

Abstract
Human DJ‐1 is a cytoprotective protein whose absence causes Parkinson's disease and is also associated with other diseases. DJ‐1 has an established role as a redox‐regulated protein that defends against oxidative stress and mitochondrial dysfunction. Multiple studies have suggested that DJ‐1 is also a protein/nucleic acid deglycase that plays a key role in the repair of glycation damage caused by methylglyoxal (MG), a reactive α‐keto aldehyde formed by central metabolism. Contradictory reports suggest that DJ‐1 is a glyoxalase but not a deglycase and does not play a major role in glycation defense. Resolving this issue is important for understanding how DJ‐1 protects cells against insults that can cause disease. We find that DJ‐1 reduces levels of reversible adducts of MG with guanine and cysteine in vitro. The steady‐state kinetics of DJ‐1 acting on reversible hemithioacetal substrates are fitted adequately with a computational kinetic model that requires only a DJ‐1 glyoxalase activity, supporting the conclusion that deglycation is an apparent rather than a true activity of DJ‐1. Sensitive and quantitative isotope‐dilution mass spectrometry shows that DJ‐1 modestly reduces the levels of some irreversible guanine and lysine glycation products in primary and cultured neuronal cell lines and whole mouse brain, consistent with a small but measurable effect on total neuronal glycation burden. However, DJ‐1 does not improve cultured cell viability in exogenous MG. In total, our results suggest that DJ‐1 is not a deglycase and has only a minor role in protecting neurons against methylglyoxal toxicity.
Keywords: deglycase, enzyme mechanism, glycation stress, glyoxalase, PARK7, Parkinson's disease
The neuroprotective protein DJ‐1 has been described as a deglycase that repairs the adducts of methylglyoxal with proteins and nucleic acids. In this study, experiments and kinetic simulation show that DJ‐1's consensus glyoxalase activity explains its in vitro activities, with no need for a deglycase activity. Isotope dilution mass spectrometry of several types of neurons shows that the absence of DJ‐1 results in minor increases in glycation products but does not make cells more vulnerable to methylglyoxal. We conclude that DJ‐1 is a glyoxalase but not a deglycase and makes a minor contribution to neuronal defense against methylglyoxal.
Abbreviations
- AMU
atomic mass unit
- BDNF
brain‐derived neurotrophic factor
- CEdG
N 2 ‐(1‐carboxyethyl)‐2′‐deoxyguanosine
- CEG
N 2 ‐(1‐carboxyethyl)‐2′‐guanosine
- CEL
Nε‐(1‐carboxyethyl)‐L‐lysine
- cMG‐dG
1,N 2 ‐(1,2‐dihydroxy‐2‐methyl)ethano‐2′‐deoxyguanosine
- CoA
coenzyme A; dG, deoxyguanosine
- DNPH
2,4‐dinitrophenylhydrazine
- DTT
dithiothreitol
- GDNF
glial cell line‐derived neurotrophic factor
- GSH
glutathione
- HEPES
4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
- HRP
horseradish peroxidase
- iPSC
induced pluripotent stem cell
- LC–MS
liquid chromatography‐mass spectrometry
- MG
methylglyoxal
- MG‐CEdG
N 2 ‐(1‐carboxyethyl)‐7‐1‐hydroxy‐2‐oxopropyl‐deoxyguanosine
- NAC
N‐acetyl‐l‐cysteine
- PBS
phosphate‐buffered cysteine
- shRNA
short hairpin RNA
1. INTRODUCTION
DJ‐1 is a 20 kDa homodimeric protein that is conserved from bacteria to humans (Bandyopadhyay & Cookson, 2004; Lucas & Marin, 2007; Wilson et al., 2005). In humans, mutations in DJ‐1 (PARK7) cause rare forms of autosomal recessive parkinsonism (Bonifati et al., 2003). Eukaryotic DJ‐1 promotes cell survival, particularly during oxidative stress or mitochondrial dysfunction, and is found in multiple cellular compartments (Cali et al., 2015; Canet‐Aviles et al., 2004; Junn et al., 2009; Shadrach et al., 2013; Taira et al., 2004). Consistent with its cytoprotective role, DJ‐1 is highly expressed in several types of cancers (Cao et al., 2017; Hod, 2004; Le Naour et al., 2001; MacKeigan et al., 2003; Nagakubo et al., 1997) and it plays important roles in maintaining normal function in lung (Bahmed et al., 2016; Bahmed et al., 2019), eye (Martin‐Nieto et al., 2019; Shadrach et al., 2013), and kidney (Cuevas et al., 2012; Eltoweissy et al., 2011). A conserved cysteine (Cys106) in DJ‐1 is both essential for its cytoprotective activity during oxidative stress (Bahmed et al., 2019; Canet‐Aviles et al., 2004; Shadrach et al., 2013; Taira et al., 2004; Waak et al., 2009; Xu et al., 2018) and is oxidation‐prone, forming cysteine sulfinate (‐SO2 −) and sulfonate (‐SO3 −) species (Canet‐Aviles et al., 2004; Kinumi et al., 2004; Mita et al., 2018). The formation of Cys106‐SO2 − has been proposed as one of several mechanisms that allow DJ‐1 to act as a sensor of cellular redox state and to activate cytoprotective responses through the PTEN/Akt (Oswald et al., 2018) and ASK1 (Cao et al., 2014; Waak et al., 2009) signaling pathways as well as by altering pathological protein aggregates (Atieh et al., 2021; Kumar et al., 2019; Zhou et al., 2006). Other activities proposed for DJ‐1 include protease (Mitsugi et al., 2013; Olzmann et al., 2004), esterase (Vazquez‐Mayorga et al., 2016), chaperone (Lee et al., 2003; Shendelman et al., 2004), transnitrosylase (Choi, Nakamura, et al., 2014), and an RNA binding protein (Hod et al., 1999; van der Brug et al., 2008). Very recently, DJ‐1 was shown to prevent protein damage by derivatives of 1,3, bisphosphoglycerate (Heremans et al., 2022). Despite years of intensive study motivated by its biomedical importance, the molecular activities of DJ‐1 that are responsible for its role in cell survival remain incompletely understood.
It has long been suspected that DJ‐1 may possess an enzymatic activity. Cys106 has a low pKa value of 5.4 and makes a hydrogen bond with a conserved protonated glutamic acid residue (Glu18) (Witt et al., 2008), which are features suggestive of an enzyme active site. These residues are conserved in the large DJ‐1/PfpI superfamily (Wei et al., 2007), which contains well‐established enzymes such as archaeal PfpI proteases (Du et al., 2000; Halio et al., 1996), isocyanide hydratases (Goda et al., 2001; Goda et al., 2002; Lakshminarasimhan et al., 2010), and the Hsp31 family of glutathione‐independent glyoxalases (Subedi et al., 2011). In all of these enzymes, the conserved cysteine residue is a catalytic nucleophile and the protonated glutamic/aspartic acid is a probable general acid/general base. In contrast to these validated enzymes, most proposed enzymatic functions of human DJ‐1 have been controversial and confounded by weak apparent activities and variable assay conditions between studies (Andres‐Mateos et al., 2007; Chen et al., 2010; Mitsugi et al., 2013; Olzmann et al., 2004).
Considerable interest has been generated by reports that human DJ‐1 is a glutathione‐independent glyoxalase that converts glyoxal to glycolate and methylglyoxal (MG) to L‐lactate (Hasim et al., 2014; Kwon et al., 2013; Lee et al., 2012; Zhao et al., 2014). In addition, DJ‐1 has been reported to be a deglycase that repairs early glycation adducts that MG forms with DNA, RNA, small molecule thiols, and proteins (Matsuda et al., 2017; Richarme et al., 2015; Richarme et al., 2017; Richarme et al., 2018; Richarme & Dairou, 2017; Sharma et al., 2019; Zheng et al., 2019). Both of these activities could protect cells by detoxifying reactive α‐ketoaldehydes produced by metabolism, but in distinct ways. A glyoxalase removes MG directly, while a deglycase acts on glycated substrates and repairs primary damage, indirectly removing MG by repairing its initial adducts. The glyoxalase activity of DJ‐1 has been widely reproduced but its reported kcat is ~104–105 lower than the primary glutathione‐dependent glyoxalase Glo1 (Marmstal et al., 1979; Thornalley, 1990). By contrast, DJ‐1's deglycase activity has been controversial, and a recent study suggests that deglycation may be a secondary effect of DJ‐1 acting on MG that is in rapid equilibrium with reversible MG adducts (Andreeva et al., 2019). Some reports indicate that DJ‐1‐mediated deglycation is important for repairing certain proteins (Galligan et al., 2018; Richarme et al., 2015; Richarme et al., 2017; Sharma et al., 2019; Zheng et al., 2019), while others have indicated that DJ‐1 has a negligible effect on total cellular glycation burden (Andreeva et al., 2019; Pfaff et al., 2017) and does not deglycate disease‐relevant proteins such as α‐synuclein (Atieh et al., 2021). In addition, several reports indicate that DJ‐1 has no effect on cell viability under glycation stress in systems ranging from cultured human cells to yeast (Andreeva et al., 2019; Natkanska et al., 2017; Pfaff et al., 2017; Zhao et al., 2014). Therefore, both the true substrate and physiological relevance of DJ‐1's anti‐glycation action are debated (Jun & Kool, 2020).
Given the importance of DJ‐1 in the etiology of several diseases, it is important to determine if a glyoxalase/deglycase activity is a major contributor to DJ‐1 cytoprotection. If true, such an activity would have wide‐reaching ramifications for the molecular etiology of several diseases. For example, a primary glyoxalase/deglycase activity for DJ‐1 implies that reactive dicarbonyl species play a key role in the etiology of parkinsonism, as somatic DJ‐1 deficiency invariably results in Parkinson's disease in humans (Bonifati et al., 2003). A dominant glyoxalase/deglycase model for DJ‐1 cytoprotection also implies that cells possessing enhanced ability to detoxify dicarbonyls are prone to neoplastic transformation, as DJ‐1 is an oncogene and its levels are elevated in multiple cancers (Cao et al., 2017). Because even low levels of glycation can cause cellular dysfunction (Moraru et al., 2018), quantitative methods for measuring macromolecular glycation such as isotope dilution mass spectrometry are needed to accurately evaluate the potential influence of DJ‐1 on cellular glycation burden.
In this work, we use in vitro assays to show that DJ‐1 cannot deglycate irreversible adducts of MG and guanine but can deglycate reversible adducts by an indirect mechanism. Steady‐state enzyme kinetics using purified MG and reversible hemithioacetal substrates support a prior proposal that DJ‐1's apparent deglycase activity is because of its glyoxalase activity acting on free MG. Our kinetic constants are in reasonable agreement with those recently reported by Andreeva et al. (Andreeva et al., 2019) and confirm that DJ‐1's in vitro glyoxalase activity is many orders of magnitude lower than that of glyoxalase I (Glo1). Kinetic modeling demonstrates that the glyoxalase activity of DJ‐1 is sufficient to explain the apparent deglycation kinetics. We use quantitative isotope‐dilution mass spectrometry to show that DJ‐1 measurably reduces the total DNA, RNA, and protein glycation burden in various neuronal cell lines and mouse brain, although the effect size is small. Cell survival in MG stress is unaffected by DJ‐1 status, suggesting that DJ‐1 plays a minor role in glycation defense.
2. MATERIAL AND METHODS
2.1. Protein expression and purification
The gene for human DJ‐1 was cloned between the NdeI and XhoI sites of Escherichia coli expression vector pET15b and the C106S, E18D, and E18N mutants were generated by site‐directed mutagenesis and previously described (Witt et al., 2008). The constructs were transformed into E. coli BL21 (DE3) cells and protein expression and purification were performed as described previously (Lin et al., 2012). Briefly, the protein was purified by Ni2+ metal affinity chromatography, the hexahistitdine tag was removed from recombinant DJ‐1 by thrombin cleavage, and the protein was passed through Hi‐Q anion exchange column (Bio‐Rad Laboratories) to remove minor nucleic acid contamination. Thrombin was removed using benazamidine‐sepharose 4 resin (Cytiva catalog number 17512310). All proteins ran as a single band on overloaded Coomassie‐stained SDS‐PAGE and were concentrated using a 10‐kDa cutoff centrifugal concentrator (Millipore) to 20 mg/ml in storage buffer (25 mM HEPES pH 7.5, 100 mM KCl, 2–5 mM DTT). DJ‐1 concentration was determined by the absorbance at 280 nm using a calculated extinction coefficient at 280 nm of 4400 M−1 cm−1. The purified protein in storage buffer was snap‐frozen in liquid nitrogen and stored at −80°C.
