

Abstract
We investigated genetic determinants of single‐dose simvastatin pharmacokinetics in a prospective study of 170 subjects and a retrospective cohort of 59 healthy volunteers. In a microarray‐based genomewide association study with the prospective data, the SLCO1B1 c.521T>C (p.Val174Ala, rs4149056) single nucleotide variation showed the strongest, genomewide significant association with the area under the plasma simvastatin acid concentration‐time curve (AUC; P = 6.0 × 10−10). Meta‐analysis with the retrospective cohort strengthened the association (P = 1.6 × 10−17). In a stepwise linear regression candidate gene analysis among all 229 participants, SLCO1B1 c.521T>C (P = 1.9 × 10−13) and CYP3A4 c.664T>C (p.Ser222Pro, rs55785340, CYP3A4*2, P = 0.023) were associated with increased simvastatin acid AUC. Moreover, the SLCO1B1 c.463C>A (p.Pro155Thr, rs11045819, P = 7.2 × 10−6) and c.1929A>C (p.Leu643Phe, rs34671512, P = 5.3 × 10−4) variants associated with decreased simvastatin acid AUC. Based on these results and the literature, we classified the volunteers into genotype‐predicted OATP1B1 and CYP3A4 phenotype groups. Compared with the normal OATP1B1 function group, simvastatin acid AUC was 273% larger in the poor (90% confidence interval (CI), 137%, 488%; P = 3.1 × 10−6), 40% larger in the decreased (90% CI, 8%, 83%; P = 0.036), and 67% smaller in the highly increased function group (90% CI, 46%, 80%; P = 2.4 × 10−4). Intermediate CYP3A4 metabolizers (i.e., heterozygous carriers of either CYP3A4*2 or CYP3A4*22 (rs35599367)), had 87% (90% CI, 39%, 152%, P = 6.4 × 10−4) larger simvastatin acid AUC than normal metabolizers. These data suggest that in addition to no function SLCO1B1 variants, increased function SLCO1B1 variants and reduced function CYP3A4 variants may affect the pharmacokinetics, efficacy, and safety of simvastatin. Care is warranted if simvastatin is prescribed to patients carrying decreased function SLCO1B1 or CYP3A4 alleles.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The SLCO1B1 c.521T>C variant increases simvastatin exposure and risk for muscle‐related adverse effects. However, marked unexplained interindividual variability exists in simvastatin pharmacokinetics.
WHAT QUESTION DID THIS STUDY ADDRESS?
With our genomewide association study of simvastatin pharmacokinetics, we comprehensively assessed the effect of genetic variation on simvastatin pharmacokinetics in 229 healthy volunteers.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
OATP1B1 no function variants SLCO1B1*5 and *15 associate with increased simvastatin acid exposure and OATP1B1 increased function variants *14 and *20 associate with reduced simvastatin acid exposure. In addition, CYP3A4*2 and *22 variants associate with increased simvastatin exposure. This is the first study to demonstrate the effects of SLCO1B1*14, SLCO1B1*20, and CYP3A4*2 on simvastatin pharmacokinetics.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Our findings indicate that SLCO1B1 and CYP3A4 genetic variants affect simvastatin exposure. Simvastatin is probably best avoided in individuals with genetically poor OATP1B1 or CYP3A4 activity, or decreased activity of both OATP1B1 and CYP3A4. Lowering of the simvastatin dose should be considered in individuals with decreased OATP1B1 or CYP3A4 activity.
Simvastatin is a 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitor widely prescribed for hypercholesterolemia. Simvastatin is usually effective and well‐tolerated, but, for some patients, it can cause muscle symptoms ranging from mild myalgia to potentially lethal rhabdomyolysis. 1 , 2 , 3 , 4 Factors increasing simvastatin exposure, such as drug interactions or genetic variation, heighten the risk for adverse effects. 1 , 2 , 4 , 5
In simvastatin pharmacokinetics, marked interindividual variability exists. 6 Simvastatin is an inactive lactone prodrug, which converts to pharmacologically active simvastatin acid. 1 , 6 Simvastatin lactone and acid are extensively metabolized by cytochrome P450 (CYP) 3A enzymes, with a minor contribution from CYP2C8 and uridine diphosphate‐glucuronosyltransferases (UGT). 7 , 8 , 9 , 10 The CYP3A4*22 (rs35599367) intron variant reduces CYP3A4 expression and associates with increased simvastatin exposure and cholesterol‐lowering efficacy. 11 , 12 Furthermore, the CYP3A5*3 (rs776746) splice site variant causing loss‐of‐function for CYP3A5, may elevate simvastatin concentrations. 12
Simvastatin acid is also a substrate for the organic anion transporting polypeptide 1B1 (OATP1B1), a hepatic influx transporter encoded by the SLCO1B1 gene. 6 The SLCO1B1 c.521T>C (p.Val174Ala, rs4149056) missense variant raises simvastatin acid exposure and increases the risk for adverse effects. 2 , 3 , 4 , 5 , 6 Some studies have suggested that simvastatin acid is also a substrate of the P‐glycoprotein, 13 , 14 an efflux transporter encoded by the ABCB1 gene, although the evidence is contradictory. 15 Evidence as to the effect of genetic variation in ABCB1 on simvastatin pharmacokinetics is unclear. 16 , 17 , 18
As the risk for muscle‐related adverse effects increases along with simvastatin exposure, 1 , 4 , 5 understanding the genetic variants affecting simvastatin pharmacokinetics is important. Studies on simvastatin pharmacokinetics have identified several important pharmacogenetic variants, however, these studies have investigated candidate‐gene associations, and yet‐unrecognized variants may exist. The aim of this study was to comprehensively characterize the effect of genetic variation on simvastatin pharmacokinetics by use of genomewide methods in a relatively large cohort.
METHODS
Subjects and study design
The study sample comprises a prospective simvastatin pharmacokinetic study cohort and a retrospective cohort with earlier published pharmacokinetic data on simvastatin. A total of 170 (83 men and 87 women, age 24.6 ± 4.2 years (mean ± SD), height 174.4 ± 9.4 cm, weight 70.3 ± 12.7 kg, and body mass index 23.0 ± 2.8 kg/m2) unrelated, healthy Finnish White volunteers gave written informed consent and participated in the prospective study. Medical history, physical examination, and routine laboratory tests confirmed their health prior to their entering the study. None of the participants used any continuous medication, including hormonal contraception, and all were nonsmokers.
The Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District (record number 86/13/03/00/2015) and the Finnish Medicines Agency (EudraCT number 2015‐000540‐41) approved the study protocol. Following an overnight fast, the volunteers ingested a single oral dose of 40 mg simvastatin (Zocor tablet; Merck Sharp & Dohme B.V., Haarlem, The Netherlands). The volunteers had a standardized warm meal 4 hours, and light meals 7 and 10 hours after simvastatin administration. The volunteers provided timed venous EDTA blood samples of 4 or 9 mL prior to and for up to 24 hours after the ingestion of simvastatin. We placed the tubes on ice immediately after sampling, separated plasma from the samples within 30 minutes, and stored the plasma aliquots at −70°C until analysis. Use of any other drugs was forbidden 1 week before and 3 days after simvastatin administration. No consumption of alcohol was allowed 1 day before and 2 days after simvastatin, and also no grapefruit products 2 days before and 2 days after simvastatin.
The retrospective cohort comprised 59 healthy volunteers (32 men and 27 women, age 22.8 ± 2.8 years (mean ± SD), height 174.4 ± 9.8 cm, weight 68.2 ± 11.1 kg, and body mass index 22.3 ± 2.4 kg/m2) who had participated in earlier single‐dose pharmacokinetic studies 6 , 16 , 19 on either 20 mg (24 participants) or 40 mg (35 participants) simvastatin. The volunteers ingested simvastatin tablets after an overnight fast and received a standardized warm meal 4 hours and standardized light meals 7 and 10 hours after simvastatin intake. Competent ethics committees and national authority (Fimea) approved the study protocols, including genetic analyses. Each participant had given written informed consent.
