

Abstract
We studied the clinical, histologic, and molecular features distinguishing DSA‐negative from DSA‐positive molecularly defined antibody‐mediated rejection (mABMR). We analyzed mABMR biopsies with available DSA assessments from the INTERCOMEX study: 148 DSA‐negative versus 248 DSA‐positive, compared with 864 no rejection (excluding TCMR and Mixed). DSA‐positivity varied with mABMR stage: early‐stage (EABMR) 56%; fully developed (FABMR) 70%; and late‐stage (LABMR) 58%. DSA‐negative patients with mABMR were usually sensitized, 60% being HLA antibody‐positive. Compared with DSA‐positive mABMR, DSA‐negative mABMR was more often C4d‐negative; earlier by 1.5 years (average 2.4 vs. 3.9 years); and had lower ABMR activity and earlier stage in molecular and histology features. However, the top ABMR‐associated transcripts were identical in DSA‐negative versus DSA‐positive mABMR, for example, NK‐associated (e.g., KLRD1 and GZMB) and IFNG‐inducible (e.g., PLA1A). Genome‐wide class comparison between DSA‐negative and DSA‐positive mABMR showed no significant differences in transcript expression except those related to lower intensity and earlier time of DSA‐negative ABMR. Three‐year graft loss in DSA‐negative mABMR was the same as DSA‐positive mABMR, even after adjusting for ABMR stage. Thus, compared with DSA‐positive mABMR, DSA‐negative mABMR is on average earlier, less active, and more often C4d‐negative but has similar graft loss, and genome‐wide analysis suggests that it involves the same mechanisms.
Summary Sentence
In 398 kidney transplant biopsies with molecular antibody‐mediated rejection, the 150 DSA‐negative cases are earlier, less intense, and mostly C4d‐negative, but use identical molecular mechanisms and have the same risk of graft loss as the 248 DSA‐positive cases.
Keywords: basic (laboratory) research/science, biopsy, kidney transplantation/nephrology, microarray/gene array, rejection: antibody‐mediated (ABMR), rejection
Short abstract
In a population of kidney transplant indication biopsies with a molecular diagnosis of antibody‐mediated rejection, those without compared to those with donor‐specific antibody typically occur earlier, are less active, and are more often C4d‐negative but have similar graft loss risk and, according to genome‐wide analyses, involve the same mechanisms.
Abbreviations
- ABMR
antibody‐mediated rejection
- AKI
acute kidney injury
- ABMRProb
ABMR classifier (ABMR, [C4d‐negative and C4d‐positive] vs. everything else)
- C4d
Complement factor d
- cg > 0Prob
glomerular double contours probability classifier
- DSA
donor‐specific HLA antibody
- eGFR
estimated glomerular filtration rate
- g > 1Prob
glomerulitis classifier
- HLA
human leukocyte antigen
- i > 1Prob
interstitial infiltrate classifier
- IgG
immunoglobulin G
- INTERCOMEX
Diagnostic and Therapeutic Applications of Microarrays in Organ Transplantation (ClinicalTrials.gov Identifier: NCT01299168)
- EABMR
early‐stage mABMR
- FABMR
fully developed mABMR
- LABMR
late‐stage mABMR
- mABMR
molecularly defined ABMR (by archetypes)
- MFI
mean fluorescence intensity
- MVI
microvascular inflammation
- MMDx
Molecular Microscope® Diagnostic System
- NK
natural killer
- ptc > 1Prob
peritubular capillaritis classifier
- SOC
standard‐of‐care
- t > 1Prob
tubulitis classifier
- TCMR
t cell‐mediated rejection
- TCMRProb
TCMR classifier (TCMR vs. everything else)
1. INTRODUCTION
In kidney transplant biopsies, antibody‐mediated rejection (ABMR) is characterized by a constellation of molecular features, histologic microcirculation changes, donor‐specific human leukocyte antigen (HLA) antibody (DSA), and complement factor d (C4d) deposition. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 Recently, considerable attention has focused on ABMR lacking detectable circulating DSA. When we defined ABMR molecularly (mABMR), we found that mABMR was often DSA‐negative, although usually HLA antibody (“PRA”)‐positive 11 : even fully developed mABMR (FABMR) was only 76% DSA positive. 12 Senev et al. 13 showed that 85 DSA‐positive ABMR and 123 DSA‐negative ABMR were histologically similar, but DSA‐negative ABMR was more often C4D‐negative, more transient, and had superior graft survival. Callemeyn et al. 14 found that 26 kidney allografts with histologic microvascular inflammation (MVI), but no DSA had typical ABMR‐induced transcript changes. Natural killer (NK) cell recognition of missing‐self may be involved in DSA‐negative ABMR: missing‐self increases risk of MVI independent of demonstrable HLA‐DSA 15 and could interact with NK Fc receptor recognition of bound DSA. 15 , 16 , 17 Absorption of the highest affinity DSA by the kidney could be involved in DSA‐negative ABMR, but DSA eluted from kidney biopsies is similar to circulating DSA rather than displaying unique specificities. 18 , 19 A possible role for autoantibodies such as anti‐AT1R has been raised, 20 , 21 potentially interacting with alloantibody mechanisms. 22 , 23
Although many previous reports have focused on histologically defined DSA‐negative ABMR, the present study sought to define the clinical, histologic, and molecular features of DSA‐negative ABMR when ABMR is defined exclusively by automatically assigned molecular archetype classes using the Molecular Microscope algorithms. This allows us to study the clinical and histologic features of DSA without using those features for diagnosis. In the present study, we used biopsies collected in the INTERCOMEX study (ClinicalTrials.gov NCT01299168) classified by molecular archetypes to study how histology lesions, DSA details, C4d, and outcomes distribute in DSA‐negative versus DSA‐positive mABMR. Acknowledging that histologic DSA‐negative and DSA‐positive ABMR are transcriptionally similar, 14 we focused on genome‐wide comparisons to discover transcripts that might reveal distinct mechanisms operating in DSA‐negative mABMR.
2. MATERIALS AND METHODS
2.1. Population and demographics
All 1679 biopsies were prospectively collected at participating international centers collaborating on the INTERCOMEX study (Table S1) as previously described. 24 , 25 Biopsy and patient demographics of the full population are shown in Table S1. Table S1 shows a list of abbreviations. Selected transcript sets are described in Table S1. Table S1 shows histology and Molecular Microscope Diagnostic System (MMDx) diagnoses along with DSA status. DSA status was defined as the standard‐of‐care (SOC) diagnosis by the local HLA laboratory, as required by the Banff guidelines (see Discussion). When patients had more than one indication biopsy, all were included because we found that there was no effect of exclusion of repeat biopsies.
The research plan is summarized in Figure 1.
FIGURE 1.
Study design flowchart
2.2. Selection of biopsies with recorded donor‐specific HLA antibody status
We studied the biopsies in the 1679 cohort with recorded DSA status per SOC at the local center. In DSA‐positive versus DSA‐negative comparisons, only mABMR biopsies with available status were used (N = 398). As a baseline, we used biopsies called No rejection by molecular archetypes with available DSA status (N = 854).
2.3. Archetypal analysis
Archetypal analysis is an unsupervised method that generates clusters based on the multidimensional distribution of input variables. The dominant input features define the “archetype” locations—hypothetical samples representing extreme phenotypes within the distribution. As published, 12 , 26 rejection archetypes used g > 1Prob, ptc > 1Prob, cg > 0Prob, ABMRProb, TCMRProb, i > 1Prob, and t > 1Prob classifier scores as input. These represent molecular estimates of histologic rejection and the key lesion scores used in those diagnoses.
The number of clusters is user‐chosen, guided by the trade‐off between complexity (more clusters) and model quality, visualized as a scree plot (not shown). Based on this, and the biological interpretability of the clusters, six archetypes were chosen. Their names were based on their average histologic and molecular characteristics: no rejection, TCMR1 (often mixed and associated with nonadherence), TCMR2, early‐stage ABMR (EABMR), FABMR, and late‐stage ABMR (LABMR). Each biopsy gets a score from 0.0 to 1.0 for each archetype, summing to 1.0. Scores are interpreted as the proportional contribution of each of the six archetypes. A biopsy's “cluster” was defined as the highest of its six archetype scores.
Biopsies in any of EABMR, FABMR, or LABMR were defined as “mABMR.” Because the archetypes were assigned independent of their DSA or C4d status, an examination of the mABMR relationships with variables such as DSA and C4d was possible.
For the present study, we have emphasized the archetypal clustering because it is completely automatic and avoids subjectivity. However, the biopsies are always signed out in MMDx based on both the archetypal scores and the binary classifiers provided on the MMDx report. This is because archetypes are proportions and therefore can be misleading when more than one score is elevated.
2.4. Moving average plots
Moving averages (window size 150) were plotted by first sorting the biopsies by their time posttransplant, then plotting the mean of biopsies 1–150 y‐variable values against the mean of the biopsies 1–150 days posttransplant. The window is then slid to biopsies 2–151, 3–152, etc. with the process repeated and the mean score plotted each time. This method was selected to permit comparisons with past analyses. 12
2.5. Statistical analysis
All analyses used version 4.1.0 of R. 27 We performed t‐tests, Chi‐squared analyses, and Fisher's exact test when comparing different histologic and DSA groups within the ABMR archetype population. Differential gene expression was done using a Bayesian t‐test from R's “limma” package. 28
3. RESULTS
3.1. Rederiving the archetype classes
The archetype classifications developed in the first 1208 biopsies 12 were completely rederived in 1679 biopsies using the same strategies. As in previous analyses, biopsy groups were assigned automatically based on highest archetype score. This established the following rejection classes, similar to earlier analysis 12 : 1040 No rejection, 175 TCMR (including mixed: 75 TCMR1 and 100 TCMR2), and 464 mABMR (210 EABMR, 182 FABMR, and 72 LABMR). The TCMR/mixed biopsies will not be discussed in this paper and will be presented in detail in a separate analysis (in preparation).