DJ‐1 was oxidized at Cys106 as described previously (Lin et al., 2012). Briefly, 0.25 mM DJ‐1 in storage buffer was dialyzed against PBS (pH 7.4), followed by the addition of H2O2 (Invitrogen) to a final concentration of 1.75 mM (i.e., a 7:1 molar ratio of H2O2 to DJ‐1 monomer) and the mixture was incubated on ice for 45 min. Unreacted H2O2 was removed by Bio‐Gel P‐6 desalting resin. Prior work has shown that this procedure selectively oxidizes Cys106 in human DJ‐1 (Blackinton et al., 2009; Lin et al., 2012; Zhou et al., 2006). Electrospray mass spectrometry using an Agilent 1200 LC system (Agilent Technologies) with the electrospray ionization (ESI) source of a Bruker Solarix—70 hybrid Fourier transform mass spectrometer (Redox Biology Center [RBC] Mass Spectrometry Core Facility, the University of Nebraska‐Lincoln) confirmed that DJ‐1 was purified in the fully reduced form and that H2O2 oxidation increased the intact mass of DJ‐1 by 32 a.m.u, consistent with Cys106‐SO2 − formation. In addition, preparations of DJ‐1 were tested for proper folding by crystallization and X‐ray crystal structure analysis. All preparations of the protein crystallize readily and produce crystal structures that are essentially identical (Cα RMSD ~0.05 Å) to prior human DJ‐1 structures deposited in the Protein Data Bank (e.g., accession code 1P5F, 5SY6).
2.2. In vitro deglycase and glyoxalase assays
Commercial methylglyoxal (MG) is known to be contaminated with several species that may interfere with kinetic measurements. Therefore, MG was purified from pyruvaldehyde‐1‐dimethyl acetal as previously reported (Shuck et al., 2018) and stored frozen in small aliquots at −80°C until needed. 10 mM N‐acetyl‐L‐cysteine (NAC; Alfa Aesar catalog number A15409), glutathione (GSH; Acros Organics catalog number 120000250), or coenzyme A (CoA; Affymetrix catalog number 13787) were mixed with 10 mM of freshly thawed methyglyoxal and incubated at 25°C until the absorption at 288 nm was stable (~1 h), indicating that hemithioacetal formation was at equilibrium.
DJ‐1 was dialyzed against degased PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 pH 7.4) immediately before the kinetic measurements. This buffer was chosen because it was used by both Richarme et al. (Richarme et al., 2015) and Pfaff et al. (Pfaff et al., 2017), thereby facilitating direct comparison of our results to prior reports. We note that DJ‐1 is reported to be less active in PBS than in other buffers (Mulikova et al., 2021), which might be relevant for reconciling the low glyoxalase activity of DJ‐1 in vitro with its observed effects in vivo. Andreeva et al. recently reported new equilibrium constants for the formation of MG‐hemithioacetals, which are substantially lower than prior values (Andreeva et al., 2019). Therefore, not all of the added MG and thiol react to form the hemithioacetal species (Mulikova et al., 2021). Initial concentrations of the NAC‐MG and GSH‐MG hemithioacetal substrates were determined using the updated molar extinction coefficient of 300 and 250 M−1 cm−1, respectively (Andreeva et al., 2019). The CoA‐MG hemithioacetal molar extinction coefficient has not been reported, so it was presumed to be the same as NAC‐MG. Initial concentrations of the hemithioacetal substrates were determined using A288. The reaction was initiated by the addition of DJ‐1 (wild‐type, Cys106‐SO2 −, C106S, E18N, and E18D) to 10 μM final concentration. The A288 was measured for 6 min at 25°C using a Cary50 UV–visible spectrophotometer (Varian) and always showed a linear decrease over this timeframe. The slope of the best‐fit line to the measured decrease in A288 signal was used to calculate the initial rate (V0) of hemithioacetal consumption. Because the hemithioacetal spontaneously degrades in aqueous solutions, a buffer‐alone baseline was measured in order to correct for non‐catalytic loss of 288 nm signal. The difference between the buffer‐alone and buffer‐enzyme curve was taken to reflect DJ‐1 activity. All measurements were repeated at least three times and average values with associated standard deviations are reported.
DJ‐1 glyoxalase activity was measured using a coupled assay of L‐lactate oxidase and Amplex Red/horseradish peroxidase (HRP). In this assay, DJ‐1 glyoxalase activity generates L‐lactate (Choi, Kim, et al., 2014), which is oxidized by L‐lactate oxidase and molecular oxygen to generate pyruvate and H2O2. The liberated H2O2 and Amplex Red are co‐substrates for HRP, generating fluorescent resorufin. The rate of H2O2 generation was monitored using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen). Various concentrations of purified MG were added to air‐saturated Amplex Red working solution (including the Amplex Red reagent and HRP) and 0.4 units of L‐lactate oxidase (Sigma, catalog number L9795) in reaction buffer (50 mM sodium phosphate, pH 7.4, as provided in the kit) at 25°C. The reaction was initiated by adding human DJ‐1 to a final concentration 0.1 μM. H2O2 generation was measured for five min using a Cary Eclipse spectrofluorimeter (Varian) with an excitation wavelength at 540 nm and emission wavelength of 590 nm. The measured rates are linear in DJ‐1 concentration and do not change significantly when the concentration of the coupling enzymes is increased, confirming that the coupling enzymes were not rate‐limiting. All reaction rates were linear over this timeframe and the slope of the best‐fit line was converted to H2O2 concentration using a standard curve generated with known concentrations of H2O2. All rates were measured a minimum of three times per substrate concentration.
2.3. DJ‐1 glyoxalase/deglycase kinetic simulations and data fitting
Data were fit using KinTek global kinetic explorer software (version 10.1.8.beta). The KinTek software simulates reactions by automatically deriving the corresponding system of rate equations, solving them using numerical integration, and fitting kinetic data using nonlinear least squares (Johnson et al., 2009b). Initial conditions were set using the experimental values. Loss of MG‐NAC hemithioacetal was simulated using a molar extinction value at 288 nm of 300 M−1 cm−1, along with the mechanism (Scheme 1) and rate constants described in Table 2 using experimental conditions described above. The simulation recapitulated the experimental protocol, simulating initial hemoithioacetal formation by mixing 10 mM of MG and 10 mM NAC for 2 h, diluted as indicated in Figure 4, and mixed with DJ‐1 (10 μM) for 2 min prior to data collection at 288 nm. The equilibrium constant for Scheme 1 was fixed using a previously determined value of 500 M−1 (Andreeva et al., 2019). The rate constants in Scheme 1 were adjusted to globally fit all progress curve data, where best‐fit rate constants can be found in Table 2. Confidence intervals for parameters were obtained by the KinTek software (Johnson et al., 2009a) by finding rates constants that produce a 0.83 relative error or chi‐squared ratio (to the best‐fit found in Table 2. DJ‐1‐catalyzed L‐lactate formation was monitored through a coupled lactate oxidase and Amplex red assay as described above. Lactate oxidation and subsequent formation of H2O2 to produce fluorescence were made non‐rate limiting in the simulation by increasing the rate constant for formation of fluorescent product to an arbitrary value of 3.9 × 104 min−1. Simulation of the fluorescent product using a linear fluorescence coefficient was based on a H2O2 Amplex Red standard curve but then optimized to 3.63 × 107 M−1 in the fitting.
SCHEME 1.
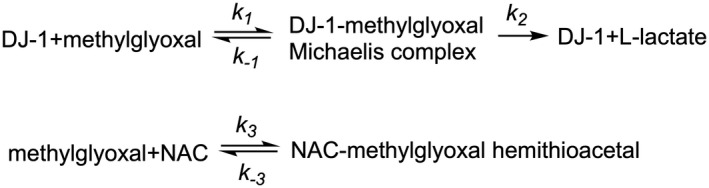
Coupled equilibria for DJ‐1 action on methylglyoxal and the methylglyoxal adduct with N‐acetyl cysteine (NAC).
TABLE 2.
DJ‐1 kinetic parameters with MG substrate from kinetic simulation
| Rate constant | Best‐fit value | a Lower boundary | a Upper boundary |
|---|---|---|---|
| k 1 (M−1 s−1) | 360 | 123.83 | 11466.67 |
| k −1 (s−1) | 7.82 × 10−2 | 2.62 × 10−12 | 4.13 |
| k 2 (k cat ) (s−1) | 4.02 × 10−2 | 3.82 × 10−2 | 4.27 × 10−2 |
| b Calculated K M (M) | 3.29 × 10−4 | c 2.76 × 10−4 | c 3.63 × 10−4 |
| k 3 (M−1 s−1) | 3.25 | 1.98 | 104.17 |
| k −3 (s−1) | 6.5 × 10−3 | d ND | d ND |
Lower and upper boundaries were determined by a published algorithm that varies the rate constants to find values that fall within a 0.83 relative error to the best‐fit (82).
The K M was calculated using the definition .
Lower and upper boundaries are not determined directly in KinTek global software for steady‐state constants therefore they were calculated from the collection of rate constants that fit the data within a 0.83 relative error from the best‐fit.
KinTek global explorer software only estimates one rate constant when the ratio is being constrained by the equilibrium constant (500 M−1).
FIGURE 4.
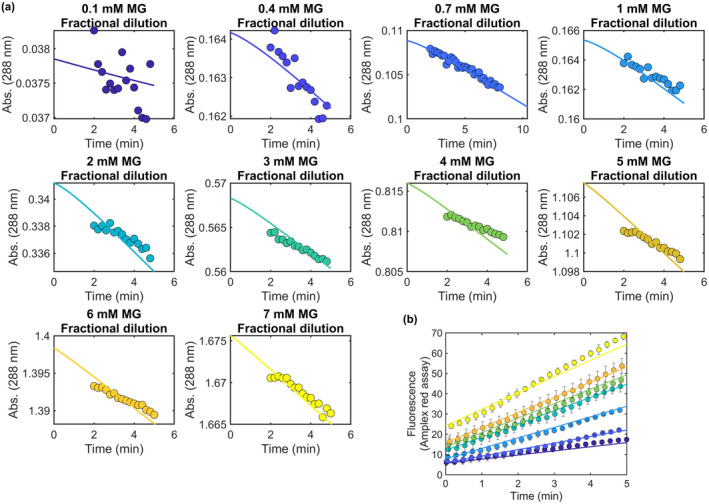
Kinetic simulation of DJ‐1 deglycase and glyoxalase activities. (a) Experimental progress curves for loss of MG‐NAC hemithioacetal signal at 288 nm (colored dots) are superimposed with kinetic modeling results (solid lines) for various concentrations of total MG concentration at initial dilution. The simulation requires only a DJ‐1 glyoxalase activity to produce good fits to the apparent deglycase activity. (b) Experimentally measured DJ‐1 glyoxalase activity against various concentrations of MG (colored dots) is well‐modeled by the kinetic simulation (solid lines) whose rate constants also explain the apparent deglycase activities in (a).
2.4. Culture of M17 neuroblastoma cell lines
Clonal M17 cell lines stably expressing different control and DJ‐1 shRNA sequences were prepared as described previously (van der Brug et al., 2008). The efficiency of shRNA knockdown was determined using western blotting (Figure S1). Proteins from stable M17 cell lines were extracted with lysis buffer (1x cell lysis buffer (Cell Signaling Technology, catalog number 9803) and then separated on 4–20% Criterion TGX gels (Bio‐Rad) and transferred to PVDF membranes using a Trans‐Blot Turbo system (Bio‐Rad). Antibodies to DJ‐1 (Abcam catalog number ab76008, 1:4000 dilution) and β‐actin (Sigma catalog number A1978, 1:40 000 dilution) were applied overnight at 4°C, followed by secondary antibodies for 1 h at room temperature (22°C). Blots were imaged on a Li‐Cor Odyssey CL‐x imager. This line is not known to be a commonly misidentified line and cells were used within 10 passages of stable shRNA modification. Cells were cultured to confluency in Opti‐MEM (ThermoFisher, catalog number 31985088) + 10% fetal bovine serum (Gemini Bio‐Products, catalog number 900–108) + 5 μg/ml Blasticidin (ThermoFisher, catalog number A1113903). For treatment experiments, control and DJ‐1 shRNA M17 cells (n = 4) were treated with either regular media or buthionine sulfoximine (43 mM) for 8 h. Following treatment, cells were processed for DNA, RNA, and protein isolation (see below). Treatment concentrations used were the IC50 determined by toxicity following 24 h treatment exposure using 0, 0.1, 1, 10, and 100 mM for buthionine sulfoximine (Zepeta‐Flores et al., 2018).
2.5. Murine primary neuronal cultures
Cortical neuron‐enriched primary cultures were prepared from wild‐type and DJ‐1 knockout mouse pups (n = 18–24) between postnatal days 1–2 and cultured in Basal medium eagle (+ 1x B‐27 minus vitamin A, 1x N2 supplement, 0.5 mM glutamine, 45% glucose, 1x Penicillin–Streptomycin). Pups were sacrificed using rapid decapitation. Pups were not sexed at the time of sacrifice. Cultures were made by pooling samples from six brains and then plating cells.