Determination of drug concentrations
Simvastatin lactone, simvastatin acid, 3″‐hydroxy simvastatin acid, 3′,5′‐dihydrodiol simvastatin lactone, 3′,5′‐dihydrodiol simvastatin acid, simvastatin‐D6, and simvastatin acid‐D6 were purchased from Toronto Research Chemicals (Toronto, Ontario, Canada).
Prior to analysis, plasma samples were pre‐treated using a VersaPlate supported liquid extraction system in 96‐Well format (Agilent Technologies, Waldbronn, Germany). A volume of 150 μL of plasma was mixed with 100 μL of ice‐cold ammonium acetate (100 mM, pH 4.5), containing the internal standards (10 ng/mL both). The sample mixture was then transferred to extraction plate, incubated for 10 minutes, and eluted three times with 0.5 mL of methyl tert‐butyl ether. The sample extract was evaporated and reconstituted in 80 μL of 40% acetonitrile, and a volume of 5 μL was delivered to the analytical column. The chromatographic separation of analytes was carried out on a Luna Omega polar C18 column (100 × 2.1 mm I.D., 1.6 μm particle size; Phenomenex, Torrance, CA) using 5 mM ammonium formate (pH 3.9, adjusted with 98% formic acid) as mobile phase A and acetonitrile as mobile phase B. The flow rate and the column temperature were maintained at 300 μL/min and 40°C. The mobile phase gradient conditions were as follows: 1 minute at 20% B on hold, then a linear ramp from 20% B to 40% B over 3 minutes followed by a second linear ramp to 90% B over 2 minutes, and 1 minute at 90% B before a re‐equilibration step back to the initial conditions (20% B). The drug concentrations were determined using a Sciex 5500Qtrap mass spectrometer (ABSciex, Toronto, Ontario, Canada) interfaced with an electrospray ion source. The characteristic multiple reaction monitoring mass‐to‐charge (m/z) ion transitions were applied for each analyte. Simvastatin lactone (436–285), simvastatin acid (437–303), and 3′,5′‐dihydrodiol simvastatin lactone (470–319) were detected in positive mode, and 3″‐hydroxy simvastatin acid (451–335) and 3′,5′‐dihydrodiol simvastatin acid (469–353) were detected using negative mode. Isotope labeled simvastatin lactone served as an internal standard for simvastatin lactone and isotope labeled simvastatin acid for all other analytes. The limits of quantification (ng/mL) for simvastatin lactone, simvastatin acid, 3″‐hydroxy simvastatin acid, 3′,5′‐dihydrodiol simvastatin lactone, and 3′,5′‐dihydrodiol simvastatin acid were 0.05, 0.05, 0.25, 0.25, and 0.5, respectively. The day‐to‐day precision values for all compounds were below 15% (expressed as percent coefficient of variation) and the accuracy within +/−15%, except for the lower limit of quantification, for which both precision and accuracy were within ±20%.
Pharmacokinetics
In the prospective study, we calculated the area under the plasma concentration‐time curve (AUC), peak plasma concentration (C max), and elimination half‐life (t ½) values for simvastatin, simvastatin acid, 3′,5′‐dihydrodiol simvastatin lactone, 3′,5′‐dihydrodiol simvastatin acid, and 3″‐hydroxy simvastatin by standard noncompartmental methods (Phoenix WinNonlin, version 6.4; Certara, Princeton, NJ). We calculated the AUC value from 0 hours to infinity (AUC0–∞) for all metabolites, with the exception of 3′,5′‐dihydrodiol simvastatin acid and 3″‐hydroxy simvastatin acid, for which AUC from 0 to 24 hours (AUC0–24 hours) was determined instead, because the t ½ could not be reliably determined for all participants. In our retrospective cohort, concentration data were available only for simvastatin lactone and simvastatin acid, and pharmacokinetic variables were recalculated from the original concentration data.
Genotyping
We extracted genomic DNA from whole blood samples using the Maxwell 16 LEV Blood DNA Kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI). The Institute for Molecular Medicine Finland (FIMM) Technology Centre, University of Helsinki, performed the genomewide genotyping. The volunteers in the prospective study were genotyped with the Illumina HumanCoreExome‐24v1‐1_A BeadChip (Illumina, San Diego, CA). Quality thresholds for including genotype data in statistical analysis were Hardy–Weinberg equilibrium P value >10−5 and proportion of missing ≤0.03. The genomewide statistical analysis included 261,851 single‐nucleotide variants (SNVs) with minor‐allele frequency (MAF) of ≥0.05. The retrospective cohort was genotyped with Illumina GSAMD‐24v2‐0_A1 beadchip (Illumina). Data for the genomewide association study meta‐analysis included 134,431 SNVs present in both genotyping chips. In order to supplement missing genotypes for candidate‐gene analysis, we used TaqMan genotyping assays on a QuantStudio 12 K Flex Real‐Time PCR system (Thermo Fisher Scientific, Waltham, MA) to genotype the participants for selected variants in ABCB1, CYP3A4, CYP3A5, SLCO1B1, SLCO1B3, UGT1A1, and UGT1A3. For the candidate‐gene analysis, we selected missense or suspected functional variants with MAF of ≥0.01 (Table S1 ).
We computed the SLCO1B1 haplotypes from the c.388A>G (p.Asn130Asp, rs2306283), c.463C>A (p.Pro155Thr, rs11045819), c.521T>C, and c.1929A>C (p.Leu643Phe, rs34671512) SNVs with PHASE version 2.1.1. 20 , 21 Haplotypes were defined according to Pharmacogene Variation Consortium. 22 , 23 By combining variants with similar effect, it is possible to achieve greater statistical power. Therefore, we divided the participants into genotype‐predicted OATP1B1 phenotype groups, as described previously 24 (Figure S1 ). As an exploratory analysis, we then classified heterozygous carriers of either CYP3A4 c.664T>C (p.Ser222Pro, rs55785340, and CYP3A4*2) or rs35599367 (CYP3A4*22) as intermediate CYP3A4 metabolizers. One participant was compound heterozygote for these alleles and classified as a poor CYP3A4 metabolizer. CYP3A4*22 was included as it reduces CYP3A4 expression in the liver 11 and has been associated with higher plasma concentrations of simvastatin acid and simvastatin lactone. 12
Statistical analysis
The data were analyzed with the statistical programs JMP Genomics 8.0 (SAS Institute, Cary, NC) and IBM SPSS Statistics for Windows 27.0 (Armonk, NY). Pharmacokinetic variables and body weight were log‐transformed before statistical analysis. We investigated the possible effects of demographic covariates (sex and body weight) on the pharmacokinetics of simvastatin lactone and its metabolites using a forward stepwise linear regression analysis. The P value thresholds of 0.05 and 0.10 were used for entry into and removal from the model. Possible effects of genetic variants on pharmacokinetic variables were investigated using stepwise linear regression analysis. Significant demographic covariates, weight for simvastatin acid, and 3′,5′‐dihydrodiol simvastatin acid, sex for 3′,5′‐dihydrodiol simvastatin lactone, were set as fixed factors. Genomewide meta‐analysis was carried out using the inverse variance fixed effect model. A P value of below 5 × 10−8 was considered genomewide significant. The candidate gene analysis was performed without correction for multiple testing and with Bonferroni correction for the number of tested genetic variants. For SLCO1B1 and CYP3A4 genotype‐predicted phenotype groups, analysis of variance adjusting for significant demographic and appropriate genotype covariates was carried out with pairwise comparisons with the Fisher’s Least Significant Difference method. A P value of below 0.05 was considered statistically significant.
