

Abstract
Background and Aims
Recent development of multiple treatments for human hepatocellular carcinoma (HCC) has allowed for the selection of combination therapy to enhance the effectiveness of monotherapy. Optimal selection of therapies is based on both HCC and its microenvironment. Therefore, it is critical to develop and validate preclinical animal models for testing clinical therapeutic solutions.
Approach and Results
We established cell line–based or patient‐derived xenograft–based humanized‐immune‐system mouse models with subcutaneous and orthotopic HCC. Mice were injected with human‐specific antibodies (Abs) to deplete human immune cells. We analyzed the transcription profiles of HCC cells and human immune cells by using real‐time PCR and RNA sequencing. The protein level of HCC tumor cells/tissues or human immune cells was determined by using flow cytometry, western blotting, and immunohistochemistry. The HCC tumor size was measured after single, dual‐combination, and triple‐combination treatment using N‐(1ʹ,2‐Dihydroxy‐1,2ʹ‐binaphthalen‐4ʹ‐yl)‐4‐methoxybenzenesulfonamide (C188‐9), bevacizumab, and pembrolizumab. In this study, human immune cells in the tumor microenvironment were strongly selected and modulated by HCC, which promoted the activation of the IL‐6/Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling pathway in tumor cells and led to augmented HCC proliferation and angiogenesis by releasing angiogenic cytokines in humanized‐immune‐system mice with HCC. In particular, intratumor human cluster of differentiation–positive (hCD14+) cells could produce IL‐33 through damage‐associated molecular pattern/Toll‐like receptor 4/activator protein 1, which up‐regulated IL‐6 in other intratumor immune cells and activated the JAK2/STAT3 pathway in HCC. Specific knockdown of the CD14 gene in human monocytes could impair IL‐33 production induced by cell lysates. Subsequently, we evaluated the in vivo anti‐HCC effect of C188‐9, bevacizumab, and pembrolizumab. The results showed that the anti‐HCC effect of triple‐combination therapy was superior to that of single or dual treatments.
Conclusions
Humanized‐immune‐system HCC mouse models are suitable for identifying targets from cancer and immune components and for testing combinational therapies.
Abbreviations
- Ab
antibody
- AKT
protein kinase B
- AP‐1
activator protein 1
- BM
bone marrow
- CCL
C‐C motif chemokine ligand
- CXCL
C‐X‐C motif chemokine ligand
- DAMP
damage‐associated molecular pattern
- DE
differentially expressed
- EDU
5‐ethynyl‐2′‐deoxyuridine
- HCC
hepatocellular carcinoma
- hCD
human cluster of differentiation
- HLA
human leukocyte antigen
- HMGB1
high mobility group box 1
- HSC
hematopoietic stem cell
- IFN‐gamma
interferon‐gamma
- ILC
innate lymphoid cell
- JAK2
Janus kinase 2
- LPS
lipopolysaccharide
- MAPK
mitogen‐activated protein kinase
- MDSC
myeloid‐derived suppressor cell
- M‐MDSC
mononuclear MDSC
- MMP
matrix metalloproteinase
- MØ1
type 1 macrophage
- MØ2
type 2 macrophage
- MVD
microvessel density
- NK
natural killer
- NSG
NOD.Cg‐Prkdc scid Il2rg tm 1 Wjl/SzJ
- p‐
phospho‐
- PCA
principal component analysis
- PD‐1
programmed death 1
- PDGF
platelet‐derived growth factor
- PD‐L1
programmed death ligand 1
- PDX
patient‐derived xenograft
- shRNA
short‐hairpin RNA
- STAT
signal transducer and activator of transcription
- TAM
tumor‐associated macrophage
- Tc
T cytotoxic
- TcCM
central memory Tc cell
- TcEM
effector memory Tc cell
- Th
T‐helper
- ThCM
central memory Th cell
- ThEM
effector memory Th cell
- TIL
tumor‐infiltrating leukocyte
- TLR
Toll‐like receptor
- VCSA
microvessel cross‐sectional area
- VD3
1,25‐dihydroxyvitamin D3
Hepatocellular carcinoma (HCC) remains one of the most common causes of cancer‐related deaths and has a high recurrence rate.( 1 ) HCC tumorigenesis is impacted by chronic inflammation induced by liver injury,( 2 , 3 , 4 ) which promotes HCC progression and development.( 5 , 6 , 7 , 8 ) However, the mechanisms that underlie the crosstalk between human HCC and the human immune system remain poorly understood. Therefore, examination of the interactions between the human immune system and HCC, which may improve existing therapy and help to identify strategies for HCC treatment, is warranted.
HCC cells attract various immune cells into the tumor microenvironment by releasing multiple chemokines, including C‐X‐C motif chemokine ligand 12 (CXCL12), C‐C motif chemokine ligand 1 (CCL1), CCL22, CXCL16, CCL5, CXCL10, and CXCL11.( 9 , 10 , 11 , 12 , 13 ) Furthermore, HCC cells can modulate the activities and functions of immune cells and drive the acquisition of tumor‐associated phenotypes in these cells.( 14 , 15 , 16 , 17 ) Tumor‐associated immune cells, including T‐helper (Th) type 2 (Th2) cells, type 2 innate lymphoid cells (ILC2s), tumor‐associated regulatory T cells, tumor‐associated macrophages (TAMs), tumor‐associated neutrophils, and myeloid‐derived suppressor cells (MDSCs) are responsible for producing various cytokines (IL‐1, IL‐6, IL‐8, and IL‐10), growth factors (VEGF, platelet‐derived growth factor [PDGF], EGF, HGF, FGF, and insulin‐like growth factor), matrix metalloproteinase 2 (MMP‐2) and MMP‐9, and tissue inhibitor of metalloproteinase 1 (TIMP‐1) and TIMP‐2 to create an optimal environment for the proliferation, survival, and metastasis of tumor cells.( 18 , 19 , 20 , 21 ) Previous studies have demonstrated that numerous signaling pathways dysregulated in HCC were associated with these HCC tumor–associated immune cells.( 22 , 23 , 24 ) For example, IL‐8 released from TAMs may enhance the epithelial–mesenchymal transition (EMT) in HCC through the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3)/Snail pathway.( 22 ) Similarly, activation of the JAK2/STAT3 pathway, induced by IL‐6 from TAMs, also played a critical role in HCC stem‐cell expansion.( 23 ) In addition, increased functional activity in the rat sarcoma/rapidly accelerated fibrosarcoma/mitogen‐activated protein kinase (MAPK), phosphoinositide 3‐kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin and JAK/STAT pathways, caused by multiple growth factors released from MDSCs, were also observed in human HCC.( 24 ) Although this evidence demonstrated the interactions between the human immune system and HCC, countless other mechanisms remain unclear, particularly because of the lack of a robust in vivo human cancer–immune system model. The absence of sufficient models also limits the development of therapeutics targeting tumor‐associated immune cells and their modulation pathways.
Over time, various monotherapies that target HCC proliferation, angiogenesis, and immune resistance have been developed.( 25 , 26 ) These include, for example, treatments counteracting elevated IL‐6 signaling. Serum IL‐6 levels are elevated in patients with HCC, compared with healthy individuals, which might enhance HCC proliferation and angiogenesis through activation of the JAK2/STAT3 signaling pathway.( 27 , 28 ) Hence, tocilizumab (IL‐6 receptor blockade) and N‐(1ʹ,2‐Dihydroxy‐1,2ʹ‐binaphthalen‐4ʹ‐yl)‐4‐methoxybenzenesulfonamide (C188‐9) (STAT3 inhibitor) were developed and achieved optimistic results in reducing HCC growth in vivo by impairing IL‐6 signaling from TAMs to HCC.( 6 , 29 )
VEGF is essential for tumor growth and angiogenesis. Bevacizumab, an anti‐VEGF monoclonal antibody (Ab), has been applied to patients with HCC and has proven to be effective in clinical trials.( 30 , 31 ) Similarly, immune checkpoint blockade therapies, such as nivolumab and pembrolizumab (programmed death 1 [PD‐1] blockade) and atezolizumab (programmed death ligand 1 [PD‐L1] blockade) also exerted certain levels of anti‐HCC effects by protecting and recovering T‐cell function in animal models and patients.( 32 ) Despite the development of different monotherapies, patients with HCC only exhibit partial responses to these treatments.( 33 ) In recent years, combination therapy (polytherapy) has been considered for synergistic efficacy, which may improve the overall survival of patients with HCC by reducing their resistance to monotherapy.( 33 ) Previous studies have demonstrated that the combination of anti–IL‐6 and anti–PD‐L1 treatment could ameliorate the anti‐HCC efficacy in vivo.( 34 ) In clinical settings, a combination of bevacizumab and atezolizumab treatment led to effective antitumor immune response in patients with HCC.( 35 ) Furthermore, combined blockade of IL‐6 and VEGF receptor also enhanced antitumor activity, as was reported in another study.( 36 ) Therefore, it is anticipated that the combined therapy of anti–IL‐6–or IL‐6–related signaling pathways, anti‐VEGF, and immune checkpoint blockade may be more efficacious.