2.6. Monolayer forebrain neuronal differentiation from human iPSC
A commercial iPSC line (ThermoFisher, catalog number A18945) and the DJ‐1 knockout iPSC line HT‐188 (Mazza et al., 2021) were grown on a layer of MEFs (MTI‐GlobalStem, catalog number GSC‐6201G) in Essential 8TM media (ThermoFisher, catalog number A1517001) until 100% confluent. Confluent cells were transitioned to N3 media (50% DMEM/F12, 50% Neurobasal with 1x Penicillin–Streptomycin, 0.5x B‐27 minus vitamin A, 0.5x N2 supplement, 1x Glutamax, 1x NEAA, 0.055 mM 2‐mercaptoethanol and 1 μg/ml insulin) plus 1.5 μM Dorsomorphin (Tocris Bioscience) and 10 μM SB431542 (Stemgent) daily for 11 days. On days 12–15, cells were fed each day with N3 without Dorsomorphin and SB431542. On days 16–20, N3 was supplemented with 0.05 μM retinoic acid. On day 20, cells were split 1:2 with trypsin and seeded with ROCK inhibitor onto poly‐l‐ornithine (Sigma), fibronectin, and laminin‐coated plates. From days 21 to time of extraction, cells were fed with N4 media (same as N3 plus 0.05 μM retinoic acid, 2 ng/ml BDNF and 2 ng/ml GDNF). The maximum passage used for both iPSC lines was p11. Neither of the iPSC lines used is listed as commonly misidentified cell lines by the International Cell Line Authentication Committee. The characterization, karyotyping, sequencing, and confirmation of DJ‐1 ablation for the iPSC lines used in the current study is described in (Mazza et al., 2021).
2.7. DJ‐1 knockout mice
All animal work performed in this study was performed after approval of the National Institute on Aging (NIA) Animal Care and Use Committee, Animal Study Protocol number 463‐LNG‐2024 in compliance with the ARRIVE guidelines. This study was not pre‐registered. Mice were not treated so no randomization was performed to allocate subjects. Male and female C57Bl6/J (wild‐type [WT], n = 5 animals) and DJ‐1 knockout (RRID:MMRRC_032090‐MU from Dr. Huaibin Cai, n = 6 animals) mice were given access to food and water ad libitum and housed in a facility with 12‐h light/dark cycles. Mice were sacrificed by CO2 inhalation between 20 and 22 months of age by and brains were flash‐frozen and stored at −80°C. Brains were homogenized into a frozen powder using liquid nitrogen and separated for DNA, RNA, and protein extractions. No exclusion criteria were pre‐determined, and all mice were included in the analysis.
2.8. RNA extraction for glycation analysis
All cell lines were seeded at ~1 × 106 per 12 well with n = 4 wells used for RNA, DNA, or protein extraction. Cells were lysed using 0.4 ml TRIzol™ reagent (ThermoFisher, catalog number 15596026) and 0.08 ml chloroform. Following centrifugation for 15 min at 4°C at 12 000 g, the top aqueous layer was transferred to a new Eppendorf tube. RNA was precipitated with the addition of 0.2 ml isopropanol followed by centrifugation for 10 min. RNA pellet was washed with 0.4 ml 75% ethanol and centrifuged for 5 min at 7500 g. Supernatant was discarded and pellet was air‐dried then resuspended in 20 μl RNase‐free water. To complete solubilizing the RNA, incubation in water bath at 55°C occurred for 15 min. RNA from brain samples was processed using the same protocol. RNA concentration was quantified using spectrophotometer.
2.9. DNA extraction for glycation analysis
Cells were rinsed, scraped, and pelleted using sterile PBS. Cells were resuspended in equal volumes PBS and phenol:chloroform:isoamyl (25:24:1) and spun for 5 min at 16000xg. The top aqueous layer was transferred to a new Eppendorf tube. DNA was precipitated by adding 0.5× volume of 7.5 M ammonium acetate and 3x volume of 100% ethanol then stored at −20°C overnight. DNA was pelleted by centrifugation for 30 min at 4°C at 16 000 g. Supernatant was removed and pellet was washed with 70% ethanol. Following centrifugation, supernatant was removed, pellet was air‐dried and resuspended in 100 μl autoclaved water. DNA from brain samples was isolated using the DNeasy Blood and Tissue kit (Qiagen, catalog number 69504). DNA concentration was quantified using spectrophotometer.
2.10. Protein extraction for glycation analysis
Cells were scraped and lysed with lysis buffer (1x cell lysis buffer (Cell Signaling Technology, catalog number 9803), 1x protease inhibitor (Sigma, catalog number 4693159001), and 1x phosphatase inhibitor (ThermoFisher, catalog number 78427)). Lysed cells were set to rotate for 30 min at 4°C then spun at >20 000 g for 8 min at 4°C. Supernatant was transferred to a new Eppendorf tube and protein concentration was measured using the Pierce™ 660 nm protein assay. Protein from brain samples was extracted using a similar protocol.
2.11. HPLC analysis of MG reaction with dG in the presence of DJ‐1
MG was synthesized as previously described (Jaramillo et al., 2017) and reacted (5 mM) with dG (0.5 mM) in 50 mM sodium phosphate, pH 7.5 for 1 h at 37°C. DJ‐1 (5 μM) was added for an additional 24 h. Reactions were also performed in which DJ‐1 was added concomitantly with MG and dG. Following incubation, reactions were analyzed on an Agilent 1100 HPLC as previously described (Shuck et al., 2018). Reactions with dinitrophenylhydrazine (DNPH) were performed by making a 10 mM DNPH stock in 2.5 M HCl with dilution to 5 mM in H2O. DNPH (5 mM) was added to reactions for 15 min followed by the addition of 2.5 M NaOH prior to injection on HPLC. Absorbance from 210 to 510 nm was monitored.
2.12. Mass spectrometric analysis of CEdG, CEG, and CEL from M17, IPSC cells, and mouse tissue
All analytical measurements were not blinded to M17 cell line (Control vs. DJ‐1 shRNA), genotype of IPSC cells, or genotype of mice or primary neurons derived from these animals. Standards were synthesized as previously described (Shuck et al., 2018). CEL‐d4 standard was obtained from Santa Cruz Biotechnology (catalog number sc219424). CEL was obtained from Abcam (catalog number ab145095). DNA and RNA were isolated as described above, spiked with 5 ng/ml 15N5‐CEdG or 15N5‐CEG and digested as previously described (Jaramillo et al., 2017). Protein was isolated as described above and up to 50 μg of protein per sample was spiked with 5 ng/ml of CEL‐d4 and Lys‐d4. Samples were incubated in 6 M HCl for 18 h at 110°C and then dried under nitrogen with incubation at 80°C, resuspended in 50 μl 0.5% HFBA, and then analyzed using LC–MS/MS. CEdG and CEG were analyzed by injection onto an Agilent Zorbax SB‐C18 column (2.1 × 50 mm, 1.8 μm) with mobile phases A: H2O + 0.1% formic acid and B: acetonitrile +0.1% formic acid at 0.4 ml/min. The following gradient was used to separate analytes: 3%–10% B from 0–4.5 min, 10%–97% B from 4.5–4.8 min, 97% B from 4.8 to 5.2 min, and 97% to 3% B from 5.2 to 5.7 min. CEL was analyzed on this same column with an isocratic flow of 10% ACN + 0.1% formic acid at 0.2 ml/min. The following mass transitions were monitored: CEdG m/z 340 to 224, 15N5‐CEdG m/z 345 to 229, CEG m/z 356 to 224, and 15N5‐CEG m/z 361 to 229. CEL m/z 219 to 130 CEL‐d4 m/z 223–134, Lys m/z 147–84, and Lys‐d4 m/z 151 to 88. Analytes were quantified by fitting to a standard curve and the normalization to dG or guanosine (measured as described below), or lysine, as appropriate.
2.13. HPLC quantitation of dG and guanosine
Following mass spectrometric quantitation of CEdG and CEG, dG and guanosine were quantified on an Agilent 1100 HPLC using an Atlantis T3 column 4.6 × 150 mm, 5 μm. The mobile phases were A: H2O + 0.1% formic acid and B: ACN + 0.1% formic acid with a flow rate of 0.426 ml/min. The following gradient was used: 0–9% B, 0–20 min; 9–9.5% B, 20–55 min; 9.5%–90% B, 55–60 min and hold 60–70 min. 90%–0% B, 70–75 min; 0% B, 75–80 min. The area under the curve was manually integrated and then fit to a standard curve to determine dG or G concentration.
2.14. Statistical analysis
No power calculations were used to predetermine sample sizes, although post hoc power calculations for the relevant figures were performed as described in the Supplemental Material, Tables S1 and S2. Normality was tested using the Shapiro–Wilk test for small datasets and the D'Agostino and Pearson test with higher n datasets (e.g., n = 8). Datasets passed normality when these tests are applied in this way, although the larger n = 8 datasets did not pass normality when using Shapiro–Wilk instead of D'Agostino and Pearson tests. We note that the Shapiro–Wilk test performs less well with tied values, perhaps explaining this result. No outliers were detected. Comparisons between various groups were performed using two‐way ANOVA with Tukey's multiple comparisons test and an alpha value of 0.05 as implemented in Prism 9.3.0 (GraphPad software). ANOVA is robust to modest deviations from normality and was therefore appropriate for analysis of these data.
3. RESULTS
3.1. DJ‐1 deglycates only reversible guanine‐MG adducts
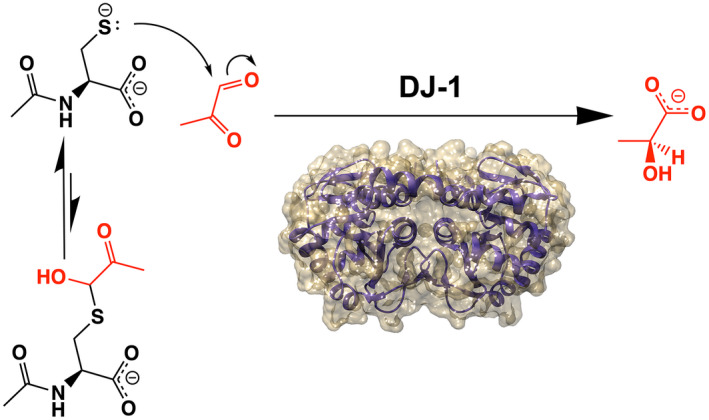
DJ‐1 and its homologs have been reported to deglycate a range of MG adducts including lysine residues in proteins (Richarme et al., 2015) and guanine bases in DNA and RNA (Richarme et al., 2017) (Figure 1a). While the role of DJ‐1 on glycated amino acids have been studied by several groups with conflicting results (Andreeva et al., 2019; Jun & Kool, 2020; Pfaff et al., 2017; Richarme et al., 2015; Richarme et al., 2018; Richarme & Dairou, 2017), DJ‐1's influence on nucleotide glycation has been less studied (Richarme et al., 2017). We investigated the role of DJ‐1 in reversing MG‐modified guanine nucleosides because this reaction has been proposed to play an essential role in maintaining genomic integrity in several organisms (Richarme et al., 2017). MG glycation of deoxyguanosine (dG) in DNA can form either a reversible 1,N 2 ‐(1,2‐dihydroxy‐2‐methyl)ethano‐2′‐dG (cMG‐dG) adduct or irreversible N 2 ‐(1‐carboxyethyl)‐2′‐dG (CEdG) adduct via Schiff base formation and hydrolysis of an initial cMG‐dG adduct (Figure 1b). In situations where high concentrations of MG are present, CEdG can be further glycated to the stable MG‐CEdG adduct (Figure 1b). Glycation of RNA forms the corresponding guanosine adducts. The ability of recombinant DJ‐1 to catalyze the deglycation of various guanine adducts was tested by mixing DJ‐1 with a pre‐equilibrated mixture of MG and dG and analyzing the resulting products by HPLC and mass spectrometry. We used MG purified by vacuum distillation (see Methods), as the purity of commercial MG is low (Shuck et al., 2018). The addition of DJ‐1 at the beginning of the reaction prevents formation of both reversible (cMG‐dG) and irreversible (CEdG, MG‐CEdG) adducts more effectively than when MG and dG are incubated together for 1 h before DJ‐1 addition (Figure 2a; Figure S2). This reduction in MG adducts requires the catalytic nucleophile C106, as the C106S DJ‐1 mutation abrogates this effect (Figure 2b; Figure S2). In contrast to DJ‐1's ability to prevent the formation of MG adducts when added early, incubation of purified R‐CEdG and DJ‐1 shows no change in R‐CEdG chromatographic retention time on a C18 reverse phase column (Shuck et al., 2018), indicating that this irreversible adduct is not a substrate for DJ‐1 (Figure 2c).
FIGURE 1.