RESULTS
In the prospective study, the AUC0–∞ of simvastatin lactone varied 20.1‐fold, that of simvastatin acid 21.3‐fold, and that of 3′,5′‐dihydrodiol simvastatin lactone 21.1‐fold, whereas the AUC0–24 hours of 3′,5′‐dihydrodiol simvastatin acid varied 58.5‐fold and that of 3″‐hydroxy simvastatin acid 39.5‐fold. Body weight was a significant covariate for simvastatin acid AUC0–∞ and 3′,5′‐dihydrodiol simvastatin acid AUC0–24 hours, whereas sex was a significant covariate for 3′,5′‐dihydrodiol simvastatin lactone AUC0–∞. In the retrospective cohort, the dose‐adjusted AUC0–∞ of simvastatin lactone varied 14.5‐fold and that of simvastatin acid 20.6‐fold.
Genomewide association study
In the prospective study, we investigated the associations of 261,030 SNVs on AUC0–∞ or AUC0–24 hours of simvastatin lactone, simvastatin acid, 3′,5′‐dihydrodiol simvastatin lactone, 3′,5′‐dihydrodiol simvastatin acid, and 3″‐hydroxy simvastatin acid. Two SNVs in the SLCO1B1 gene were genomewide significantly associated with increased simvastatin acid AUC0–∞. The strongest association was observed with the SLCO1B1 c.521T>C missense variant, with a 61% (90% confidence interval (CI), 43%, 82%; P = 6.0 × 10−10) larger AUC0–∞ per copy of the variant allele (Figure 1 ). The SLCO1B1 intron variant, rs4363657, was associated with a 45% (90% CI, 31%, 62%; P = 3.3 × 10−8) increase per variant allele. In the prospective study, no other genomewide significant associations were observed.
Figure 1.
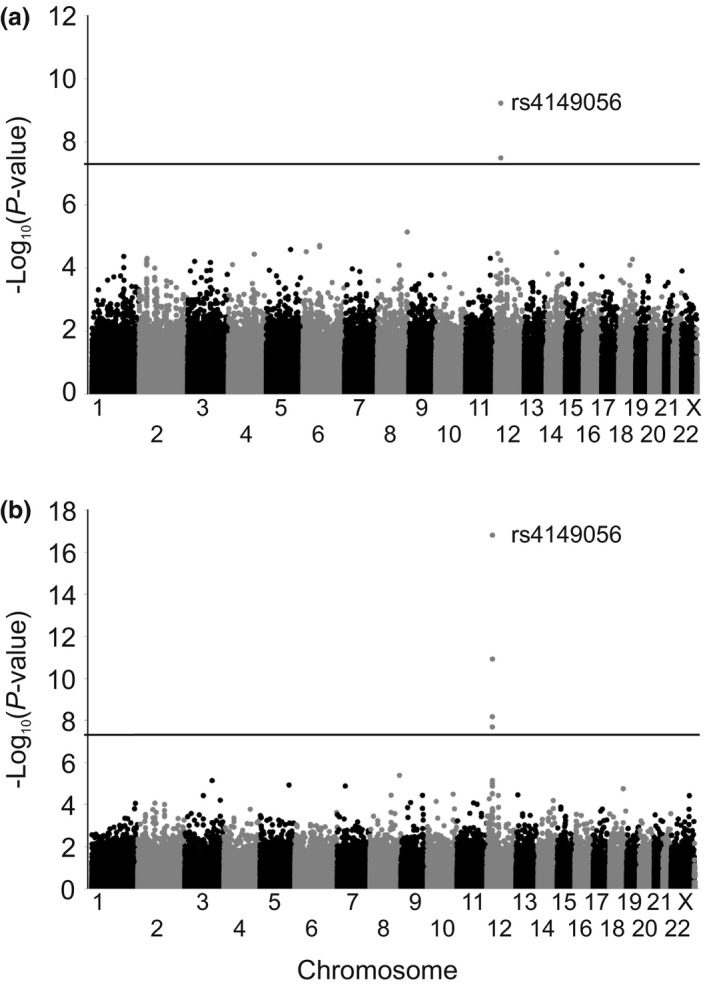
Manhattan plots of area under the plasma simvastatin acid concentration‐time curve from 0 hours to infinity (AUC0–∞) (a) genomewide association study in 170 healthy volunteers of the prospective study and (b) genomewide association meta‐analysis of the prospective study and retrospective cohort in altogether 229 participants. Horizontal lines indicate the genomewide significance level of 5 × 10−8.
In the genomewide association study meta‐analysis including the retrospective cohort, we investigated the effects of 136,688 SNVs on simvastatin lactone and simvastatin acid AUC0–∞. Only simvastatin acid AUC0–∞ showed genomewide significant associations. Five SNVs in the SLCO1B1‐SLCO1B3 locus were associated with an increased simvastatin acid AUC0–∞ (Table 1 , Figure 1 ). The SLCO1B1 c.521T>C SNV showed the strongest association with a 68% (90% CI, 52%, 86%; P = 1.6 × 10−17) increase in simvastatin acid AUC0–∞ per variant allele. The SLCO1B1 intronic variant rs4363657 was associated with a 46% (90% CI, 33%, 60%; P = 1.2 × 10−11) increase per variant allele. In addition, two SLCO1B3 missense variants c.334G>T (p.Ala112Ser, rs4149117) and c.699A>G (p.Ile233Met, rs7311358) were associated with a 43% (90% CI, 29%, 58%; P = 6.9 × 10−9) increase in simvastatin acid AUC0–∞ per variant allele. These variants were in a complete linkage (r 2 = 1, P = 9.8 × 10−52) with each other and the intronic SLCO1B3 variant rs1304539 (r 2 = 0.99, P = 3.9 × 10−51). All SNVs showing genomewide significant associations were in a strong linkage disequilibrium with SLCO1B1 c.521T>C: rs4363657 (r 2 = 0.53, P = 4.2 × 10−28), SLCO1B3 c.334G>T, and c.699A > G (r 2 = 0.35, P = 3.1 × 10−19).
Table 1.
Genomewide significant associations with simvastatin acid AUC0–∞ from genome‐wide meta‐analysis of the prospective study and retrospective cohort in altogether 229 participants
| dbSNP rs number | Gene | Nucleotide change | Amino acid change | Effecta | 90% CI | P value | MAF | Linkage disequilibrium with rs4149056 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| D′ | r 2 | P value | ||||||||
| rs4149056 | SLCO1B1 | c.521T>C | p.Val174Ala | 68% | 52%, 86% | 1.6 × 10−17 | 0.19 | |||
| rs4363657 | SLCO1B1 | Intron variant | 46% | 33%, 60% | 1.2 × 10−11 | 0.30 | 0.98 | 0.53 | 4.2 × 10−28 | |
| rs4149117 | SLCO1B3 | c.334G>T | p.Ala112Ser | 43% | 29%, 59% | 6.9 × 10−9 | 0.24 | 0.68 | 0.35 | 3.1 × 10−19 |
| rs7311358 | SLCO1B3 | c.699A>G | p.Ile233Met | 43% | 29%, 59% | 6.9 × 10−9 | 0.24 | 0.68 | 0.35 | 3.1 × 10−19 |
| rs1304539 | SLCO1B3 | Intron variant | 42% | 28%, 58% | 2.1 × 10−8 | 0.23 | 0.69 | 0.36 | 1.7 × 10−19 | |
AUC0–∞, area under the plasma simvastatin acid concentration‐time curve from 0 hours to infinity; CI, confidence interval; dbSNP, National Center for Biotechnology Information Short Genetic Variations database; MAF, minor allele frequency; rs, reference single nucleotide variation.
Per variant allele copy.