We have shown that an HCC mouse model can be generated through the engraftment of human HCC cell lines or patient‐derived xenografts (PDXs) into immunodeficient mice bearing human leukocyte antigen (HLA)‐matched human immune systems (humanized mice).( 37 ) Intriguingly, some of the HCC‐PDX tumors regressed in the humanized mice following pembrolizumab and ipilimumab treatment, suggesting that the HCC humanized mice may represent an emerging platform for the evaluation of cancer monotherapy or combination therapy.( 37 ) In the present study, we demonstrate that the human immune system in humanized mice may enhance HCC proliferation and angiogenesis through a network involving damage‐associated molecular patterns (DAMPs), Toll‐like receptors (TLRs), cytokines, and intracellular protein phosphorylation. More importantly, triple‐combination therapy targeting STAT3‐induced HCC proliferation, VEGF‐related angiogenesis, and the PD‐1 immune checkpoint resulted in a combinational anti‐HCC effect in our humanized mouse model.
Materials and Methods
MICE
NOD.Cg‐Prkdc scid Il2rg tm 1 Wjl/SzJ (NSG) mice (The Jackson Laboratory) and humanized mice were bred and generated, respectively, as described earlier.( 37 , 38 ) All manipulations with mice were approved by Institutional Animal Care and Use Committee in Agency for Science, Technology and Research. All animal experimental procedures were conducted in accordance to the approved protocols and Guide for the Care and Use of Laboratory Animals. All human fetal liver and HCC Patient tumor tissues with written consent were obtained from guardians of donors, and in accordance with the ethical guidelines of Kandang Kerbau (KK) Women’s and Children’s Hospital (Singapore) and National University Hospital (Singapore). HCC humanized mice were generated by engrafting different HCC cell lines or PDXs subcutaneously or orthotopically (intrasplenic injection with 2 × 106 purified and live human tumor cells in 50 µL of saline) according to HLA‐A (HCC‐PDX tumor 1# [A*02 and A*11] matched with hematopoietic stem cell [HSC] 1# [A*02 and A*11]; HCC‐PDX tumor 2# [A*02 and A*11] matched with HSC 1# [A*02 and A*11]; HCC‐PDX tumor 3# [A*02 and A*01] matched with HSC 2# [A*02 and A*01]; HCC‐PDX tumor 4# [A*02 and A*11] matched with HSC 1# [A*02 and A*11]; and HepG2 and LM3 cell lines [A*02 and A*24] matched with HSC 3# [A*02 and A*24]) (Supporting Table S1). To deplete specific immune‐cell subsets in immune humanized mice, anti–human cluster of differentiation 45 (anti‐hCD45) (a human leukocyte marker), anti‐hCD4 (a human Th cell marker), anti‐hCD8 (a human Tc cell maker), anti‐hCD14 (a human monocyte marker), anti‐hCD19 (a human B‐cell marker), and anti‐hCD56 (a human natural killer [NK] cell marker) (50 µg/mouse) were given i.v. weekly. For the evaluation of drug efficacy, small molecules or Abs were injected into the mice either alone or in combination. Briefly, C188‐9 dissolved in DMSO was injected intraperitoneally (75 mg/kg) on a daily basis, whereas anti–human IL‐33 Ab (50 µg/mouse), bevacizumab, and pembrolizumab (5 mg/kg) were injected i.v. on a weekly basis. Further methodological details can be found in the Supporting Information.
Results
The Human Immune System Promotes HCC Proliferation and Angiogenesis
To explore the role of the humanized immune system with respect to HCC, we engrafted HCC‐PDX tumor tissue into NSG humanized mice and hCD45‐depleted humanized mice that were treated with anti‐hCD45 Ab to remove the humanized immune system 2 weeks after engraftment. Interestingly, the size and weight of the HCC tumors and spleens from humanized mice increased compared with those from the NSG and hCD45‐depleted humanized mice (Fig. 1A). Similar results were also observed in HCC‐PDX tumors from different donors, in both subcutaneous and orthotopic models (Supporting Figs. S1A‐S6A). The expression of MKI67 (Ki67) on the HCC‐PDX tumor was quantified by using immunohistochemical analysis, which indicated a significant increase in the number of Ki67+ cells in the HCC tumor from humanized mice (Fig. 1B and Supporting Figs. S1B‐S6B). Moreover, the 5‐ethynyl‐2′‐deoxyuridine (EDU) levels in isolated and purified HCC‐PDX tumor cells from the three groups were analyzed using flow cytometry. Similarly, we observed that the HCC‐PDX tumor cells isolated from humanized mice had the highest EDU level among these groups (Fig. 1C). Furthermore, angiogenesis was quantified in the vascular endothelium of the HCC‐PDX tumor by counting the vessels with a lumen (CD31 immunohistochemical staining that was both negative and positive) (Fig. 1D and Supporting Figs. S1C‐S6C). Both the microvessel density (MVD) and the microvessel cross‐sectional area (VCSA) were calculated (Fig. 1D and Supporting Figs. S1D‐S6D). More vascular invasion was observed in HCC tumors in humanized mice than in both NSG and hCD45‐depleted humanized mice, suggesting that the humanized immune system could augment HCC tumor proliferation and angiogenesis. To dissect the underlying molecular mechanisms that regulated the proliferation and angiogenesis affected by the humanized immune system, the LM3 (HCC) cell line was selected because of its sharing the HLA‐A type with the HCC‐PDX tumor (all are HLA‐A type 2). In concordance with the results from HCC‐PDX tumors, LM3 tumors in humanized mice were larger than those in NSG and humanized mice treated with anti‐hCD45 Abs (Fig. 1E). After 3 weeks of engraftment, the LM3 tumors were harvested, and the purified LM3 HCC cells were subjected to RNA sequencing (Fig. 1F). Principal component analysis (PCA) of all expressed genes (Fig. 1G) and heatmap analysis of differentially expressed (DE) genes (Fig. 1H and Supporting Table S2) revealed that the transcriptomic profiles among these groups were distinct and well separated, indicating that LM3 cells were strongly influenced by the immune system. Results from the hCD45‐depleted group indicated that the immune system had already exerted some impact on the tumors even though it had been removed 2 weeks after tumor implantation. Ingenuity pathway analysis revealed that the acute‐phase‐response signaling pathways were highly modulated in LM3 cells from humanized mice, including triggering receptor expressed on myeloid cells 1, IL‐6, high mobility group box 1 (HMGB1), and IL‐10 signaling pathways, compared with NSG and hCD45‐depleted humanized mice (Fig. 1I). Immunohistochemistry staining of HCC‐PDX tumors and western blot analysis of isolated HCC‐PDX tumor cells indicated that the JAK2/STAT3 of the tumor was highly phosphorylated in humanized mice (Fig. 1J,K). Hence, we anticipated that the humanized immune system in our model may enhance HCC‐PDX proliferation through the activation of the JAK2/STAT3 intracellular signaling pathways in the HCC cells.
FIG. 1.
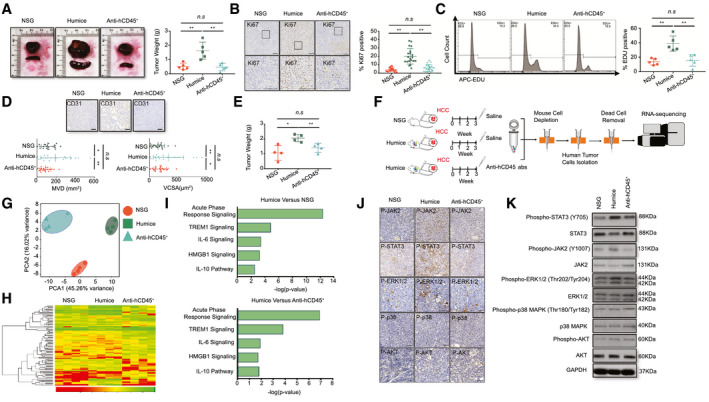
Human immune system–promoted HCC proliferation and angiogenesis in the Humice model. (A) Representative images of HCC‐PDX tumor 1# and spleens, and the statistical analysis of HCC‐PDX tumor 1# weight from NSG (n = 5), Humice (n = 5), and Humice treated with anti–hCD45 Ab (50 ug/mouse/week for 4 weeks; n = 5). (B) Immunohistochemistry staining of Ki67 in HCC‐PDX tumor 1# tissues and the statistical analysis of the proportion of Ki67+ cells (n = 20, four randomly selected photographs/each sample [total of five samples]). (C) The EDU staining of the HCC‐PDX tumor 1# cells isolated from NSG Humice and Humice treated with anti‐hCD45 Ab and the statistical analysis of EDU+ cells (n = 5). (D) Immunohistochemistry staining of CD31 in tumor tissues and the statistical analysis of the MVD and VCSA (n = 5 samples/group; four images were randomly selected for each sample for quantification [total of 20]). (E) Five million cells from the HCC cell line LM3 were engrafted into different groups. The tumor weight was analyzed after 3 weeks (n = 4). (F) The methodology and workflow for RNA sequencing of isolating LM3 tumor cells from different mice groups. (G) PCA of LM3 tumor cells from different groups. (H) The heatmap of top DE genes in RNA‐sequencing results. (I) The signal pathway analysis of DE genes from LM3 tumor cells between Humice and NSG mice or Humice and Humice treated with anti‐hCD45 Abs. (J) Immunohistochemistry staining of p‐STAT3, p‐JAK2, p‐ERK1/2, p‐p38, and p‐AKT using HCC‐PDX tumor 1#. (K) Western blot analysis of p‐STAT3, p‐JAK2, p‐ERK1/2, p‐p38, and p‐AKT from isolated HCC‐PDX tumor cells from different groups. Abbreviations: APC, allophycocyanin; ERK, extracellular signal‐regulated kinases; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Humice, humanized mice; n.s, not significant; TREM1, triggering receptor expressed on myeloid cells 1. *P < 0.05; **P < 0.01.