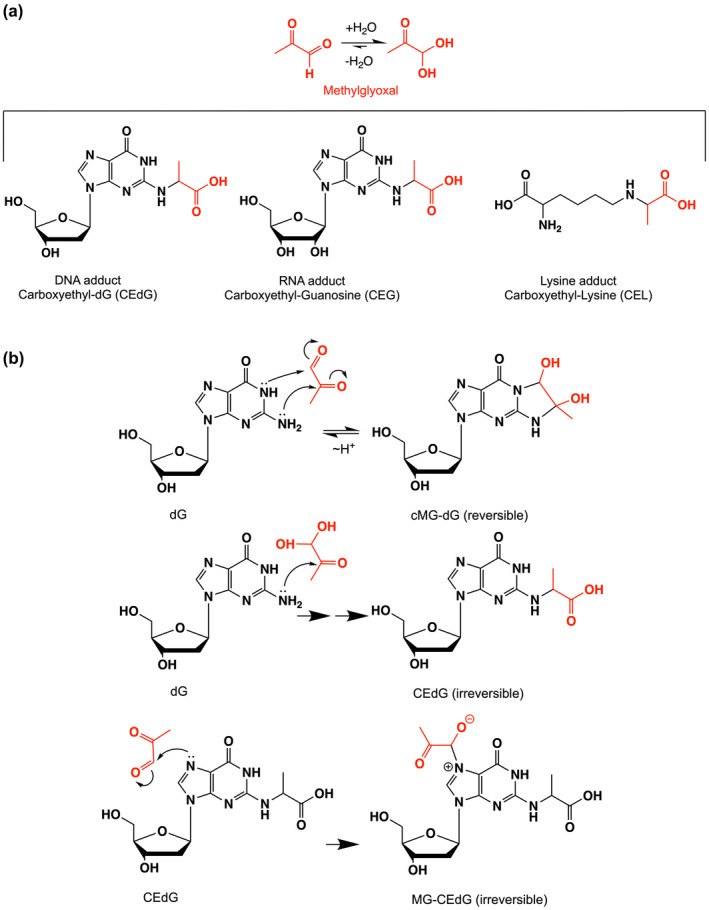
Adduct species generated by methylglyoxal (MG). (a) Shows the hydration equilibrium reaction of MG (red), which lies to the right in aqueous solution. MG can form stable adducts with both guanine and lysine. (b) Shows abbreviated mechanisms for MG modification of deoxyguanosine to form three adducts. Only cMG‐dG is reversible, as indicated.
FIGURE 2.
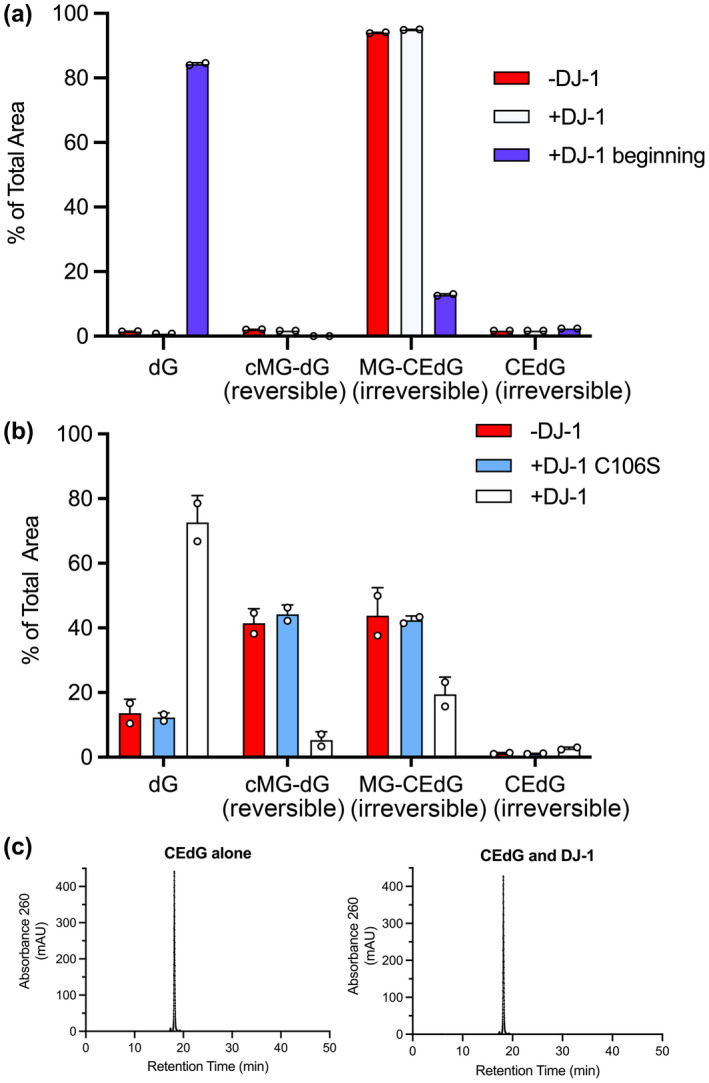
DJ‐1 can only repair reversible adducts of MG and guanine in vitro. (a) the addition of DJ‐1 at the same time as MG and dG (+DJ‐1 beginning; blue) reduces levels of both reversible cMG‐dG and irreversible MG‐CEdG adducts, while the addition of DJ‐1 after 1 h of preincubation of MG and dG (+DJ‐1; white) results in an accumulation of irreversible MG‐CEdG and cannot restore unmodified dG. (b) the C106S mutant (cyan), which removes the catalytic thiolate nucleophile, is inactive and cannot prevent dG modification. Only catalytically active DJ‐1 prevents the formation of dG adducts when added at the same time as MG. (c) the addition of DJ‐1 results in no change in the HPLC retention time of purified CEdG, an irreversible adduct. In all panels, each measurement is shown as a circle with a standard error of the mean shown in error bars.
By definition, reversible adducts of MG are in equilibrium with free MG. Our observations suggest that DJ‐1 may act on free MG directly rather than on the early adducts of MG and guanine. If true, this predicts that a similar reduction of only the reversible MG adducts should be observed when small molecule aldehyde scavengers are added. We confirmed this by adding the aldehyde‐reactive compounds DNPH and aminoguanidine, which behave similarly to DJ‐1 in reducing the levels of reversible MG adducts with dG (Figure S3) and form adducts with DNPH described in (Gilbert & Brandt, 1975). In aggregate, these findings are consistent with DJ‐1 acting on free MG as the primary substrate, which indirectly reduces the level of only the reversible MG‐nucleoside adducts.
3.2. DJ‐1 has weak glyoxalase and apparent deglycase activities in vitro
Measurement of the putative deglycase activity of DJ‐1 has produced variable and contradictory results, although the groups measuring this activity have used a similar in vitro protocols (Pfaff et al., 2017; Richarme et al., 2015; Richarme & Dairou, 2017; Witt et al., 2008). We measured the apparent deglycase activity of DJ‐1 using a well‐established assay that follows the decay of the 288 nm absorbance of the reversible hemithioacetal formed by incubation of MG with free thiols (Lo et al., 1994)(Figure 3a). MG‐hemithioacetals were formed by reaction of MG with NAC, GSH, or CoA (see Methods). NAC is a standard model thiol, GSH is the most abundant small molecule thiol in the cell, and CoA has been used previously as a physiologically relevant thiol whose glycation DJ‐1 can reduce (Matsuda et al., 2017). Tris‐containing buffers were not used to avoid previously reported buffer artifacts (Pfaff et al., 2017). The addition of DJ‐1 reduces the concentrations of each of these three hemithioacetals with initial steady‐state rates that are well‐fitted using the Michaelis–Menten kinetics model (Figure 3b,c). For the three hemithioacetal substrates, the apparent k cat values were ~0.007–0.009 s−1 and KM values were ~60–150 μM (Table 1). These activities are markedly lower than some previously reported values (Richarme et al., 2015). DJ‐1 apparent deglycase catalytic efficiency (k cat/K M) values are ~50–100 M−1 s−1, approximately 1 × 105 fold lower than that of glyoxalase 1 acting on GSH‐MG hemithioacetal as substrate (1.2 × 107 M−1 s−1 for E. coli GlxI) (Clugston et al., 1998).
FIGURE 3.
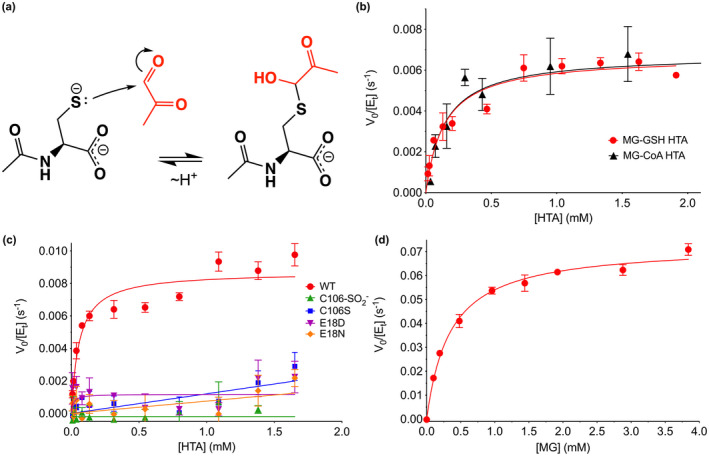
DJ‐1 has apparent hemithioacetal deglycase and glyoxalase activities in vitro. (a) Mechanism of formation of hemithioacetal formation by NAC (black) and MG (red). The hemithioacetal is reversible, as indicated by the arrows. (b) Steady‐state enzyme kinetics of DJ‐1 acting on the MG‐glutathione (MG‐GSH; red) and MG‐coenzyme a (MG‐CoA; black) hemitioacetal substrates. (c) Steady‐state kinetics of DJ‐1 acting on the MG‐N‐acetyl cysteine (MG‐NAC) hemithioacetal (HTA) substrate. Wild‐type (WT) enzyme is in red, and mutants and oxidative modification are shown in the inset legend with the indicated symbols and colors. Only WT DJ‐1 is active enough to be reliably fitted using the Michaelis–Menten model (solid lines). (d) Steady‐state kinetics of DJ‐1 glyoxalase activity against MG as substrate. In panels (b–d), initial velocity (V 0) is divided by total enzyme concentration ([Et]) on the Y‐axis, rates were measured a minimum of three times with standard deviation shown in bars, and the fitted Michaelis–Menten curves are shown as solid lines.
TABLE 1.
DJ‐1 kinetic parameters from experiment
| Substrate | k cat (s−1) | K M (M) | k cat/K M (M−1 s−1) |
|---|---|---|---|
| MG | 7.3(2) × 10−2 | 3.47(34) × 10−4 | 210 |
| MG‐NAC | 8.7(3) × 10−3 | 5.80(11) × 10−5 | 150 |
| MG‐GSH | 6.7(3) × 10−3 | 1.42(28) × 10−4 | 47 |
| MG‐CoA | 6.8(7) × 10−3 | 1.44(64) × 10−4 | 47 |
The reactive Cys106 residue is the presumed catalytic nucleophile in human DJ‐1. We confirmed that DJ‐1's ability to consume MG‐NAC hemithioacetal is Cys106‐dependent by showing that neither the C106S mutant nor the Cys106‐sulfinate (C106‐SO2 −) oxidized form of the protein has significant apparent deglycase activity (Figure 3c). The active site environment of Cys106 features a protonated Glu18 residue that makes a hydrogen bond to the Cys106 thiolate (Witt et al., 2008) and was identified as catalytically important in prior reports of DJ‐1 glyoxalase activity (Lee et al., 2012). Consistent with these prior reports, mutations at the nearby protonated Glu18 ablated DJ‐1‐mediated consumption of MG‐NAC hemithioacetal (Figure 3c). Our prior structural studies showed that E18D, E18N, and wild‐type DJ‐1 have nearly identical crystal structures and that Glu18 mutations only slightly perturb the steric environment of Cys106 (Blackinton et al., 2009; Witt et al., 2008), suggesting that these mutations do not eliminate DJ‐1's apparent deglycation activity via major changes in protein structure. Instead, it is likely that mutations at Glu18 remove an important general acid/base in the proposed DJ‐1 glyoxalase mechanism (Figure S4).
Because hemithioacetals are obligatorily reversible MG adducts that cannot be converted to irreversible glycated products via the Amadori rearrangement, they always exist in equilibrium with free MG. Therefore, DJ‐1's apparent deglycation activity may be because of its action on free MG, as we proposed above for guanine MG adducts and was also proposed by others for other reversible MG adducts (Andreeva et al., 2019). DJ‐1 has an established glyoxalase activity, although estimates for its kinetic parameters have varied widely (Andreeva et al., 2019; Choi, Kim, et al., 2014; Hasim et al., 2014; Lee et al., 2012; Pfaff et al., 2017; Richarme et al., 2015). Unlike some other glyoxalases in the DJ‐1 superfamily, several studies indicate that human DJ‐1 produces exclusively L‐lactate as a product when acting on MG (Choi, Kim, et al., 2014; Hasim et al., 2014), although one report indicates that DJ‐1 produces a minor fraction of D‐lactate when acting on MG‐NAC hemithioacetal (Richarme et al., 2015). The dominant production of L‐lactate allows us to measure DJ‐1 catalytic activity using an L‐lactate‐coupled assay. We measured the rate at which DJ‐1 converts MG to L‐lactate in real time using L‐lactate oxidase, horseradish peroxidase, and resorufin in a coupled assay that is similar to one used previously (Andreeva et al., 2019). Varying the concentration of the coupling enzymes L‐lactate oxidase and horseradish peroxidase has little effect on the initial rate measurements, confirming that DJ‐1's activity is rate‐limiting in this coupled assay. DJ‐1's glyoxalase activity is well‐fitted using the Michaelis–Menten model (Figure 3d) and produces kinetic constants (Table 1) which are in reasonable agreement with the k cat ~ 0.02 s−1 reported by Andreeva et al. (Andreeva et al., 2019). DJ‐1's glyoxalase k cat is ~10‐fold greater than its apparent deglycase k cat.