We analyzed the features of all 464 mABMR biopsies (regardless of DSA status) in terms of diagnoses, lesions, transcript sets, and genes (Table 1), including 1040 No rejection for comparison. Within mABMR, there was a progression in mean time of biopsy posttransplant: EABMR 2.1 years; FABMR 4.1 years; LABMR 7.6 years. The mean eGFR (cc/min/M2) declined from 50 in EABMR to 43 in FABMR and 32 in LABMR. The histology ptc‐ and g‐scores and v‐lesions, and the arterial fibrous intimal thickening (cv‐score) scores, were highest in FABMR. The cg‐ and hyalinosis scores were highest in LABMR.
TABLE 1.
Clinical variables and histologic lesion scores in the No rejection and mABMR archetype clusters
| Variable | Mean a value or score in each archetype b | |||||
|---|---|---|---|---|---|---|
| No rejection (N = 1040) | All mABMR (N = 464) | EABMR (N = 210) | FABMR (N = 182) | LABMR (N = 72) | ||
| Clinical | Median time of biopsy posttransplant (days) | 371 | 1159 | 724 | 1482 | 2744 |
| GFR (cc/min) | 44 | 45 | 50 | 43 | 32 | |
| Proteinuria c | 0.55 | 0.69 | 0.58 | 0.78 | 0.77 | |
| Donor age (years) | 46 | 42 | 46 | 38 | 39 | |
| Recipient age (years) | 52 | 49 | 51 | 47 | 47 | |
| ABMR lesions | g (glomerulitis) | 0.27 | 1.28 | 1.07 | 1.70 | 0.87 |
| ptc (capillaritis) | 0.25 | 1.35 | 1.08 | 1.81 | 1.03 | |
| cg (double contours) | 0.18 | 0.99 | 0.47 | 1.39 | 1.59 | |
| TCMR lesions | i (interstitial infiltrate) | 0.32 | 0.55 | 0.54 | 0.51 | 0.64 |
| t (tubulitis) | 0.30 | 0.41 | 0.42 | 0.38 | 0.44 | |
| Rejection lesions | v (vasculitis) | 0.01 | 0.08 | 0.04 | 0.14 | 0.05 |
| Atrophy‐fibrosis‐related | ci (fibrosis) | 1.12 | 1.44 | 1.24 | 1.64 | 1.57 |
| ct (atrophy) | 1.03 | 1.21 | 0.98 | 1.40 | 1.45 | |
| cv (fibrous intimal thickening) | 0.90 | 0.97 | 0.81 | 1.18 | 0.88 | |
| ah (hyalinosis) | 1.00 | 1.19 | 1.00 | 1.25 | 1.69 | |
Note: Shading and bolding indicate the highest value per row.
Main table entries indicate means, except for time posttransplant which are medians.
TCMR archetypes are not shown.
Proteinuria is coded as positive = 1, negative = 0. Therefore, the means for these variables indicate the fraction of biopsies that were positive. Missing values were excluded from the calculations.
(The use of automated archetype assignments means that some biopsies will have ambiguous features e.g. biopsies with ABMR‐like features below the diagnostic thresholds, i.e., boundary discrepancies. 29 Twelve of 1040 NR biopsies by archetype assignment had positive scores [above the threshold] for the binary ABMR and Rejection classifiers and eight were signed out as ABMR by MMDx on the report. Of 83 biopsies called NR by archetype assignment but called ABMR by histology, 52 were DSA‐positive, 33 were C4d‐positive; 22 had an ABMRProb classifier score above the threshold [0.20]; 23 were signed out as MMDx ABMR; and 17 failed.)
3.2. Relationships between time posttransplant and the findings within the 464 mABMR biopsies
Figure 2A shows that within mABMR, the ABMRProb molecular classifier score rose then plateaued after 500 days. Other ABMR activity classifier scores (ptc > 0Prob, and g > 0Prob) rose slightly in the first 500 days but were high throughout the posttransplant period. The cg > 0Prob classifier was low initially and rose steadily over time.
FIGURE 2.
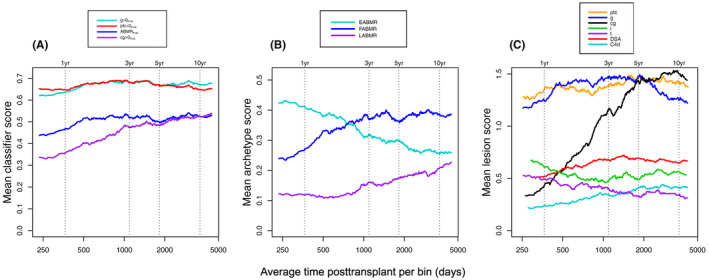
Moving averages showing relationships between ABMR‐associated molecular and histologic features and time posttransplant in 464 archetype‐assigned mABMR biopsies. (A) Molecular classifier scores over time, (B) ABMR‐associated archetypes over time, and (C) ABMR‐associated histology features, DSA, and C4d over time. Moving averages (window size 150) were plotted by first sorting the biopsies by their time posttransplant, then plotting the mean of biopsies 1–150 y‐variable values against the mean of the biopsies 1–150 days posttransplant. The window is then slid to biopsies 2–151, 3–152, etc. with the process repeated. Consecutive window “bins” are created, and the moving averages calculated for each bin
Among archetype scores (Figure 2B), EABMR scores were high early and declined steadily as the FABMR score increased, with the LABMR score increasing after 500 days.
In Figure 2C, the mean histologic ptc‐ and g‐scores in mABMR were high throughout the posttransplant period. The histologic cg‐score rose sharply, peaked at 5 years, and plateaued. DSA and C4d positivity gradually increased in frequency over time then plateaued. The i‐scores (interstitial infiltrate) and t‐scores (tubulitis)—canonical lesions of TCMR—were higher in the early period (although far below the diagnostic TCMR levels) probably reflecting resolving mild injury‐induced changes from transplantation (see Discussion).
We examined the transcript set and classifier scores in mABMR biopsies before and after 1 year posttransplant (Table S1). The most significant differences were related to changes that increase with time and atrophy‐fibrosis—immunoglobulin transcripts (IGTs) 30 , 31 and the atrophy‐fibrosis classifier (ciProb). Some ABMR scores were also higher in biopsies after >1 year posttransplant. Among individual transcripts (Table S1), most transcripts differentially expressed between mABMR biopsies before and after 1 year were IGTs, which were increased after 1 year.
3.3. DSA‐negative mABMR is earlier posttransplant than DSA‐positive mABMR
Table 2 shows that DSA‐negative mABMR biopsies had an earlier time posttransplant than DSA‐positive mABMR by ~1.5 years (mean 2.4 vs. 3.9 years). This difference was most significant in early‐stage mABMR (EABMR). There was a similar trend in FABMR (not significant).
TABLE 2.
Time of biopsy posttransplant in DSA‐negative versus DSA‐positive mABMR
| Biopsy group | Time of biopsy posttransplant | |||
|---|---|---|---|---|
| Median and mean | DSA‐negative (N = 150) | DSA‐positive (N = 248) | p value for DSA‐positive vs DSA‐negative | |
| All mABMR | Median days (years) (range in days) | 867 [2.4 year](8–12, 371) | 1408 [3.9 year] (3–9889) | 1.2 × 10 −3 |
| Mean days (years) | 1698 (4.7 year) | 2219 (6.1 year) | 6.7 × 10 −4 | |
| EABMR | Median days (years) (range in days) | 484 [1.3 year](8–8030) | 998 [2.7 year] (3–9466) | 4.0 × 10 −3 |
| Mean days (years) | 1035 (2.8 year) | 1846 (5.1 year) | 3.3 × 10 −3 | |
| FABMR | Median (years) (range in days) | 1260 [3.5 year](72–9525) | 1610 [4.4 year](9–9889) | 0.31 |
| Mean days (years) | 1904 (5.2 year) | 2213 (6.1 year) | 0.32 | |
| LABMR | Median days (years) (range in days) | 2763 [7.6 year](14–12, 371) | 2690 [7.4 year](107–8252) | 0.66 |
| Mean days (years) | 3262 (8.9 year) | 3274 (9.0 year) | 0.29 | |
Note: t‐test and Wilcoxon sign rank test was used for time of biopsy posttransplant. The p value column is in italics as it compares the previous two columns.
Differences p < .01 are shaded and the larger fraction is bolded.
3.4. Clinical and histologic features of DSA‐negative and DSA‐positive mABMR
Table 3 compares the features of 150 DSA‐negative and 248 DSA‐positive mABMR biopsies, plus 854 No rejection biopsies.
TABLE 3.
