

Abstract
Green tea (GT) alters the disposition of a number of drugs, such as nadolol and lisinopril. However, it is unknown whether GT affects disposition of hydrophilic anti‐allergic drugs. The purpose of this study was to investigate whether pharmacokinetics of fexofenadine and pseudoephedrine are affected by catechins, major GT components. A randomized, open, 2‐phase crossover study was conducted in 10 healthy Japanese volunteers. After overnight fasting, subjects were simultaneously administered fexofenadine (60 mg) and pseudoephedrine (120 mg) with an aqueous solution of green tea extract (GTE) containing (−)‐epigallocatechin gallate (EGCG) of ~ 300 mg or water (control). In vitro transport assays were performed using HEK293 cells stably expressing organic anion transporting polypeptide (OATP)1A2 to evaluate the inhibitory effect of EGCG on OATP1A2‐mediated fexofenadine transport. In the GTE phase, the area under the plasma concentration‐time curve and the amount excreted unchanged into urine for 24 hours of fexofenadine were significantly decreased by 70% (P < 0.001) and 67% (P < 0.001), respectively, compared with control. There were no differences in time to maximum plasma concentration and the elimination half‐life of fexofenadine between phases. Fexofenadine was confirmed to be a substrate of OATP1A2, and EGCG (100 and 1,000 μM) and GTE (0.1 and 1 mg/mL) inhibited OATP1A2‐mediated uptake of fexofenadine. On the contrary, the concomitant administration of GTE did not influence the pharmacokinetics of pseudoephedrine. These results suggest that intake of GT may result in a markedly reduced exposure of fexofenadine, but not of pseudoephedrine, putatively by inhibiting OATP1A2‐mediated intestinal absorption.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Green tea (GT) and its catechin components interact with hydrophilic drugs, such as nadolol and lisinopril. A combination of fexofenadine and pseudoephedrine is clinically available for treatment of allergic rhinitis.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study evaluated whether pharmacokinetics of fexofenadine and pseudoephedrine are affected when orally administered with an aqueous solution of green tea extract (GTE) in healthy volunteers.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Plasma concentrations and urinary excretions of fexofenadine were markedly decreased when coadministered with GTE, containing ~ 300 mg of (−)‐epigallocatechin gallate. GTE and (−)‐epigallocatechin gallate significantly inhibited OATP1A2‐mediated fexofenadine uptake. No differences were observed in the pharmacokinetics of pseudoephedrine between water and GTE.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ GT and its catechin supplement reduce oral bioavailability of fexofenadine, but not of pseudoephedrine. The inhibition of intestinal OATP1A2 by (−)‐epigallocatechin gallate is a likely mechanism underlying this interaction.
Green tea (GT; Camellia sinensis) and its main components, catechins, are perpetrators of clinically relevant food‐drug interactions with hydrophilic and nonmetabolized drugs, such as nadolol and lisinopril. 1 , 2 , 3 , 4 Among naturally occurring catechins, (−)‐epigallocatechin gallate (EGCG) plays a major role in GT–drug interactions. 2 , 4 Orally ingested EGCG likely inhibits membrane permeation of drugs into enterocytes mediated by uptake transporters expressed in the apical membrane, such as organic anion transporting polypeptide (OATP)s. 1 , 2 , 5 , 6 , 7 These findings led to the hypothesis that pharmacokinetics of hydrophilic drugs which are generally categorized as class 3 drugs according to the biopharmaceutics classification system (BCS) and the biopharmaceutics drug disposition classification system (BDDCS), are affected when co‐administered with GT.
The H1 blocker fexofenadine has intensively been investigated with respect to pharmacokinetic characteristics and transporter‐mediated drug interactions. 8 , 9 Due to its hydrophilicity, fexofenadine is not a substrate of CYP enzymes and is largely excreted unchanged with minimal hepatic metabolism. 10 Both uptake and efflux transporters are involved in the disposition of fexofenadine including P‐glycoprotein and OATPs. 11 , 12 , 13 Pharmacokinetics after oral administration of fexofenadine are influenced not only by drugs, such as itraconazole and rifampicin, 13 , 14 , 15 but also by concomitant food or beverages, such as grapefruit juice and apple juice. 16 , 17 , 18 To date, however, no studies have examined the effects of GT or catechins on the pharmacokinetics of fexofenadine.