3.3. Kinetic modeling indicates that DJ‐1 glyoxalase activity is sufficient to explain its apparent deglycase activity
To determine if the low apparent deglycase activity for DJ‐1 could be explained by its higher glyoxalase activity, we used kinetic modeling. We simulated the DJ‐1‐catalyzed glyoxalase reaction using a simple Michaelis–Menten mechanism (reaction 1) coupled to hemiothioacetal production via the reaction of methylglyoxal with N‐acetylcysteine (Scheme 1).
The measured progress curve for DJ‐1‐mediated loss of NAC‐MG hemithioacetal can be adequately modeled using a kinetic scheme that includes only a glyoxalase activity for DJ‐1 and an equilibrium of free MG and MG‐NAC hemithioacetal, without need for a DJ‐1 deglycase activity (Figure 4a). The modeled kinetic parameters for DJ‐1 glyoxalase activity also agree well with the measured glyoxalase activity progress curves of the enzyme (Figure 4b) and produce glyoxalase kcat and Km values that agree well with the experimentally determined ones (Table 2). The ability to fit the data in Figure 4 without needing to invoke a postulated DJ‐1 deglycase activity supports the conclusion (Andreeva et al., 2019) that DJ‐1 is not a primary deglycase but reduces the concentrations of reversible MG adducts through its action on MG and the equilibria in Scheme 1.
3.4. DJ‐1 reduces total protein and nucleic acid glycation burden in cultured neuronal cells and mice
DJ‐1's contribution to reducing the overall glycation burden in cells is contentious, with some studies indicating major effects (Richarme et al., 2015; Richarme et al., 2017; Sharma et al., 2019) and others detecting no changes (Andreeva et al., 2019; Pfaff et al., 2017). In these studies, protein glycation has been measured primarily using western blotting. Western‐based detection of glycated macromolecules is complicated by the need for specific antibodies against the modification of interest and the difficulty of quantifying the blots. To address these limitations, we used quantitative isotope dilution mass spectrometry with internal 15N5‐CEdG,15N5‐CEG, CEL‐d4, and Lys‐d4 standards for quantifying glycated guanine in DNA, RNA, and glycated lysine in proteins. Using dopaminergic M17 neuroblastoma cells, we knocked down DJ‐1 using shRNA (see Methods, Figure S1) and observed a trend of increased CEdG, CEG, and CEL levels when DJ‐1 levels are decreased, although these effects are small (Figure 5a).
FIGURE 5.
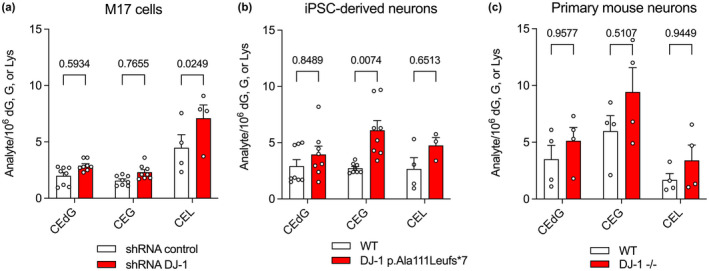
DJ‐1 has a modest impact on cellular concentrations of irreversible glycation products in neurons. In all panels, isotope‐dilution mass spectrometry was used to obtain relative concentrations of modified versus unmodified dG, G, or Lys. Replicate independent culture sample is shown as a circle with standard error of the mean shown in error bars (n = 4–9 independent cultures as indicated). Two‐way ANOVA with multiple comparisons was used for statistical analysis with Tukey's honest significant post hoc test for individual comparisons with p‐values indicated. Detailed two‐way ANOVA values are shown in Tables S3–S5. Small but consistent elevations in glycated products were observed in (a) shRNA knockdown of DJ‐1 in immortalized M17 neuroblastoma, (b) iPSC‐derived neurons derived from wild‐type (WT) cells and cell bearing a DJ‐1 missense mutation that eliminates protein (DJ‐1 p.Ala111Leufs*7), and (c) primary neurons from WT and DJ‐1−/− mice. Each replicate's independent culture sample is shown as a circle with standard error of the mean shown in error bars (n = 4–9 independent cultures as indicated).
Although M17 cells are biochemically similar to the vulnerable neurons in Parkinson's Disease, they are immortalized and thus have potential alterations in metabolism that could affect glycation. To address this, we also measured glycated products in two distinct neuronal lineages: induced pluripotent stem cell‐derived forebrain neurons created from fibroblasts donated by a patient bearing a A111L missense mutation that eliminates steady‐state DJ‐1 (Mazza et al., 2021) and primary neurons cultured from DJ‐1−/− mice. In both DJ‐1‐deficient neuronal cultures, glycation products are slightly elevated (Figure 5a–c), although this increase only reaches the p < 0.05 significance threshold for CEL in M17 cells and CEG in iPSC‐derived neurons (Figure 5a,b). A similar effect is seen in overall mouse brain tissue, where DJ‐1 knockout elevates all three classes of glycated products by 1–2 modified species/106 unmodified, which is statistically significant for CEG and CEL (Figure S5).
The glutathione‐dependent glyoxalase I/II (GloI/II) system is the primary means for detoxifying MG in the cell. Because the in vitro catalytic rate of GloI is several orders of magnitude greater than that of DJ‐1, it is possible that DJ‐1 would make a larger contribution to glycation defense when the GloI/II is impaired. We tested this using buthionine sulfoximine (BSO), an inhibitor of γ‐glutamyl cysteine ligase, the rate‐limiting enzyme in glutathione (GSH) biosynthesis (Griffith & Meister, 1979). As GSH is a co‐substrate for GloI, reduction in the GSH pool should reduce GloI activity in cells. BSO administration to M17 neuroblastoma cells markedly increases CEL levels when compared with vehicle alone, although the effect on CEG and CEdG levels is not significant. When BSO is applied to DJ‐1 knockdown cells, it leads to significant increases in all three glycated metabolites (CEdG, CEG, and CEL) and this effect is again most pronounced for CEL (Figure 6c). The enhancement of glycation when BSO is applied to DJ‐1 knockdown cells suggests that Glo1's stronger glyoxalase activity dominates the cellular MG defense compared to DJ‐1's weaker glyoxalase activity.
FIGURE 6.
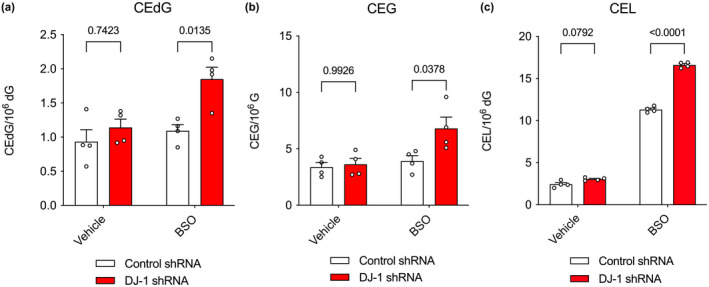
Chemical reduction in cellular glutathione enhances the glycation burden of DJ‐1 knockdown M17 cells. In all panels, isotope‐dilution mass spectrometry was used to obtain relative concentrations of modified versus unmodified dG, G, or Lys in cultured M17 neuroblastoma cells. Two‐way ANOVA with multiple comparisons was used for statistical analysis with Tukey's honest significant post hoc test for individual comparisons with p‐values indicated. Detailed two‐way ANOVA values are shown in Tables S6–S8. BSO was used to reduce cellular levels of GSH, which is a co‐substrate for the dominant glyoxalase Glo1. Administration of BSO enhances the effect of DJ‐1 deficiency on CEdG (a), CEG (b), and CEL (c) levels. Replicate independent culture sample is shown as a circle with standard error of the mean shown in error bars (n = 4 independent cultures per cell line and treatment).
3.5. DJ‐1 does not protect cells against MG toxicity
DJ‐1 decreases total cellular CEdG, CEG, and CEL levels in some instances, although the absolute magnitude of this decrement is relatively small (~1–2 glycated products/106 unmodified). To determine if this reduction in glycation corresponds to increased cellular protection against MG toxicity by DJ‐1, we measured the effect of DJ‐1 on the survival of both M17 neuroblastoma cells and iPSC‐derived neurons in 10–10 000 μM exogenously added MG. M17 cells show a clear loss of viability with an IC50 of 809.2 μM MG, but DJ‐1 siRNA knockdown has no effect on cell survival (Figure 7). Sensitization to MG, resulting in a decrease of IC50, would be expected if DJ‐1 made a significant contribution to cellular viability during MG challenge of these cells. Neurons derived from iPSCs show no loss of viability across a 10–1000 μM MG range, and the inactivating A111L DJ‐1 mutation does not sensitize them to MG (Figure 7). These negative results are consistent with prior reports that loss of DJ‐1 has no effect on MG sensitivity in Drosophila and HEK293 cells (Andreeva et al., 2019; Pfaff et al., 2017).
FIGURE 7.
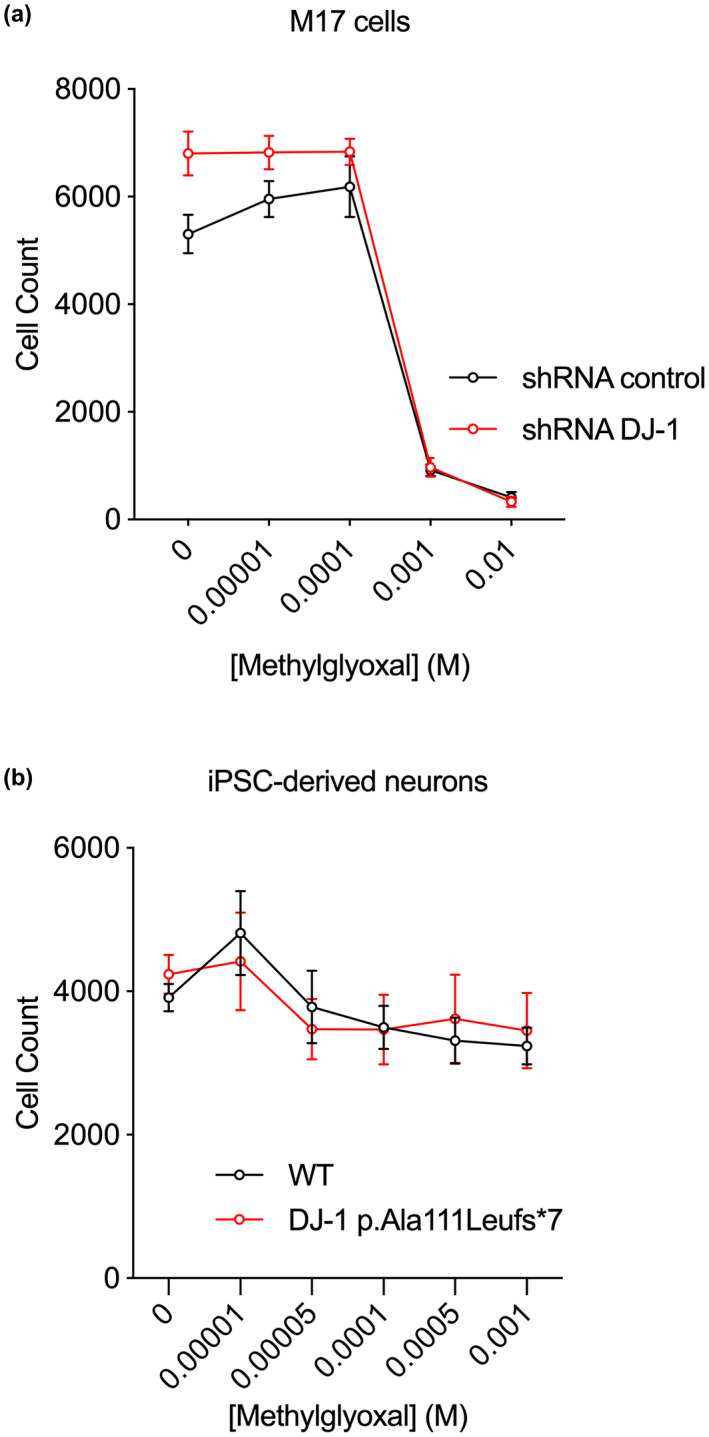
DJ‐1 deficiency does not enhance cell death in the presence of exogenous MG. Exogenous MG was added to cultured M17 cells (a) and iPSC‐derived neurons (b). No enhancement of cell death was observed when DJ‐1 was knocked down with shRNA (a) or absent at the protein level because of mutation (b) (n = 6 replicate cultures per cell line and treatment).
