

Abstract
Proteases are fundamental for a plethora of biological processes, including signalling and tissue remodelling, and dysregulated proteolytic activity can result in pathogenesis. In this review, we focus on a subclass of membrane‐bound and soluble proteases that are defined as channel‐activating proteases (CAPs), since they induce Na+ ion transport through an autocrine mechanism when co‐expressed with the highly amiloride‐sensitive epithelial sodium channel (ENaC) in Xenopus oocytes. These experiments first identified CAP1 (channel‐activating protease 1, prostasin) followed by CAP2 (channel‐activating protease 2, TMPRSS4) and CAP3 (channel‐activating protease 3, matriptase) as in vitro mediators of ENaC current. Since then, more serine‐, cysteine‐ and metalloproteases were confirmed as in vitro CAPs that potentially cleave and regulate ENaC, and thus this nomenclature was not further followed, but is accepted as functional term or alias. The precise mechanism of ENaC modulation by proteases has not been fully elucidated. Studies in organ‐specific protease knockout models revealed evidence for their role in increasing ENaC activity, although the proteases responsible for ENaC activation are yet to be identified. We summarize recent findings in animal models of these CAPs with respect to their implication in ENaC activation. We discuss the consequences of dysregulated CAPs underlying epithelial phenotypes in pathophysiological conditions, and the role of selected protease inhibitors. We believe that these proteases may present interesting therapeutic targets for diseases with aberrant sodium homoeostasis.
Keywords: epithelial phenotype, epithelial sodium channel, homoeostasis, kidney disease
1. INTRODUCTION
Epithelia form barriers that are essential for life by lining surfaces of organs and body cavities to maintain homoeostasis. At the same time, they need to restrict free passage of water, ions, and larger solutes. In an intact epithelium, specialized cell junctions that confer strength and selective permeability determine the epithelial barrier function and its integrity, although multiple regulatory proteins as matrix components, adhesion molecules and/or proteases are implicated and their composition may change during development. Within the last years, the list of factors that change epithelial barrier function and its integrity is steadily increasing, and the role of proteases here just starts to emerge. Amongst those, serine proteases belong to the largest class of proteolytic enzymes, and, here, the catalytic triad of the amino acids histidine (H), aspartate (D) and serine (S) functions as a site for enzymatic activity. A manually curated proteolytic enzyme database called MEROPS offers detailed information on serine protease families and subfamilies in the human degradome. 1 These proteases are further classified either as membrane‐anchored (type‐I, type‐II or glycosylphosphatidyinositol‐anchored), secreted or as intracellular serine proteases (Figure 1). In the Xenopus oocyte co‐expression assay where cRNAs of all ENaC subunits were co‐injected with candidate proteases, an increased inward amiloride‐sensitive Na+ current was observed. Using this approach or whole‐cell patch‐clamp techniques on cellular systems, several channel activating proteases have been identified. 2 , 3 This includes the membrane‐bound channel activating proteases, the soluble serine proteases, the cysteine proteases and the metalloproteases (Figure 1).
FIGURE 1.
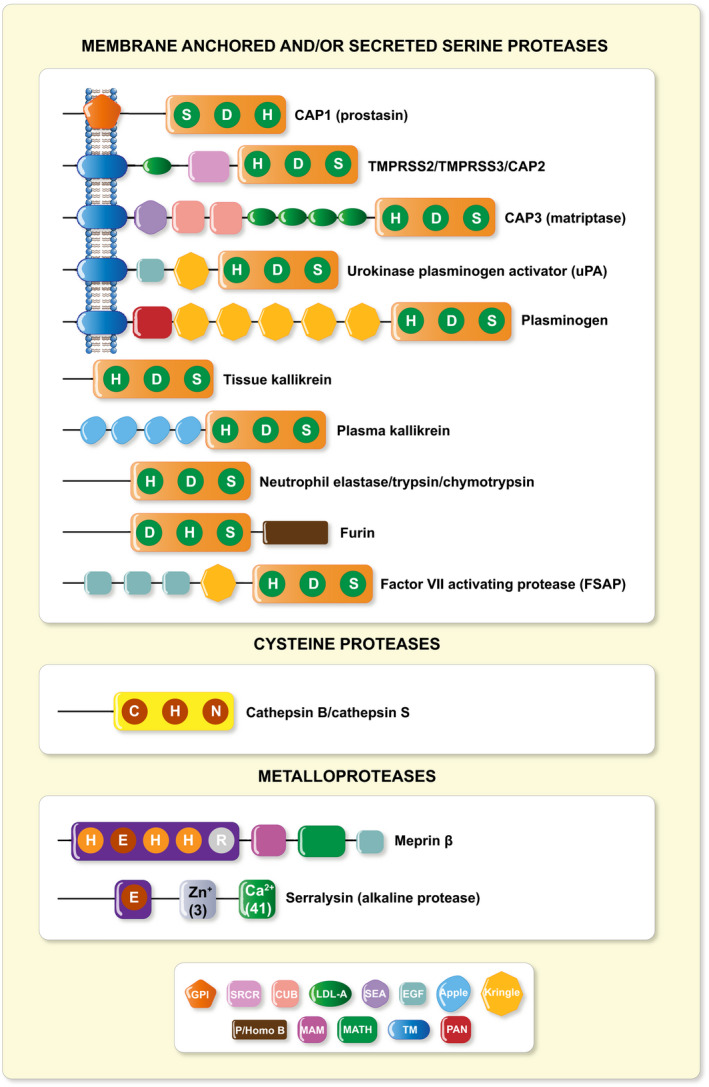
Structural schematic of identified ENaC CAPs. CAP1 (Prss8, prostasin), 4 , 5 CAP2 (Tmprss4), 6 CAP3 (St14/Matriptase), 6 Tmprss3, 7 Tmprss2, 8 , 9 uPA (urokinase‐type plasminogen activator), 10 , 11 plasminogen, 12 , 13 , 14 , 15 trypsin, 16 chymotrypsin, 14 tissue 17 , 18 and plasma kallikrein, 19 elastase, 20 furin, 21 factor VII activating protease, 22 cathepsin B, 23 , 24 cathepsin S, 14 meprin β 25 and serralysin. 26 Predicted structural domains of mouse proteases are indicated 27 : CUB, complement C1r/C2s, urchin embryonic growth factor, bone morphogenic protein 1; EGF, epidermal growth factor‐like; apple; kringle; GPI, glycophosphatidylinositol anchor; LDL‐A, low density lipoprotein A; MAM, meprin, A5 protein, receptor protein phosphatase μ; MATH, meprin and TRAF‐C homology; P/Homo B, paired basic amino acid residue‐cleaving enzyme/homo sapiens B; PAN, PAN/apple; SEA, sperm protein, enterokinase and agrin; SRCR, scavenger receptor cysteine‐rich; TM, transmembrane
Activation of ENaC is needed for Na+ reabsorption across epithelia such as kidney, lung and intestine, and thus, any dysregulation may result in disease, eg, cystic fibrosis, salt‐sensitive hypertension, liver cirrhosis 28 or nephrotic syndrome. 29 In this review, we explore several questions: how these proteases modulate ENaC function mechanistically, whether ENaC activation is dependent on its proteolytic cleavage, whether this proteolytic activity is essential in vivo and to which extent a loss‐ or gain‐of‐function of proteases in mice determines ENaC‐mediated Na+ losing or retaining phenotypes.