Clinical variables and histologic lesion scores in the No rejection (N = 854) and mABMR (N = 398) biopsies with defined DSA statuswith No rejection (N = 854) shown for reference
| Variable | Mean a value or score in each archetype b | ||||
|---|---|---|---|---|---|
| No rejection (N = 854) | DSA‐negative mABMR (N = 150) | DSA‐positive mABMR (N = 248) | p value DSA‐positive vs DSA‐negative | ||
| Clinical | eGFR (cc/min/M2) | 45 | 43 | 49 | 0.03 |
| Proteinuria c | 0.53 | 0.65 | 0.69 | 0.46 | |
| Donor age (years) | 45 | 42 | 40 | 0.64 | |
| Recipient age (years) | 51 | 48 | 49 | 0.83 | |
| ABMR lesions | g (glomerulitis) | 0.28 | 1.22 | 1.39 | 0.14 |
| ptc (capillaritis) | 0.26 | 1.15 | 1.50 | 3 × 10 −3 | |
| cg (double contours) | 0.19 | 0.86 | 1.09 | 0.04 | |
| TCMR lesions | i (interstitial infiltrate) | 0.30 | 0.72 | 0.38 | 1 × 10 −4 |
| t (tubulitis) | 0.31 | 0.47 | 0.33 | 0.42 | |
| Rejection lesions | v (vasculitis) | 0.01 | 0.04 | 0.10 | 0.16 |
| Atrophy‐fibrosis‐related | ci (fibrosis) | 1.14 | 1.38 | 1.52 | 0.24 |
| ct (atrophy) | 1.04 | 1.12 | 1.24 | 0.26 | |
| cv (fibrous intimal thickening) | 0.87 | 0.78 | 1.06 | 0.03 | |
| ah (hyalinosis) | 1.01 | 0.97 | 1.35 | 6 × 10 −3 | |
Note: Differences at p < .05 are bolded, and at p < .01 are bolded and shaded. The p value column is in italics as it compares the previous two columns.
Main table entries indicate means, except for time posttransplant which are medians.
TCMR archetypes are not shown.
Proteinuria is coded as positive = 1, negative = 0. Therefore, the means for this variables indicate the fraction of biopsies that were positive. Missing values were excluded from the calculations.
Among clinical features, DSA‐negative mABMR biopsies had a slightly lower eGFR than DSA‐positive mABMR.
Among histologic lesions, DSA‐negative mABMR showed lower ptc‐lesions (p = 3 × 10−3) and cg‐lesions (p = .04) and lower g‐lesions (not significant). However, the i‐scores (p = 1 × 10−4) and the t‐scores (not significant) were higher in DSA‐negative mABMR. DSA‐negative mABMR had somewhat lower atrophy‐fibrosis‐related (ci‐ and ct‐) scores and significantly lower scores for arterial fibrous intimal thickening (cv‐score, p = .03) and hyalinosis (ah‐score, p = 6 × 10−3).
Table 4 examines molecular features of DSA‐negative versus DSA‐positive mABMR, again including No rejection controls. Among transcript sets, DSA‐negative mABMR had lower mean ABMR‐related transcript set scores: DSA‐associated transcripts (DSAST) and NK‐associated transcripts (NKB) but high fibrillar collagen transcripts (FICOL), which are increased by injury. 31 TCMR‐related, atrophy‐fibrosis‐related, and most injury‐related transcript sets were not significantly different.
TABLE 4.
Differences in mean scores for transcript sets and molecular classifiers between DSA‐negative and DSA‐positive mABMR archetype biopsies with available DSA status (N = 398) with No rejection (N = 854) biopsies shown for reference
| Variable | Mean value or score in each group b | |||||
|---|---|---|---|---|---|---|
| No rejection (N = 854) | DSA‐negative mABMR (N = 150) | DSA‐positive mABMR (N = 248) | p value a DSA‐positive vs DSA‐negative mABMR | p value a DSA‐negative vs No rejection | ||
| Mean transcript set scores | ||||||
| TCMR‐related | Cytotoxic CD8 T cell‐associated (QCAT) | 0.60 | 1.30 | 1.32 | 0.68 | 6 × 10 −50 |
| ABMR‐related | DSA‐selective transcripts (DSAST) | 0.07 | 0.55 | 0.68 | 2 × 10 −5 | 2 × 10 −61 |
| NK cell burden (NKB) | 0.36 | 1.10 | 1.25 | 9 × 10 −4 | 6 × 10 −64 | |
| Recent injury‐related | Fibrillar collagen (FICOL) | 1.06 | 1.08 | 0.89 | 0.01 | 0.10 |
| Injury‐repair induced, day 3 (IRITD3) | 0.03 | 0.10 | 0.09 | 0.32 | 5 × 10 −9 | |
| Injury‐repair induced, day 5 (IRITD5) | 0.31 | 0.45 | 0.41 | 0.11 | 6 × 10 −13 | |
| Injury‐repair associated transcripts (IRRAT) | 0.22 | 0.46 | 0.41 | 0.28 | 4 × 10 −7 | |
| Atrophy‐fibrosis | Immunoglobulin (IGT) | 0.60 | 1.17 | 1.32 | 0.16 | 5 × 10 −10 |
| Mean molecular classifier scores | ||||||
| TCMR‐related | i‐score (i > 1 Prob ) | 0.06 | 0.12 | 0.13 | 0.11 | 1 × 10 −25 |
| t‐score (t > 1 Prob ) | 0.06 | 0.10 | 0.11 | 0.11 | 6 × 10 −20 | |
| TCMR (TCMR Prob ) | 0.03 | 0.05 | 0.05 | 0.14 | 2 × 10 −15 | |
| ABMR‐related | ABMR (ABMR Prob ) | 0.08 | 0.42 | 0.54 | 8 × 10 −6 | 1 × 10 −68 |
| Glomerular double contours (cg > 0 Prob ) | 0.09 | 0.39 | 0.49 | 2.10 −4 | 3 × 10 −54 | |
| Peritubular capillaritis (ptc > 0 Prob ) | 0.15 | 0.62 | 0.68 | 5 × 10 −4 | 7 × 10 −80 | |
| Glomerulitis (g > 0 Prob ) | 0.18 | 0.62 | 0.69 | 2 × 10 −4 | 1 × 10 −76 | |
| Rejection‐related | Rejection (Rej Prob ) | 0.13 | 0.60 | 0.69 | 7 × 10 −5 | 1 × 10 −76 |
| Recent injury‐related | lowGFRProb | 0.30 | 0.34 | 0.30 | 0.13 | 9 × 10 −3 |
| Atrophy‐fibrosis | ci‐score (ci > 1Prob) | 0.31 | 0.43 | 0.42 | 0.78 | 3 × 10 −9 |
Shading and bold highlight significant p values, that is, scores that were significantly different (p < .05) between DSA‐positive and DSA‐negative mABMR biopsies. Wilcoxon sign rank test was used for histology lesion scores, and t‐test was used for all other scores.
TCMR archetypes are not shown.
The main p value column is in italics as it compares the previous two columns.
Among classifier scores, DSA‐negative mABMR had lower scores for ABMR and rejection activity classifiers and the ABMR‐stage (cg >0) classifier compared with DSA‐positive mABMR, but no difference in scores related to TCMR or atrophy‐fibrosis.
As expected, ABMR‐related scores were increased in ABMR (DSA‐negative or positive) versus No rejection.
Thus, DSA‐negative mABMR had typical ABMR changes but lower ABMR activity and earlier stage compared with DSA‐positive mABMR.
3.5. Detailed HLA antibody and DSA assessment
In 1394 biopsies with available DSA status, 42% were DSA‐positive (Table 5). The mABMR biopsies were 60% DSA‐positive, peaking in FABMR: No rejection 34%; EABMR 56%; FABMR 70%; and LABMR 58%. The DSA positivity increased with ABMR activity and stage. Anti‐class II was common (315/389, 81%) across all archetypal biopsy groups, even in No rejection.
TABLE 5.
DSA detail in population with available DSA status (1394 tested of 1679 total) and in No rejection and mABMR
| Biopsy group | Number of biopsies broken down by DSA status | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Total DSA‐negative | DSA‐negative/PRA‐negative a | DSA‐negative/PRA‐positive | Total DSA‐positive (% of column total) | DSA Class I | DSA Class I/II | DSA Class II | DSA‐class not recorded | |
| All | 1394 | 807 (58%) | 402 | 277 | 587 (42%) | 74 | 113 | 202 | 198 |
| No rejection | 854 | 566 (66%) | 309 | 169 | 288 (34%) | 38 | 45 | 103 | 102 |
| All mABMR | 398 | 150 (40%) | 50 | 75 b | 248 (60%) | 31 | 56 | 74 | 56 |
| EABMR | 177 | 77 (44%) | 23 | 40 | 100 (56%) | 13 | 19 | 26 | 42 |
| FABMR | 159 | 47 (30%) | 17 | 23 | 112 (70%) | 13 | 31 | 34 | 3 |
| LABMR | 62 | 26 (42%) | 10 | 12 | 36 (58%) | 5 | 6 | 14 | 11 |
The totals for the PRA‐negative plus PRA‐positive are less than the total DSA‐negative because not all DSA‐negative results had PRA results reported.
Chi‐square for fraction DSA‐negative/PRA‐positive with No rejection (169/478 or 35%) versus DSA‐negative/PRA‐positive with mABMR (75/125 or 60%): p = 5.79 × 10−7.
The DSA‐negative mABMR patients were usually sensitized: among DSA‐negative patients, HLA antibody (“panel‐reactive antibody” or “PRA”) was more frequent in mABMR (60%) than in No rejection (35%, p = 5.8 × 10−7).
3.6. C4d positivity is strongly related to DSA status
As in earlier analyses, 14 C4d positivity was strongly associated with DSA positivity across all biopsy groups, even No rejection (Table 6, showing biopsies where both C4d and DSA were tested). DSA‐negative mABMR was 86% C4d‐negative.
TABLE 6.