Candidate gene association study
Due to the risk of false negative associations with the conservative P value threshold in the genomewide analysis, we next performed a candidate gene analysis on simvastatin acid AUC0–∞ among all the 229 participants. In this analysis, we included missense and suspected functional variants with MAF of ≥ 0.01 in genes suggested to be involved in simvastatin pharmacokinetics (Table S1 ). In a stepwise linear regression analysis, the strongest association was observed with SLCO1B1 c.521T>C, with a 57% increase in simvastatin acid AUC0–∞ per variant allele (90% CI, 43%, 73%; P = 1.9 × 10−13; Table 2 ). In addition, two SLCO1B1 missense variants were associated with decreased simvastatin acid AUC0–∞: c.463C>A with a 32% (90% CI, 22%, 41%; P = 7.2 × 10−6) and c.1929A>C with a 36% (90% CI, 21%, 48%; P = 5.3 × 10−4) decrease per variant allele. CYP3A4*2 allele was associated with a 60% (90% CI, 14%, 125%; P = 0.023) increase in simvastatin acid AUC0–∞ per variant allele.
Table 2.
Results of candidate gene analysis on simvastatin acid AUC0–∞ in 229 healthy volunteers
| Variable | Effecta | 90% CI | P value | Bonferroni P value | r 2 | Heterozygotes | Homozygotes |
|---|---|---|---|---|---|---|---|
| n (%) | n (%) | ||||||
| Weight (per 10% increase) | −7.7% | −10.5%, −4.8% | 2.3 × 10−5 | 4.8 × 10−4 | 0.02 | ||
| SLCO1B1 c.521T>C | 57.4% | 43.1%, 73.2% | 1.9 × 10−13 | 4.0 × 10−12 | 0.25 | 64 (27.9) | 11 (4.8) |
| SLCO1B1 c.463C>A | −32.4% | −41.3%, −22.2% | 7.2 × 10−6 | 1.5 × 10−4 | 0.31 | 37 (16.2) | 1 (0.4) |
| SLCO1B1 c.1929A>C | −35.9% | −47.9%, −21.0% | 5.3 × 10−4 | 0.011 | 0.35 | 17 (7.4) | 0 |
| CYP3A4 c.664T>C | 60.3% | 14.1%, 125.3% | 0.023 | 0.48 | 0.36 | 6 (2.6) | 0 |
AUC0–∞, area under the plasma simvastatin acid concentration‐time curve from 0 hours to infinity; CI, confidence interval.
Per variant allele copy or 10% increase in weight.
Effects of OATP1B1 and CYP3A4 phenotype classes on simvastatin acid and lactone
To further elucidate the roles of SLCO1B1 and CYP3A4 in simvastatin pharmacokinetics, we then grouped the participants as an exploratory post hoc analysis into SLCO1B1 and CYP3A4 genotype‐predicted phenotype groups (Figure 2 ). Only one participant was classified as poor CYP3A4 metabolizer and was therefore excluded from these analyses. In addition, the participant had also decreased OATP1B1 function. This participant had the third largest AUC0–∞ of simvastatin acid (76.1 ng/mL × hours), and the second largest AUC0–∞ of simvastatin lactone (202.6 ng/mL × hours).
Figure 2.
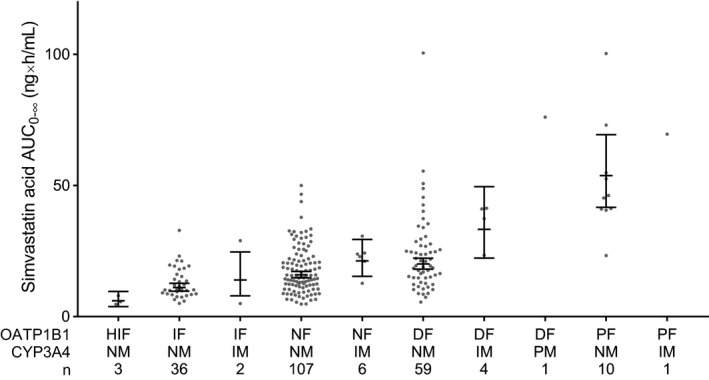
Area under the plasma simvastatin acid concentration‐time curve from 0 hours to infinity (AUC0–∞) values grouped by genotype predicted organic anion transporting polypeptide 1B1 (OATP1B1) and cytochrome P‐450 3A4 (CYP3A4) phenotypes. Horizontal lines indicate estimated marginal means, whiskers indicate 90% confidence intervals, and gray dots indicate individual AUC0–∞ values. DF, decreased function; HIF, highly increased function; IF, increased function; IM, intermediate metabolizer; NF, normal function; NM, normal metabolizer; PF, poor function; PM, poor metabolizer.
When compared with the normal OATP1B1 function group, the AUC0–∞ of simvastatin acid was 273% (90% CI, 137%, 488%; P = 3.1 × 10−6) larger in the poor function group, 40% (90% CI, 8%, 83%; P = 0.036) larger in the decreased function group, borderline significantly 32% (90% CI, 5%, 52%; P = 0.056) smaller in the increased function group, and 67% (90% CI, 46%, 80%; P = 2.4 × 10−4) smaller in the highly increased function group (Table 3 ). The AUC0–∞ of simvastatin acid in the poor OATP1B1 function group was 1,029% (90% CI, 505%, 2007%; P = 8.3 × 10−10) larger than in the highly increased group, 451% (90% CI, 231%, 817%; P = 9.3 × 10−8) larger than in the increased function group, and 166% (90% CI, 66%, 326%; P = 7.2 × 10−4) larger than in the decreased function group. The simvastatin acid C max was 273% (90% CI, 141%, 477%; P = 3.1 × 10−6) higher in the poor function group and 73% (90% CI, 57%, 83%; P = 6.1 × 10−6) lower in the highly increased function group than in the normal function group.
Table 3.
Pharmacokinetic variables of simvastatin lactone and simvastatin acid in individuals with different genotype‐predicted OATP1B1 phenotypes
| OATP1B1 phenotype (n) | Geometric meana (90% CI) | Ratio to normal function (90% CI) | P value |
|---|---|---|---|
| Simvastatin lactone C max | |||
| Highly increased function (3) | 5.8 (3.3, 10.2) | 0.59 (0.33, 1.08) | 0.15 |
| Increased function (38) | 13.5 (9.4, 19.2) | 1.37 (0.91, 2.06) | 0.21 |
| Normal function (113) | 9.9 (8.0, 12.1) | ||
| Decreased function (63) | 15.3 (11.9, 19.7) | 1.55 (1.12, 2.15) | 0.026 |
| Poor function (11) | 15.0 (9.0, 25.1) | 1.52 (0.88, 2.64) | 0.21 |
| Simvastatin lactone t ½ | |||
| Highly increased function (3) | 4.1 (2.2, 7.5) | 0.96 (0.50, 1.83) | 0.91 |
| Increased function (38) | 4.1 (2.8, 6.0) | 0.96 (0.61, 1.49) | 0.87 |
| Normal function (113) | 4.2 (3.4, 5.3) | ||
| Decreased function (63) | 5.0 (3.8, 6.6) | 1.18 (0.83, 1.67) | 0.45 |
| Poor function (11) | 3.9 (2.2, 6.7) | 0.91 (0.59, 1.66) | 0.80 |
| Simvastatin lactone AUC0–∞ | |||
| Highly increased function (3) | 23.9 (13.2, 43.3) | 0.64 (0.34, 1.21) | 0.25 |
| Increased function (38) | 51.5 (35.4, 75.0) | 1.38 (0.90, 2.13) | 0.22 |
| Normal function (113) | 37.3 (30.0, 46.3) | ||
| Decreased function (63) | 47.2 (36.1, 61.6) | 1.27 (0.90, 1.78) | 0.26 |
| Poor function (11) | 48.0 (27.9, 82.4) | 1.29 (0.72, 2.31) | 0.47 |
| Simvastatin acid C max | |||
| Highly increased function (3) | 0.6 (0.4, 1.0) | 0.27 (0.17, 0.43) | 6.1 × 10–6 |
| Increased function (38) | 1.3 (1.0, 1.8) | 0.56 (0.41, 0.78) | 0.0036 |
| Normal function (113) | 2.4 (2.0, 2.8) | ||
| Decreased function (63) | 3.1 (2.5, 3.8) | 1.30 (1.01, 1.68) | 0.086 |
| Poor function (11) | 8.9 (5.9, 13.3) | 3.73 (2.41, 5.77) | 1.3 × 10−6 |
| Simvastatin acid t ½ | |||
| Highly increased function (3) | 3.9 (2.6, 6.0) | 1.14 (0.73, 1.8) | 0.62 |
| Increased function (38) | 4.2 (3.2, 5.5) | 1.23 (0.90, 1.67) | 0.27 |
| Normal function (113) | 3.5 (3.0, 4.0) | ||
| Decreased function (63) | 4.2 (3.4, 5.0) | 1.20 (0.94, 1.54) | 0.21 |
| Poor function (11) | 3.7 (2.5, 5.5) | 1.08 (0.71, 1.63) | 0.76 |
| Simvastatin acid AUC0–∞ | |||
| Highly increased function (3) | 6.1 (3.8, 9.6) | 0.33 (0.20, 0.54) | 2.4 × 10−4 |
| Increased function (38) | 12.5 (9.3, 16.7) | 0.68 (0.48, 0.95) | 0.056 |
| Normal function (113) | 18.4 (15.6, 21.8) | ||
| Decreased function (63) | 25.8 (21.0, 31.7) | 1.40 (1.08, 1.83) | 0.036 |
| Poor function (11) | 68.7 (45.1, 104.8) | 3.73 (2.37, 5.88) | 3.1 × 10−6 |
| Simvastatin acid / lactone AUC0–∞ ratio | |||
| Highly increased function (3) | 0.25 (0.15, 0.42) | 0.51 (0.29, 0.87) | 0.040 |
| Increased function (38) | 0.23 (0.17, 0.32) | 0.46 (0.32, 0.67) | 8.1 × 10−4 |
| Normal function (113) | 0.50 (0.41, 0.60) | ||
| Decreased function (63) | 0.56 (0.45, 0.71) | 1.13 (0.84, 1.52) | 0.49 |
| Poor function (11) | 1.21 (0.76, 1.93) | 2.43 (1.47, 4.01) | 0.0039 |
AUC0–∞, area under the plasma concentration‐time curve from 0 hours to infinity; CI, confidence interval; C max, peak plasma concentration; OATP1B1, organic anion transporting polypeptide 1B1; t ½ , terminal half‐life.