2. ENAC REGULATION BY PROTEASES
ENaC is expressed in the tight epithelia of various organs and plays an essential role in regulating fluid volume and sodium homoeostasis. It belongs to the ENaC/degenerin family of ion channels and is highly Na+‐selective and voltage‐insensitive. 2 ENaC subunits are encoded by different genes, SCNN1A (alpha/α), SCNN1B (beta/β), SCNN1G (gamma/γ), and, in human, there is a less expressed fourth subunit SCNN1D (delta/δ). The functional unit of ENaC consists of a trimeric assembly of α, β and γ subunits. 30 , 31 ENaC is expressed in the epithelia such as the distal colon, airways, sweat and salivary glands, skin, placenta and female reproductive tract (Figure 2A). 32 In the kidney, ENaC is expressed in the aldosterone‐sensitive distal part of the nephron (ASDN) that includes the distal convoluted tubule (DCT2), the connecting tubule (CNT) and the collecting duct (CCD) (Figure 2B). 33 Other extracellular factors, such as ions, mechanical forces and proteases may affect single‐channel open probability. The latter might occur by sequential cleavage steps carried out by at least two proteases targeting unique regions termed ‘inhibitory tract’ in the extracellular domains. Furin cuts twice on the α‐ and once on the γ‐ENaC subunit. This is followed by an additional proteolytic cleavage on the γ‐ENaC subunit. 33 Up to now, several potential candidate proteases were identified in mouse and tested in vitro, and their corresponding predicted consensus cleavage sites were determined (Figure 3). Many proteases have been identified to cleave human and rat ENaC in vitro, and given the high degree of sequence homology between human, rat and mouse, those proteases would be predicted to also cleave mouse ENaC (Figure 4). 3 Nevertheless, the responsible protease(s) for proteolytic activation of ENaC in vivo is yet to be identified. To summarize the current in vivo findings exploring ENaC activation by CAPs, different aspects are detailed below based on data obtained from genetically modified mouse models (Table 1).
FIGURE 2.

ENaC and CAP transcriptional expression in male C57BL/6 mouse organs and nephron segments shown as transcripts per million. A, Data according to the EMBL‐EBI expression atlas data. 34 , 35 B, RNA expression data across 14 mouse renal tubule segments from 6‐ to 8‐week‐old mouse microdissected tubules. 36 ATL, thin ascending limb of the loop of Henle; CCD, cortical collecting duct; CNT, connecting tubule; CTAL, cortical thick ascending limb of the loop of Henle; DCT, distal convoluted tubule; DTL1, short descending limb of the loop of Henle; DTL2, long descending limb of the loop of Henle in the outer medulla; DTL3, long descending limb of the loop of Henle in the inner medulla; IMCD, inner medullary collecting duct; MTAL, medullary thick ascending limb of the loop of Henle; OMCD, outer medullary collecting duct; PST1, initial segment of the proximal tubule; PST2, proximal straight tubule in cortical‐medullary rays; PST3, last segment of the proximal straight tubule in the outer stripe of outer medulla
FIGURE 3.
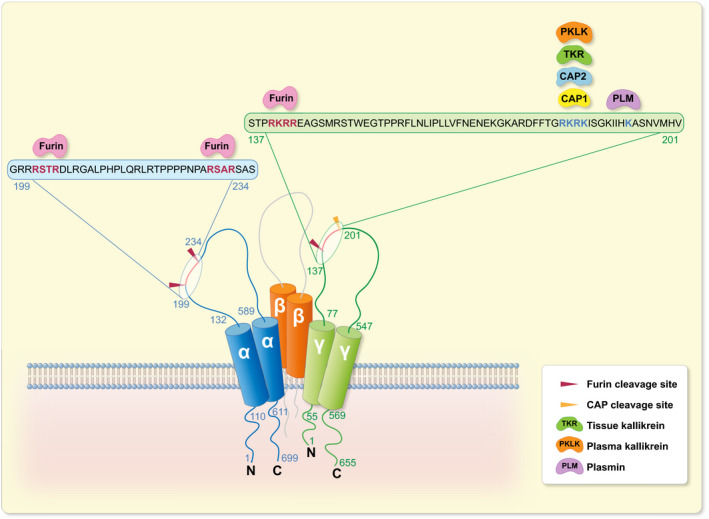
Identified consensus sites for proteolytic ENaC cleavage by CAPs in mouse. Illustration of the mouse α‐, β‐ and γ‐ENaC subunits depicting in vitro identified mouse CAP cleavage sites. α‐ENaC amino acid residues numbered in blue and γ‐ENaC in green. Furin cleaves at two sites in the α subunit and at one site in the γ subunit. 21 CAP1, 75 CAP2, 76 tissue kallikrein, 17 plasma kallikrein 19 and plasmin 15 cleave mouse γ‐ENaC distal to the furin cleavage site
FIGURE 4.
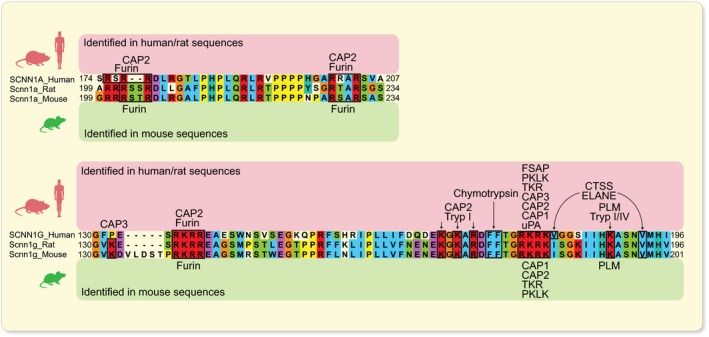
In vitro identified consensus sites for proteolytic ENaC cleavage by CAPs. Alignment of human, rat and mouse α‐ and γ‐ENaC subunits showing a high degree of conservation between species at cleavage sites. Amino acid numbering at the end of transcripts indicates the portion of sequence analysed. CAPs shown to cleave human/rat (indicated above) and mouse (indicated below) ENaC consensus sites in vitro are specified. 3 , 37 , 38 , 39 CTSS, cathepsin S; ELANE, neutrophil elastase; FSAP, factor VII‐activating protease; PKLK, plasma kallikrein; PLM, plasmin; TKR, tissue kallikrein; Tryp, trypsin; uPA, urokinase‐type plasminogen
TABLE 1.