C4d status in relation to DSA status in biopsies with DSA test results
| Number of biopsies C4d‐positive as fraction (%) of biopsies tested for both C4d and DSA | |||
|---|---|---|---|
| DSA‐negative | DSA‐positive | Fisher's test p value of DSA status versus C4d status | |
| All biopsies | 48/632 (8%) | 151/493 (31%) | 7 × 10−24 |
| No rejection | 29/444 (7%) | 42/234 (18%) | 9 × 10−6 |
| All mABMR | 16/116 (14%) a | 98/224 (44%) | 1 × 10−8 |
| EABMR | 7/64 (11%) | 36/92 (40%) | 0.0001 |
| FABMR | 6/32 (19%) | 53/100 (53%) | 0.0009 |
| LABMR | 3/20 (15%) | 9/32 (28%) | 0.33 |
DSA‐negative mABMR was more often C4d‐positive than DSA‐negative No rejection (p = .02).
3.7. Genome‐wide class comparisons between DSA‐negative mABMR versus DSA‐positive mABMR
We used genome‐wide class comparisons to compare gene expression between DSA‐negative and DSA‐positive mABMR. There were essentially no differences in top‐ranked genes at the rigorous adjusted p < .05: only two of 49, 495 probe sets were significantly lower in DSA‐negative mABMR (Table 7), and none were significantly higher in DSA‐negative mABMR (Table S1). There were small differences significant at unadjusted p values, all consistent with lower ABMR activity and earlier stage of DSA‐negative mABMR.
TABLE 7.
Top 20 increased genes differing between DSA‐positive and DSA‐negative mABMR
| Gene symbol | Gene name | Transcript set annotation | p value | FDR | DSA‐negative | DSA‐positive | No rejection |
|---|---|---|---|---|---|---|---|
| P2RX7 | Purinergic receptor P2X, ligand‐gated ion channel, 7 | IFNG‐inducible (GRIT) | 1 × 10−6 | 0.03 | 29 | 33 | 26 |
| TM4SF18 | Transmembrane 4 L six family member 18 | ABMR‐RAT | 1 × 10−6 | 0.03 | 423 | 497 | 331 |
| ITGAL | Integrin, alpha L (antigen CD11A [p180], lymphocyte function‐associated antigen 1; alpha polypeptide) | ABMR‐RAT | 4 × 10−6 | 0.05 | 36 | 39 | 32 |
| STAT2 | Signal transducer and activator of transcription 2, 113 kDa | IFNG‐inducible (GRIT) | 4 × 10−6 | 0.05 | 63 | 68 | 59 |
| UVSSA | UV‐stimulated scaffold protein A | 7 × 10−6 | 0.07 | 50 | 55 | 51 | |
| ROBO4 | Roundabout, axon guidance receptor, homolog 4 (Drosophila) | ABMR‐RAT | 3 × 10−5 | 0.14 | 844 | 964 | 597 |
| TNFRSF14 | Tumor necrosis factor receptor superfamily, member 14 | IFNG‐inducible (GRIT) | 4 × 10−5 | 0.17 | 475 | 513 | 433 |
| SEMA3D | Sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3D | 6 × 10−5 | 0.17 | 84 | 101 | 80 | |
| COL13A1 | Collagen, type XIII, alpha 1 | ABMR‐RAT | 7 × 10−5 | 0.17 | 50 | 58 | 38 |
| CACTIN | Cactin, spliceosome C complex subunit | 8 × 10−5 | 0.18 | 211 | 224 | 213 | |
| LILRA1 | Leukocyte immunoglobulin‐like receptor, subfamily A (with TM domain), member 1 | ABMR‐RAT | 9 × 10−5 | 0.18 | 77 | 85 | 69 |
| KRT36 | Keratin 36 | 1 × 10−4 | 0.19 | 19 | 20 | 20 | |
| CX3CR1 | Chemokine (C‐X3‐C motif) receptor 1 | ABMR‐RAT | 1 × 10−4 | 0.19 | 442 | 531 | 212 |
| ALKBH6 | alkB, alkylation repair homolog 6 (E. coli) | 1 × 10−4 | 0.21 | 120 | 128 | 129 | |
| THEMIS2 | Thymocyte selection associated family member 2 | IFNG‐inducible (GRIT) | 2 × 10−4 | 0.21 | 118 | 127 | 104 |
| HLA‐B | Major histocompatibility complex, class I, B | IFNG‐inducible (GRIT) | 2 × 10−4 | 0.21 | 81 | 93 | 67 |
| LYPD2 | LY6 | 2 × 10−4 | 0.23 | 47 | 51 | 48 | |
| AURKAPS1 | Aurora kinase A pseudogene 1 | 2 × 10−4 | 0.23 | 541 | 570 | 563 | |
| SPN | Sialophorin | 2 × 10−4 | 0.23 | 96 | 103 | 83 | |
| ARAP3 | ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 3 | IFNG‐inducible (GRIT) | 3 × 10−4 | 0.23 | 115 | 124 | 111 |
Note: Gray shading signifies p < .05 FDR.
Abbreviation: FDR, false discovery rate.
3.8. Comparing the top ABMR‐increased transcripts in DSA‐negative ABMR and in DSA‐positive mABMR
We separately identified the top genes increased in DSA‐negative mABMR versus No rejection and in DSA‐positive mABMR versus No rejection.
The top genes increased by ABMR were virtually identical in DSA‐negative and DSA‐positive mABMR, both by fold change and p value (Figure 3). Figure 3A plots the top genes by fold changes in DSA‐negative mABMR on the y‐axes, against those in DSA‐positive mABMR on the x‐axis. Each dot represents a probe set. IFNG‐inducible chemokines CXCL9/10/11, as well as NK genes such as CCL4 and GNLY, were most strongly increased in both DSA‐negative and DSA‐positive mABMR. The fold changes are slightly lower in DSA‐negative i.e. lower intensity. Figure 3B plots the top genes by p value. The most significantly increased genes in both DSA‐negative and DSA‐positive mABMR were NK genes and IFNG‐inducible genes. The p values are lower in DSA‐negative mABMR, probably reflecting fewer biopsies and lower intensity.
FIGURE 3.
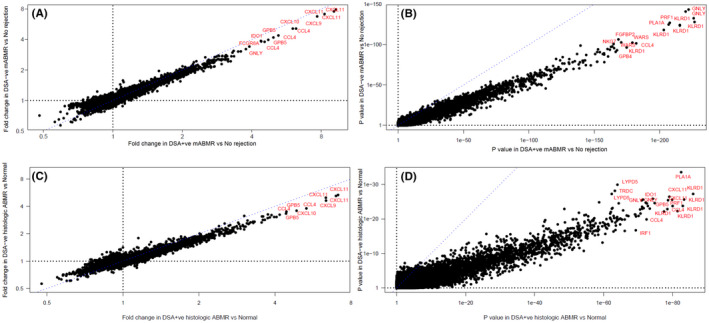
Scatterplots showing probe set (A) fold change in DSA‐negative mABMR biopsies versus No rejection biopsies (y‐axis) and DSA‐positive mABMR biopsies versus No rejection biopsies (x‐axis); (B) p values for the same class comparisons; (C) fold change in DSA‐negative histologic ABMR biopsies versus histologic Normal/AKI biopsies (y‐axis) and DSA‐positive histologic ABMR biopsies versus histologic Normal/AKI biopsies (x‐axis); and (D) p values for the same class comparisons. Blue dashes show the 1:1 line in each plot
The findings were similar when we used histologic diagnoses of ABMR (Figure 3C and D).
The top 10 ABMR‐associated genes by p value were listed for DSA‐positive mABMR in Table 8 and DSA‐negative mABMR in Table 9. In both DSA‐negative and DSA‐positive mABMR, 13 of the top 20 genes were NK expressed.
TABLE 8.
Top 20 genes by p value increased in DSA‐positive mABMR (E, F, L, N = 248) versus No rejection
| Gene symbol | Gene name | Transcript set annotation | p value | FDR | No rejection | DSA‐positive mABMR |
|---|---|---|---|---|---|---|
| KLRD1 | Killer cell lectin‐like receptor subfamily D, member 1 | NK; ABMR‐RAT | 2 × 10−227 | 1 × 10−222 | 41 | 96 |
| GNLY | Granulysin | NK; ABMR‐RAT | 8 × 10−223 | 1 × 10−218 | 53 | 208 |
| PLA1A | Phospholipase A1 member A | IFNG‐inducible (GRIT) | 4 × 10−216 | 3 × 10−212 | 231 | 623 |
| PRF1 | Perforin 1 (pore forming protein) | NK; ABMR‐RAT | 2 × 10−208 | 1 × 10−204 | 87 | 180 |
| CCL4 | Chemokine (C‐C motif) ligand 4 | NK; ABMR‐RAT | 8 × 10−183 | 4 × 10−179 | 73 | 331 |
| WARS | Tryptophanyl‐tRNA synthetase | IFNG‐inducible (GRIT) | 7 × 10−180 | 3 × 10−176 | 557 | 1108 |
| FGFBP2 | Fibroblast growth factor binding protein 2 | NK; ABMR‐RAT | 3 × 10−169 | 1 × 10−165 | 40 | 102 |
| GBP4 | Guanylate binding protein 4 | IFNG‐inducible (GRIT) | 4 × 10−167 | 1 × 10−163 | 225 | 570 |
| S1PR5 | Sphingosine‐1‐phosphate receptor 5 | NK; ABMR‐RAT | 7 × 10−166 | 2 × 10−162 | 19 | 30 |
| NKG7 | Natural killer cell group 7 sequence | NK; ABMR‐RAT | 2 × 10−165 | 5 × 10−162 | 164 | 293 |
| TRDV3 | T cell receptor delta variable 3 | NK; ABMR‐RAT | 2 × 10−164 | 4 × 10−161 | 13 | 31 |
| CXCL11 | Chemokine (C‐X‐C motif) ligand 11 | IFNG‐inducible (GRIT) | 2 × 10−163 | 4 × 10−160 | 20 | 165 |
| TRDC | T cell receptor delta constant | NK; ABMR‐RAT | 3 × 10−161 | 6 × 10−158 | 52 | 131 |
| LYPD5 | LY6 | ABMR‐RAT | 2 × 10−158 | 4 × 10−155 | 16 | 30 |
| CD160 | CD160 molecule | NK; ABMR‐RAT | 1 × 10−153 | 2 × 10−150 | 25 | 63 |
| CCL4L1 | Chemokine (C‐C motif) ligand 4‐like 1 | NK; ABMR‐RAT | 5 × 10−153 | 8 × 10−150 | 28 | 92 |
| KLRF1 | Killer cell lectin‐like receptor subfamily F, member 1 | ABMR‐RAT, NKB | 6 × 10−153 | 9 × 10−150 | 22 | 41 |
| IDO1 | Indoleamine 2,3‐dioxygenase 1 | IFNG‐inducible (GRIT) | 2 × 10−151 | 3 × 10−148 | 103 | 452 |
| SH2D1B | SH2 domain containing 1B | NK; ABMR‐RAT, NKB | 3 × 10−144 | 4 × 10−141 | 12 | 24 |
| GBP1 | Guanylate binding protein 1, interferon‐inducible | IFNG‐inducible (GRIT) | 1 × 10−143 | 2 × 10−140 | 305 | 870 |
Note: Gray shading signifies NK cell‐expressed genes.