Intratumor hCD14 Cells Contribute to Promoting HCC Proliferation and Angiogenesis
To validate the functions of human immune cells from different organs on HCC in vitro, hCD45+ cells were isolated from blood, spleen, bone marrow (BM), and tumor‐infiltrating leukocytes (TILs) of humanized mice bearing HepG2 cells and LM3 cells (with HLA‐A2, similar to HCC‐PDX tumors 1#‐4#), and the average composition of immune‐cell subsets was analyzed (Fig. 2A). A colony formation assay was performed by two‐dimensional co‐culturing of hCD45+ cells and HepG2 cells or LM3 cells for 10 days. The results showed that only TILs could strongly stimulate HepG2 cell and LM3 cells proliferation when they were compared with immune cells from other origins (Fig. 2B). Subsequently, hCD45+ cells were isolated from blood and TILs from humanized mice bearing HCC‐PDX tumor 1#. The mRNA expression levels of various chemokines, cytokines, and growth factors were analyzed by RT–quantitative real‐time PCR; IL‐33, IL‐6, CCL15, VEGF, CCL16, PDGF, IL‐8, IL‐10, and CXCR7 were highly expressed in hCD45+ TILs (Fig. 2C and Supporting Table S3). To further identify the subtypes of hCD45+ TILs that influenced HCC proliferation and angiogenesis, different immune‐cell subsets were depleted in vivo using humanized mice bearing HCC‐PDX tumor 1# (Supporting Fig. S7). The weight of each HCC‐PDX tumor 1# was measured 4 weeks after treatment, and the results revealed that hCD14+ or CD19+ cell depletion could reduce HCC tumor weight. In contrast, hCD8+ cell depletion increased tumor weight (Fig. 2D). Immunohistochemical staining of Ki67 and EDU flow cytometry assays showed a decrease in HCC proliferation after hCD14+ cell depletion (Fig. 2E,F and Supporting Fig. S8). Moreover, angiogenesis in the HCC tumor, indicated by the MVD and VCSA, was impaired by hCD14+ or hCD19+ cell depletion (Fig. 2G,H and Supporting Fig. S8). The presence and protein expression levels of phospho‐JAK2 (p‐JAK2) and p‐STAT3 in the HCC tumor and the purified HCC cells were examined by using immunohistochemistry (Fig. 2I,J and Supporting Fig. S8) and western blotting (Fig. 2K), respectively. Our results showed that only hCD14+ cell depletion could significantly diminish the expression of JAK2 and STAT3 in the tumor. In addition, hCD14+ or CD19+ cell depletion also altered the composition of human immune‐cell subsets of TILs (Fig. 2L,M). After the depletion of hCD14+ cells, the proportion of total CD8+ cells, central memory Th cells (ThCMs), effector memory cytotoxic T (Tc) cells (TcEMs), and central memory Tc cells (TcCMs) increased, whereas the proportion of effector memory Th cells (ThEMs), type 1 macrophages (MØ1s) or type 2 macrophages (MØ2s), and mononuclear MDSCs (M‐MDSCs) decreased (Fig. 2L). The total number of CD8+ cells, CD19+ cells, ThCMs, TcEMs, and TcCMs increased per gram of tumor tissue, whereas effector memory Th cells (ThEMs), MØ1s or MØ2s, and M‐MDSCs decreased in hCD14+ cell–depleted tumor tissues (Supporting Fig. S9A). Similarly, a depletion in hCD19+ cells also led to an increase in the percentage of total CD8+ cells and TcCMs and a decrease in the proportion of MØ2s (Fig. 2M). In terms of the cell number per gram of tumor tissue, total CD8+ cells, ThCMs, TcEMs, TcCMs, hCD14+hCD16− cells, and early‐stage MDSCs increased after hCD19+‐cell depletion (Supporting Fig. S9B). Taken together, the intratumor hCD14+ cells could stimulate HCC tumor proliferation and angiogenesis by modulating the profile of the immune cells in the tumor and plausibly through the activation of the IL6/JAK2/STAT3 signaling pathway.
FIG. 2.
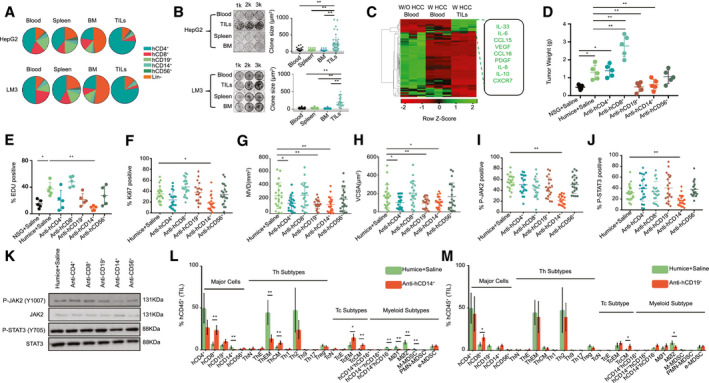
hCD14+ TILs are one of the major players in HCC proliferation and angiogenesis. (A) The average proportion of multiple human immune cells (hCD45+) in blood, spleen, BM, and TILs from HepG2 or LM3‐engrafted Humice, which were used in an HepG2 or LM3 co‐culture in vitro experiment. (B) The HCC clone‐formation analysis. HepG2 or LM3 cells are co‐cultured with human immune cells from blood, spleen, BM, and TILs of HepG2‐engrafted Humice for 10 days. The ratio was 1:10, exchanging new medium and adding new human immune cells every two days. The statistical analysis of the size of clone is shown (n = 50). (C) Total RNA was extracted from blood or hCD45+ TILs from Humice without the HCC‐PDX tumor and HCC‐PDX tumor 1#–bearing Humice. Gene expression profiles are analyzed using RT–quantitative real‐time PCR (n = 3). A heat map of the gene expression is shown. (D) HCC‐PDX tumor 1# was engrafted into NSG and Humice, which were treated with saline, anti–hCD4 Abs, anti‐hCD8 Abs, anti‐hCD19 Abs, anti‐hCD14 Abs, and anti‐hCD56 Abs. (E) The HCC‐PDX tumor 1# cells from different groups were analyzed using EDU staining (n = 5). (F‐J) Immunohistochemistry staining of Ki67, CD31, p‐JAK2 and p‐STAT3 were performed in HCC‐PDX tumor tissues. The statistical results of (F) Ki67+, (G) MVD, (H) VCSA, (I) p‐JAK2, and (J) p‐STAT3 are shown (n = 5 samples/group; four images were randomly selected for each sample for quantification [total of 20]). (K) Western blot analysis of the p‐STAT3 and p‐JAK2 level in isolated HCC‐PDX tumor cells from different groups. (L‐M) The changes in human immune cells’ proportion in TILs after saline and (L) anti‐hCD14 Ab or (M) anti‐hCD19 Ab injection (n = 5). Abbreviations: e‐MDSC, early‐stage MDSC; Humice, humanized mice; k, thousand; PMN‐MDSC, polymorphonuclear leukocyte–MDSC; TcE, effector Tc cell; TcN, naive cytotoxic T cells; ThE, effector Th cell; ThN, naive T helper cells; Treg, regulatory T cell; W, with; W/O, without. *P < 0.05; **P < 0.01.