ENaC channel‐activating proteases tested in animal models and their associated epithelial phenotypes
| Serine proteases | Rodent model & study condition | Phenotype (effect) | Identified in vivo substrate(s) | Ref. |
|---|---|---|---|---|
| CAP1/Prss8 (prostasin) | Constitutive KO, unchallenged | Placenta – syncytialization defect (impaired differentiation and signal transduction) | Not reported; CAP3/St14 | 40, 41 |
| Epidermal‐specific KO, unchallenged | Skin – orthokeratotic hyperkeratosis, hair follicle dysmaturation, tight junction leakiness (impaired barrier function/integrity) | Profilaggrin, occludin | 42 | |
| Colon‐specific KO, unchallenged, low Na+ diet | Colon – colonic pseudohypoaldosteronism type 1 (impaired ion transport) | ENaC | 43 | |
| Colon‐specific KO, DSS‐induced colitis | Colon – inflammation (altered signal transduction) | TLR4 | 44 | |
| Alveolar‐specific KO, unchallenged, acute volume overload | Lung – decreased alveolar fluid clearance hydrostatic oedema (impaired ion transport) | ENaC | 45 | |
| Liver‐specific KO, high fat diet | Liver – insulin resistance | TLR4 | 46 | |
| Spontaneous mutation frV170D, unchallenged | Reduced embryonic vitality; skin – dehydration, hyperkeratosis; colon – reduced ENaC activity (impaired ion transport) | ENaC (colon) | 47 | |
| Spontaneous mutation frCR (rats), unchallenged | Reduced embryonic vitality; skin – baldness, dehydration, hyperkeratosis; colon – reduced ENaC activity, diarrhea (impaired ion transport) | ENaC (colon) | 47 | |
| Spontaneous mutation frCR (rats), DSS‐induced colitis | Colon – epithelial remodelling; intestinal inflammation (impaired signal transduction and differentiation) | ENaC not confirmed | 48 | |
| Knockin Prss8R44Q (zymogen‐locked), unchallenged | Skin – impaired/delayed whisker and pelage hair formation (altered signal transduction) | CAP3/St14 suspected | 49 | |
| Knockin Prss8R44Q (zymogen‐locked), low Na+ (high K+) diet, triamterene |
Kidney – normal Na+ conservation; hypokalaemia; hyperaldosteronism Na+ wasting, weight loss (impaired ion transport) |
ENaC not confirmed, ENaC suspected |
50 | |
| Knockin Prss8S238A (catalytically inactive), unchallenged | Skin – delayed whisker and pelage hair formation (altered signal transduction) | Not reported | 51 | |
| Knockin Prss8S238A (catalytically inactive), low Na+ (high K+) diet, triamterene | Kidney – normal Na+ conservation; no obvious phenotype | ENaC not confirmed | 50 | |
| Adenovirus‐induced Prss8wt overexpression, unchallenged | Kidney – induced mineralocorticoid production (hypertension, impaired electrolyte homoeostasis) | Kallikrein | 52 | |
| Epidermal‐specific transgenic mice (Prss8wt), unchallenged | Skin – hyperkeratosis, dehydration, inflammation (altered signal transduction) | PAR2; nexin‐1 | 53, 54 | |
| Epidermal‐specific transgenic mice (Prss8 S238A), unchallenged | Skin – hyperkeratosis, dehydration, inflammation (altered signal transduction) | PAR2, nexin‐1 | 53 | |
| CAP2/ TMPRSS4 | Constitutive KO, low Na+ diet | No obvious phenotype; kidney | ENaC excluded | 55 |
| Constitutive KO, low K+ diet | Skin – ichthyosis; impaired water handling | HKA2, Nr3c1, AC6 | 56 | |
| CAP3/ St14 (matriptase) | Constitutive KO, unchallenged | Skin, thymus – postnatal lethality, ichthyosis, thymocyte apoptosis (impaired epithelial barrier function and thymus development) | Not reported | 57, 58 |
| Tamoxifen‐induced | Skin, intestine – loss of tight junction, ichthyosis, enlarged colon (impaired integrity of tight junctions) | Occludin, ZO‐1, claudin‐1 | 59 | |
| Adenoviral‐induced salivary gland KO, virus‐induced | Salivary glands – altered tight junction distribution (Sjögren's syndrome‐like disease) | Claudin‐3 | 60 | |
| Salivary‐gland KO, unchallenged | (Impaired gland function) | Not reported | 59 | |
| Intestinal‐specific KO, unchallenged | Colon – failed terminal differentiation, colitis, adenocarcinoma, (altered signal transduction, impaired epithelial integrity) | E‐cadherin, ZO‐1, occluding, β‐catenin and laminin suspected | 59 | |
| Hypomorphic mice, unchallenged | Skin – ichthyosis with hypotrichosis‐like syndrome (impaired epidermal barrier) | CAP1/Prss8, profilaggrin, claudin‐2 | 61 | |
| Epidermal‐specific transgenic mice, unchallenged, DMBA‐induced | Skin – carcinogenesis (malignant transformation, altered differentiation) | Ras | 62 | |
| uPA (uro‐kinase‐type plasminogen activator) | Anti‐uPA targeting anti‐body; induced podocin KO, tamoxifen‐induced, ± amiloride | Kidney – attenuation of sodium retention (impaired ion transport) | ENaC | 63 |
| Constitutive KO, amiloride, doxorubicin‐induced nephrotic syndrome | Kidney – phenotype not different from control | ENaC suspected | 64 | |
| Plasminogen | Constitutive KO; inducible podocin KO, doxycycline‐induced nephrotic syndrome | Kidney – phenotype not different from control | ENaC not confirmed | 65 |
| Tissue Kallikrein | KO, aldosterone infusion or low Na+ diet | Kidney, colon, lung – decreased ENaC activity in kidney and colon, but not in lung (partly impaired ion transport) | ENaC | 18 |
| Plasma kallikrein | KO, doxorubicin‐induced nephrotic syndrome | Kidney – phenotype not different from control | ENaC not confirmed | 19 |
| Tmprss3 | Constitutive KO, unchallenged | Ear – organ of Corti and hair cell degeneration, deafness (impaired ion transport) | ENaC not confirmed | 66, 67 |
| Tmprss2 | Constitutive KO, unchallenged | No obvious phenotype | Not reported | 68 |
| FSAP | Constitutive KO, doxorubicin‐induced nephrotic syndrome | Kidney – no obvious phenotype | ENaC not confirmed | 22 |
Unchallenged, no specific pretreatment.
Abbreviations: AC6, adenylate cyclase 6; FSAP, factor VII‐activating protease.; HKA2, H+, K+‐ATPase type 2; KO, knockout; Nr3c1, nuclear receptor subfamily 3 group C member 1; TLR4, toll‐like receptor 4; ZO‐1, zona occludens 1.
2.1. CAP1/Prss8 (prostasin)
CAP1/Prss8 belongs to the GPI (glycophosphatidylinositol)‐anchored class of serine proteases, 4 and it was the first identified CAP shown to activate ENaC in vitro. 4 , 5 In the mouse, the constitutive knockout of CAP1 caused embryonic lethality due to placental failure. 40 Epiblast‐specific CAP1 knockout mice survived until birth, but then died due to rapid and severe dehydration caused by ichthyosis indicating a severe impairment of the epidermal barrier function. 40 Surprisingly, zymogen‐locked CAP1/Prss8 knock‐in (KIR44Q) mice, in which the activation site was rendered cleavage‐resistant, developed only a minor epidermal phenotype comparable to mice carrying the CAP1/Prss8 mutant frizzy (fr/fr). 47 , 49 Mice carrying a mutation at the CAP1 catalytic site (S238A) displayed normal tissue development and homoeostasis. 51 Sodium homoeostasis was preserved in these mice during Na+‐deprivation, while zymogen‐locked CAP1 mice developed a compromised triamterene tolerance. Interestingly, in both models, proteolysis of α‐ and γ‐ENaC subunits as well as their subcellular localization were conserved in kidney (Table 1). 50
The adult epithelial phenotype caused by tissue‐specific gene deletion clearly confirmed an implication of CAP1 in ENaC‐mediated sodium transport in lung and colon 43 , 45 , 47 but not in skin. 42 , 47 , 53 , 54 Alveolar‐specific CAP1 knockout mice displayed a 40% reduction in the ENaC‐mediated Na+ current and impairment in alveolar fluid clearance. However, alveolar oedema, change in lung morphology, or tight junction protein abundance was not observed in unchallenged mice, but only under increased hydrostatic pressure. 45 Likewise, reduced ENaC activity was observed in colon‐specific CAP1 knockout mice. 43 , 47 Using an experimental rat colitis model, CAP1 was found to preserve colonic integrity, and to protect against dextran sodium sulphate (DSS)‐induced inflammation, and likely against tissue remodelling. 48 In skin, the tight junction protein occludin was completely missing in epidermis‐specific CAP1 knockout mice resulting in an impaired barrier function leading to fatal dehydration. 42 In this context, it is interesting to note that Gong and coworkers revealed an effect of CAP1 on paracellular chloride permeation that is regulated through tight junctions in renal epithelia and therefore may participate in blood pressure regulation. 69 Interestingly, adenovirus‐induced CAP1 overexpression was linked to increased aldosterone production and hypertension in rats 52 (Table 1). Overall, in vivo studies suggest a link between CAP1, epithelial Na+ transport and barrier function, although the direct interaction and the mechanism of proteolytic ENaC activation are still not completely determined.