Abbreviation: FDR, false discovery rate.
TABLE 9.
Top 20 genes by p value increased in DSA‐negative mABMR (E, F, L, N = 150) versus No rejection
| Gene symbol | Gene name | Transcript set annotation | p value | FDR | No rejection | DSA‐negative mABMR |
|---|---|---|---|---|---|---|
| GNLY | Granulysin | ABMR‐RAT | 4.17 × 10−144 | 2.07 × 10−139 | 53 | 182 |
| KLRD1 | Killer cell lectin‐like receptor subfamily D, member 1 | ABMR‐RAT | 2.18 × 10−133 | 3.60 × 10−129 | 33 | 75 |
| PRF1 | Perforin 1 (pore forming protein) | ABMR‐RAT | 8.66 × 10−128 | 8.57 × 10−124 | 87 | 165 |
| PLA1A | Phospholipase A1 member A | IFNG‐inducible (GRIT) | 4.87 × 10−125 | 3.44 × 10−121 | 231 | 549 |
| FGFBP2 | Fibroblast growth factor binding protein 2 | ABMR‐RAT | 3.78 × 10−107 | 1.87 × 10−103 | 40 | 93 |
| WARS | Tryptophanyl‐tRNA synthetase | IFNG‐inducible (GRIT) | 1.80 × 10−103 | 8.11 × 10−100 | 640 | 1196 |
| CCL4 | Chemokine (C‐C motif) ligand 4 | ABMR‐RAT | 2.77 × 10−102 | 1.05 × 10−98 | 73 | 276 |
| NKG7 | Natural killer cell group 7 sequence | ABMR‐RAT | 1.61 × 10−101 | 5.70 × 10−98 | 164 | 275 |
| KLRC3 | Killer cell lectin‐like receptor subfamily C, member 3 | ABMR‐RAT | 1.67 × 10−99 | 5.51 × 10−96 | 7 | 14 |
| TRDV3 | T cell receptor delta variable 3 | ABMR‐RAT | 1.91 × 10−98 | 5.91 × 10−95 | 13 | 27 |
| CXCL11 | Chemokine (C‐X‐C motif) ligand 11 | IFNG‐inducible (GRIT) | 6.10 × 10−98 | 1.78 × 10−94 | 20 | 142 |
| S1PR5 | Sphingosine‐1‐phosphate receptor 5 | ABMR‐RAT | 2.38 × 10−96 | 6.21 × 10−93 | 19 | 27 |
| CD160 | CD160 molecule | ABMR‐RAT | 7.45 × 10−95 | 1.76 × 10−91 | 25 | 56 |
| GBP4 | Guanylate binding protein 4 | IFNG‐inducible (GRIT) | 9.55 × 10−92 | 1.97 × 10−88 | 225 | 507 |
| KLRF1 | Killer cell lectin‐like receptor subfamily F, member 1 | ABMR‐RAT, NKB | 1.46 × 10−91 | 2.90 × 10−88 | 22 | 38 |
| GZMB | Granzyme B (granzyme 2, cytotoxic T‐lymphocyte‐associated serine esterase 1) | ABMR‐RAT | 4.12 × 10−89 | 7.29 × 10−86 | 56 | 149 |
| TRDC | T cell receptor delta constant | ABMR‐RAT | 4.58 × 10−89 | 7.82 × 10−86 | 39 | 120 |
| IDO1 | Indoleamine 2,3‐dioxygenase 1 | IFNG‐inducible (GRIT) | 4.97 × 10−89 | 8.20 × 10−86 | 103 | 400 |
| KLRC1 | Killer cell lectin‐like receptor subfamily C, member 1 | ABMR‐RAT | 3.27 × 10−85 | 4.76 × 10−82 | 15 | 41 |
| GBP1 | Guanylate binding protein 1, interferon‐inducible | IFNG‐inducible (GRIT) | 3.19 × 10−81 | 4.16 × 10−78 | 305 | 780 |
Note: Gray shading signifies NK cell‐expressed genes.
Abbreviation: FDR, false discovery rate.
The findings were similar in fully developed mABMR: the top genes in DSA‐positive FABMR (Table S1) and DSA‐negative FABMR (Table S1) were nearly identical, dominated by the same NK cell‐associated transcripts (shaded and bolded) and IFNG‐inducible transcripts.
The top 10 increased transcripts shared between DSA‐positive and DSA‐negative mABMR and FABMR are summarized and compared in Table 10, showing that many of the top 10 are shared, and most are NK cell‐associated.
TABLE 10.
Top genes increased in expression in all mABMR (E, F, L) and in FABMR, compared with No rejection a
| Biopsy groups compared | Top 10 genes (by p value) increased b | |
|---|---|---|
| All mABMR | All DSA‐positive mABMR versus No rejection |
KLRD1 , GNLY , PLA1A , PFR1 , CCL4 , WARS , FGFBP2 , GBP4, S1PR5, NKG7 Of top 10 genes in DSA‐positive mABMR, all 10 are in top 20 in DSA‐negative mABMR. |
| All DSA‐negative mABMR versus No rejection |
GNLY , KLRD1 , PRF1 , PLA1A , FGFBP2 , WARS , CCL4 , NKG7 , KLRC3, TRDV3 Of top 10 genes in DSA‐negative mABMR, 9 are in top 20 in DSA‐positive mABMR. |
|
| Full‐developed mABMR | DSA‐positive FABMR versus No rejection |
KLRD1 , LYPD5 , GNLY , WARS , PLA1A , PRF1 , S1PR5 , TRDV3, KLRC3 , KLRF1 Of top 10 genes in DSA‐positive mABMR, all 10 are in top 20 in DSA‐negative |
| DSA‐negative FABMR versus No rejection |
LYPD5 , PLA1A , WARS , KLRD1 , PRF1 , GNLY , KLRC3 , S1PR5 , SH2D1B, KLRF1 Of top 10 genes in DSA‐negative, all 10 are in top 20 in DSA‐positive |
|
Note: NK‐associated transcripts are bolded and underlined.
The top decreased genes were less significant but two endothelial cell genes (ESM1 and F8) were highly ranked in the top 10 decreased in all.
Genes common between DSA‐positive top 10 and DSA‐negative top 10 mABMR versus No rejection genes are underlined, bolded, and italicized.
3.9. Survival
We calculated death‐censored graft survival at 3 years post‐biopsy based on one random biopsy per transplant (Figure 4). Figure 4A shows that EABMR had better survival than FABMR or LABMR, as in earlier analyses. 12 Within each mABMR group or in all mABMR, DSA status (negative, positive, or unknown) had little impact on graft survival: in EABMR (Figure 4B), FABMR (Figure 4C), LABMR (Figure 4D), or all mABMR (Figure 4E). Similarly DSA status did not predict survival in No rejection (Figure 4F), similar to our previous report. 24 Thus, the presence of mABMR and the stage of mABMR affect survival after biopsy, but DSA status does not.
FIGURE 4.
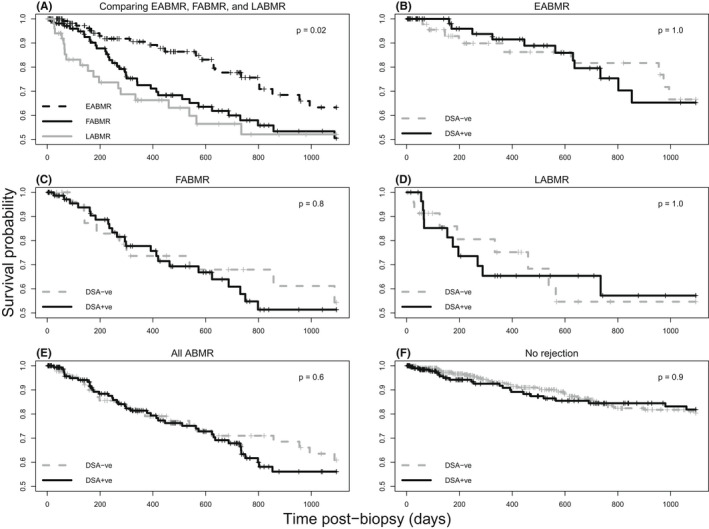
Kaplan–Meier curves comparing survival probabilities among biopsy groups in mABMR or in No rejection biopsies. One random biopsy per patient was selected for the analysis, and biopsies were not used if they did not have a valid follow‐up time or status. Survival probabilities are compared among (A) mABMR biopsy groups: EABMR, FABMR, and LABMR (N = 305); (B) DSA‐positive and DSA‐negative EABMR biopsies (N = 122); (C) DSA‐positive and DSA‐negative FABMR biopsies (N = 111); (D) DSA‐positive and DSA‐negative LABMR biopsies (N = 54); (E) DSA‐positive and DSA‐negative all‐ABMR biopsies (N = 268); and (F) DSA‐positive and DSA‐negative No rejection biopsies (N = 639)
3.10. How does DSA‐negative mABMR transition over time?
Establishing whether DSA‐negative mABMR transitions to DSA‐positive mABMR (and vice versa) or no rejection cannot be formally answered in this cross‐sectional study of indication biopsies because follow‐up biopsies are not generally indicated as SOC. The fact that DSA‐positivity rises in the ABMR population from EABMR to FABMR before falling in LABMR complicates this question.