4. DISCUSSION
DJ‐1 defends multiple types of cells against oxidative stress and mitochondrial damage. One model of DJ‐1‐mediated cytoprotection is that it is a redox‐responsive protein that uses cysteine oxidation to sense changes in redox homeostasis and enhances cell survival through the PTEN/Akt (Kim et al., 2005), Nrf2 (Bahmed et al., 2016; Clements et al., 2006), and ASK1 (Cao et al., 2014; Waak et al., 2009) signaling pathways. However, DJ‐1 has more recently been reported to have Cys106‐dependent glyoxalase and deglycase activities, indicating that the protein may also play important roles in defense against reactive carbonyl species such as MG. DJ‐1's glyoxalase activity is widely corroborated, although it is significantly less efficient than Glo1 and there is considerable disagreement regarding DJ‐1's kinetic constants. The deglycase activity for DJ‐1 is more controversial, with some groups reporting that it is either an artifact (Pfaff et al., 2017) or an apparent activity of DJ‐1 acting on methylglyoxal (Andreeva et al., 2019), while others report that deglycation is the most important activity of DJ‐1 (Richarme et al., 2015; Richarme et al., 2017).
Our results help resolve this dispute, as we find that DJ‐1 is a weak glyoxalase with a k cat ~ 0.07 s−1 in PBS buffer. This is ~100x lower than the average enzyme kcat value (Bar‐Even et al., 2011), but is still 10x higher than the maximal rate of DJ‐1's apparent deglycase activity. There has been disagreement in the literature about kinetic constants for DJ‐1's glyoxalase activity (Jun & Kool, 2020), and we note that our results agree reasonably well with those of Andreeva et al. measured in a similar buffer (Andreeva et al., 2019). We observe that DJ‐1 can only reduce the levels of freely reversible MG adducts in vitro, and our kinetic modeling shows that DJ‐1's glyoxalase activity is sufficient to explain its apparent activity on MG adducts. Here again, these new quantitative kinetic modeling results support the conclusions of Andreeva et al. (Andreeva et al., 2019) that the apparent deglycase activity of DJ‐1 is likely because of its action on free MG regardless of the reversibly glycated species: aminocarbinol, hemithioacetal, cyclic dihydroimidazolone, or others. This conclusion contradicts several reports from the Richarme group (Richarme et al., 2015; Richarme et al., 2017; Richarme et al., 2018; Richarme & Dairou, 2017) but is chemically sensible, as there is no clear mechanism by which a cysteine nucleophile‐dependent enzyme could deglycate these diverse species, all of which lack a plausible electrophilic center for Cys106 thiolate attack. In addition, we find that both the E18N and E18D mutants eliminate DJ‐1's ability to revert reversibly glycated species, consistent with some prior reports (Lee et al., 2012; Matsuda et al., 2017) and demonstrating that Glu18 is catalytically essential for DJ‐1 glyoxalase activity. Considering these results in the context of the DJ‐1 crystal structure, we propose a mechanism where the protonated Glu18 is the general acid that protonates the oxyanion formed upon Cys106 attack at the electrophilic aldehyde carbon atom of MG (Figure S4).
Prior studies show that E18N and E18Q DJ‐1 can protect cells from mitochondrial damage induced by rotenone, preserve normal mitochondrial morphology (Blackinton et al., 2009), and are functional in other assays of DJ‐1 activity (Cao et al., 2014; Xu et al., 2018). These results indicate that aspects of DJ‐1 cytoprotection related to oxidative stress response and mitochondrial dysfunction are not explained by DJ‐1's glyoxalase activity. Furthermore, any cytoprotective enzymatic activity that requires Cys106 as the catalytic nucleophile will be abrogated by oxidation of Cys106 that occurs during oxidative stress, suggesting that DJ‐1's known enzymatic and redox‐sensing protective mechanisms are orthogonal and require different cellular pools of the protein.
The low catalytic efficiency of DJ‐1's glyoxalase activity (k cat/K M = 210 M−1 s−1) raises questions about its physiological relevance, as it is much lower than the ~107 M−1 s−1 k cat/K M of Glo1 (Clugston et al., 1998; Inoue & Kimura, 1996). Glo1, a highly conserved and widely expressed enzyme that converts the MG‐glutathione hemithioacetal to S‐lactoylglutathione, is the dominant mechanism of MG detoxification in most cells. It seems implausible that DJ‐1 could contribute significantly to the cellular defense against MG against a background of more catalytically proficient Glo1 unless Glo1 activity is diminished by glutathione depletion or other mechanisms. Despite these considerations, we find using sensitive and quantitative isotope dilution mass spectrometry that the absence of DJ‐1 results in small elevations in neuronal CEdG, CEG, and CEL, three surrogate biomarkers for MG reaction with DNA, RNA, and protein, respectively. These elevated levels are found in immortalized M17 neuroblastoma cells, iPSC‐derived neurons, primary mouse neurons, and whole mouse brain, establishing that this is observed across multiple neuronal types. Moreover, the effect is observed both in vivo and under standard cell culture conditions without external (and unphysiological) administration of bolus MG. Although the effect size is modest, it is nonetheless surprising given the low catalytic activity of DJ‐1. We offer two speculative hypotheses that may explain this result. First, DJ‐1 may have substantially greater glyoxalase activity in the cell than observed in vitro using recombinant protein, perhaps owing to the presence of yet‐unknown modifications or regulators in the cell that enhance its activity. This explanation may seem contrived, however this effect has been observed for the related Hsp31 glyoxalase in Saccharomyces cerevisiae, although the cause of endogenous Hsp31's enhanced activity remains unknown (Aslam & Hazbun, 2016). Further supporting the sensitivity of DJ‐1's glyoxalase activity to the details of the solution reaction environment, the in vitro DJ‐1 glyoxalase activity is markedly higher in phosphate buffer than in PBS (Mulikova et al., 2021). Second, it is possible that DJ‐1's glyoxalase activity is not directly responsible for its ability to reduce cellular glycation burden. In this model, DJ‐1 would enhance the activities of other pathways that are more effective at reducing steady‐state levels of MG. This model could be tested using mutations such as E18N that eliminate DJ‐1 glyoxalase activity but preserve its protective activity against oxidative and mitochondrial damage stressors and warrant further investigation.
DJ‐1 does not improve cellular viability against MG toxicity in our experiments, supporting some prior reports (Andreeva et al., 2019; Pfaff et al., 2017) while contradicting others (Lee et al., 2012; Richarme et al., 2015). It is possible that DJ‐1's modest ability to reduce glycation may be more important for the survival of certain types of cells, although the studies reporting negative results used human and mouse neurons (this work), HEK293 cells (Andreeva et al., 2019), Drosophila (Pfaff et al., 2017), ectopic expression of DJ‐1 in Glo1‐deficient S. cerevisiae (Natkanska et al., 2017), and S. pombe (Zhao et al., 2014). The yeast studies are informative, as they show that over‐expressed human or S. pombe DJ‐1 does not rescue the greatly increased sensitivity to exogenous MG that results from knockout of Glo1. By contrast, these same studies showed that Hsp31 proteins, which are more active glutathione‐independent glyoxalases from a different clade of the DJ‐1 superfamily, can rescue the viability of Glo1‐deficient yeasts (Natkanska et al., 2017; Zhao et al., 2014). Hsp31 has a k cat/K M for MG of ~103–104 M−1 s−1, which is 10–100‐fold higher than DJ‐1 but still 1000–10 000‐fold lower than Glo1. Therefore, the ability of Hsp31 to rescue MG sensitivity in ΔGLO1 yeast shows that these complementation experiments are sensitive enough to detect glyoxalase activities that are several orders of magnitude lower than Glo1. In light of our present results and prior independent negative reports, DJ‐1's glyoxalase activity does not appear to be physiologically relevant for overall cell viability when challenged with exogenous MG. Our results do not directly address a potential role for DJ‐1 in protecting specific proteins from glycation, which has been reported for histones (Galligan et al., 2018; Zheng et al., 2019). It is speculatively possible that DJ‐1's weak glyoxalase activity may be especially important for sensitive proteins or specific cellular compartments that are not addressed in this global study of cellular glycation.
Very recently, DJ‐1 and its close homologs were shown to strongly protect proteins and metabolites against glycerate and phosphoglycerate modifications (Heremans et al., 2022). In that study, DJ‐1 was proposed to use Cys106 to open a reactive cyclic 1,3 phosphoglycerate metabolite that may be spontaneously formed from 1,3 bisphosphoglycerate (i.e., DJ‐1 is a cyclic 1,3 phosphoglycerate phosphodiesterase). This enzymatic activity is difficult to directly assay owing to the instability of the presumptive substrate so we did not investigate it in this study. However, we note that such an activity may provide a functional explanation for the tendency of the Arg28/Arg48 motif near the DJ‐1 active site to bind tetrahedral anions (Witt et al., 2008). The presence of this anion‐binding di‐arginine motif is more consistent with DJ‐1 acting on a phosphate‐containing substrate than with MG being its primary substrate. As we discussed above for DJ‐1 glyoxalase activity, mutation of Glu18 could be used to interrogate the importance of a DJ‐1 cyclic 1,3 phosphoglycerate phosphodiesterase activity relative to other potential roles of oxidized isoforms of the protein, which we predict would be affected differently by E18N/D/Q mutations. Finally, it is intriguing (though possibly coincidental) that both glyoxalase and cyclic 1,3 phosphoglycerate phosphodiesterase activities act on triose phosphate‐derived metabolites resulting from glycolysis, which is restricted to the cytosol in eukaryotic cells.
AUTHOR CONTRIBUTIONS
J.T., M.R.C., and M.A.W. conceived the study, M.A.M, J.L., S.C.S., and M.C.M. designed and performed experiments. M.A.M, J.L., S.C.S., M.C.M., M.R.C., J.T., and M.A.W. analyzed the data and wrote the manuscript.
CONFLICT OF INTEREST
The other authors declare no conflicts of interest that would affect the objectivity of this study.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank Dr. Javier Seravalli (Redox Biology Center Mass Spectrometry Core Facility, University of Nebraska) for assistance with mass spectrometry of recombinant DJ‐1. This research was supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Institute on Aging. MAW acknowledges support from NIH R01GM139978 and JT acknowledges R01CA176611. Work performed in the Mass Spectrometry Shared Resource at City of Hope is supported by the National Cancer Institute of the National Institute of Health under award number P30 CA033572. Mark Cookson is a former Senior Editor of the Journal of Neurochemistry. Open access funding enabled and organized by ProjektDEAL.
Mazza, M. C. , Shuck, S. C. , Lin, J. , Moxley, M. A. , Termini, J. , Cookson, M. R. , & Wilson, M. A. (2022). DJ‐1 is not a deglycase and makes a modest contribution to cellular defense against methylglyoxal damage in neurons. Journal of Neurochemistry, 162, 245–261. 10.1111/jnc.15656
Melissa Conti Mazza and Sarah C. Shuck contributed equally to this work.
Contributor Information
John Termini, Email: jtermini@coh.org.
Mark R. Cookson, Email: cookson@mail.nih.gov.