The data are geometric estimated marginal means adjusted for weight (simvastatin acid AUC0–∞ and C max) and genotype‐predicted cytochrome P‐450 3A4 (CYP3A4) phenotype.
In the decreased OATP1B1 function group, the C max of simvastatin lactone was 55% (90% CI, 12%, 115%; P = 0.026) higher compared with the normal function group, but there were no other significant associations between OATP1B1 function groups and pharmacokinetic variables of simvastatin lactone (Table 3 ). Simvastatin acid / simvastatin lactone AUC0–∞ ratio was 49% (90% CI, 13%, 71%; P = 0.04) lower in the highly increased, 54% (90% CI, 33%, 68%; P = 8.1 × 10−4) lower in the increased, and 143% (90% CI, 47%, 301%; P = 0.0039) higher in the poor function group than in the normal function group. We also analyzed the associations of OATP1B1 phenotype classes with simvastatin pharmacokinetics without adjusting for CYP3A4 classes, and the associations were similar but stronger (Table S2 ).
In the CYP3A4 intermediate metabolizer group, simvastatin lactone AUC0–∞ was 63% (90% CI, 11%, 140%; P = 0.036) higher, and simvastatin acid AUC0–∞ was 87% (90% CI, 39%, 152%; P = 6.4 × 10−4) higher than in the normal metabolizer group (Table 4 ). The intermediate CYP3A4 metabolizers had 54% (90% CI, 7%, 122%; P = 0.05) higher simvastatin lactone C max, and 79% (90% CI, 35%, 139%; P = 9.0 × 10−4) higher simvastatin acid C max than normal metabolizers. CYP3A4 phenotype was not associated with the half‐life of either simvastatin acid or simvastatin lactone, or with the simvastatin acid/lactone AUC0–∞ ratio.
Table 4.
Pharmacokinetic variables of simvastatin lactone and simvastatin acid in individuals with different genotype‐predicted CYP3A4 phenotypes
| CYP3A4 phenotype (n) | Geometric meana (90% CI) | Ratio to normal function (90% CI) | P value |
|---|---|---|---|
| Simvastatin lactone C max | |||
| Normal metabolizer (215) | 9.9 (8.7, 11.4) | ||
| Intermediate metabolizer (13) | 15.4 (11, 21.5) | 1.54 (1.07, 2.22) | 0.050 |
| Simvastatin lactone t ½ | |||
| Normal metabolizer (215) | 4.4 (3.8, 5.1) | ||
| Intermediate metabolizer (13) | 4.1 (2.8, 5.9) | 0.92 (0.62, 1.37) | 0.74 |
| Simvastatin lactone AUC0–∞ | |||
| Normal metabolizer (215) | 34.1 (29.6, 39.4) | ||
| Intermediate metabolizer (13) | 55.8 (39.0, 79.8) | 1.63 (1.11, 2.40) | 0.036 |
| Simvastatin acid C max | |||
| Normal metabolizer (215) | 2.0 (1.8, 2.2) | ||
| Intermediate metabolizer (13) | 3.6 (2.7, 4.6) | 1.79 (1.35, 2.39) | 9.0 × 10−4 |
| Simvastatin acid t ½ | |||
| Normal metabolizer (215) | 3.8 (3.4, 4.2) | ||
| Intermediate metabolizer (13) | 4.0 (3.1, 5.2) | 1.06 (0.81, 1.40) | 0.72 |
| Simvastatin acid AUC0–∞ | |||
| Normal metabolizer (215) | 16.3 (14.6, 18.3) | ||
| Intermediate metabolizer (13) | 30.5 (23.1, 40.3) | 1.87 (1.39, 2.52) | 6.4 × 10−4 |
| Simvastatin acid /simvastatin lactone AUC0–∞ ratio | |||
| Normal metabolizer (215) | 0.47 (0.42, 0.54) | ||
| Intermediate metabolizer (13) | 0.51 (0.37, 0.69) | 1.07 (0.77, 1.49) | 0.73 |
AUC0–∞, area under the plasma concentration‐time curve from 0 hours to infinity; CI, confidence interval; C max, peak plasma concentration; CYP3A4, cytochrome P‐450 3A4; t ½ , terminal half‐life.
The data are geometric estimated marginal means adjusted for weight (simvastatin acid AUC0–∞ and C max) and genotype‐predicted organic anion transporting polypeptide 1B1 (OATP1B1) phenotype.
Effects of OATP1B1 and CYP3A4 phenotype classes on other simvastatin metabolites
We next investigated the associations of OATP1B1 and CYP3A4 phenotype classes with simvastatin oxidative metabolite pharmacokinetics from the prospective study. As there were only two participants with highly increased OATP1B1 function and only one poor CYP3A4 metabolizer, they were excluded from the analyses. In the poor OATP1B1 function group, 3′,5′‐dihydrodiol simvastatin lactone AUC0–∞ was 80% (90% CI, 29%, 151%; P = 0.0040) higher, 3′,5′‐dihydrodiol simvastatin acid AUC0‐24hours was 105% (90% CI, 27%, 229%; P = 0.014) higher, and 3″‐hydroxy simvastatin acid AUC0‐24hours was 133% (90% CI, 57%, 246%; P = 5.4 × 10−4) higher than in the normal OATP1B1 function group (Figure 3 , Table S3 ). The 3′,5′‐dihydrodiol simvastatin lactone / simvastatin lactone AUC0–∞ ratio was 43% (90% CI, 10%, 64%; P = 0.046) lower in the increased function group than in the normal function group, but OATP1B1 phenotype was not associated with differences in metabolite / parent compound AUC ratios of other metabolites. CYP3A4 phenotype was not significantly associated with the AUC of the oxidative simvastatin metabolites. However, intermediate CYP3A4 metabolizers had 49% (90% CI, 26%, 65%; P = 0.0034) lower 3′,5′‐dihydrodiol simvastatin lactone / simvastatin lactone AUC0–∞ ratio than normal CYP3A4 metabolizers (Table S4 ).