2.2. CAP2/Tmprss4
In Xenopus oocytes, CAP2/Tmprss4 increased ENaC activity via its open probability. 70 It cleaves at the identified furin consensus cleavage site of the γ‐ENaC subunit which, when mutated, completely abolished Na+ current therefore strongly supporting proteolytic activation of ENaC by CAP2. 37 The epithelial phenotype in CAP2/Tmprss4 constitutive knockout mice, however, did not reveal any impairment of ENaC‐mediated Na+ transport even under Na+‐deprived diet. No difference was observed in the protein abundance of full‐length and cleaved γ‐ENaC subunit (Table 1). 55 However, we identified a dysregulation of renal water handling upon dietary potassium depletion with upregulation of adenylate cyclase 6, cAMP overproduction and increased protein expression of aquaporin 2 (AQP2) and the Na+‐K+‐2Cl− cotransporter 2 (NKCC2). 56 Interestingly, nephron‐specific deletion of the glucocorticoid receptor (GR) in mice led to the “mirrored” phenotype including increased water intake and urine output, urinary alkalinization and downregulation of HKA2, AQP2 and NKCC2 therefore unveiling a novel role of this serine protease and the GR in renal water handling. Under Na+ restriction, amiloride‐sensitive renal potential difference and ENaC‐mediated sodium balance remained unchanged in vivo. 56 Overall, CAP2/Tmprss4 knockout mice did not seem to be directly implicated in the regulation and/or proteolytic ENaC activation.
2.3. CAP3/St14 (matriptase)
CAP3/St14 is known to induce a 6.8‐fold higher INa+ when co‐expressed with rat α‐, β‐ and γ‐ENaC subunits in Xenopus oocytes. 70 In vitro analyses by Kota and coworkers identified a region near the rat γ‐ENaC furin site as a critical cleavage site for CAP3‐mediated ENaC stimulation. 38 Contrary to the constitutive CAP1/Prss8 knockout, mice with constitutive lack of CAP3/St14 survived until birth, but then died due to severe ichthyosis. All epidermal surfaces were grossly abnormal which consequently compromised the epidermal barrier function (Table 1). 57 Deficiency of matriptase in adult mice (using tamoxifen‐induced deletion) predominantly resulted in impaired epithelial barrier function in the salivary gland epithelium with a near‐complete loss of saliva production 59 and led to a Sjögren's syndrome‐like disease. 60 In the large intestine, a disruption of normal tissue architecture was documented and accompanied by an increased intestinal permeability and oedema of crypt and submucosa cells. 59 Surprisingly, further matriptase‐expressing epithelia eg the upper digestive tract, small intestine, kidney, liver, lungs, spleen and pancreas showed normal macroscopic appearance. 59 CAP3 hypomorphic mice displayed a ~30% reduction in intestinal transepithelial electrical resistance likely via dysregulated claudin‐2 expression at intercellular junctions. 58 Intestine‐specific matriptase deficiency induced malignant transformation of colonic epithelium. 71 Overall, although a role of CAP3 in epidermal barrier function was confirmed, its implication in the proteolytic activation of ENaC has not yet been demonstrated.
2.4. Urokinase‐type plasminogen activator
In human, nephrotic syndrome characterized by severe peripheral oedema and ENaC‐mediated Na+ retention is thought to be due to aberrantly filtered plasminogen which is cleaved into plasmin. 63 , 72 In Xenopus oocytes, preincubation with plasminogen and uPA increased ENaC current, whereas uPA alone showed only a marginal effect. 12 Experimentally induced nephrotic mouse models with chronic versus acute inhibition of uPA resulted in opposite conclusions. 72 Indeed, constitutive uPA knockout mice in which the nephrotic syndrome was induced by doxorubicin showed no phenotypic difference when compared with controls, ie no implication in ENaC‐mediated Na+ retention, 64 whereas an antagonistic uPA treatment of podocin knockout mice led to a marked attenuation of Na+ retention. 63 Furthermore, plasminogen deficiency did not prevent ENaC‐mediated sodium retention in an induced experimental nephrotic mouse model, and thus, the uPA/plasmin‐mediated pathway might not be the only player for ENaC activation (Table 1). 65 uPA, like tissue‐type plasminogen activator, has a divergent role in fibrinolysis and macrophage function and further affects complex biological processes. 73
2.5. Tissue and plasma kallikrein
Tissue kallikrein has been proposed to be the physiologically relevant protease modulating ENaC activity. 17 It is highly expressed in the colon and kidney (Figure 2A), and in the kidney, it localizes to the distal portion of the nephron including the principal cells of the connecting tubule (CNT) where it is also secreted into the urine. It cleaves kininogen to bradykinin and activates the B2 bradykinin receptor to increase sodium excretion. This is facilitated by the inhibition of sodium reabsorption in the collecting duct. 74 Tissue kallikrein‐deficient mice adapt normally to dietary sodium restriction despite defective γ‐ENaC activity and reduced renal ENaC activity (Table 1). 18 However, the authors did not exclude that this effect on ENaC may be indirect. Absence of plasma kallikrein (PKLK), an aprotinin‐sensitive serine protease, did not protect nephrotic mice from oedema formation, suggesting that ENaC‐mediated Na+ retention is independent of this protease (Table 1). 19
2.6. Tmprss3
Proteolytic processing of ENaC by Tmprss3 was associated with increased ENaC‐mediated current in vitro, and Tmprss3 mutants causing human deafness failed to proteolytically cleave and activate ENaC in the Xenopus oocyte expression system. 7 Constitutive Tmprss3 knockout mice and ENU (ethyl‐nitrosourea)‐induced mutant Tmprss3 mice carrying a protein‐truncating nonsense mutation both exhibited a cochlear hair cell degeneration, 66 , 67 although direct experimental evidence of in vivo ENaC activation by Tmprss3 is still missing (Table 1). Further epithelial phenotypes of the Tmprss3 knockout mice were not yet reported.