Mark A. Wilson, Email: mwilson13@unl.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. A preprint of this article was posted on February 19th, 2022 on BioRxiv: https://www.biorxiv.org/content/10.1101/2022.02.18.481064v2
REFERENCES
- Andreeva, A. , Bekkhozhin, Z. , Omertassova, N. , Baizhumanov, T. , Yeltay, G. , Akhmetali, M. , Toibazar, D. , & Utepbergenov, D. (2019). The apparent deglycase activity of DJ‐1 results from the conversion of free methylglyoxal present in fast equilibrium with hemithioacetals and hemiaminals. The Journal of Biological Chemistry, 294, 18863–18872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres‐Mateos, E. , Perier, C. , Zhang, L. , Blanchard‐Fillion, B. , Greco, T. M. , Thomas, B. , Ko, H. S. , Sasaki, M. , Ischiropoulos, H. , Przedborski, S. , Dawson, T. M. , & Dawson, V. L. (2007). DJ‐1 gene deletion reveals that DJ‐1 is an atypical peroxiredoxin‐like peroxidase. Proceedings of the National Academy of Sciences of the United States of America, 104, 14807–14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam, K. , & Hazbun, T. R. (2016). Hsp31, a member of the DJ‐1 superfamily, is a multitasking stress responder with chaperone activity. Prion, 10, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atieh, T. B. , Roth, J. , Yang, X. , Hoop, C. L. , & Baum, J. (2021). DJ‐1 acts as a scavenger of alpha‐synuclein oligomers and restores monomeric glycated alpha‐synuclein. Biomolecules, 11, 1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahmed, K. , Boukhenouna, S. , Karim, L. , Andrews, T. , Lin, J. , Powers, R. , Wilson, M. A. , Lin, C. R. , Messier, E. , Reisdorph, N. , Powell, R. L. , Tang, H. Y. , Mason, R. J. , Criner, G. J. , & Kosmider, B. (2019). The effect of cysteine oxidation on DJ‐1 cytoprotective function in human alveolar type II cells. Cell Death & Disease, 10, 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahmed, K. , Messier, E. M. , Zhou, W. , Tuder, R. M. , Freed, C. R. , Chu, H. W. , Kelsen, S. G. , Bowler, R. P. , Mason, R. J. , & Kosmider, B. (2016). DJ‐1 modulates nuclear erythroid 2‐related Factor‐2‐mediated protection in human primary alveolar type II cells in smokers. American Journal of Respiratory Cell and Molecular Biology, 55, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay, S. , & Cookson, M. R. (2004). Evolutionary and functional relationships within the DJ1 superfamily. BMC Evolutionary Biology, 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Even, A. , Noor, E. , Savir, Y. , Liebermeister, W. , Davidi, D. , Tawfik, D. S. , & Milo, R. (2011). The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry, 50, 4402–4410. [DOI] [PubMed] [Google Scholar]
- Blackinton, J. , Lakshminarasimhan, M. , Thomas, K. J. , Ahmad, R. , Greggio, E. , Raza, A. S. , Cookson, M. R. , & Wilson, M. A. (2009). Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ‐1. The Journal of Biological Chemistry, 284, 6476–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati, V. , Rizzu, P. , van Baren, M. J. , Schaap, O. , Breedveld, G. J. , Krieger, E. , Dekker, M. C. , Squitieri, F. , Ibanez, P. , Joosse, M. , van Dongen, J. W. , Vanacore, N. , Swieten, J. C. , Brice, A. , Meco, G. , van Duijn, C. M. , Oostra, B. A. , & Heutink, P. (2003). Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset parkinsonism. Science, 299, 256–259. [DOI] [PubMed] [Google Scholar]
- Cali, T. , Ottolini, D. , Soriano, M. E. , & Brini, M. (2015). A new split‐GFP‐based probe reveals DJ‐1 translocation into the mitochondrial matrix to sustain ATP synthesis upon nutrient deprivation. Human Molecular Genetics, 24, 1045–1060. [DOI] [PubMed] [Google Scholar]
- Canet‐Aviles, R. M. , Wilson, M. A. , Miller, D. W. , Ahmad, R. , McLendon, C. , Bandyopadhyay, S. , Baptista, M. J. , Ringe, D. , Petsko, G. A. , & Cookson, M. R. (2004). The Parkinson's disease protein DJ‐1 is neuroprotective due to cysteine‐sulfinic acid‐driven mitochondrial localization. Proceedings of the National Academy of Sciences of the United States of America, 101, 9103–9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Chen, X. , Ying, M. , He, Q. , & Yang, B. (2017). DJ‐1 as a therapeutic target against cancer. Advances in Experimental Medicine and Biology, 1037, 203–222. [DOI] [PubMed] [Google Scholar]
- Cao, J. , Ying, M. , Xie, N. , Lin, G. , Dong, R. , Zhang, J. , Yan, H. , Yang, X. , He, Q. , & Yang, B. (2014). The oxidation states of DJ‐1 dictate the cell fate in response to oxidative stress triggered by 4‐hpr: Autophagy or apoptosis? Antioxidants & Redox Signaling, 21, 1443–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Li, L. , & Chin, L. S. (2010). Parkinson disease protein DJ‐1 converts from a zymogen to a protease by carboxyl‐terminal cleavage. Human Molecular Genetics, 19, 2395–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, D. , Kim, J. , Ha, S. , Kwon, K. , Kim, E. H. , Lee, H. Y. , Ryu, K. S. , & Park, C. (2014). Stereospecific mechanism of DJ‐1 glyoxalases inferred from their hemithioacetal‐containing crystal structures. The FEBS Journal, 281, 5447–5462. [DOI] [PubMed] [Google Scholar]
- Choi, M. S. , Nakamura, T. , Cho, S. J. , Han, X. , Holland, E. A. , Qu, J. , Petsko, G. A. , Yates, J. R. 3rd , Liddington, R. C. , & Lipton, S. A. (2014). Transnitrosylation from DJ‐1 to PTEN attenuates neuronal cell death in parkinson's disease models. The Journal of Neuroscience, 34, 15123–15131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements, C. M. , McNally, R. S. , Conti, B. J. , Mak, T. W. , & Ting, J. P. (2006). DJ‐1, a cancer‐ and Parkinson's disease‐associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proceedings of the National Academy of Sciences of the United States of America, 103, 15091–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clugston, S. L. , Barnard, J. F. , Kinach, R. , Miedema, D. , Ruman, R. , Daub, E. , & Honek, J. F. (1998). Overproduction and characterization of a dimeric non‐zinc glyoxalase I from Escherichia coli: Evidence for optimal activation by nickel ions. Biochemistry, 37, 8754–8763. [DOI] [PubMed] [Google Scholar]
- Cuevas, S. , Zhang, Y. , Yang, Y. , Escano, C. , Asico, L. , Jones, J. E. , Armando, I. , & Jose, P. A. (2012). Role of renal DJ‐1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension, 59, 446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X. , Choi, I. G. , Kim, R. , Wang, W. , Jancarik, J. , Yokota, H. , & Kim, S. H. (2000). Crystal structure of an intracellular protease from Pyrococcus horikoshii at 2‐a resolution. Proceedings of the National Academy of Sciences of the United States of America, 97, 14079–14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltoweissy, M. , Muller, G. A. , Bibi, A. , Nguye, P. V. , Dihazi, G. H. , Muller, C. A. , & Dihazi, H. (2011). Proteomics analysis identifies PARK7 as an important player for renal cell resistance and survival under oxidative stress. Molecular BioSystems, 7, 1277–1288. [DOI] [PubMed] [Google Scholar]
- Galligan, J. J. , Wepy, J. A. , Streeter, M. D. , Kingsley, P. J. , Mitchener, M. M. , Wauchope, O. R. , Beavers, W. N. , Rose, K. L. , Wang, T. , Spiegel, D. A. , & Marnett, L. J. (2018). Methylglyoxal‐derived posttranslational arginine modifications are abundant histone marks. Proceedings of the National Academy of Sciences of the United States of America, 115, 9228–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, R. P. , & Brandt, R. B. (1975). Spectrometric determination of methylglyoxal with 2,4‐dinitrophenylhydrazine. Analytical Chemistry, 47, 2418–2422. [DOI] [PubMed] [Google Scholar]
- Goda, M. , Hashimoto, Y. , Shimizu, S. , & Kobayashi, M. (2001). Discovery of a novel enzyme, isonitrile hydratase, involved in nitrogen‐carbon triple bond cleavage. The Journal of Biological Chemistry, 276, 23480–23485. [DOI] [PubMed] [Google Scholar]
- Goda, M. , Hashimoto, Y. , Takase, M. , Herai, S. , Iwahara, Y. , Higashibata, H. , & Kobayashi, M. (2002). Isonitrile hydratase from pseudomonas putida N19‐2. Cloning, sequencing, gene expression, and identification of its active acid residue. The Journal of Biological Chemistry, 277, 45860–45865. [DOI] [PubMed] [Google Scholar]
- Griffith, O. W. , & Meister, A. (1979). Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S‐n‐butyl homocysteine sulfoximine). The Journal of Biological Chemistry, 254, 7558–7560. [PubMed] [Google Scholar]
- Halio, S. B. , Blumentals, I. I. , Short, S. A. , Merrill, B. M. , & Kelly, R. M. (1996). Sequence, expression in Escherichia coli, and analysis of the gene encoding a novel intracellular protease (PfpI) from the hyperthermophilic archaeon Pyrococcus furiosus. Journal of Bacteriology, 178, 2605–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasim, S. , Hussin, N. A. , Alomar, F. , Bidasee, K. R. , Nickerson, K. W. , & Wilson, M. A. (2014). A glutathione‐independent glyoxalase of the DJ‐1 superfamily plays an important role in managing metabolically generated methylglyoxal in Candida albicans. The Journal of Biological Chemistry, 289, 1662–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heremans, I. P. , Caligiore, F. , Gerin, I. , Bury, M. , Lutz, M. , Graff, J. , Stroobant, V. , Vertommen, D. , Teleman, A. A. , Van Schaftingen, E. , & Bommer, G. T. (2022). Parkinson's disease protein PARK7 prevents metabolite and protein damage caused by a glycolytic metabolite. Proceedings of the National Academy of Sciences of The United States of America, 119, e2111338119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hod, Y. (2004). Differential control of apoptosis by DJ‐1 in prostate benign and cancer cells. Journal of Cellular Biochemistry, 92, 1221–1233. [DOI] [PubMed] [Google Scholar]
- Hod, Y. , Pentyala, S. N. , Whyard, T. C. , & El‐Maghrabi, M. R. (1999). Identification and characterization of a novel protein that regulates RNA‐protein interaction. Journal of Cellular Biochemistry, 72, 435–444. [PubMed] [Google Scholar]
- Inoue, Y. , & Kimura, A. (1996). Identification of the structural gene for glyoxalase I from Saccharomyces cerevisiae. The Journal of Biological Chemistry, 271, 25958–25965. [PubMed] [Google Scholar]
- Jaramillo, R. , Shuck, S. C. , Chan, Y. S. , Liu, X. , Bates, S. E. , Lim, P. P. , Tamae, D. , Lacoste, S. , O'Connor, T. R. , & Termini, J. (2017). DNA advanced glycation end products (DNA‐AGEs) are elevated in urine and tissue in an animal model of type 2 diabetes. Chemical Research in Toxicology, 30, 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, K. A. , Simpson, Z. B. , & Blom, T. (2009a). FitSpace explorer: An algorithm to evaluate multidimensional parameter space in fitting kinetic data. Analytical Biochemistry, 387, 30–41. [DOI] [PubMed] [Google Scholar]
- Johnson, K. A. , Simpson, Z. B. , & Blom, T. (2009b). Global kinetic explorer: A new computer program for dynamic simulation and fitting of kinetic data. Analytical Biochemistry, 387, 20–29. [DOI] [PubMed] [Google Scholar]
- Jun, Y. W. , & Kool, E. T. (2020). Small substrate or large? Debate over the mechanism of glycation adduct repair by DJ‐1. Cell Chemical Biology, 27, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn, E. , Jang, W. H. , Zhao, X. , Jeong, B. S. , & Mouradian, M. M. (2009). Mitochondrial localization of DJ‐1 leads to enhanced neuroprotection. Journal of Neuroscience Research, 87, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, R. H. , Peters, M. , Jang, Y. , Shi, W. , Pintilie, M. , Fletcher, G. C. , DeLuca, C. , Liepa, J. , Zhou, L. , Snow, B. , Binari, R. C. , Manoukian, A. A. , Bray, M. R. , Liu, F. F. , Tsao, M. S. , & Mak, T. W. (2005). DJ‐1, a novel regulator of the tumor suppressor PTEN. Cancer Cell, 7, 263–273. [DOI] [PubMed] [Google Scholar]
- Kinumi, T. , Kimata, J. , Taira, T. , Ariga, H. , & Niki, E. (2004). Cysteine‐106 of DJ‐1 is the most sensitive cysteine residue to hydrogen peroxide‐mediated oxidation in vivo in human umbilical vein endothelial cells. Biochemical and Biophysical Research Communications, 317, 722–728. [DOI] [PubMed] [Google Scholar]
- Kumar, R. , Kumar, S. , Hanpude, P. , Singh, A. K. , Johari, T. , Majumder, S. , & Maiti, T. K. (2019). Partially oxidized DJ‐1 inhibits alpha‐synuclein nucleation and remodels mature alpha‐synuclein fibrils in vitro. Communications Biology, 2, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, K. , Choi, D. , Hyun, J. K. , Jung, H. S. , Baek, K. , & Park, C. (2013). Novel glyoxalases from Arabidopsis thaliana. The FEBS Journal, 280, 3328–3339. [DOI] [PubMed] [Google Scholar]