Intratumor Human Immune Cells are Strongly Modulated by HCC Tumors
To characterize the phenotypes and functions of the humanized immune system in the absence or presence of an HCC tumor burden, we isolated multiple immune‐cell subsets, including CD4+, CD8+, CD14+, CD19+, and CD56+ cells, from the spleen and from among the TILs of humanized mice bearing an HCC‐PDX tumor and analyzed their RNA profiles (Fig. 3A). Through PCA and heatmap analysis, it was found that the profiles of CD4+ (Fig. 3B), CD8+ (Fig. 3C), CD14+ (Fig. 3D), CD19+ (Fig. 3E), and CD56+ (Fig. 3F) cells among the groups were distinct and well separated. The list of DE genes was detected and calculated for CD4+, CD8+, CD19+, CD14+, and CD56+ cells (Supporting Table S2). Despite minor differences between splenic immune cells from humanized mice with and without HCC, the intratumor human immune cells were significantly different, including in terms of the expression of surface markers, chemokines, cytokines, and functional proteins. For example, cell migration (CCL13 and CCL18), angiogenesis and invasion (CCL2, CCL7, CCL9, CCL10, IL‐8, IL‐10, PDGFA, VEGFA, EGF, MMP7, MMP9, and MMP19), and MØ2 type markers (CD163 and peroxisome proliferator–activated receptor gamma genes) were found to be up‐regulated in hCD14+ TILs, whereas MØ1 type maker (CD86) and phagocytosis markers (neutrophil cytosol factor 1 [NCF‐1], phosphatidylinositol 4‐phosphate 5‐kinase type‐1 gamma [PIP5K1C], vasodilator‐stimulated phosphoprotein [VASP], hematopoietic cell kinase [HCK], phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit gamma [PIK3CG], spleen tyrosine kinase [SYK], actin related protein 2/3 complex subunit 4 [ARPC4], AKT1, WASP family member 2 [WASF2], vav guanine nucleotide exchange factor 1 [VAV1], actin related protein 2/3 complex subunit 1B [ARPC1B], P21 (RAC1) activated kinase 1 [PAK1], cofilin 1 [CFL1] and AKT serine/threonine kinase 2 [AKT2]) were down‐regulated. These results suggested that HCC tumors could alter human immune cells in humanized mice, particularly on intratumor human immune cells.
FIG. 3.
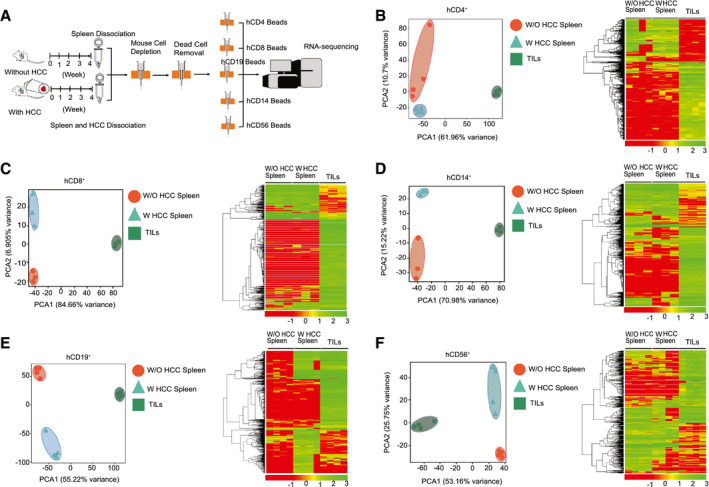
Human immune cells from TILs were modulated by HCC tumors. Multiple kinds of human immune cells are isolated from spleen (in HCC‐PDX tumor 1#–bearing humanized mice and non–HCC humanized mice) and HCC‐PDX tumor 1# tissue. PCA and heatmap analysis are performed using RNA‐sequencing results. (A) The workflow of isolation of multiple kinds of human immune cells for RNA sequencing. (B) Results of hCD4+ cells (human Th cells). (C) Results of hCD8+ cells (human Tc cells). (D) Results of hCD14+ cells (human monocytes). (E) Results of hCD19+ cells (human B cells). (F) Results of hCD56+ cells (human NK cells). Abbreviations: W, with; W/O, without.
IL‐33 Secreted From Intratumor hCD14+ Cells Up‐Regulates IL‐6 Expression in Multiple Intratumor Immune Cells and Enhances HCC Proliferation
Previous studies have shown that serum IL‐33 levels are higher in patients with HCC than in healthy donors.( 39 ) Consistent with these clinical findings, our results also verified the elevation of IL‐6 and IL‐33 expression in intratumor hCD45+ cells (Fig. 2C). In our HCC‐PDX humanized mouse model, IL‐33+ cells were only present in intratumor hCD14+ cells, and these HCC cells did not express IL‐33 (data not shown), whereas IL‐6+ cells were detected among the CD4+, CD14+, and lineage (Lin)− TILs (Fig. 4A‐C). Therefore, the correlation between the IL‐6 level in the captioned immune cells and the IL‐33 level in intratumor hCD14+ cells was analyzed in HCC samples from 10 patients. The results showed that the IL‐33 level in intratumor hCD14+ cells was closely correlated with the IL‐6 level in intratumor hCD4+ and hCD14+ cells but was weakly correlated with the intratumor hCD19+ cells and Lin− cells (Fig. 4A). Interestingly, ILC groups from Lin− cells were found in our model, particularly in BM and among TILs (Supporting Fig. S10A). The major ILC subtype in the BM and among TILs were ILC1 and ILC2, respectively (Supporting Fig. S10B). Meanwhile, the majority of the ILC2s could express IL‐6, whereas other ILC subsets, such as ILC1, NCR+ILC3, and NCR−ILC3, did not express IL‐6 (Fig. 4E). Interleukin 1 receptor‐like 1 (ST2) is a member of the IL‐1 receptor family, which is the receptor of IL‐33. We found that ST2 was highly expressed in intratumor CD4+, CD14+, and Lin− cells (Supporting Fig. S11A). Furthermore, the proportion of ST2+ IL‐6+ cells was much higher than that of ST2− IL‐6+ cells in CD4+, CD14+, and Lin− cells (Supporting Fig. S11B), suggesting that IL‐33 could modulate IL‐6 expression through the ST2 receptor. To confirm the regulatory effect of IL‐33 on HCC in vivo, anti–human IL‐33 Ab was injected into HCC‐PDX humanized mice to neutralize IL‐33. After 4 weeks of treatment, the tumor weight for these mice was drastically reduced compared with that of their saline‐injected counterparts (Fig. 4F). Meanwhile, human IL‐33 neutralization led to a decrease in the IL‐6 level in intratumor CD4+, CD14+, and Lin− cells (Fig. 4G) and led to lower activation of JAK2 and STAT3 in HCC cells (Fig. 4H). Furthermore, the MVD and VCSA suggested that IL‐33 was also involved in angiogenesis (Fig. 4I,J). After human IL‐33 neutralization, the proportions of intratumor Th2 and regulatory T cells decreased, whereas the proportion of TcEMs increased, suggesting that CD14+ monocyte–derived IL‐33 may also be involved in regulating other immune‐cell types in the tumor environment (Fig. 4K). These results suggested that IL‐33 from intratumor hCD14+ cells could up‐regulate IL‐6 expression in multiple cell types, thereby enhancing HCC proliferation.
FIG. 4.
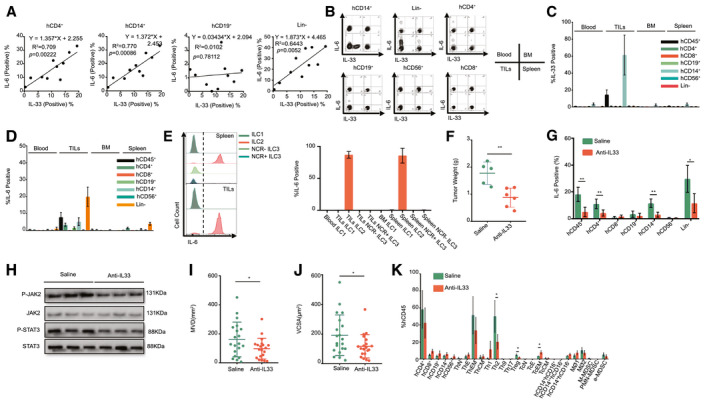
IL‐33 from CD14+ TILs could up‐regulate IL‐6 expression in TILs’ multiple immune cells and enhance HCC proliferation and angiogenesis. (A) Correlation of IL‐33 levels in monocytes and IL‐6 levels in CD4+, CD14+, CD19+, and Lin− TILs of patients with HCC. (B‐D) Expression of IL‐33 and IL‐6 in hCD4+, CD8+, CD19+, CD14+, and Lin− cells from blood, spleen, and BM and from among TILs in HCC‐PDX tumor 1# humanized mice models. The statistical results of (C) IL‐33 and (D) IL‐6 are shown. (E) IL‐6 expression in ILCs from spleen and TILs. The statistical result is shown. (F) HCC‐PDX tumor 1# weight after 4 weeks of treatment with saline and anti–IL‐33 Abs. (G) Changes in IL‐6 level in TILs’ multiple human immune cells after anti–human IL‐33 Ab treatment. (H) Western blot analysis of p‐STAT3 and p‐JAK2 from isolated HCC cells from saline and anti‐IL33 Ab treatment groups. (I‐J) Tumor angiogenesis analysis in HCC‐PDX tumor 1# tissues from saline and anti‐IL33 Ab treatment groups. The statistical results of (I) MVD and (J) VCSA are shown (n = 5 samples/group, four images were randomly selected for each sample for quantification [total of 20]). (K) Changes in the proportion of multiple human immune cells after anti‐IL33 Ab treatment in TILs. Abbreviations: e‐MDSC, early‐stage MDSC; PMN‐MDSC, polymorphonuclear leukocyte–MDSC; TcE, effector Tc cell; TcN, naive cytotoxic T cells; ThE, effector Th cell; ThN, naive T helper cells; Treg, regulatory T cell. *P < 0.05; **P < 0.01.