2.7. Tmprss2
Early functional experiments investigating the impact of TMPRSS2 on ENaC in Xenopus oocytes showed a significant decrease in ENaC current and protein levels which was not prevented by the addition of aprotinin. 8 A later study reported the opposite effect when both TMPRSS2 and ENaC cRNAs were injected into Xenopus oocytes, since an increase in ENaC current was observed similar to that of other serine proteases. 9 RNA sequencing data revealed that TMPRSS2 is highly expressed in the distal portion of the kidney (Figure 2B), but little is known about a physiological role, since Tmprss2 KO mice did not present with an observable phenotype (Table 1). 68
2.8. FSAP‐SPD
Active factor VII activating protease (FSAP) is excreted in urine of nephrotic patients and doxorubicin‐induced nephrotic mice, and found to activate ENaC in vitro in the Xenopus oocyte expression system. 22 However, in nephrotic FSAP‐deficient mice, the proteolytic cleavage pattern of α‐ and γ‐ENaC was similar to untreated animals and these mice were not protected from Na+ retention, rendering it unlikely that this protease is responsible for proteolytic ENaC activation. 3 , 22
3. SERINE PROTEASE INHIBITORS OF ENAC‐MEDIATED NA+ ABSORPTION
Serpins or serine protease inhibitors belong to a family of proteins that were first identified for their protease inhibition activity. 77 Most serpins are substrates and suicide inhibitors of serine proteases. They irreversibly inhibit their target protease by a conformational change that disrupts and blocks access to its active site. Some proteins with serpin function lack the enzyme inhibitory function. 78 The serine protease inhibitor nexin‐1 (PN‐1) antagonized CAP1/Prss8 (prostasin) activity in mice overexpressing CAP1/Prss8 in the epidermis independent of its catalytic site. 53 Thereby, co‐expression of either the wildtype or a catalytic triad‐mutant CAP1/Prss8 with nexin significantly reduced protease‐induced ENaC current in Xenopus oocytes. 53 In the kidney, a reciprocal regulation of the expression of nexin‐1 by TGF βand of CAP1/Prss8 by aldosterone has been proposed that might result in Na+ retention or natriuresis, respectively, 79 although the experimental in vivo proof of such an interaction is still missing. Experiments in mice revealed, that embryonic lethality of either hepatocyte growth factor activator inhibitor (HAI)‐1 or ‐2 was rescued by simultaneous inactivation of CAP3/St14 (matriptase). 80 Hypomorphic CAP1/Prss8 (frizzy; fr/fr) mice restored placentation and development in HAI‐1 (Spint1)‐deficient embryos. However, these defects seemed not to be caused by aberrant activity of ENaC, since neither the pharmacological block by ENaC inhibitor amiloride, nor ENaC inactivation did rescue the Spint1‐deficient mice. 80 Indeed, depending on the concentration used, amiloride can block u‐PA. 81 In human, mutations in the serine protease inhibitor SPINT2 were associated with congenital tufting enteropathy characterized by severe intestinal dysfunction. 82 Organoid crypt cultures indicated that Spint2 ablation induced decreased claudin‐7 expression in tight junctions that resulted in organoid rupture. 82 These clinical features could be prevented by intestinal‐specific inactivation of CAP3/St14 encoding matriptase. 83 The authors proposed that excessive matriptase activity might be causative for this genetic disorder. 20
In mouse M‐1 cortical collecting duct cells, aprotinin inhibited the amiloride sensitive current and transepithelial resistance. 84 A mathematical model using renal epithelial A6 cells from Xenopus laevis predicted that the serine protease inhibitors might affect intracellular trafficking and reduce the residency time of ENaC at the apical membrane. 85 Treatment of doxorubicin‐induced nephrotic mice with the serine protease inhibitor aprotinin normalized urinary serine protease activity and prevented Na+ retention, 86 and γ‐ENaC cleavage was reduced. 87 Hence, inhibition of urinary serine protease activity was proposed as treatment for proteinuria and volume retention. In Xenopus oocytes, aprotinin had no direct inhibitory effect on channel activity. 86 In a venom‐induced acute kidney injury (AKI) rat model, aprotinin prevented glomerular injury and a decrease in glomerular filtration rate, thereby restoring fluid and electrolyte homoeostasis by inhibition of augmented serine protease kallikrein levels. Pretreatment with aprotinin restored fluid homoeostasis and protected from kidney injury. 88 However, inhibition of ENaC activity was not reported here. Increased levels of proteases in the tubular fluid contributed to enhanced ENaC activity and thus Na+ retention as commonly seen in patients with chronic heart failure. 89 Two weeks of aprotinin treatment of a myocardial infarct rat model abrogated the enhanced diuretic and natriuretic responses to ENaC inhibitor benzamil. 89 In this context, it is interesting to note that aprotinin exerted nephrotoxic effects in healthy mice by inhibiting proximal tubular function and unexpectedly led to increased proteolytic ENaC activation. This was explained by a counterregulatory stimulation of ENaC‐mediated sodium transport. 90 Further studies are needed to unveil the in vivo effect of aprotinin on sodium transport independent of the proteolytic ENaC activation.
Amongst the chemical inhibitors, camostat mesylate (CM) inhibited several serine proteases and was used as an inhibitor of progressive chronic renal failure. 91 , 92 Use of CM resulted in reduced proteinuria and oedema, showing a beneficial effect on diabetic nephropathy that is associated with progressive loss in kidney function due to the high blood glucose levels. 93 In Dahl salt‐sensitive rats fed a high Na+ diet, systolic blood pressure and urinary protein excretion were reduced by oral administration of CM. 94 However, systemic treatment of ENaC inhibitors has not been reported in this experiment. Treatment of CM in nephrectomy‐induced kidney disease inhibited the progression of chronic renal failure by decreasing serum creatinine and proteinuria. 91 Similarly, in rats that developed an adenine‐induced chronic kidney disease, CM attenuated the progression of the disease through its antioxidant effects and decreased blood pressure, serum creatinine and fibrotic markers. 92 A water‐soluble irreversible serine protease inhibitor, AEBSF (4‐(2‐aminoethyl) benzenesulphonyl fluoride), has been recently shown to inhibit serine protease activity in nephrotic mice and also in nephrotic patients. 95
4. CONCLUDING REMARKS
Proteases might be the important therapeutic targets for epithelial disorders due to their pivotal role in the maintenance of epithelial homoeostasis in a variety of tissues. The mechanism by which these proteases modulate ENaC function is still not completely understood. The proteolytic activation of ENaC was well demonstrated in vitro, although less clear in vivo. In this context, loss‐ or gain‐of‐function mutations of these proteases in rodent models had nearly no effect or resulted in only mild ENaC‐mediated Na+ losing or retaining phenotypes. CAP1/Prss8 (prostasin)‐deficient mice exhibited a reduced ENaC‐mediated Na+ transport when exposed to challenging conditions in lung and colon, but this was not associated with reduced ENaC cleavage. Additionally, during Na+ deprivation, mice with catalytically inactive prostasin showed similar proteolytic ENaC activation as controls and, overall, a normal sodium balance. Tissue kallikrein‐deficiency, on the other hand, leads to a defective ENaC processing and function, but mice adapted normally to dietary changes. This could mean that the responsible protease for proteolytic ENaC activation is not yet identified and/or there is functional redundancy amongst these proteases in activating ENaC. The detailed analysis of these knockout models revealed additional substrates, which may explain the described phenotype(s) and thus elucidating pathways that may directly and/or indirectly affect ENaC activation.