Figure 3.
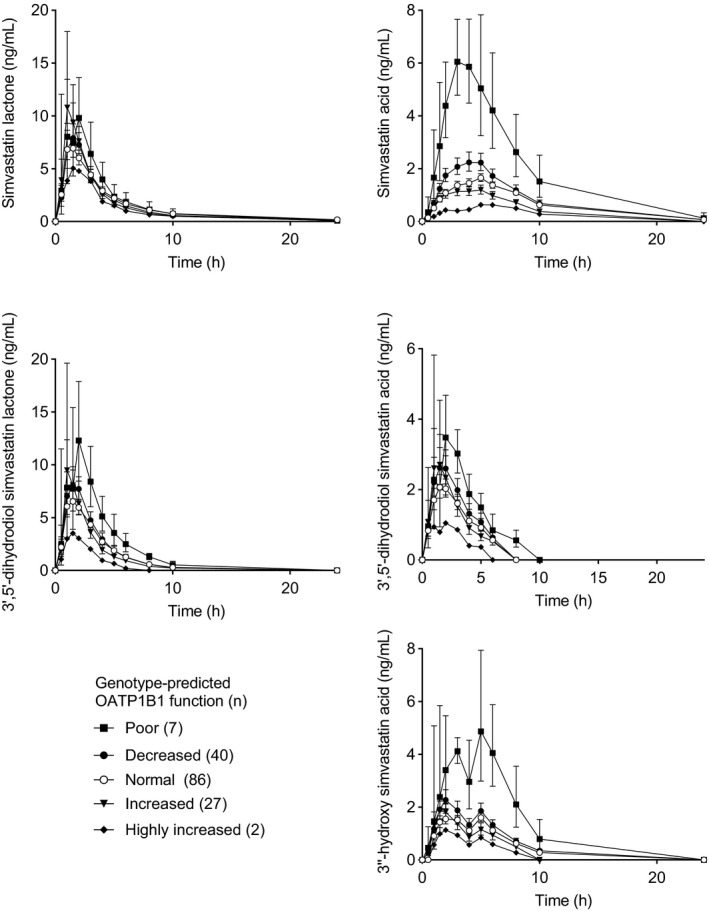
Geometric mean (90% confidence interval) plasma concentrations of simvastatin and its metabolites grouped by genotype‐predicted organic anion transporting polypeptide 1B1 (OATP1B1) phenotype. Some error bars were omitted for clarity. Carriers of CYP3A4*2 and CYP3A4*22 alleles were excluded. DF, decreased function; HIF, highly increased function; IF, increased function; NF, normal function; PF, poor function.
DISCUSSION
In this study, we investigated genetic variation of simvastatin pharmacokinetics in 229 healthy volunteers. Five SNVs in the SLCO1B1‐SLCO1B3 locus associated genomewide significantly with increased simvastatin acid AUC. The strongest association was observed with the SLCO1B1 c.521T>C, OATP1B1 no function variant. In addition, a candidate‐gene analysis suggested associations of increased function SLCO1B1 alleles and decreased function CYP3A4 alleles with simvastatin acid pharmacokinetics. Of note, simvastatin acid AUC was ~10‐fold larger in individuals with poor OATP1B1 function than in those with highly increased OATP1B1 function. To our knowledge, this is the first study to demonstrate the effects of SLCO1B1*14, SLCO1B1*20, and CYP3A4*2 on simvastatin pharmacokinetics. Taken together, our data show that SLCO1B1 and CYP3A4 are major determinants of simvastatin acid pharmacokinetics.
The only genomewide significant associations were observed in the SLCO1B1‐SLCO1B3 locus. The SLCO1B1 c.521T>C SNV is probably the causative variant underlying these associations, as it showed the strongest association and is in linkage equilibrium with the other genomewide significant SNVs. The c.521T>C SNV is known to cause nearly complete loss‐of‐function of OATP1B1, 25 , 26 , 27 , 28 and to increase simvastatin acid plasma concentrations and myopathy risk. 3 , 4 , 5 , 6 Mechanistically, the association can be explained by reduced hepatic uptake of simvastatin acid due to impaired OATP1B1 function. 26 When compared with individuals with normal OATP1B1 function, those with poor OATP1B1 function (c.521T>C homozygotes) showed 273% higher and those with decreased OATP1B1 function (c.521T>C heterozygotes) showed 40% higher simvastatin acid AUC. These data are consistent with previously published data and highlight the important role of SLCO1B1 c.521T>C SNV in simvastatin pharmacokinetics. 6
In a candidate‐gene analysis, the SLCO1B1 c.463C>A and c.1929A>C variants, which define the *14 and *20 haplotypes, were associated with a reduced simvastatin acid AUC. Consistent with our results, these haplotypes have been associated with increased OATP1B1 expression in the liver. 29 Furthermore, they have been associated with reduced OATP1B1 biomarker concentrations 24 , 28 and increased methotrexate clearance in humans. 30 In addition, SLCO1B1*14 has been associated with poor response to methotrexate 31 and enhanced response to fluvastatin. 32 Although the associations of *14 and *20 were not genomewide significant, the biological mechanism is plausible and the results are consistent with the literature on other OATP1B1 substrates. 28 , 30 Taken together, the data indicate that SLCO1B1*14 and *20 are increased function variants and enhance the hepatic uptake of simvastatin acid thereby reducing its systemic exposure.
In addition, the candidate‐gene analysis showed association of CYP3A4*2 with increased simvastatin acid AUC. However, after correction for multiple testing, the association did not remain significant. Studies on the effects of CYP3A4*2 on CYP3A4 activity in vivo are scarce, but one individual with the CYP3A4*2/*2 genotype was reported to have a low 6β‐OH‐cortisol/cortisol ratio indicating slow CYP3A4 metabolism. 33 In vitro studies suggest that the effect of CYP3A4.2 may be substrate‐specific as the clearance of some CYP3A4 substrates is reduced 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 whereas the clearance of others, including testosterone, 34 , 36 , 42 is unaffected or increased. 43 , 44 Another CYP3A4 variant, CYP3A4*22, has associated with increased plasma concentrations of simvastatin lactone and acid, and enhanced cholesterol‐lowering efficacy of atorvastatin, lovastatin, and simvastatin. 11 , 12 , 45 CYP3A4*22 is an intronic variant, which alters RNA splicing and decreases CYP3A4 expression in the liver. 46 Based on these data, we classified heterozygous carriers of either CYP3A4*2 or CYP3A4*22 as intermediate CYP3A4 metabolizers. Grouping rare variants with similar effects provides better statistical power, and a similar approach is commonly used with other CYP enzymes, such as CYP2D6. Nevertheless, as the data on these CYP3A4 variants are limited, the results should be interpreted with caution. In our exploratory analyses, the intermediate CYP3A4 metabolizers had 87% higher simvastatin acid AUC and 63% higher simvastatin lactone AUC.