Co‐receptor CD14 Is Essential for IL‐33 Production Through Damp (HMGB1)/TLR4/Activator Protein 1 Pathway
To clarify the mechanism of IL‐33 production from intratumor hCD14+ cells only, we induced IL‐33 expression using primary human immune cells and U937 cells in vitro. We attempted to detect IL‐33 expression induced by using different reagents, including co‐culture with HepG2, Hep3B, Huh7, and PLC5 cell lines and stimulation by lipopolysaccharide (LPS), IL‐33, IL‐6, HepG2 lysates, Hep3B lysates, Huh7 lysates, and PLC5 lysates. We observed that only HepG2 lysates and LPS could induce high levels of IL‐33 in primary human immune cells (Fig. 5A). Meanwhile, we stimulated U937 cells using HepG2 lysates. However, no IL‐33 expression caused by the direct addition of HepG2 lysates to U937 cells was observed (Fig. 5B). U937 is a monocytic cell line, and low levels of CD14 and 1,25‐dihydroxyvitamin D3 (VD3) may induce CD14 expression in U937; these are results we validated (Supporting Fig. S12). We observed that VD3 could induce CD14 expression and that HepG2 lysates stimulated the induction of IL‐33 in VD3‐treated U937 cells (Fig. 5B). The flow cytometry plot also indicated that only CD14+ cells expressed IL‐33 (Fig. 5B). In VD3‐treated U937 cells, HepG2 lysates, Hep3B lysates, PLC5 lysates, and LPS could induce high levels of IL‐33. We also performed specific targeted hCD14 gene knockdown in U937 cells using five short‐hairpin RNAs (shRNAs) (Supporting Fig. S13). TRC1.5 vector controls and shRNAs expressing lentiviral particles were generated using third‐generation lentivirus packaging systems. The results indicated that hCD14 shRNAs could significantly impair CD14 expression induced by VD3 in U937 cells (Supporting Fig. S13B). As a result, the intracellular level of IL‐33 induced by VD3 and HepG2 lysates was down‐regulated in hCD14‐silenced U937 cells (Supporting Fig. S13C). In conclusion, CD14 is an essential molecule for IL‐33 production. By analyzing the network of highly expressed genes in intratumor CD14+ cells, we found that activator protein 1 (AP‐1) was as a key transcription factor because of its regulation of many of the up‐regulated genes. Similar results, including activation of the AP‐1 pathway, can also be observed in the RT–quantitative real‐time PCR heatmap of CD45+ TILs (Supporting Fig. S14A), U937 cells treated with VD3, HepG2 lysates (Supporting Fig. S14B), and healthy donor CD45+ cells treated with HepG2 lysates (Supporting Fig. S14C). To further validate the role of AP‐1 signaling in IL‐33 expression, we treated primary hCD45+ cells with LPS, HCC lysates (HepG2), (R)‐Ethyl 6‐(N‐(2‐chloro‐4 fluorophenyl) sulfamoyl) cyclohex‐1‐enecarboxylate (TAK‐242) (inhibitor of TLR4) and (E,E,Z,E)‐3‐Methyl‐7‐(4‐methylphenyl)‐9‐(2,6,6‐trimethyl‐1‐cyclohexen‐1‐yl)‐2,4,6,8‐nonatetraenoic acid (SR‐11302) (inhibitor of AP‐1). The results showed that either blockage of TLR4 or inhibition of AP‐1 can suppress LPS‐ or HCC lysate–induced IL‐33 expression (Fig. 5D). Additionally, we found that LPS and HCC lysates could increase Jun family (JUNB, c‐JUN, and JUND) protein levels in hCD14+ cells. However, TAK‐242 and SR‐11302 can mitigate the elevation of Jun Proto‐Oncogene (JUN)‐family proteins (Fig. 5E). We also confirmed that AP‐1 can bind multiple motifs in the promoter region of IL‐33 in U397 cells (Supporting Fig. S15A,B). HCC lysates can enhance the binding ability of AP‐1 (JUN family) to the promoter of IL‐33. TAK‐242 and SR‐11302 can mitigate the effect of HCC lysates (Fig. 5F). Moreover, HMGB1 protein is a type of DAMP that is typically located in the nucleus as a nuclear factor. However, it can also be translocated to the cytoplasm and released into the extracellular matrix, linking inflammation and HCC. HMGB1 may activate several intracellular signaling pathways (NF‐κB or AP‐1) that regulate monocyte‐induced cytokine synthesis by binding TLRs 2, 4, 5, and 9. Earlier studies have also demonstrated that HMGB1 is highly expressed in HCC tissues compared with paratumor and normal tissues and that it also promotes HCC proliferation and angiogenesis. To determine the role of HMGB1 in IL‐33 synthesis, we treated primary hCD45+ cells with HCC lysates and anti‐HMGB1 neutralizing Abs. The results indicated that anti‐HMGB1 neutralizing Abs could attenuate HCC lysate–induced IL‐33 levels in CD14+ monocytes (Fig. 5G). Furthermore, increased supernatant HMGB1 levels were observed in interferon‐gamma (IFN‐gamma)–treated HCC cells (Fig. 5H). Overall, IL‐33 could be synthesized by CD14+ cells through the DAMP(HMGB1)/TLR4/AP‐1 pathway.
FIG. 5.
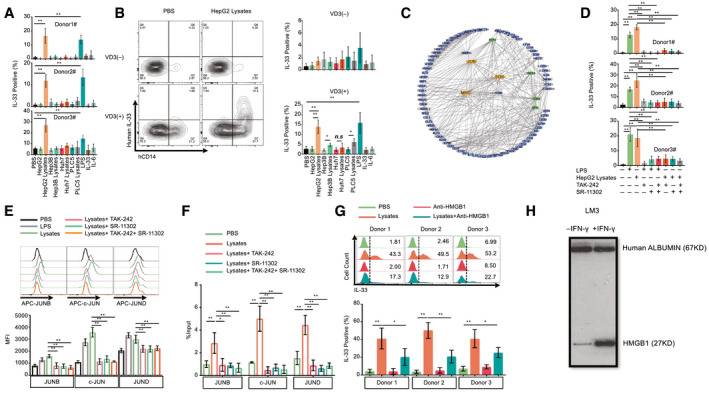
CD14 is an essential molecule for IL‐33 generation induced by DAMPs (HMGB1) from HCC through the TLR4/AP‐1 pathway. (A) IL‐33 expression levels in hCD14+ cells after stimulating hCD34− cells from three healthy donors with different reagents (n = 3). (B) The IL‐33 level in U937 cells following stimulation by different reagents with or without VD3 pretreatment (n = 3). (C) Network for highly expressed transcription factors in hCD14+ TILs. (D) IL‐33 expression levels in hCD14+ cells combinatorically treated with HepG2 lysates, TAK‐242 (TLR4 inhibitor), and SR‐11302 (AP‐1 transcription factor inhibitor). (E) JUN protein levels, including JUNB, c‐JUN, and JUND, in U937 cells (pretreated with VD3) following combinatorial treatment with HepG2 lysates, TAK‐242 and SR‐11302. (F) ChIP analysis was performed in U937 cells using anti‐JUNB Ab, anti–c‐JUN Ab, and anti‐JUND. ChIP DNA was analyzed using real‐time PCR (n = 4). (G) The effect of HMGB1 neutralizing Ab on lysate‐induced IL‐33 expression (n = 3). (H) Western blot analysis of HMGB1 levels from the supernatant after IFN‐gamma treatment of LM3 cells for 24 hours. Abbreviations: ChIP, chromatin immunoprecipitation; MFI, mean fluorescence intensity; n.s, not significant; Q, quadrant. *P < 0.05; **P < 0.01.