Many of the findings discussed in this review were obtained from animal studies which have been proven instrumental for our understanding of the role of proteases in physiology and pathophysiology. The specific origin and the developmental timing of the mutation (partial vs complete, spontaneous vs genetically engineered, constitutive vs tissue‐specific vs induced) might influence the severity of the disease, and finally explain discrepancies between findings across different studies. Further limitations of previous studies include changes of protease composition and/or activity upon physiological and pathophysiological conditions, and, therefore, their redundancy may not be reflected in single knockout mouse models. Regardless of whether altered protease expression is a cause or a consequence of the epithelial phenotype, the tight regulation of serine proteases and/or potential interaction with other proteins may be central for the maintenance of tissue/organ homoeostasis. To dissect the fine‐tuning of the regulation and downstream effects of those proteases, the development of more specific serine protease inhibitors is necessary. Their functional equilibrium is integral to many biological processes, and hence, a disturbed balance results in a wide range of epithelial pathologies.
CONFLICT OF INTEREST
The authors declare no competing interest.
ACKNOWLEDGEMENTS
The authors would like to thank Friedrich Beermann for comments on the manuscript. Due to space constraint, only a selected number of references of the field could be considered. Images were created with BioRender.com. Open access funding provided by Universite de Lausanne.
Anand D, Hummler E, Rickman OJ. ENaC activation by proteases. Acta Physiol. 2022;235:e13811. doi: 10.1111/apha.13811
Funding information
Swiss National Center of Competence in Research, NCCR; Kidney.CH: Control of Homeostasis, Switzerland; N‐403‐07‐23; Swiss National Science Foundation, grant number: 31003A_182478 to EH.
REFERENCES
- 1. Rawlings ND, Barrett AJ, Finn R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2016;44(D1):D343‐D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boscardin E, Alijevic O, Hummler E, Frateschi S, Kellenberger S. The function and regulation of acid‐sensing ion channels (ASICs) and the epithelial Na(+) channel (ENaC): IUPHAR Review 19. Br J Pharmacol. 2016;173(18):2671‐2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Althaus M, Lawong RY. Proteolytic ENaC activation in health and disease‐a complicated puzzle. Pflugers Arch. 2022;474(2):177‐179. 10.1007/s00424-021-02644-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vallet V, Chraibi A, Gaeggeler H‐P, Horisberger J‐D, Rossier BC. An epithelial serine protease activates the amiloride‐sensitive sodium channel. Nature. 1997;389(6651):607‐610. [DOI] [PubMed] [Google Scholar]
- 5. Vuagniaux G, Vallet V, Jaeger NF, et al. Activation of the amiloride‐sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J Am Soc Nephrol. 2000;11(5):828‐834. [DOI] [PubMed] [Google Scholar]
- 6. Andreasen D, Vuagniaux G, Fowler‐Jaeger N, Hummler E, Rossier BC. Activation of epithelial sodium channels by mouse channel activating proteases (mCAP) expressed in Xenopus oocytes requires catalytic activity of mCAP3 and mCAP2 but not mCAP1. J Am Soc Nephrol. 2006;17(4):968‐976. [DOI] [PubMed] [Google Scholar]
- 7. Guipponi M, Vuagniaux G, Wattenhofer M, et al. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet. 2002;11(23):2829‐2836. [DOI] [PubMed] [Google Scholar]
- 8. Donaldson SH, Hirsh A, Li DC, et al. Regulation of the epithelial sodium channel by serine proteases in human airways. J Biol Chem. 2002;277(10):8338‐8345. [DOI] [PubMed] [Google Scholar]
- 9. Faller N, Gautschi I, Schild L. Functional analysis of a missense mutation in the serine protease inhibitor SPINT2 associated with congenital sodium diarrhea. PLoS One. 2014;9(4):e94267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Z, Zhao R, Zhao M, et al. Regulation of epithelial sodium channels in urokinase plasminogen activator deficiency. Am J Physiol Lung Cell Mol Physiol. 2014;307(8):L609‐L617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ji H‐L, Zhao R, Komissarov AA, Chang Y, Liu Y, Matthay MA. Proteolytic regulation of epithelial sodium channels by urokinase plasminogen activator: cutting edge and cleavage sites. J Biol Chem. 2015;290(9):5241‐5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Svenningsen P, Bistrup C, Friis UG, et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20(2):299‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Svenningsen P, Uhrenholt TR, Palarasah Y, Skjødt K, Jensen BL, Skøtt O. Prostasin‐dependent activation of epithelial Na + channels by low plasmin concentrations. Am J Physiol Regul Integr Comp Physiol. 2009;297(6):R1733‐R1741. [DOI] [PubMed] [Google Scholar]
- 14. Haerteis S, Krappitz M, Diakov A, Krappitz A, Rauh R, Korbmacher C. Plasmin and chymotrypsin have distinct preferences for channel activating cleavage sites in the γ subunit of the human epithelial sodium channel. J Gen Physiol. 2012;140(4):375‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Passero CJ, Mueller GM, Rondon‐Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem. 2008;283(52):36586‐36591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haerteis S, Krappitz A, Krappitz M, et al. Proteolytic activation of the human epithelial sodium channel by trypsin IV and trypsin I involves distinct cleavage sites. J Biol Chem. 2014;289(27):19067‐19078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel AB, Chao J, Palmer LG. Tissue kallikrein activation of the epithelial Na channel. Am J Physiol Renal Physiol. 2012;303(4):F540‐F550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Picard N, Eladari D, El Moghrabi S, et al. Defective ENaC processing and function in tissue kallikrein‐deficient mice. J Biol Chem. 2008;283(8):4602‐4611. [DOI] [PubMed] [Google Scholar]
- 19. Haerteis S, Schork A, Dörffel T, et al. Plasma kallikrein activates the epithelial sodium channel in vitro but is not essential for volume retention in nephrotic mice. Acta Physiol. 2018;224(1):e13060. [DOI] [PubMed] [Google Scholar]
- 20. Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near‐silent epithelial Na+ channels and increases airway epithelial Na + transport. Am J Physiol Lung Cell Mol Physiol. 2005;288(5):L813‐L819. [DOI] [PubMed] [Google Scholar]
- 21. Hughey RP, Bruns JB, Kinlough CL, et al. Epithelial sodium channels are activated by furin‐dependent proteolysis. J Biol Chem. 2004;279(18):18111‐18114. [DOI] [PubMed] [Google Scholar]