Our analyses of simvastatin metabolite concentrations suggest that CYP3A4 converts simvastatin lactone to 3′,5′‐dihydrodiol simvastatin lactone as the CYP3A4 phenotype was significantly associated with the 3′,5′‐dihydrodiol simvastatin lactone / simvastatin lactone AUC ratio. In addition, poor OATP1B1 function was associated with an increased exposure to 3′,5′‐dihydrodiol simvastatin lactone, 3′,5′‐dihydrodiol simvastatin acid, and 3″‐hydroxy simvastatin acid. This suggests that these metabolites may be OATP1B1 substrates, but interconversion between the lactone and acid forms of the metabolites could also explain some of the associations. Literature regarding simvastatin metabolites is scarce, and possible contributions of the metabolites to the efficacy or toxicity of simvastatin are not known. In one study in hypercholesterolemic children and adolescents, the SLCO1B1 c.521T>C SNV showed no association with 3′,5′‐dihydrodiol simvastatin lactone pharmacokinetics, 47 but there seem to be no other pharmacogenetic studies on the oxidative metabolites of simvastatin. Taken together, our data suggest that CYP3A4 contributes to the formation of 3′,5′‐dihydrodiol simvastatin lactone, but plays a minor role only in the formation of 3″‐hydroxy simvastatin acid or 3′,5′‐dihydrodiol simvastatin acid. This knowledge may be useful for interpreting the results of drug–drug interaction studies using simvastatin as an index substrate for OATP1B1 or CYP3A4. 48
In this study, we investigated simvastatin single‐dose pharmacokinetics only. According to pharmacokinetic theory, the single‐dose AUC0–∞ should be equal to the steady‐state dose‐interval AUC. Therefore, the differences in simvastatin exposure seen in the present study between individuals with different SLCO1B1 and CYP3A4 genotypes should be similar during continuous treatment. Moreover, the present study was carried out in young healthy volunteers, whereas statin users are typically older and may use other medications concomitantly. Interindividual variability in simvastatin pharmacokinetics may therefore be even larger in patients than what was observed in the present study.
The participants of this study were White Finnish volunteers. The frequencies of SLCO1B1 and CYP3A4 variants differ between populations. 49 , 50 SLCO1B1 poor function variants are more common in the Northern hemisphere, and SLCO1B1*5 is prevalent only in Europe (MAF 2%), Middle East (5%), and North Africa (2%). 49 SLCO1B1*15 is present in other populations as well: it is more common in Europe (16%) than in East Asia (12%), and Sub‐Saharan Africa (2%). 49 SLCO1B1*14 is most common in European (16%) and Middle Eastern (15%) populations, rare in sub‐Saharan Africans (4%) and not observed in East Asians. 49 SLCO1B1*20 has its highest frequency in North Africans (15%), but it is also present in sub‐Saharan African (9%) and European (5%) populations, yet rare in East Asia (0.6%). 49 CYP3A4*2 is observed only in Europeans (1%), whereas CYP3A4*22 is most common in Europeans (5%), but also observed in South Asians (0.6%). 50 Some populations may also have pharmacogenetic variants affecting simvastatin pharmacokinetics that were not present in this study. Nevertheless, the effects of SLCO1B1 and CYP3A4 variants on simvastatin pharmacokinetics should be similar to our results in other populations, but the proportion of variability in simvastatin pharmacokinetics explained by these variants may differ.
The effect of SLCO1B1 c.521T>C on simvastatin‐induced myopathy and rhabdomyolysis risk is well‐known. 2 , 3 , 4 , 5 In order to mitigate the risk of myotoxicity, a lower simvastatin dose is advisable for patients with decreased OATP1B1 function and the use of simvastatin is best avoided in individuals with poor OATP1B1 function. The effects of SLCO1B1*14 and *20 on the clinical response to simvastatin are not known. The pharmacological target of statins, HMG‐CoA reductase, is expressed in the liver. Increased hepatic uptake caused by SLCO1B1*14 and *20 may increase simvastatin acid concentration at the active site. The higher hepatic/systemic concentration ratio may enhance the efficacy and reduce the risk for systemic adverse effects of simvastatin.
In contrast to decreased OATP1B1 function, reduced CYP3A4 activity should increase both the hepatocyte and systemic exposure to active simvastatin acid. Decreased CYP3A4 activity may therefore enhance the cholesterol‐lowering efficacy of simvastatin in addition to an increase in the adverse effect risk. Our data indicate that the effects of decreased activity CYP3A4 variants on simvastatin pharmacokinetics are quite large. Therefore, lowering of simvastatin dose should be considered for intermediate CYP3A4 metabolizers and poor CYP3A4 metabolizers should probably avoid using simvastatin. From a safety perspective, intermediate CYP3A4 metabolizers with decreased OATP1B1 function should probably also avoid the use of simvastatin as the effect may be additive.
In conclusion, this large pharmacokinetic study confirms the effects of SLCO1B1 poor and decreased function genotypes on simvastatin acid. Furthermore, it shows that SLCO1B1*14 and *20 haplotypes lower the systemic exposure to simvastatin acid. Moreover, this study is the first to suggest an association of CYP3A4*2 with increased simvastatin lactone and acid exposure. Simvastatin may be best avoided in individuals with genetically poor OATP1B1 function or CYP3A4 activity and in those with the combination of decreased OATP1B1 function and CYP3A4 activity. Reduced simvastatin doses should be considered for individuals with decreased OATP1B1 function or CYP3A4 activity.
AUTHOR CONTRIBUTIONS
A.M. and M.Ni. wrote the manuscript. S.T., J.T.B., A.T., and M.Ni. designed the research. S.T., M.Ne., M.P.‐H., E.K.T., T.L., T.T., J.T.B., A.T., and M.Ni. performed the research. A.M. and M.Ni. analyzed the data.
FUNDING
This study was supported by grants from European Research Council (Grant agreement 282106), the Sigrid Jusélius Foundation and State funding for university‐level health research (Helsinki, Finland).
CONFLICTS OF INTEREST
The authors declared no competing interests for this work.
Supporting information
Table S1
Table S2
Table S3
Table S4
Figure S1
ACKNOWLEDGMENTS
The authors thank Eija Mäkinen‐Pulli and Lisbet Partanen for their skillful technical assistance, and Minna Lehtisalo and Päivi Hirvensalo for their valuable insight in designing the statistical analyses.