However, among the relatively few ABMR patients with later biopsies, in most pairs, the DSA status remained the same in the second biopsy. DSA‐negative mABMR transitioned to DSA‐positive mABMR more often (8/32 = 25%) than the reverse (DSA‐positive mABMR to DSA‐negative mABMR 5/61 = 8.2%). In terms of transitions to no rejection, more DSA‐negative mABMR (12/32) transitioned to no rejection, compared with 10/57 DSA‐positive ABMR (p = .036, Chi‐square test), compatible with previous observations that DSA‐negative ABMR can sometimes be “transient.”
4. DISCUSSION
This study defined the relationship between DSA status and the timing, histology, C4d, molecular features, and outcomes in mABMR. DSA and C4d positivity were most frequent in the most active mABMR state – FABMR. C4d was strongly associated with DSA even in No rejection biopsies. As previously reported, 11 DSA‐negative mABMR usually occurred in sensitized patients – 60% were PRA‐positive. Compared with DSA‐positive mABMR, DSA‐negative mABMR was earlier posttransplant, mostly C4d‐negative, and had milder ABMR activity, for example, lower ptc‐lesion scores and ABMR‐related transcript set and classifier scores. In genome‐wide class comparisons, we were unable to find genes with significantly different expression in DSA‐negative versus DSA‐positive mABMR. Moreover, despite lower activity, DSA‐negative mABMR had the same top mABMR‐associated genes as DSA‐positive mABMR, suggesting that the mechanisms were the same and extending the previous finding of transcript similarity. 14 DSA‐negative mABMR had comparable effects on graft loss compared with DSA‐positive mABMR.
These results using exclusively molecular definitions of ABMR confirm some findings in studies of ABMR biopsies defined by histologic MVI. 13 , 14 Those studies also found that DSA‐negative ABMR was earlier than DSA‐positive and sometimes transient, and less frequently C4d‐positive. Unlike the present study the histologic MVI cases had a survival advantage over DSA‐positive ABMR. However, our mABMR cases were later on average than in histologic MVI cases. The differences could be influenced by the use of MVI to define ABMR. Especially in early biopsies, we have previously noted that some histologic MVI (ptc‐ and g‐lesions) may be mABMR‐negative and possibly related to other microvascular injuries. 32 The diagnosis of ABMR is challenging in the early posttransplant period 33 and more studies comparing the details of MVI and mABMR changes during this time are needed.
DSA interpretation even in expert laboratories is often challenging. The INTERCOMEX study uses SOC definitions of DSA by the local laboratory because there is no universal agreement on what technologies and cutoffs should be used. Donor‐recipient HLA genotyping may be incomplete; interpretation of DSA can differ among local laboratories; and there is no central “gold standard.” 34 , 35 Technical challenges include non‐specific bead reactivity, batch variation in bead manufacture, prozones, bead saturation, and limited reproducibility of quantitation (mean fluorescence intensity). 36 Moreover, it remains difficult to predict the pathogenicity of a DSA, in part due to the lack of understanding of the conditions that IgG must meet to be “ABMR‐effector‐competent” (see below). 37 Complement‐binding DSA assays introduced to identify pathogenic DSA 38 , 39 have not proven to be consistently superior to conventional DSA testing. 40 Moreover, mABMR can be subtle: subthreshold molecular mABMR‐like changes associated with DSA are demonstrable in some biopsies currently considered to have no rejection. 24 The future classification of ABMR is likely to be increasingly based on recognizing gradients beyond the current “binary” approaches. 41
C4d staining is negative in most DSA‐negative biopsies in both histologic 13 , 14 and molecular ABMR, and this observation invites us to reconsider the relationship of C4d staining to both the DSA status and the ABMR disease state. C4d deposition is strongly linked to DSA but can occur without HLA DSA or ABMR: for example, diffuse C4d staining occurs in ABO incompatibility without ABMR and is induced by monoclonal antibody bamlanivimab without ABMR. 42 We suggest that C4d deposition and ABMR are often associated but actually represent independent molecular processes: C4d requires widespread binding of many DSA IgGs that may or may not include ABMR‐effector‐competent IgGs, whereas ABMR is mediated by relatively small numbers of ABMR‐effector‐competent IgGs that may or may meet the requirements for C4d deposition.
Because AKI molecular changes regress and atrophy‐fibrosis changes progress over many months posttransplant, 31 DSA‐negative mABMR being 1.5 years earlier will have more unresolved AKI‐related change and less time and stage‐related features such as cg‐lesions, hyalinosis, and arterial fibrous intimal thickening. The i‐ and t‐scores are slightly higher in DSA‐negative mABMR but not close to being diagnostic for TCMR, and combined with the negative TCMRProb scores suggest the residual effects of AKI in the transplant process. The elevated FICOL transcripts, which are induced by transplant AKI and regress slowly over the first several years, 31 , 43 , 44 also support this interpretation.
Alternative explanations for DSA‐negative mABMR are not necessarily mutually exclusive because there may be several ways that ABMR can occur without DSA being identified. As listed in the introduction, these include incomplete donor genotyping, complete absorption of DSA by kidney, DSA directed against non‐HLA alloantigens, autoantibody such as AT1R, 45 and antibody‐independent NK cell recognition of missing self. This study cannot dismiss any of the proposed explanations. However, the absence of PRA in 40% of DSA‐negative mABMR cases excludes inadequate genotyping in these cases. The high protection that perfect HLA‐matching provides against mABMR, and the high success of the national distribution program for 100% sensitized people 46 argue that the targets of the missing antibody in DSA‐negative mABMR cases are mostly HLA alloantigens. But rare cases of non‐HLA‐encoded polymorphic molecules on capillary endothelium can probably evoke an ABMR‐effector‐competent alloantibody and mediate mABMR in highly sensitized patients without a DSA being detectable by existing tests. As stated earlier, the possibility that important high‐affinity DSAs are absorbed by the kidney has not been supported by elution studies. 18 , 40
Autoantibodies are common in chronic diseases causing tissue injury and their role in pathogenesis is often controversial. Autoantibodies often accompany mABMR and DSA, 23 , 47 , 48 are frequently associated with DSA, 22 and their role is being discussed in the ongoing STAR process. 35 Some issues make them unlikely to be the principal explanation of DSA‐negative mABMR: for example, it is not clear how these autoantibodies distinguish autoantigen in 150 g of donor tissue from that in the much more abundant host tissue (e.g., 70 000 g). Moreover, autoantibodies before transplant have not been shown to predict the development of ABMR posttransplant. However, synergy between DSA and autoantibodies such as AT1R in ABMR must be considered. 22 , 49
It is possible we are not detecting all the effects of antibody against donor antigens by the existing histologic and molecular approaches. 50 , 51 , 52 A different type of antibody‐mediated stress could exist in a minority of cases that evades the current definitions. ABMR‐like stress is capable of operating over a very long period at a low level, as we previously published. 24
The present study does not suggest that any unique mechanisms are operating only in DSA‐negative mABMR. For example, if DSA‐negative ABMR was mediated only by “missing self” receptor recognition, and DSA‐positive ABMR only by Fc receptor triggering by DSA, we would have expected some differences in the ABMR‐associated genes. The striking association of NK cell transcripts with all ABMR 52 is compatible with a role of missing‐self triggering of NK cells, 16 , 17 potentially augmenting Fc receptor recognition of bound DSA. Although the present class comparisons cannot exclude a unique mechanism operating only in DSA‐negative mABMR, our working hypothesis pending new evidence is that DSA‐positive and DSA‐negative ABMR use the same mechanisms i.e. Fc receptor triggering by an ABMR‐effector competent DSA (detectable or not) possibly augmented by missing self receptor triggering.
These results summarized in Table 11 suggest a model compatible with the literature. In the model, the mechanisms of all ABMR—DSA‐negative and positive—are the same, involving NK cell triggering by CD16a Fc receptors plus missing‐self recognition. ABMR is mediated by ABMR‐effector‐competent donor‐specific IgG, which assembles IgG‐antigen multimers on the endothelium that in turn assemble Fc receptor multimers for NK activation. 53 C1q binding by IgG does require IgG hexamers for activating C1q37, and we suggest that ABMR‐effector‐competence also requires multimerization. ABMR‐effector‐competent IgGs can appear early in DSA production before circulating DSA and C4d deposition is detectable. Some ABMR‐effector‐competent DSA may escape detection by bead assays because the configuration of immobilized HLA proteins on beads does not simulate the dynamic expression on endothelium required for multimerization and Fc receptor triggering. ABMR‐effector‐competent IgGs increase with time, accompanied by the larger DSA response (epitope spreading), making circulating DSA and C4d deposition detectable. Circulating DSA often accompanies ABMR‐effector‐competent DSA IgG but is neither sufficient nor necessary. 24
TABLE 11.