- 22. Artunc F, Bohnert BN, Schneider JC, et al. Proteolytic activation of the epithelial sodium channel (ENaC) by factor VII activating protease (FSAP) and its relevance for sodium retention in nephrotic mice. Pflugers Arch. 2022;474(2):217‐229. 10.1007/s00424-021-02639-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alli AA, Song JZ, Al‐Khalili O, et al. Cathepsin B is secreted apically from Xenopus 2F3 cells and cleaves the epithelial sodium channel (ENaC) to increase its activity. J Biol Chem. 2012;287(36):30073‐30083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Larionov A, Dahlke E, Kunke M, et al. Cathepsin B increases ENaC activity leading to hypertension early in nephrotic syndrome. J Cell Mol Med. 2019;23(10):6543‐6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia‐Caballero A, Ishmael SS, Dang Y, et al. Activation of the epithelial sodium channel by the metalloprotease meprin β subunit. Channels (Austin). 2011;5(1):14‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Butterworth MB, Zhang L, Liu X, Shanks RM, Thibodeau PH. Modulation of the epithelial sodium channel (ENaC) by bacterial metalloproteases and protease inhibitors. PLoS One. 2014;9(6):e100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. UniProt Consortium . UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49(D1):D480‐D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim SW. Dysregulation of ENaC in animal models of nephrotic syndrome and liver cirrhosis. Electrolyte Blood Press. 2006;4(1):23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ray EC, Rondon‐Berrios H, Boyd CR, Kleyman TR. Sodium retention and volume expansion in nephrotic syndrome: implications for hypertension. Adv Chronic Kidney Dis. 2015;22(3):179‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noreng S, Posert R, Bharadwaj A, Houser A, Baconguis I. Molecular principles of assembly, activation, and inhibition in epithelial sodium channel. Elife. 2020;9:e59038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo‐electron microscopy. Elife. 2018;7:e39340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Enuka Y, Hanukoglu I, Edelheit O, Vaknine H, Hanukoglu A. Epithelial sodium channels (ENaC) are uniformly distributed on motile cilia in the oviduct and the respiratory airways. Histochem Cell Biol. 2012;137(3):339‐353. [DOI] [PubMed] [Google Scholar]
- 33. Kleyman TR, Eaton DC. Regulating ENaC’s gate. Am J Physiol Cell Physiol. 2020;318(1):C150‐C162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huntley MA, Lou M, Goldstein LD, et al. Complex regulation of ADAR‐mediated RNA‐editing across tissues. BMC Genom. 2016;17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338(6114):1593‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen L, Chou C‐L, Knepper MA. A comprehensive map of mRNAs and their isoforms across all 14 renal tubule segments of mouse. J Am Soc Nephrol. 2021;32(4):897–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. García‐Caballero A, Dang Y, He H, Stutts MJ. ENaC proteolytic regulation by channel‐activating protease 2. J Gen Physiol. 2008;132(5):521‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kota P, García‐Caballero A, Dang H, Gentzsch M, Stutts MJ, Dokholyan NV. Energetic and structural basis for activation of the epithelial sodium channel by matriptase. Biochemistry. 2012;51(16):3460‐3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na+ channel regulation by extracellular and intracellular factors. Annu Rev Physiol. 2018;80:263‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hummler E, Dousse A, Rieder A, et al. The channel‐activating protease CAP1/Prss8 is required for placental labyrinth maturation. PLoS One. 2013;8(2):e55796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szabo R, Lantsman T, Peters DE, Bugge TH. Delineation of proteolytic and non‐proteolytic functions of the membrane‐anchored serine protease prostasin. Development. 2016;143(15):2818‐2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leyvraz C, Charles R‐P, Rubera I, et al. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170(3):487‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Malsure S, Wang Q, Charles R‐P, et al. Colon‐specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J Am Soc Nephrol. 2014;25(7):1453‐1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sugitani Y, Nishida A, Inatomi O, et al. Sodium absorption stimulator prostasin (PRSS8) has an anti‐inflammatory effect via downregulation of TLR4 signaling in inflammatory bowel disease. J Gastroenterol. 2020;55(4):408‐417. [DOI] [PubMed] [Google Scholar]
- 45. Planès C, Randrianarison NH, Charles R, et al. ENaC‐mediated alveolar fluid clearance and lung fluid balance depend on the channel‐activating protease 1. EMBO Mol Med. 2010;2(1):26‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uchimura K, Hayata M, Mizumoto T, et al. The serine protease prostasin regulates hepatic insulin sensitivity by modulating TLR4 signalling. Nat Commun. 2014;5:3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frateschi S, Keppner A, Malsure S, et al. Mutations of the serine protease CAP1/Prss8 lead to reduced embryonic viability, skin defects, and decreased ENaC activity. Am J Pathol. 2012;181(2):605‐615. [DOI] [PubMed] [Google Scholar]
- 48. Keppner A, Malsure S, Nobile A, Auberson M, Bonny O, Hummler E. Altered prostasin (CAP1/Prss8) expression favors inflammation and tissue remodeling in DSS‐induced colitis. Inflamm Bowel Dis. 2016;22(12):2824‐2839. [DOI] [PubMed] [Google Scholar]
- 49. Friis S, Madsen DH, Bugge TH. Distinct developmental functions of prostasin (CAP1/PRSS8) zymogen and activated prostasin. J Biol Chem. 2016;291(6):2577‐2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Essigke D, Ilyaskin AV, Wörn M, et al. Zymogen‐locked mutant prostasin (Prss8) leads to incomplete proteolytic activation of the epithelial sodium channel (ENaC) and severely compromises triamterene tolerance in mice. Acta Physiol (Oxf). 2021;232(1):e13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peters DE, Szabo R, Friis S, et al. The membrane‐anchored serine protease prostasin (CAP1/PRSS8) supports epidermal development and postnatal homeostasis independent of its enzymatic activity. J Biol Chem. 2014;289(21):14740‐14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang C, Chao J, Chao L. Adenovirus‐mediated human prostasin gene delivery is linked to increased aldosterone production and hypertension in rats. Am J Physiol Regul Integr Comp Physiol. 2003;284(4):R1031‐R1036. [DOI] [PubMed] [Google Scholar]
- 53. Crisante G, Battista L, Iwaszkiewicz J, et al. The CAP1/Prss8 catalytic triad is not involved in PAR2 activation and protease nexin‐1 (PN‐1) inhibition. FASEB J. 2014;28(11):4792‐4805. [DOI] [PubMed] [Google Scholar]
- 54. Frateschi S, Camerer E, Crisante G, et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat Commun. 2011;2:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Keppner A, Andreasen D, Mérillat A‐M, et al. Epithelial sodium channel‐mediated sodium transport Is not dependent on the membrane‐bound serine protease CAP2/Tmprss4. PLoS One. 2015;10(8):e0135224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Keppner A, Maric D, Sergi C, et al. Deletion of the serine protease CAP2/Tmprss4 leads to dysregulated renal water handling upon dietary potassium depletion. Sci Rep. 2019;9(1):19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. List K, Haudenschild CC, Szabo R, et al. Matriptase/MT‐SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene. 2002;21(23):3765‐3779. [DOI] [PubMed] [Google Scholar]
- 58. Buzza MS, Netzel‐Arnett S, Shea‐Donohue T, et al. Membrane‐anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc Natl Acad Sci USA. 2010;107(9):4200‐4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. List K, Kosa P, Szabo R, et al. Epithelial integrity is maintained by a matriptase‐dependent proteolytic pathway. Am J Pathol. 2009;175(4):1453‐1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yin H, Kosa P, Liu X, et al. Matriptase deletion initiates a Sjögren’s syndrome‐like disease in mice. PLoS One. 2014;9(2):e82852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. List K, Currie B, Scharschmidt TC, et al. Autosomal ichthyosis with hypotrichosis syndrome displays low matriptase proteolytic activity and is phenocopied in ST14 hypomorphic mice. J Biol Chem. 2007;282(50):36714‐36723. [DOI] [PubMed] [Google Scholar]