References
- 1. Neuvonen, P.J. , Niemi, M. & Backman, J.T. Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance. Clin. Pharmacol. Ther. 80, 565–581 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Voora, D. et al. The SLCO1B1*5 genetic variant is associated with statin‐induced side effects. J. Am. Coll. Cardiol. 54, 1609–1616 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilke, R.A. et al. The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin‐induced myopathy. Clin. Pharmacol. Ther. 92, 112–117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cooper‐DeHoff, R.M. et al. The clinical pharmacogenetics implementation consortium (CPIC) guideline for SLCO1B1, ABCG2, and CYP2C9 and statin‐associated musculoskeletal symptoms. Clin. Pharmacol. Ther. 111, 1007–1021 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. SEARCH Collaborative Group et al. SLCO1B1 variants and statin‐induced myopathy—a genomewide study. N. Engl. J. Med. 359, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 6. Pasanen, M.K. , Neuvonen, M. , Neuvonen, P.J. & Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet. Genomics 16, 873–879 (2006). [DOI] [PubMed] [Google Scholar]
- 7. Neuvonen, P.J. , Kantola, T. & Kivistö, K.T. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin. Pharmacol. Ther. 63, 332–341 (1998). [DOI] [PubMed] [Google Scholar]
- 8. Prueksaritanont, T. et al. Glucuronidation of statins in animals and humans: a novel mechanism of statin lactonization. Drug Metab. Dispos. 30, 505–512 (2002). [DOI] [PubMed] [Google Scholar]
- 9. Prueksaritanont, T. , Ma, B. & Yu, N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br. J. Clin. Pharmacol. 56, 120–124 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Filppula, A.M. et al. Comparative hepatic and intestinal metabolism and pharmacodynamics of statins. Drug Metab. Dispos. 49, 658–667 (2021). [DOI] [PubMed] [Google Scholar]
- 11. Wang, D. , Guo, Y. , Wrighton, S.A. , Cooke, G.E. & Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 11, 274–286 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitzmiller, J.P. , Luzum, J.A. , Baldassarre, D. , Krauss, R.M. & Medina, M.W. CYP3A4*22 and CYP3A5*3 are associated with increased levels of plasma simvastatin concentrations in the cholesterol and pharmacogenetics study cohort. Pharmacogenet. Genomics 24, 486–491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hochman, J.H. et al. Interactions of human P‐glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm. Res. 21, 1686–1691 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Chen, C. et al. Differential interaction of 3‐hydroxy‐3‐methylglutaryl‐coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab. Dispos. 33, 537–546 (2005). [DOI] [PubMed] [Google Scholar]
- 15. Deng, F. et al. Comparative hepatic and intestinal efflux transport of statins. Drug Metab. Dispos. 49, 750–759 (2021). [DOI] [PubMed] [Google Scholar]
- 16. Keskitalo, J.E. , Kurkinen, K.J. , Neuvoneni, P.J. & Niemi, M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin. Pharmacol. Ther. 84, 457–461 (2008). [DOI] [PubMed] [Google Scholar]
- 17. Becker, M.L. , Visser, L.E. , van Schaik, R.H.N. , Hofman, A. , Uitterlinden, A.G. & Stricker, B.H.C. Common genetic variation in the ABCB1 gene is associated with the cholesterol‐lowering effect of simvastatin in males. Pharmacogenomics 10, 1743–1751 (2009). [DOI] [PubMed] [Google Scholar]
- 18. Jiang, F. et al. The influences of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of simvastatin, in relation to CYP3A4 inhibition. Pharmacogenomics 18, 459–469 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Keskitalo, J.E. , Pasanen, M.K. , Neuvonen, P.J. & Niemi, M. Different effects of the ABCG2 c.421C>a SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics 10, 1617–1624 (2009). [DOI] [PubMed] [Google Scholar]
- 20. Stephens, M. , Smith, N.J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stephens, M. & Donnelly, P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 73, 1162–1169 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaedigk, A. et al. The Pharmacogene variation (PharmVar) consortium: incorporation of the human cytochrome P450 (CYP) allele nomenclature database. Clin. Pharmacol. Ther. 103, 399–401 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaedigk, A. et al. The evolution of PharmVar. Clin. Pharmacol. Ther. 105, 29–32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neuvonen, M. , Tornio, A. , Hirvensalo, P. , Backman, J.T. & Niemi, M. Performance of plasma coproporphyrin I and III as OATP1B1 biomarkers in humans. Clin. Pharmacol. Ther. 110, 1622–1632 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tirona, R.G. , Leake, B.F. , Merino, G. & Kim, R.B. Polymorphisms in OATP‐C: identification of multiple allelic variants associated with altered transport activity among European‐ and African‐Americans. J. Biol. Chem. 276, 35669–35675 (2001). [DOI] [PubMed] [Google Scholar]
- 26. Niemi, M. , Pasanen, M.K. & Neuvonen, P.J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63, 157–181 (2011). [DOI] [PubMed] [Google Scholar]
- 27. Hirvensalo, P. et al. Enantiospecific pharmacogenomics of fluvastatin. Clin. Pharmacol. Ther. 106, 668–680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neuvonen, M. et al. Identification of glycochenodeoxycholate 3‐O‐glucuronide and glycodeoxycholate 3‐O‐glucuronide as highly sensitive and specific OATP1B1 biomarkers. Clin. Pharmacol. Ther. 109, 646–657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nies, A.T. et al. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 5, 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramsey, L.B. et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 22, 1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramsey, L.B. et al. Association of SLCO1B1 *14 allele with poor response to methotrexate in juvenile idiopathic arthritis patients. ACR Open Rheumatol 1, 58–62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Couvert, P. et al. Association between a frequent allele of the gene encoding OATP1B1 and enhanced LDL‐lowering response to fluvastatin therapy. Pharmacogenomics 9, 1217–1227 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Shchepotina, E.G. , Vavilin, V.A. , Goreva, O.B. & Lyakhovich, V.V. Some mutations of exon‐7 in cytochrome P450 gene 3A4 and their effect on 6beta‐hydroxylation of cortisol. Bull. Exp. Biol. Med. 141, 701–703 (2006). [DOI] [PubMed] [Google Scholar]
- 34. Sata, F. et al. CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12: evidence for an allelic variant with altered catalytic activity. Clin. Pharmacol. Ther. 67, 48–56 (2000). [DOI] [PubMed] [Google Scholar]
- 35. Miyazaki, M. , Nakamura, K. , Fujita, Y. , Guengerich, F.P. , Horiuchi, R. & Yamamoto, K. Defective activity of recombinant cytochromes P450 3A4.2 and 3A4.16 in oxidation of midazolam, nifedipine, and testosterone. Drug Metab. Dispos. 36, 2287–2291 (2008). [DOI] [PubMed] [Google Scholar]
- 36. Fang, P. et al. Functional assessment of CYP3A4 allelic variants on lidocaine metabolism in vitro. Drug Des. Devel. Ther. 11, 3503–3510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu, R.‐A. et al. Functional characterization of 22 CYP3A4 protein variants to metabolize ibrutinib in vitro. Basic Clin. Pharmacol. Toxicol. 122, 383–387 (2018). [DOI] [PubMed] [Google Scholar]
- 38. Lin, Q.‐M. et al. Characterization of genetic variation in CYP3A4 on the metabolism of cabozantinib in vitro. Chem. Res. Toxicol. 32, 1583–1590 (2019). [DOI] [PubMed] [Google Scholar]
- 39. Tang, P.‐F. et al. Functional measurement of CYP2C9 and CYP3A4 allelic polymorphism on sildenafil metabolism. Drug Des. Devel. Ther. 14, 5129–5141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen, B. et al. Effects of 26 recombinant CYP3A4 variants on brexpiprazole metabolism. Chem. Res. Toxicol. 33, 172–180 (2020). [DOI] [PubMed] [Google Scholar]
- 41. Cai, Y. et al. Evaluation of recombinant CYP3A4 variants on the metabolism of oxycodone in vitro. Chem. Res. Toxicol. 34, 103–109 (2021). [DOI] [PubMed] [Google Scholar]
- 42. Akiyoshi, T. et al. Comparison of the inhibitory profiles of itraconazole and cimetidine in cytochrome P450 3A4 genetic variants. Drug Metab. Dispos. 39, 724–728 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han, M. et al. Functional assessment of the effects of CYP3A4 variants on acalabrutinib metabolism in vitro. Chem. Biol. Interact. 345, 109559 (2021). [DOI] [PubMed] [Google Scholar]
- 44. Han, M. et al. Functional evaluation of vandetanib metabolism by CYP3A4 variants and potential drug interactions in vitro. Chem. Biol. Interact. 350, 109700 (2021). [DOI] [PubMed] [Google Scholar]
- 45. Luzum, J.A. et al. Individual and combined associations of genetic variants in CYP3A4, CYP3A5, and SLCO1B1 with simvastatin and simvastatin acid plasma concentrations. J. Cardiovasc. Pharmacol. 66, 80–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang, D. & Sadee, W. CYP3A4 intronic SNP rs35599367 (CYP3A4*22) alters RNA splicing. Pharmacogenet. Genomics 26, 40–43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ogungbenro, K. , Wagner, J.B. , Abdel‐Rahman, S. , Leeder, J.S. & Galetin, A. A population pharmacokinetic model for simvastatin and its metabolites in children and adolescents. Eur. J. Clin. Pharmacol. 75, 1227–1235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tornio, A. , Filppula, A.M. , Niemi, M. & Backman, J.T. Clinical studies on drug‐drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin. Pharmacol. Ther. 105, 1345–1361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pasanen, M.K. , Neuvonen, P.J. & Niemi, M. Global analysis of genetic variation in SLCO1B1. Pharmacogenomics 9, 19–33 (2008). [DOI] [PubMed] [Google Scholar]
- 50. Zhou, Y. , Ingelman‐Sundberg, M. & Lauschke, V.M. Worldwide distribution of cytochrome P450 alleles: a meta‐analysis of population‐scale sequencing projects. Clin. Pharmacol. Ther. 102, 688–700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4
Figure S1