Summary of features of DSA‐negative mABMR compared with DSA‐positive mABMR
| Earlier on average by about 1.5 years (2.4 years versus 3.9 years for DSA‐positive) |
| Mostly sensitized: 60% PRA‐positive (higher than in No rejection). |
| Almost all (86%) are C4d‐negative (versus 56% for DSA‐positive) |
| Less histologic and molecular activity |
| Mildly elevated i‐score (not diagnostic for TCMR) and fibrillar collagen transcript set score compared to DSA‐positive mABMR probably reflecting earlier time posttransplant |
| Three‐year graft survival is similar to DSA‐positive |
Suggested model for DSA‐negative and DSA‐positive ABMR compatible with current data:
|
The goal 14 of treatment should be suppression of mABMR activity and arrest of disease progression. Suppression of measured DSA if present could also be a useful surrogate, assuming that treatments effective for suppressing DSA will also control the ABMR‐effector‐competent IgG. However, merely converting DSA‐positive ABMR to DSA‐negative ABMR will not benefit patients unless there is other evidence for disease modification. Potential strategies for reducing all DSA, including ABMR‐effector‐competent DSA, include targeting the neonatal Fc receptor, 54 , 55 proteasome inhibition, anti‐IL6 56 or anti‐IL6 receptor, 57 and plasma cell depleting monoclonals such as anti CD38 ‐ daratumumab 58 or felzartamab. (A trial of felzartamab is beginning—ClinicalTrials.gov #NCT05021484.) Direct targeting of NK cell Fc receptors or missing‐self receptors should be considered in both DSA‐positive and DSA‐negative ABMR. Complement inhibition will probably not be effective. For example, classical complement pathway inhibition by anti‐C1s monoclonal antibody BIVV009 in established mABMR reduced C4d staining but not mABMR activity. 59
In summary, the processes operating in DSA‐negative ABMR are highly similar to DSA‐positive ABMR. Examination of genome‐wide transcript expression, gene sets, classifiers, and clinical and histologic features found differences in intensity, timing, stage, and C4d but no evidence for major mechanistic differences. This strategy cannot exclude all mechanistic differences but should have revealed major differences if they existed. Therefore, we find no evidence to suggest that the management should be different for DSA‐negative versus DSA‐positive ABMR. With this in mind, DSA‐negative ABMR should be included in clinical ABMR trials (presumably stratified). Finally, definitive studies on the mechanisms operating in both DSA‐positive and DSA‐negative ABMR remain an important target for the development of new interventions to improve management and outcomes.
DISCLOSURE
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. P.F. Halloran holds shares in Transcriptome Sciences Inc., a University of Alberta research company dedicated to developing molecular diagnostics, supported in part by a licensing agreement between TSI and Thermo Fisher, and by a research grant from Natera. P.F. Halloran is a consultant to Natera. The other authors have declared no conflict of interest exists.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
This research has been principally supported by grants from Genome Canada, Canada Foundation for Innovation, the University of Alberta Hospital Foundation, the Alberta Ministry of Advanced Education and Technology, the Mendez National Institute of Transplantation Foundation, and Industrial Research Assistance Program. Partial support was also provided by funding from a licensing agreement with the One Lambda division of Thermo Fisher. Dr. Halloran held a Canada Research Chair in Transplant Immunology until 2008 and currently holds the Muttart Chair in Clinical Immunology.
We thank our valued clinicians in the INTERCOMEX study group who partnered with us for this study by contributing biopsies and feedback (Michael Picton, Timm Heinbokel, Harold Yang, Seth Narins, Carmen Lefaucheur, Alexandre Loupy, Marek Myslak, Bertram Kasiske, Arthur Matas, and Arjang Djamali).
Halloran PF, Madill‐Thomsen KS, Pon S, et al. Molecular diagnosis of ABMR with or without donor‐specific antibody in kidney transplant biopsies: Differences in timing and intensity but similar mechanisms and outcomes. Am J Transplant. 2022;22:1976‐1991. doi: 10.1111/ajt.17092
DATA AVAILABILITY STATEMENT
CEL files are available on the Gene Expression Omnibus website (GSE124203).
REFERENCES
- 1. Haas M, Loupy A, Lefaucheur C, et al. The Banff 2017 kidney meeting report: revised diagnostic criteria for chronic active T cell‐mediated rejection, antibody‐mediated rejection, and prospects for integrative endpoints for next‐generation clinical trials. Am J Transplant. 2018;18(2):293‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Loupy A, Haas M, Roufosse C, et al. The Banff 2019 kidney meeting report (I): updates on and clarification of criteria for T cell‐ and antibody‐mediated rejection. Am J Transplant. 2020;20(9):2318‐2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Halloran PF, Wadgymar A, Ritchie S, Falk J, Solez K, Srinivasa NS. The significance of the anti‐class I antibody response. I. Clinical and pathologic features of anti‐class I‐mediated rejection. Transplantation. 1990;49(1):85‐91. [DOI] [PubMed] [Google Scholar]
- 4. Bohmig GA, Exner M, Habicht A, et al. Capillary C4d deposition in kidney allografts: a specific marker of alloantibody‐dependent graft injury. J Am soc Nephrol. 2002;13(4):1091‐1099. [DOI] [PubMed] [Google Scholar]
- 5. Regele H, Bohmig GA, Habicht A, et al. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: a contribution of humoral immunity to chronic allograft rejection. J Am soc Nephrol. 2002;13(9):2371‐2380. [DOI] [PubMed] [Google Scholar]
- 6. Collins AB, Schneeberger EE, Pascual MA, et al. Complement activation in acute humoral renal allograft rejection: diagnostic significance of C4d deposits in peritubular capillaries. J Am soc Nephrol. 1999;10(10):2208‐2214. [DOI] [PubMed] [Google Scholar]
- 7. Mauiyyedi S, Pelle PD, Saidman S, et al. Chronic humoral rejection: identification of antibody‐mediated chronic renal allograft rejection by C4d deposits in peritubular capillaries. J Am soc Nephrol. 2001;12(3):574‐582. [DOI] [PubMed] [Google Scholar]
- 8. Einecke G, Sis B, Reeve J, et al. Antibody‐mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant. 2009;9(11):2520‐2531. [DOI] [PubMed] [Google Scholar]
- 9. Halloran PF, Chang J, Famulski K, et al. Disappearance of T cell‐mediated rejection despite continued antibody‐mediated rejection in late kidney transplant recipients. J Am soc Nephrol. 2015;26(7):1711‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sis B, Allanach K, Bunnag S, Mueller TFF, Halloran PF. Microarrays detect deteriorating C4d(−) human renal allografts with ongoing antibody‐mediated injury. Am J Transplant. 2008;8:275.18211504 [Google Scholar]
- 11. Halloran PF, Famulski KS, Chang J. A probabilistic approach to histologic diagnosis of antibody‐mediated rejection in kidney transplant biopsies. Am J Transplant. 2017;17(1):129‐139. [DOI] [PubMed] [Google Scholar]
- 12. Reeve J, Bohmig GA, Eskandary F, et al. Assessing rejection‐related disease in kidney transplant biopsies based on archetypal analysis of molecular phenotypes. JCI Insight. 2017;2(12):e94197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Senev A, Coemans M, Lerut E, et al. Histological picture of antibody‐mediated rejection without donor‐specific anti‐HLA antibodies: clinical presentation and implications for outcome. Am J Transplant. 2019;19(3):763‐780. [DOI] [PubMed] [Google Scholar]
- 14. Callemeyn J, Lerut E, de Loor H, et al. Transcriptional changes in kidney allografts with histology of antibody‐mediated rejection without anti‐HLA donor‐specific antibodies. J Am soc Nephrol. 2020;31(9):2168‐2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Callemeyn J, Senev A, Coemans M, et al. Missing self‐induced microvascular rejection of kidney allografts: a population‐based study. J Am soc Nephrol. 2021;32(8):2070‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koenig A, Mezaache S, Callemeyn J, et al. Missing self‐induced activation of NK cells combines with non‐complement‐fixing donor‐specific antibodies to accelerate kidney transplant loss in chronic antibody‐mediated rejection. J Am soc Nephrol. 2021;32(2):479‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koenig A, Chen CC, Marcais A, et al. Missing self triggers NK cell‐mediated chronic vascular rejection of solid organ transplants. Nat Commun. 2019;10(1):5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bachelet T, Couzi L, Lepreux S, et al. Kidney intragraft donor‐specific antibodies as determinant of antibody‐mediated lesions and poor graft outcome. Am J Transplant. 2013;13(11):2781‐3051. [DOI] [PubMed] [Google Scholar]
- 19. Cahan A. Diagnosis is driven by probabilistic reasoning: counter‐point. Diagnosis (Berl). 2016;3(3):99‐101. [DOI] [PubMed] [Google Scholar]
- 20. Dragun D, Catar R, Philippe A. Non‐HLA antibodies in solid organ transplantation: recent concepts and clinical relevance. Curr Opin Organ Transplant. 2014;18(4):430‐435. [DOI] [PubMed] [Google Scholar]
- 21. Tanaka T, Ebata T, Tajima A, Kinoshita K, Okumura K, Yagita H. Beta2‐microglobulin required for cell surface expression of blastocyst MHC. Biochem Biophys Res Commun. 2005;332(1):311‐317. [DOI] [PubMed] [Google Scholar]
- 22. Lefaucheur C, Viglietti D, Bouatou Y, et al. Non‐HLA agonistic anti‐angiotensin II type 1 receptor antibodies induce a distinctive phenotype of antibody‐mediated rejection in kidney transplant recipients. Kidney Int. 2019;96(1):189‐201. [DOI] [PubMed] [Google Scholar]
- 23. Jackson AM, Wiebe C, Hickey MJ. The role of non‐HLA antibodies in solid organ transplantation: a complex deliberation. Curr Opin Organ Transplant. 2020;25(6):536‐542. [DOI] [PubMed] [Google Scholar]
- 24. Madill‐Thomsen KS, Bohmig GA, Bromberg J, et al. Donor‐specific antibody is associated with increased expression of rejection transcripts in renal transplant biopsies classified as no rejection. J Am soc Nephrol. 2021;32(11):2743‐2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Halloran PF, Reeve J, Akalin E, et al. Real time central assessment of kidney transplant indication biopsies by microarrays: the INTERCOMEX study. Am J Transplant. 2017;17(11):2851‐2862. [DOI] [PubMed] [Google Scholar]
- 26. Reeve J, Madill‐Thomsen KS, Halloran PF, INTERCOMEX Study Group . Using ensembles of machine learning classifiers to maximize the accuracy and stability of molecular biopsy interpretation. Am J Transplant. 2019;19(S3):452‐453. [DOI] [PubMed] [Google Scholar]
- 27. RCT . (2019) R: A language and environment for statistical computing. R Foundation for statistical Computing. http://www.r‐project.org/. Published 2019. Updated 2019. Accessed.