- Lakshminarasimhan, M. , Madzelan, P. , Nan, R. , Milkovic, N. M. , & Wilson, M. A. (2010). Evolution of new enzymatic function by structural modulation of cysteine reactivity in Pseudomonas fluorescens isocyanide hydratase. The Journal of Biological Chemistry, 285, 29651–29661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Naour, F. , Misek, D. E. , Krause, M. C. , Deneux, L. , Giordano, T. J. , Scholl, S. , & Hanash, S. M. (2001). Proteomics‐based identification of RS/DJ‐1 as a novel circulating tumor antigen in breast cancer. Clinical Cancer Research, 7, 3328–3335. [PubMed] [Google Scholar]
- Lee, J. Y. , Song, J. , Kwon, K. , Jang, S. , Kim, C. , Baek, K. , Kim, J. , & Park, C. (2012). Human DJ‐1 and its homologs are novel glyoxalases. Human Molecular Genetics, 21, 3215–3225. [DOI] [PubMed] [Google Scholar]
- Lee, S. J. , Kim, S. J. , Kim, I. K. , Ko, J. , Jeong, C. S. , Kim, G. H. , Park, C. , Kang, S. O. , Suh, P. G. , Lee, H. S. , & Cha, S. S. (2003). Crystal structures of human DJ‐1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. The Journal of Biological Chemistry, 278, 44552–44559. [DOI] [PubMed] [Google Scholar]
- Lin, J. , Prahlad, J. , & Wilson, M. A. (2012). Conservation of oxidative protein stabilization in an insect homologue of parkinsonism‐associated protein DJ‐1. Biochemistry, 51, 3799–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, T. W. , Westwood, M. E. , McLellan, A. C. , Selwood, T. , & Thornalley, P. J. (1994). Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha‐acetylarginine, N alpha‐acetylcysteine, and N alpha‐acetyllysine, and bovine serum albumin. The Journal of Biological Chemistry, 269, 32299–32305. [PubMed] [Google Scholar]
- Lucas, J. I. , & Marin, I. (2007). A new evolutionary paradigm for the Parkinson disease gene DJ‐1. Molecular Biology and Evolution, 24, 551–561. [DOI] [PubMed] [Google Scholar]
- MacKeigan, J. P. , Clements, C. M. , Lich, J. D. , Pope, R. M. , Hod, Y. , & Ting, J. P. (2003). Proteomic profiling drug‐induced apoptosis in non‐small cell lung carcinoma: Identification of RS/DJ‐1 and RhoGDIalpha. Cancer Research, 63, 6928–6934. [PubMed] [Google Scholar]
- Marmstal, E. , Aronsson, A. C. , & Mannervik, B. (1979). Comparison of glyoxalase I purified from yeast (Saccharomyces cerevisiae) with the enzyme from mammalian sources. The Biochemical Journal, 183, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Nieto, J. , Uribe, M. L. , Esteve‐Rudd, J. , Herrero, M. T. , & Campello, L. (2019). A role for DJ‐1 against oxidative stress in the mammalian retina. Neuroscience Letters, 708, 134361. [DOI] [PubMed] [Google Scholar]
- Matsuda, N. , Kimura, M. , Queliconi, B. B. , Kojima, W. , Mishima, M. , Takagi, K. , Koyano, F. , Yamano, K. , Mizushima, T. , Ito, Y. , & Tanaka, K. (2017). Parkinson's disease‐related DJ‐1 functions in thiol quality control against aldehyde attack in vitro. Scientific Reports, 7, 12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza, M. C. , Beilina, A. , Roosen, D. A. , Hauser, D. , & Cookson, M. R. (2021). Generation of iPSC line from a Parkinson patient with PARK7 mutation and CRISPR‐edited Gibco human episomal iPSC line to mimic PARK7 mutation. Stem Cell Research, 55, 102506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita, Y. , Kataoka, Y. , Saito, Y. , Kashi, T. , Hayashi, K. , Iwasaki, A. , Imanishi, T. , Miyasaka, T. , & Noguchi, N. (2018). Distribution of oxidized DJ‐1 in Parkinson's disease‐related sites in the brain and in the peripheral tissues: Effects of aging and a neurotoxin. Scientific Reports, 8, 12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsugi, H. , Niki, T. , Takahashi‐Niki, K. , Tanimura, K. , Yoshizawa‐Kumagaye, K. , Tsunemi, M. , Iguchi‐Ariga, S. M. , & Ariga, H. (2013). Identification of the recognition sequence and target proteins for DJ‐1 protease. FEBS Letters, 587, 2493–2499. [DOI] [PubMed] [Google Scholar]
- Moraru, A. , Wiederstein, J. , Pfaff, D. , Fleming, T. , Miller, A. K. , Nawroth, P. , & Teleman, A. A. (2018). Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metabolism, 27(926–934), e928. [DOI] [PubMed] [Google Scholar]
- Mulikova, T. , Bekkhozhin, Z. , Abdirassil, A. , & Utepbergenov, D. (2021). A continuous spectrophotometric assay for glutathione‐independent glyoxalases. Analytical Biochemistry, 630, 114317. [DOI] [PubMed] [Google Scholar]
- Nagakubo, D. , Taira, T. , Kitaura, H. , Ikeda, M. , Tamai, K. , Iguchi‐Ariga, S. M. , & Ariga, H. (1997). DJ‐1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochemical and Biophysical Research Communications, 231, 509–513. [DOI] [PubMed] [Google Scholar]
- Natkanska, U. , Skoneczna, A. , Sienko, M. , & Skoneczny, M. (2017). The budding yeast orthologue of Parkinson's disease‐associated DJ‐1 is a multi‐stress response protein protecting cells against toxic glycolytic products. Biochimica et Biophysica Acta ‐ Molecular Cell Research, 1864, 39–50. [DOI] [PubMed] [Google Scholar]
- Olzmann, J. A. , Brown, K. , Wilkinson, K. D. , Rees, H. D. , Huai, Q. , Ke, H. , Levey, A. I. , Li, L. , & Chin, L. S. (2004). Familial Parkinson's disease‐associated L166P mutation disrupts DJ‐1 protein folding and function. The Journal of Biological Chemistry, 279, 8506–8515. [DOI] [PubMed] [Google Scholar]
- Oswald, M. C. , Brooks, P. S. , Zwart, M. F. , Mukherjee, A. , West, R. J. , Giachello, C. N. , Morarach, K. , Baines, R. A. , Sweeney, S. T. , & Landgraf, M. (2018). Reactive oxygen species regulate activity‐dependent neuronal plasticity in drosophila. eLife, 7, e39393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff, D. H. , Fleming, T. , Nawroth, P. , & Teleman, A. A. (2017). Evidence against a role for the parkinsonism‐associated protein DJ‐1 in methylglyoxal detoxification. The Journal of Biological Chemistry, 292, 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richarme, G. , Abdallah, J. , Mathas, N. , Gautier, V. , & Dairou, J. (2018). Further characterization of the Maillard deglycase DJ‐1 and its prokaryotic homologs, deglycase 1/Hsp31, deglycase 2/YhbO, and deglycase 3/YajL. Biochemical and Biophysical Research Communications, 503, 703–709. [DOI] [PubMed] [Google Scholar]
- Richarme, G. , & Dairou, J. (2017). Parkinsonism‐associated protein DJ‐1 is a bona fide deglycase. Biochemical and Biophysical Research Communications, 483, 387–391. [DOI] [PubMed] [Google Scholar]
- Richarme, G. , Liu, C. , Mihoub, M. , Abdallah, J. , Leger, T. , Joly, N. , Liebart, J. C. , Jurkunas, U. V. , Nadal, M. , Bouloc, P. , Dairou, J. , & Lamouri, A. (2017). Guanine glycation repair by DJ‐1/Park7 and its bacterial homologs. Science, 357, 208–211. [DOI] [PubMed] [Google Scholar]
- Richarme, G. , Mihoub, M. , Dairou, J. , Bui, L. C. , Leger, T. , & Lamouri, A. (2015). Parkinsonism‐associated protein DJ‐1/Park7 is a major protein deglycase that repairs methylglyoxal‐ and glyoxal‐glycated cysteine, arginine, and lysine residues. The Journal of Biological Chemistry, 290, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadrach, K. G. , Rayborn, M. E. , Hollyfield, J. G. , & Bonilha, V. L. (2013). DJ‐1‐dependent regulation of oxidative stress in the retinal pigment epithelium (RPE). PLoS One, 8, e67983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, N. , Rao, S. P. , & Kalivendi, S. V. (2019). The deglycase activity of DJ‐1 mitigates alpha‐synuclein glycation and aggregation in dopaminergic cells: Role of oxidative stress mediated downregulation of DJ‐1 in Parkinson's disease. Free Radical Biology & Medicine, 135, 28–37. [DOI] [PubMed] [Google Scholar]
- Shendelman, S. , Jonason, A. , Martinat, C. , Leete, T. , & Abeliovich, A. (2004). DJ‐1 is a redox‐dependent molecular chaperone that inhibits alpha‐synuclein aggregate formation. PLoS Biology, 2, e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuck, S. C. , Wuenschell, G. E. , & Termini, J. S. (2018). Product studies and mechanistic analysis of the reaction of methylglyoxal with deoxyguanosine. Chemical Research in Toxicology, 31, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi, K. P. , Choi, D. , Kim, I. , Min, B. , & Park, C. (2011). Hsp31 of Escherichia coli K‐12 is glyoxalase III. Molecular Microbiology, 81, 926–936. [DOI] [PubMed] [Google Scholar]
- Taira, T. , Saito, Y. , Niki, T. , Iguchi‐Ariga, S. M. , Takahashi, K. , & Ariga, H. (2004). DJ‐1 has a role in antioxidative stress to prevent cell death. EMBO Reports, 5, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley, P. J. (1990). The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. The Biochemical Journal, 269, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Brug, M. P. , Blackinton, J. , Chandran, J. , Hao, L. Y. , Lal, A. , Mazan‐Mamczarz, K. , Martindale, J. , Xie, C. , Ahmad, R. , Thomas, K. J. , Beilina, A. , Gibbs, J. R. , Ding, J. , Myers, A. J. , Zhan, M. , Cai, H. , Bonini, N. M. , Gorospe, M. , & Cookson, M. R. (2008). RNA binding activity of the recessive parkinsonism protein DJ‐1 supports involvement in multiple cellular pathways. Proceedings of the National Academy of Sciences of the United States of America, 105, 10244–10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez‐Mayorga, E. , Diaz‐Sanchez, A. G. , Dagda, R. K. , Dominguez‐Solis, C. A. , Dagda, R. Y. , Coronado‐Ramirez, C. K. , & Martinez‐Martinez, A. (2016). Novel redox‐dependent esterase activity (EC 3.1.1.2) for DJ‐1: Implications for Parkinson's disease. International Journal of Molecular Sciences, 17, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waak, J. , Weber, S. S. , Gorner, K. , Schall, C. , Ichijo, H. , Stehle, T. , & Kahle, P. J. (2009). Oxidizable residues mediating protein stability and cytoprotective interaction of DJ‐1 with apoptosis signal‐regulating kinase 1. The Journal of Biological Chemistry, 284, 14245–14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Y. , Ringe, D. , Wilson, M. A. , & Ondrechen, M. J. (2007). Identification of functional subclasses in the DJ‐1 superfamily proteins. PLoS Computational Biology, 3, e10. [DOI] [PubMed] [Google Scholar]
- Wilson, M. A. , Ringe, D. , & Petsko, G. A. (2005). The atomic resolution crystal structure of the YajL (ThiJ) protein from Escherichia coli: A close prokaryotic homologue of the parkinsonism‐associated protein DJ‐1. Journal of Molecular Biology, 353, 678–691. [DOI] [PubMed] [Google Scholar]
- Witt, A. C. , Lakshminarasimhan, M. , Remington, B. C. , Hasim, S. , Pozharski, E. , & Wilson, M. A. (2008). Cysteine pKa depression by a protonated glutamic acid in human DJ‐1. Biochemistry, 47, 7430–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S. , Yang, X. , Qian, Y. , & Xiao, Q. (2018). Parkinson's disease‐related DJ‐1 modulates the expression of uncoupling protein 4 against oxidative stress. Journal of Neurochemistry, 145, 312–322. [DOI] [PubMed] [Google Scholar]
- Zepeta‐Flores, N. , Valverde, M. , Lopez‐Saavedra, A. , & Rojas, E. (2018). Glutathione depletion triggers Actin cytoskeleton changes via Actin‐binding proteins. Genetics and Molecular Biology, 41, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. , Su, Y. , Wang, Z. , Chen, C. , Wu, T. , & Huang, Y. (2014). Identification of glutathione (GSH)‐independent glyoxalase III from Schizosaccharomyces pombe. BMC Evolutionary Biology, 14, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Q. , Omans, N. D. , Leicher, R. , Osunsade, A. , Agustinus, A. S. , Finkin‐Groner, E. , D'Ambrosio, H. , Liu, B. , Chandarlapaty, S. , Liu, S. , & David, Y. (2019). Reversible histone glycation is associated with disease‐related changes in chromatin architecture. Nature Communications, 10, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, W. , Zhu, M. , Wilson, M. A. , Petsko, G. A. , & Fink, A. L. (2006). The oxidation state of DJ‐1 regulates its chaperone activity toward alpha‐synuclein. Journal of Molecular Biology, 356, 1036–1048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. A preprint of this article was posted on February 19th, 2022 on BioRxiv: https://www.biorxiv.org/content/10.1101/2022.02.18.481064v2