hCD14+ Monocytes Among TILs are Crucial for Combinatorial Immunotherapy on HCC
The above data proved that the presence of intratumor CD14+ cells enhances HCC proliferation and angiogenesis by activating the JAK2/STAT3 pathway or releasing angiogenic cytokines (VEGF and so on). HCC‐PDX humanized mice were treated with C188‐9 (STAT3 inhibitor) or bevacizumab (VEGF inhibitor) alone or in combination with pembrolizumab, and the additive effects were observed following treatment (Fig. 6A,B). Next, the efficacy and safety of triple‐combination therapy using C188‐9, bevacizumab, and pembrolizumab were evaluated and showed a more significant anti‐HCC effect (Fig. 6C). Similar results were consistently observed from subcutaneous (Supporting Figs. S1G, S2G and S3G) and orthotopic mouse models reconstituted with PDXs from different donors (Supporting Figs. S4D, S5D and S6D). The Ki‐67 immunohistochemical staining revealed that HCC tumors treated with triple‐combination therapy had the lowest level of proliferation (Fig. 6D and Supporting Fig. S15). Besides this, MVD and VCSA results also indicated that triple‐combination therapy could reduce angiogenesis (Fig. 6E,F and Supporting Fig. S15). Interestingly, the phosphorylation of STAT3 in HCC tumors was reduced after triple‐combination therapy, albeit with an increase in the tumor size following pembrolizumab treatment (Fig 6G,I and Supporting Fig. S16). Meanwhile, the expression of JAK2 was unchanged after triple‐combination therapy (Fig. 6H,I and Supporting Fig. S16). To further investigate the role of individual human immune‐cell types in this triple‐combination therapy, we compared the anti‐HCC effect of triple‐combination therapy when hCD14 and hCD8 cells were depleted separately in both subcutaneous (HCC‐PDX tumor 1#) (Supporting Fig. S17) and orthotopic models (HCC‐PDX tumors 1# and 2#) (Supporting Figs. S18 and S19). The results revealed that the anti‐HCC effect of triple‐combination therapy were greatly hampered without hCD8 or hCD14 cells in HCC humanized mice. It confirmed that these two human immune‐cell types were both critical in this combination therapy. Taken together, the results showed that triple‐combination therapy using C188‐9, bevacizumab, and pembrolizumab could significantly increase the efficacy of the anti‐HCC response in vivo compared with monotherapy or dual‐therapy.
FIG. 6.
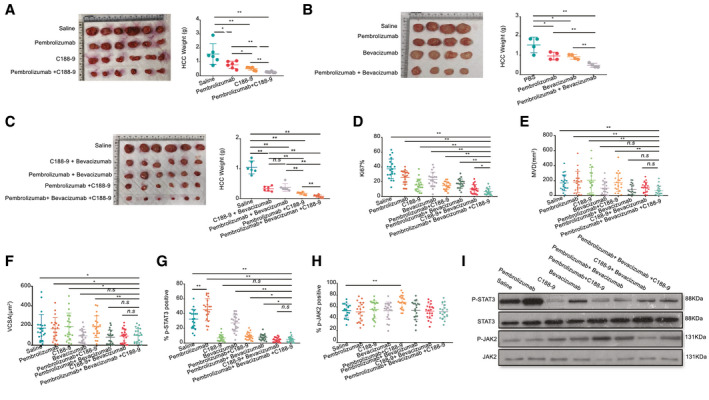
hCD14+ myeloid cells from TILs are one of the potential target cells for developing combinatorial HCC immunotherapy. (A) The HCC‐PDX tumor weight after 4 weeks of treatment with C188‐9 (n = 6), pembrolizumab (n = 6), and combination treatment (n = 6). (B) The HCC‐PDX tumor weight after 4 weeks of treatment with bevacizumab (n = 4), pembrolizumab (n = 4), and combination treatment (n = 4). (C) The HCC‐PDX tumor weight after 3.5 weeks of treatment with saline (n = 6), C188‐9 + bevacizumab (n = 6), pembrolizumab + bevacizumab (n = 6), pembrolizumab + C188‐9 (n = 6) and combination treatment (n = 6). (D‐G) Immunohistochemistry staining of Ki67, CD31, p‐STAT3 and p‐JAK2. The statistical results of (D) Ki67+, (E) MVD, (F) VCSA, (G) p‐STAT3, and (H) p‐JAK2 are shown (n = 5 samples/group, four images were randomly selected for each sample for quantification [total of 20]). (I) Western blot analysis of p‐STAT3 and p‐JAK2 levels from different groups. Abbreviations: APC, allophycocyanin; n.s, not significant. *P < 0.05; **P < 0.01.
In summary, our humanized‐immune‐system HCC mouse model provides a useful in vivo platform for testing HCC combinational immunotherapy on the basis of the dissection of molecular features of the human immune microenvironment and human tumors. A general picture of tumor–immune‐cell interaction is proposed in our study (Fig. 7). Initially, when the human immune system is exposed to an HCC tumor, it becomes activated and releases proinflammatory cytokines, such as IFN‐gamma. The HCC tumor senses the immune responses and generates resistant reactions by releasing DAMPs, such as HMGB1, to immunomodulate CD14+ monocytes. For example, this occurs through the binding of DAMPs to TLR4 and the subsequent synthesis of IL‐33 through the TLR4/AP‐1 signaling pathway and VEGF, and so on. IL‐33 stimulates CD4+ cells, CD14+ cells, and ILC2s to release IL‐6, stimulating HCC proliferation through the JAK2/STAT3 pathway and VEGF‐related angiogenesis to overcome the killing activity of the immune system. In this context, we demonstrated that a triple therapy comprising C188‐9 to inhibit STAT3‐related HCC proliferation induced by IL‐6, bevacizumab to block VEGF‐induced angiogenesis from intratumor monocytes, and pembrolizumab to block PD‐1–triggered T‐cell exhaustion induced by PD‐L1/2 has a combinational effect.
FIG. 7.
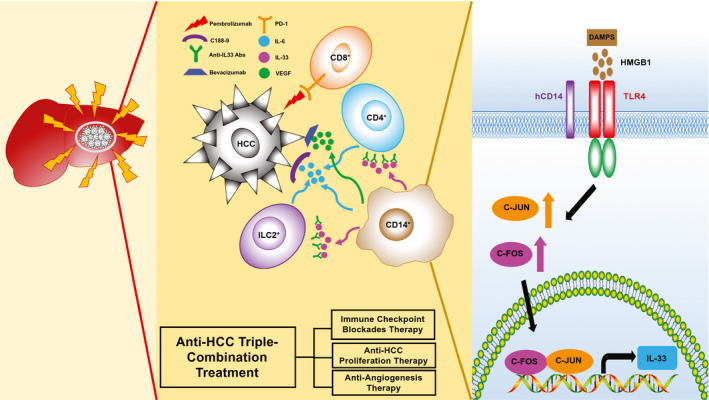
The DAMP/TLR4/AP‐1/IL‐33/IL‐6 model and combinational anti‐HCC effect of triple‐combination treatment (immune checkpoint blockades, anti‐HCC proliferation therapy, and angiogenesis inhibitors).
Discussion
The human immune system is instrumental in modulating HCC progression. On the one hand, cytotoxic effector cells eliminate HCC cells, whereas tumor‐associated immune cells stimulate the proliferation, angiogenesis, EMT, invasion, and metastasis of the HCC.( 40 , 41 , 42 ) In addition, several inflammatory signaling pathways in HCC, namely JAK/STAT, MAPK, PI3K/AKT, and extracellular signal‐regulated kinases1/2 pathways are induced by the immune cells.( 22 , 23 , 24 , 43 , 44 ) In our humanized‐immune‐system HCC mouse model, increased HCC proliferation was associated with activation of the JAK/STAT pathway in the tumor cells, and VEGF‐induced angiogenesis was also triggered by the human immune system. These results suggest that the human immune system could alter HCC progression by changing the signaling pathways in our humanized‐immune‐system HCC mouse model, in a way similar to that observed in patients with HCC.