- 28. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Madill‐Thomsen K, Perkowska‐Ptasinska A, Bohmig GA, et al. Discrepancy analysis comparing molecular and histology diagnoses in kidney transplant biopsies. Am J Transplant. 2020;20(5):1341‐1350. [DOI] [PubMed] [Google Scholar]
- 30. Einecke G, Reeve J, Mengel M, et al. Expression of B cell and immunoglobulin transcripts is a feature of inflammation in late allografts. Am J Transplant. 2008;8(7):1434‐1443. [DOI] [PubMed] [Google Scholar]
- 31. Venner JM, Famulski KS, Reeve J, Chang J, Halloran PF. Relationships among injury, fibrosis, and time in human kidney transplants. JCI Insight. 2016;1(1):e85323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Halloran PF, Sellares J. Microcirculation lesions alone are not reliable for identifying antibody‐mediated rejection. Am J Transplant. 2013;13(7):1931‐1932. [DOI] [PubMed] [Google Scholar]
- 33. Cornell LD. Histopathologic features of antibody mediated rejection: the Banff classification and beyond. Front Immunol. 2021;12(3918):718122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reed EF, Rao P, Zhang Z, et al. Comprehensive assessment and standardization of solid phase multiplex‐bead arrays for the detection of antibodies to HLA. Am J Transplant. 2013;13(7):1859‐1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tambur AR, Campbell P, Claas FH, et al. Sensitization in transplantation: assessment of risk (STAR) 2017 working group meeting report. Am J Transplant. 2018;18(7):1604‐1614. [DOI] [PubMed] [Google Scholar]
- 36. Reed EF, Rao P, Zhang Z, et al. Comprehensive assessment and standardization of solid phase multiplex‐bead arrays for the detection of antibodies to HLA‐drilling down on key sources of variation. Am J Transplant. 2013;13(11):3050‐3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diebolder CA, Beurskens FJ, de Jong RN, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343(6176):1260‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loupy A, Lefaucheur C, Vernerey D, et al. Complement‐binding anti‐HLA antibodies and kidney‐allograft survival. N Engl J Med. 2013;369(13):1215‐1226. [DOI] [PubMed] [Google Scholar]
- 39. Chin C, Chen G, Sequeria F, et al. Clinical usefulness of a novel C1q assay to detect immunoglobulin G antibodies capable of fixing complement in sensitized pediatric heart transplant patients. J Heart Lung Transplant. 2011;30(2):158‐163. [DOI] [PubMed] [Google Scholar]
- 40. Courant M, Visentin J, Linares G, et al. The disappointing contribution of anti‐human leukocyte antigen donor‐specific antibodies characteristics for predicting allograft loss. Nephrol Dial Transplant. 2018;33(10):1853‐1863. [DOI] [PubMed] [Google Scholar]
- 41. Callemeyn J, Ameye H, Lerut E, et al. Revisiting the changes in the Banff classification for antibody‐mediated rejection after kidney transplantation. Am J Transplant. 2021;21(7):2413‐2423. [DOI] [PubMed] [Google Scholar]
- 42. Klomjit N, El Ters M, Adam BA, et al. Diffuse C4d staining of peritubular capillaries in renal allograft following bamlanivimab therapy. Am J Transplant. 2022;22(1):289‐293. [DOI] [PubMed] [Google Scholar]
- 43. Halloran PF, Bohmig GA, Bromberg JS, et al. Discovering novel injury features in kidney transplant biopsies associated with TCMR and donor aging. Am J Transplant. 2021;21(5):1725‐1739. [DOI] [PubMed] [Google Scholar]
- 44. Kreepala C, Famulski KS, Halloran PF. Fundamental concepts regarding graft injury and regeneration: Tissue injury, tissue quality and recipient factors. In: Kirk AD, Knechtle SJ, Larsen CP, Madsen JC, Pearson TC, Webber SA, eds. Textbook of organ transplantation. 1st ed. John Wiley & Sons, Ltd.; 2014:99‐118. [Google Scholar]
- 45. Crespo M, Llinas‐Mallol L, Redondo‐Pachon D, et al. Non‐HLA antibodies and epitope mismatches in kidney transplant recipients with histological antibody‐mediated rejection. Front Immunol. 2021;12:703457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jackson KR, Segev DL. Rethinking incompatibility in kidney transplantation. Am J Transplant. 2021;22:1031‐1036. [DOI] [PubMed] [Google Scholar]
- 47. Jackson AM, Delville M, Lamarthee B, Anglicheau D. Sensitization to endothelial cell antigens: unraveling the cause or effect paradox. Hum Immunol. 2019;80(8):614‐620. [DOI] [PubMed] [Google Scholar]
- 48. Delville M, Lamarthee B, Pagie S, et al. Early acute microvascular kidney transplant rejection in the absence of anti‐HLA antibodies is associated with preformed IgG antibodies against diverse glomerular endothelial cell antigens. J Am soc Nephrol. 2019;30(4):692‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sorohan BM, Ismail G, Leca N, et al. Angiotensin II type 1 receptor antibodies in kidney transplantation: an evidence‐based comprehensive review. Transplant Rev (Orlando). 2020;34(4):100573. [DOI] [PubMed] [Google Scholar]
- 50. Sellares J, Reeve J, Loupy A, et al. Molecular diagnosis of antibody‐mediated rejection in human kidney transplants. Am J Transplant. 2013;13(4):971‐983. [DOI] [PubMed] [Google Scholar]
- 51. Halloran PF, Pereira AB, Chang J, et al. Microarray diagnosis of antibody‐mediated rejection in kidney transplant biopsies: an international prospective study (INTERCOM). Am J Transplant. 2013;13(11):2865‐2874. [DOI] [PubMed] [Google Scholar]
- 52. Venner JM, Hidalgo LG, Famulski KS, Chang J, Halloran PF. The molecular landscape of antibody‐mediated kidney transplant rejection: evidence for NK involvement through CD16a fc receptors. Am J Transplant. 2015;15(5):1336‐1348. [DOI] [PubMed] [Google Scholar]
- 53. Zhang Y, Boesen CC, Radaev S, et al. Crystal structure of the extracellular domain of a human fc gamma RIII. Immunity. 2000;13(3):387‐395. [DOI] [PubMed] [Google Scholar]
- 54. Jordan SC, Ammerman N, Vo A. Implications of fc neonatal receptor (FcRn) manipulations for transplant Immunotherapeutics. Transplantation. 2020;104(1):17‐23. [DOI] [PubMed] [Google Scholar]
- 55. Kiessling P, Lledo‐Garcia R, Watanabe S, et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: a randomized phase 1 study. Sci Transl Med. 2017;9(414):eaan1208. [DOI] [PubMed] [Google Scholar]
- 56. Bohmig GA, Durr M, Jilma B, et al. Three‐month results of a phase 2 trial evaluating Clazakizumab in late antibody‐mediated rejection ‐ early impact of Interleukin‐6 blockade on donor‐specific antibody levels, rejection morphology and gene expression. Am J Transplant. 2020;20(S3):404. [Google Scholar]
- 57. Choi J, Aubert O, Vo A, et al. Assessment of tocilizumab (anti‐Interleukin‐6 receptor monoclonal) as a potential treatment for chronic antibody‐mediated rejection and transplant glomerulopathy in HLA‐sensitized renal allograft recipients. Am J Transplant. 2017;17(9):2381‐2389. [DOI] [PubMed] [Google Scholar]
- 58. Doberer K, Klager J, Gualdoni GA, et al. CD38 antibody daratumumab for the treatment of chronic active antibody‐mediated kidney allograft rejection. Transplantation. 2021;105(2):451‐457. [DOI] [PubMed] [Google Scholar]
- 59. Eskandary F, Jilma B, Muhlbacher J, et al. Anti‐C1s monoclonal antibody BIVV009 in late antibody‐mediated kidney allograft rejection‐results from a first‐in‐patient phase 1 trial. Am J Transplant. 2018;18(4):916‐926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
CEL files are available on the Gene Expression Omnibus website (GSE124203).