- 62. List K, Szabo R, Molinolo A, et al. Deregulated matriptase causes ras‐independent multistage carcinogenesis and promotes ras‐mediated malignant transformation. Genes Dev. 2005;19(16):1934‐1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hinrichs GR, Weyer K, Friis UG, et al. Urokinase‐type plasminogen activator contributes to amiloride‐sensitive sodium retention in nephrotic range glomerular proteinuria in mice. Acta Physiol (Oxf). 2019;227(4):e13362. [DOI] [PubMed] [Google Scholar]
- 64. Bohnert BN, Daiminger S, Wörn M, et al. Urokinase‐type plasminogen activator (uPA) is not essential for epithelial sodium channel (ENaC)‐mediated sodium retention in experimental nephrotic syndrome. Acta Physiol (Oxf). 2019;227(4):e13286. [DOI] [PubMed] [Google Scholar]
- 65. Xiao M, Bohnert BN, Aypek H, et al. Plasminogen deficiency does not prevent sodium retention in a genetic mouse model of experimental nephrotic syndrome. Acta Physiol. 2021;231(1):13512. 10.1111/apha.13512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fasquelle L, Scott HS, Lenoir M, et al. Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J Biol Chem. 2011;286(19):17383‐17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tang T, Li L, Tang J, et al. A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol. 2010;28(7):749‐755. [DOI] [PubMed] [Google Scholar]
- 68. Kim TS, Heinlein C, Hackman RC, Nelson PS. Phenotypic analysis of mice lacking the Tmprss2‐encoded protease. Mol Cell Biol. 2006;26(3):965‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gong Y, Yu M, Yang J, et al. The Cap1‐claudin‐4 regulatory pathway is important for renal chloride reabsorption and blood pressure regulation. Proc Natl Acad Sci USA. 2014;111(36):E3766‐E3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane‐bound channel‐activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum‐ and glucocorticoid‐regulated kinase (Sgk1) in Xenopus Oocytes. J Gen Physiol. 2002;120(2):191‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kosa P, Szabo R, Molinolo AA, Bugge TH. Suppression of Tumorigenicity‐14, encoding matriptase, is a critical suppressor of colitis and colitis‐associated colon carcinogenesis. Oncogene. 2012;31(32):3679‐3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ehmke H. Sodium retention by uPA in nephrotic syndrome? Acta Physiol (Oxf). 2020;228(1):e13393. [DOI] [PubMed] [Google Scholar]
- 73. Carmeliet P, Schoonjans L, Kieckens L, et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368(6470):419‐424. [DOI] [PubMed] [Google Scholar]
- 74. Tomita K, Pisano JJ, Knepper MA. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest. 1985;76(1):132‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bruns JB, Carattino MD, Sheng S, et al. Epithelial Na+ channels are fully activated by furin‐ and prostasin‐dependent release of an inhibitory peptide from the gamma‐subunit. J Biol Chem. 2007;282(9):6153‐6160. [DOI] [PubMed] [Google Scholar]
- 76. Passero CJ, Mueller GM, Myerburg MM, Carattino MD, Hughey RP, Kleyman TR. TMPRSS4‐dependent activation of the epithelial sodium channel requires cleavage of the γ‐subunit distal to the furin cleavage site. Am J Physiol Renal Physiol. 2012;302(1):F1‐F8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Silverman GA, Bird PI, Carrell RW, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. 2001;276(36):33293‐33296. [DOI] [PubMed] [Google Scholar]
- 78. Law RHP, Zhang Q, McGowan S, et al. An overview of the serpin superfamily. Genome Biol. 2006;7(5):216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kitamura K, Tomita K. Regulation of renal sodium handling through the interaction between serine proteases and serine protease inhibitors. Clin Exp Nephrol. 2010;14(5):405‐410. [DOI] [PubMed] [Google Scholar]
- 80. Szabo R, Uzzun Sales K, Kosa P, et al. Reduced prostasin (CAP1/PRSS8) activity eliminates HAI‐1 and HAI‐2 deficiency–associated developmental defects by preventing matriptase activation. PLoS Genet. 2012;8(8):e1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Buckley BJ, Aboelela A, Minaei E, et al. 6‐Substituted hexamethylene amiloride (HMA) derivatives as potent and selective inhibitors of the human urokinase plasminogen activator for use in cancer. J Med Chem. 2018;61(18):8299‐8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kawaguchi M, Yamamoto K, Takeda N, et al. Hepatocyte growth factor activator inhibitor‐2 stabilizes Epcam and maintains epithelial organization in the mouse intestine. Commun Biol. 2019;2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Szabo R, Callies LK, Bugge TH. Matriptase drives early‐onset intestinal failure in a mouse model of congenital tufting enteropathy. Development. 2019;146(22):dev183392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu L, Hering‐Smith KS, Schiro FR, Hamm LL. Serine protease activity in m‐1 cortical collecting duct cells. Hypertension. 2002;39(4):860‐864. [DOI] [PubMed] [Google Scholar]
- 85. Sasamoto K, Marunaka R, Niisato N, et al. Analysis of aprotinin, a protease inhibitor, action on the trafficking of epithelial Na+ channels (ENaC) in renal epithelial cells using a mathematical model. Cell Physiol Biochem. 2017;41(5):1865‐1880. [DOI] [PubMed] [Google Scholar]
- 86. Bohnert BN, Menacher M, Janessa A, et al. Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int. 2018;93(1):159‐172. [DOI] [PubMed] [Google Scholar]
- 87. Bohnert BN, Essigke D, Janessa A, et al. Experimental nephrotic syndrome leads to proteolytic activation of the epithelial Na+ channel in the mouse kidney. Am J Physiol Renal Physiol. 2021;321(4):F480‐F493. [DOI] [PubMed] [Google Scholar]
- 88. Berger M, de Moraes JA, Beys‐da‐Silva WO, et al. Renal and vascular effects of kallikrein inhibition in a model of Lonomia obliqua venom‐induced acute kidney injury. PLoS Negl Trop Dis. 2019;13(2):e0007197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zheng H, Liu X, Sharma NM, Li Y, Pliquett RU, Patel KP. Urinary proteolytic activation of renal epithelial Na+ channels in chronic heart failure. Hypertension. 2016;67(1):197‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wörner S, Bohnert BN, Wörn M, et al. Renal effects of the serine protease inhibitor aprotinin in healthy conscious mice. Acta Pharmacol Sin. 2022;43(1):111‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hayata M, Kakizoe Y, Uchimura K, et al. Effect of a serine protease inhibitor on the progression of chronic renal failure. Am J Physiol Renal Physiol. 2012;303(8):F1126‐F1135. [DOI] [PubMed] [Google Scholar]
- 92. Ueda M, Uchimura K, Narita Y, et al. The serine protease inhibitor camostat mesilate attenuates the progression of chronic kidney disease through its antioxidant effects. Nephron. 2015;129(3):223‐232. [DOI] [PubMed] [Google Scholar]
- 93. Onbe T, Makino H, Kumagai I, Haramoto T, Murakami K, Ota Z. Effect of proteinase inhibitor camostat mesilate on nephrotic syndrome with diabetic nephropathy. J Diabet Complications. 1991;5(2–3):167‐168. [DOI] [PubMed] [Google Scholar]
- 94. Maekawa A, Kakizoe Y, Miyoshi T, et al. Camostat mesilate inhibits prostasin activity and reduces blood pressure and renal injury in salt‐sensitive hypertension. J Hypertens. 2009;27(1):181‐189. [DOI] [PubMed] [Google Scholar]
- 95. Wörn M, Bohnert BN, Alenazi F, et al. Proteasuria in nephrotic syndrome‐quantification and proteomic profiling. J Proteomics. 2021;230:103981. [DOI] [PubMed] [Google Scholar]