Different human immune‐cell subsets, such as T cells, B cells, monocytes, NK cells, and NK T cells, were present in the HCC tumor microenvironment.( 45 ) These human immune cells may promote or inhibit HCC growth, depending on the immune responses elicited. For example, depletion of CD4+ T cells and CD8+ T cells could accelerate HCC progression,( 46 , 47 ) which was also seen in our model. By contrast, we also demonstrated that depletion of B cells, monocytes, and macrophages could attenuate the growth and angiogenesis of HCC, consistent with findings in relation to other models.( 23 , 48 ) In our study, intratumor hCD14+ cells triggered HCC proliferation through IL‐33 and triggered angiogenesis through VEGF. The angiogenesis seen in this model is likely of mouse origin; however, human VEGF is highly conserved in mice and could thus stimulate mouse endothelial cells and be validated in our model.( 49 ) CD14 is a co‐receptor of TLR4 and the TLR4/AP‐1 pathway, which has been reported to up‐regulate IL‐33 expression.( 50 , 51 ) Consistent with findings from earlier studies, we found that CD14+ cells could up‐regulate IL‐33 after stimulation with HCC lysates in vitro, and we further proved that intratumor CD14+ cells are the only immune‐cell type responsible for IL‐33–induced HCC proliferation in vivo. The AP‐1 (JUN and FOS [Fos proto‐oncogene]) signaling pathway was activated in intratumor CD14+ cells and HCC lysate–treated primary CD14+ cells, in which the expression of IL‐33 was reduced after TLR4 and AP‐1 inhibition. Another study demonstrated that HMGB1 could induce IL‐33 expression by binding with TLR4.( 52 ) Our results indicated that the increased IL‐33 expression level induced by HCC lysates was hampered after HMGB1 neutralization. On the basis of these results, we demonstrated that the DAMP (HMGB1)/TLR4/AP‐1/IL‐33 pathway was critical for HCC survival and proliferation. It also appeared that intratumor hCD14+ cells and their associated pathways could serve as targets for anticancer treatment. Besides CD14+ cells, other immune‐cell types in the tumor microenvironment all exhibited significant differential RNA‐sequencing profiles compared with their counterparts in peripheral organs, such as blood and spleen, suggesting that HCC tumors have robust selection and learning capabilities regarding immune cells, enabling them to escape immune surveillance, which is an interesting avenue for further study of the individual functions of various immune subsets in tumors. However, HCC intracellular signaling pathways were also significantly impacted by the immune system, as evidenced by the comparison of tumors from mice with and without the human immune system. These changes may contribute to the evolution of the tumor, selection of mutations, and development of drug resistance. Study of the pool of molecular changes in this model, whereby different conditions could be created by manipulating immune components, could facilitate enhanced understanding of the complexity of heterogeneity, mutation burden, and drug resistance and may be further developed into potential therapeutic targets. When combining the discoveries from the angles of the tumor and immune system, our model may subsequently be used to validate the targets and the interfering strategies developed against these targets in single or combined approaches.
In addition to target identification, this model also demonstrated its usefulness in testing existing anticancer and repurposed drugs without putting patients at risk, particularly in combination‐therapy settings. A combination therapy that targets HCC and the tumor microenvironment showed a synergistic effect and has been identified as a promising treatment regimen for patients with HCC.( 25 , 26 , 33 ) Recently, the results of a phase III IMbrave150 trial (a combination of atezolizumab [Tecentriq] and bevacizumab [Avastin]) indicated that the therapy could markedly decrease the risk of progression or death in patients with HCC, compared with treatment with sorafenib (Nexavar).( 26 , 35 ) Hence, there is an urgent need to validate existing combination therapies and to explore therapies with the aid of preclinical animal models in HCC‐PDX and HCC microenvironments that are similar to those encountered in patients with HCC. However, the risk and cost of combining different drugs that possess various toxicity levels have increased significantly. As such, it is difficult and costly to conduct combinational clinical trials. Our results indicate that monotherapy, dual therapy, and triple therapy could be used to validate the anti‐HCC effects and side effects in humanized mice. Monotherapy, such as the depletion of hCD14+ cells, neutralization of human IL‐33, and the use of C188‐9, bevacizumab, and pembrolizumab, resulted in differential anti‐HCC effects. Dual therapy using the combination of C188‐9 and pembrolizumab and the combination of bevacizumab and pembrolizumab in humanized mice showed the additive effects compared with the aforementioned monotherapy. Furthermore, triple therapy using pembrolizumab, bevacizumab, and C188‐9 was evaluated in our HCC, immune system–humanized mouse model and resulted in an improved combinational effect compared with the monotherapy and dual therapy. This suggested that triple‐targeting of HCC proliferation, angiogenesis, and the immune checkpoint together may offer a combination therapy scheme for HCC treatment. In conclusion, our HCC humanized‐immune‐system mouse model provides a platform for studying the interaction between the HCC tumor and human immune cells and for testing combination therapy that targets the molecular features of HCC and the HCC immune‐system microenvironment.
Authors Contributions
Y.Z. designed and performed the experiments, analyzed and interpreted data, and prepared the manuscript. J.W., S.Y.F., S.Y.T., J.Y.C., W.W.S.T., and M.L. performed the experiments and analyzed data; W.N.L., T.W.H.S., S.H., J.K.Y.C., C.E.C., G.H.L., H.C.T., S.G.L., and Y.W. contributed research tools and reagents and prepared the manuscript. Q.C. conceived the study, designed the experiments, supervised the project, and prepared the manuscript.
Supporting information
Supporting Information
Acknowledgment
We thank the KK Women’s and Children’s Hospital and National University Hospital for provision of human fetal liver/cord blood and HCC tumor samples.
Supported by the National Medical Research Council Singapore, the Virus‐Induced Cancer: Translational Oncology Research & immunologY Programme (NMRC/OFLCG/003/2018), the Senior Clinician Scientist Award (NMRC/CSASI16nov006), the Eradication of HBV T‐cell receptor Program (NMRC/TCR/014‐NUHS/2015), a Clinician–Scientist Individual Research Grant (CIRG19may‐0051), and the National Research Foundation Singapore (NRF) Fellowship (NRF‐NRFF2017‐03), an NRF–Israel Science Foundation joint grant (NRF2019–NRF‐ISF003‐3127), an NRF–Competitive Research Programme grant (NRF2016‐CRP001‐103), and the Agency for Science, Technology and Research Industry Alignment Fund‐Pre‐positioning (H18/01/a0/022) to Q.C.
Potential conflict of interest: Dr. Chee is on the speakers’ bureau for Roche. Dr. Lim advises, is on the speakers’ bureau for, and received grants from Gilead, Abbott, and Roche. He advises AbbVie, Arbutus, Assembly, and Eisai. He received grants from Merck Sharp & Dohme Corp and Grifols.
References
Author names in bold designate shared co–first authorship.
- 1. Villanueva A. Hepatocellular carcinoma. N Engl J Med 2019;380:1450‐1462. [DOI] [PubMed] [Google Scholar]
- 2. Bakiri L, Hamacher R, Graña O, Guío‐Carrión A, Campos‐Olivas R, Martinez L, et al. Liver carcinogenesis by FOS‐dependent inflammation and cholesterol dysregulation. J Exp Med 2017;214:1387‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation‐induced IgA+ cells dismantle anti‐liver cancer immunity. Nature 2017;551:340‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Todoric J, Antonucci L, Karin M. Targeting inflammation in cancer prevention and therapy. Cancer Prev Res 2016;9:895‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang W, Chen Z, Zhang L, Tian D, Wang D, Fan D, et al. Interleukin‐8 induces expression of FOXC1 to promote transactivation of CXCR1 and CCL2 in hepatocellular carcinoma cell lines and formation of metastases in mice. Gastroenterology 2015;149:1053‐1067, e14. [DOI] [PubMed] [Google Scholar]
- 6. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor‐associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014;147:1393‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma S, Cheng Q, Cai Y, Gong H, Wu Y, Yu X, et al. IL‐17A produced by gammadelta T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res 2014;74:1969‐1982. [DOI] [PubMed] [Google Scholar]
- 8. Shao Y, Lo CM, Ling CC, Liu XB, Ng K‐P, Chu ACY, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett 2014;355:264‐272. [DOI] [PubMed] [Google Scholar]
- 9. Ghanem I, Riveiro ME, Paradis V, Faivre S, de Parga PM, Raymond E. Insights on the CXCL12‐CXCR4 axis in hepatocellular carcinoma carcinogenesis. Am J Transl Res 2014;6:340‐352. [PMC free article] [PubMed] [Google Scholar]
- 10. Wiedemann GM, Rohrle N, Makeschin MC, Fesseler J, Endres S, Mayr D, et al. Peritumoural CCL1 and CCL22 expressing cells in hepatocellular carcinomas shape the tumour immune infiltrate. Pathology 2019;51:586‐592. [DOI] [PubMed] [Google Scholar]
