rate of Mendelian disorders. medRxiv. 2021. doi: 10.1101/2021.03.09.21253187. [DOI] [Google Scholar]
- 127. Coene KLM, Kluijtmans LAJ, van der Heeft E, et al. Next‐generation metabolic screening: targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J Inherit Metab Dis. 2018;41:337‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. van Karnebeek CD, Bonafe L, Wen XY, et al. NANS‐mediated synthesis of sialic acid is required for brain and skeletal development. Nat Genet. 2016;48:777‐784. [DOI] [PubMed] [Google Scholar]
- 129. van Outersterp RE, Moons SJ, Engelke UFH, et al. Amadori rearrangement products as potential biomarkers for inborn errors of amino‐acid metabolism. Commun Biol. 2021;4:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Martens J, Berden G, Bentlage H, et al. Unraveling the unknown areas of the human metabolome: the role of infrared ion spectroscopy. J Inherit Metab Dis. 2018;41:367‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ching‐Lopez A, Martinez‐Gonzalez LJ, Arrabal L, et al. Combined genome, transcriptome and metabolome analysis in the diagnosis of childhood cerebellar ataxia. Int J Mol Sci. 2021;22:2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Graham Linck EJ, Richmond PA, Tarailo‐Graovac M, et al. metPropagate: network‐guided propagation of metabolomic information for prioritization of metabolic disease genes. NPJ Genom Med. 2020;5:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kerkhofs M, Haijes HA, Willemsen AM, et al. Cross‐omics: integrating genomics with metabolomics in clinical diagnostics. Metabolites. 2020;10:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Alaimo JT, Glinton KE, Liu N, et al. Integrated analysis of metabolomic profiling and exome data supplements sequence variant interpretation, classification, and diagnosis. Genet Med. 2020;22:1560‐1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Vaz FM, McDermott JH, Alders M, et al. Mutations in PCYT2 disrupt etherlipid biosynthesis and cause a complex hereditary spastic paraplegia. Brain. 2019;142:3382‐3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. van Outersterp RE, Engelke UFH, Merx J, et al. Metabolite identification using infrared ion spectroscopy horizontal line novel biomarkers for pyridoxine‐dependent epilepsy. Anal Chem. 2021;93:15340‐15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Feichtinger RG, Hullen A, Koller A, et al. A spoonful of L‐fucose‐an efficient therapy for GFUS‐CDG, a new glycosylation disorder. EMBO Mol Med. 2021;13:e14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wortmann SB, Vaz FM, Gardeitchik T, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. 2012;44:797‐802. [DOI] [PubMed] [Google Scholar]
- 139. Oberle I, Rousseau F, Heitz D, et al. Instability of a 550‐base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097‐1102. [DOI] [PubMed] [Google Scholar]
- 140. Staps P, Rizzo WB, Vaz FM, et al. Disturbed brain ether lipid metabolism and histology in Sjogren‐Larsson syndrome. J Inherit Metab Dis. 2020;43:1265‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Herzog K, Pras‐Raves ML, Ferdinandusse S, et al. Functional characterisation of peroxisomal beta‐oxidation disorders in fibroblasts using lipidomics. J Inherit Metab Dis. 2018;41:479‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wanders RJA, Vaz FM, Ferdinandusse S, et al. Translational metabolism: a multidisciplinary approach towards precision diagnosis of inborn errors of metabolism in the omics era. J Inherit Metab Dis. 2019;42:197‐208. [DOI] [PubMed] [Google Scholar]
- 143. Post MA, Lefeber DJ. Clinical glycomics in the diagnostic laboratory. Ann Transl Med. 2019;7:S220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Reijngoud DJ. Flux analysis of inborn errors of metabolism. J Inherit Metab Dis. 2018;41:309‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. van Harskamp D, Garrelfs SF, Oosterveld MJS, Groothoff JW, van Goudoever JB, Schierbeek H. Development and validation of a new gas chromatography‐tandem mass spectrometry method for the measurement of enrichment of glyoxylate metabolism analytes in hyperoxaluria patients using a stable isotope procedure. Anal Chem. 2020;92:1826‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ferdinandusse S, McWalter K, Te Brinke H, et al. An autosomal dominant neurological disorder caused by de novo variants in FAR1 resulting in uncontrolled synthesis of ether lipids. Genet Med. 2021;23:740‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hipgrave Ederveen AL, de Haan N, Baerenfaenger M, Lefeber DJ, Wuhrer M. Dissecting total plasma and protein‐specific glycosylation profiles in congenital disorders of glycosylation. Int J Mol Sci. 2020;21:7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Barbosa M, Joshi RS, Garg P, et al. Identification of rare de novo epigenetic variations in congenital disorders. Nat Commun. 2018;9:2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Gueant JL, Chery C, Oussalah A, et al. APRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat Commun. 2018;9:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yamazawa K, Ogata T, Ferguson‐Smith AC. Uniparental disomy and human disease: an overview. Am J Med Genet C Semin Med Genet. 2010;154C:329‐334. [DOI] [PubMed] [Google Scholar]
- 151. Aref‐Eshghi E, Kerkhof J, Pedro VP, et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. Am J Hum Genet. 2020;106:356‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sadikovic B, Levy MA, Kerkhof J, et al. Clinical epigenomics: genome‐wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med. 2021;23:1065‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Aref‐Eshghi E, Bend EG, Colaiacovo S, et al. Diagnostic utility of genome‐wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet. 2019;104:685‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome‐wide consortium meta‐analysis. Am J Hum Genet. 2016;98:680‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Cobben JM, Krzyzewska IM, Venema A, et al. DNA methylation abundantly associates with fetal alcohol spectrum disorder and its subphenotypes. Epigenomics. 2019;11:767‐785. [DOI] [PubMed] [Google Scholar]
- 156. Lussier AA, Morin AM, MacIsaac JL, et al. DNA methylation as a predictor of fetal alcohol spectrum disorder. Clin Epigenetics. 2018;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gawande A. The Checklist Manifesto: How to Get Things Right. Metropolitan Books; 2009. [Google Scholar]
- 158. De Bie AJR, Mestrom E, Compagner W, et al. Intelligent checklists improve checklist compliance in the intensive care unit: a prospective before‐and‐after mixed‐method study. Br J Anaesth. 2021;126:404‐414. [DOI] [PubMed] [Google Scholar]
- 159. Spector JM, Agrawal P, Kodkany B, et al. Improving quality of care for maternal and newborn health: prospective pilot study of the WHO safe childbirth checklist program. PLoS One. 2012;7:e35151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ramsay G, Haynes AB, Lipsitz SR, et al. Reducing surgical mortality in Scotland by use of the WHO surgical safety checklist. Br J Surg. 2019;106:1005‐1011. [DOI] [PubMed] [Google Scholar]
- 161. Turro E, Astle WJ, Megy K, et al. Whole‐genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583:96‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Brockman DG, Austin‐Tse CA, Pelletier RC, et al. Randomized prospective evaluation of genome sequencing versus standard‐of‐care as a first molecular diagnostic test. Genet Med. 2021;23:1689‐1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting information
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.