- 11. Chew V, Lee YH, Pan LU, Nasir NJM, Lim CJ, Chua C, et al. Immune activation underlies a sustained clinical response to Yttrium‐90 radioembolisation in hepatocellular carcinoma. Gut 2019;68:335‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, et al. Chemokine‐driven lymphocyte infiltration: an early intratumoural event determining long‐term survival in resectable hepatocellular carcinoma. Gut 2012;61:427‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang CM, Chen L, Hu H, Ma HY, Gao LL, Qin J, et al. Chemokines and their receptors play important roles in the development of hepatocellular carcinoma. World J Hepatol 2015;7:1390‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sachdeva M, Chawla YK, Arora SK. Immunology of hepatocellular carcinoma. World J Hepatol 2015;7:2080‐2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Flores‐Borja F, Irshad S, Gordon P, Wong F, Sheriff I, Tutt A, et al. Crosstalk between innate lymphoid cells and other immune cells in the tumor microenvironment. J Immunol Res 2016;2016:7803091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi C, Chen Y, Chen Y, Yang Y, Bing W, Qi J. CD4+ CD25+ regulatory T cells promote hepatocellular carcinoma invasion via TGF‐beta1‐induced epithelial‐mesenchymal transition. Onco Targets Ther 2019;12:279‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou SL, Yin D, Hu ZQ, Luo CB, Zhou ZJ, Xin HY, et al. A Positive feedback loop between cancer stem‐like cells and tumor‐associated neutrophils controls hepatocellular carcinoma progression. Hepatology 2019;70:1214‐1230. [DOI] [PubMed] [Google Scholar]
- 18. Budhu A, Wang XW. The role of cytokines in hepatocellular carcinoma. J Leukoc Biol 2006;80:1197‐1213. [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Ding J, Feng Y, Weng L, Zhao G, Xiang J, et al. Targeting of growth factors in the treatment of hepatocellular carcinoma: the potentials of polysaccharides. Oncol Lett 2017;13:1509‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen R, Cui J, Xu C, Xue T, Guo K, Gao D, et al. The significance of MMP‐9 over MMP‐2 in HCC invasiveness and recurrence of hepatocellular carcinoma after curative resection. Ann Surg Oncol 2012;19(Suppl. 3):S375‐S384. [DOI] [PubMed] [Google Scholar]
- 21. Giannelli G, Bergamini C, Marinosci F, Fransvea E, Quaranta M, Lupo L, et al. Clinical role of MMP‐2/TIMP‐2 imbalance in hepatocellular carcinoma. Int J Cancer 2002;97:425‐431. [DOI] [PubMed] [Google Scholar]
- 22. Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ, Zhou SL, et al. Macrophage‐secreted IL‐8 induces epithelial–mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol 2015;46:587‐596. [DOI] [PubMed] [Google Scholar]
- 23. Kong L, Zhou Y, Bu H, Lv T, Shi Y, Yang J. Deletion of interleukin‐6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J Exp Clin Cancer Res 2016;35:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trikha P, Carson WE, III . Signaling pathways involved in MDSC regulation. Biochim Biophys Acta 2014;1846:55‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stotz M, Gerger A, Haybaeck J, Kiesslich T, Bullock MD, Pichler M. Molecular targeted therapies in hepatocellular carcinoma: past, present and future. Anticancer Res 2015;35:5737‐5744. [PubMed] [Google Scholar]
- 26. Kudo M. Targeted and immune therapies for hepatocellular carcinoma: predictions for 2019 and beyond. World J Gastroenterol 2019;25:789‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bergmann J, Müller M, Baumann N, Reichert M, Heneweer C, Bolik J, et al. IL‐6 trans‐signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017;65:89‐103. [DOI] [PubMed] [Google Scholar]
- 28. Shakiba E, Ramezani M, Sadeghi M. Evaluation of serum interleukin‐6 levels in hepatocellular carcinoma patients: a systematic review and meta‐analysis. Clin Exp Hepatol 2018;4:182‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jung KH, Yoo W, Stevenson HL, Deshpande D, Shen H, Gagea M, et al. Multifunctional effects of a small‐molecule STAT3 inhibitor on NASH and hepatocellular carcinoma in mice. Clin Cancer Res 2017;23:5537‐5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siegel AB, Cohen EI, Ocean A, Lehrer D, Goldenberg A, Knox JJ, et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J Clin Oncol 2008;26:2992‐2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fang P, Hu JH, Cheng ZG, Liu ZF, Wang JL, Jiao SC. Efficacy and safety of bevacizumab for the treatment of advanced hepatocellular carcinoma: a systematic review of phase II trials. PLoS One 2012;7:e49717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scheiner B, Kirstein MM, Hucke F, Finkelmeier F, Schulze K, von Felden J, et al. Programmed cell death protein‐1 (PD‐1)‐targeted immunotherapy in advanced hepatocellular carcinoma: efficacy and safety data from an international multicentre real‐world cohort. Aliment Pharmacol Ther 2019;49:1323‐1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pinter M, Peck‐Radosavljevic M. Review article: systemic treatment of hepatocellular carcinoma. Aliment Pharmacol Ther 2018;48:598‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu H, Shen J, Lu K. IL‐6 and PD‐L1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem Biophys Res Commun 2017;486:239‐244. [DOI] [PubMed] [Google Scholar]
- 35. Medavaram S, Zhang Y. Emerging therapies in advanced hepatocellular carcinoma. Exp Hematol Oncol 2018;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eichten A, Su J, Adler AP, Zhang LI, Ioffe E, Parveen AA, et al. Resistance to anti‐VEGF therapy mediated by autocrine IL6/STAT3 signaling and overcome by IL6 blockade. Cancer Res 2016;76:2327‐2339. [DOI] [PubMed] [Google Scholar]
- 37. Zhao Y, Shuen TWH, Toh TB, Chan XY, Liu M, Tan SY, et al. Development of a new patient‐derived xenograft humanised mouse model to study human‐specific tumour microenvironment and immunotherapy. Gut 2018;67:1845‐1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao Y, Liu M, Chan XY, Tan SY, Subramaniam S, Fan Y, et al. Uncovering the mystery of opposite circadian rhythms between mouse and human leukocytes in humanized mice. Blood 2017;130:1995‐2005. [DOI] [PubMed] [Google Scholar]
- 39. Zhang P, Liu XK, Chu Z, Ye JC, Li KL, Zhuang WL, et al. Detection of interleukin‐33 in serum and carcinoma tissue from patients with hepatocellular carcinoma and its clinical implications. J Int Med Res 2012;40:1654‐1661. [DOI] [PubMed] [Google Scholar]
- 40. Makarova‐Rusher OV, Medina‐Echeverz J, Duffy AG, Greten TF. The yin and yang of evasion and immune activation in HCC. J Hepatol 2015;62:1420‐1429. [DOI] [PubMed] [Google Scholar]
- 41. Chen Y, Hao X, Sun R, Wei H, Tian Z. Natural killer cell‐derived interferon‐gamma promotes hepatocellular carcinoma through the epithelial cell adhesion molecule‐epithelial‐to‐mesenchymal transition axis in hepatitis B virus transgenic mice. Hepatology 2019;69:1735‐1750. [DOI] [PubMed] [Google Scholar]
- 42. Disis ML. Immune regulation of cancer. J Clin Oncol 2010;28:4531‐4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor‐associated macrophages. Biomed Res Int 2013;2013:187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nikolaou K, Sarris M, Talianidis I. Molecular pathways: the complex roles of inflammation pathways in the development and treatment of liver cancer. Clin Cancer Res 2013;19:2810‐2816. [DOI] [PubMed] [Google Scholar]
- 45. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour‐infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017;66:342‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma C, Kesarwala AH, Eggert T, Medina‐Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Endig J, Buitrago‐Molina LE, Marhenke S, Reisinger F, Saborowski A, Schütt J, et al. Dual role of the adaptive immune system in liver injury and hepatocellular carcinoma development. Cancer Cell 2016;30:308‐323. [DOI] [PubMed] [Google Scholar]
- 48. He H, Wu J, Zang M, Wang W, Chang X, Chen X, et al. CCR6+ B lymphocytes responding to tumor cell‐derived CCL20 support hepatocellular carcinoma progression via enhancing angiogenesis. Am J Cancer Res 2017;7:1151‐1163. [PMC free article] [PubMed] [Google Scholar]
- 49. Gerber HP, Wu X, Yu L, Wiesmann C, Liang XH, Lee CV, et al. Mice expressing a humanized form of VEGF‐A may provide insights into the safety and efficacy of anti‐VEGF antibodies. Proc Natl Acad Sci U S A 2007;104:3478‐3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He Z, Riva M, Björk P, Swärd K, Mörgelin M, Leanderson T, et al. CD14 is a co‐receptor for TLR4 in the S100A9‐induced pro‐inflammatory response in monocytes. PLoS One 2016;11:e0156377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ho JE, Chen WY, Chen MH, Larson MG, McCabe EL, Cheng S, et al. Common genetic variation at the IL1RL1 locus regulates IL‐33/ST2 signaling. J Clin Invest 2013;123:4208‐4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chang J, Xia Y, Wasserloos K, Deng M, Blose KJ, Vorp DA, et al. Cyclic stretch induced IL‐33 production through HMGB1/TLR‐4 signaling pathway in murine respiratory epithelial cells. PLoS One 2017;12:e0184770. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information