At present, a combined tablet of fexofenadine and pseudoephedrine, a nasal decongestant, is available for the treatment of allergic rhinitis in several countries, such as the United States (Allegra‐D) and Japan (Dellegra). 19 It has been reported that the pharmacokinetics of fexofenadine/pseudoephedrine combination formulation are bioequivalent to that of the individual drugs. 20 Of note, pseudoephedrine is also hydrophilic and a weak base compound, and is classified as BCS and BDDCS class 3 drug. 21 , 22 Indeed, pseudoephedrine is metabolized in the liver to a minor extent and is mainly excreted unchanged in the urine. 23 However, contrarily to fexofenadine, orally administered pseudoephedrine is nearly completely absorbed from the gastrointestinal tract. 23 Previous studies showed a negligible food effect on pseudoephedrine pharmacokinetics in humans. 24 Nevertheless, it remains to be elucidated whether plasma concentrations of pseudoephedrine are influenced by concomitant beverages as it is the case for other hydrophilic drugs such as fexofenadine.
Taking into account the anti‐allergic potential of GT and catechins, 25 the opportunity of drinking GT during the treatment with anti‐allergic drugs will arise in expectation of its beneficial effect on allergic symptoms. Therefore, the objective of this study was to assess whether GT catechins affect the pharmacokinetics of fexofenadine and pseudoephedrine in humans using a commercially available EGCG‐concentrated green tea extract (GTE). We also performed in vitro assays to investigate possible molecular mechanisms of the interaction between GTE and fexofenadine.
METHODS
Subjects
Ten healthy nonsmoking Japanese volunteers (8 men and 2 women) participated in this study with an age range of 21–45 years and body mass index of 18.4–26.0 kg/m2. All volunteers provided a written informed consent for study participation. The volunteers were ascertained to be healthy by medical history, physical examination, and routine laboratory tests. They were prohibited from consuming GT and fruit products, including apple, grapefruit, and orange juices, for 7 days before each trial day. The participants were genotyped for SLCO2B1*3 (c.1457C>T) single nucleotide variation (SNV), as stated in Methods S1 .
Clinical study design
The study protocol was approved by the ethics committee of the Fukushima Medical University (approval number: RK29037) and was registered at the UMIN Clinical Trials Registry (UMIN000032828). The study was conducted in compliance with the principles of the Declaration of Helsinki. In a single‐center, open‐label, randomized 2‐way crossover study with a washout period of 2 weeks, the participants ingested a single oral dose of 2 tablets of fexofenadine (30 mg × 2) and (+)‐pseudoephedrine (sustained‐release formulation, 60 mg × 2; Dellegra; LTL Pharma, Tokyo, Japan) simultaneously with 150 mL of an aqueous solution of a commercial GTE (Sunphenon‐EGCG, Taiyo Kagaku, Yokkaichi, Japan) or with 150 mL of water after overnight fasting. The GTE contained 92.5% (w/w) of EGCG, and 325 mg of which was dissolved in water with stirring. Subjects had a snack at 1 hour and a standardized meal at 4 hours after administration. In each study period, 5 mL venous blood samples were collected from an indwelling catheter placed in an antecubital vein or by direct venipuncture into EDTA‐treated tubes at 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 24 hours after administration. Blood samples were immediately centrifuged at 2,000 g for 10 minutes at 4°C. Urine was collected during periods of 0–4, 4–8, and 8–24 hours after administration. Plasma and urine samples were stored at −80°C until analysis.
Determinations of drug concentration in plasma and urine
The concentrations of fexofenadine and pseudoephedrine in plasma and urine were determined using ultra‐performance liquid chromatography (UPLC) with fluorescence detection (UPLC, Waters, Milford, MA), as summarized in Methods S1 . Diphenhydramine and phenylephrine were used as internal standards for fexofenadine and pseudoephedrine, respectively. The limit of quantification for fexofenadine and pseudoephedrine was both 10 ng/mL. The inter‐day coefficients of variation of fexofenadine and pseudoephedrine were 9.9% and 7.3%, respectively.
Transport assays
To test whether GTE and EGCG affect OATP1A2‐ and OATP2B1‐mediated uptake of fexofenadine, we performed in vitro transport assays using OATP1A2‐ and OATP2B1‐stably expressing HEK cells and the respective HEK‐VC vector control cells. Sulfobromophthalein (BSP) was used as a typical substrate for OATP2B1. The transporter‐expressing cells and the respective vector control cells were cultured according to previous studies. 1 , 26 The experimental and quantitation methods of fexofenadine in the in vitro samples using liquid chromatography–tandem mass spectrometry (Thermo Fisher Scientific, Waltham, MA) are described in Methods S1 .
Chemical binding assays
To examine the possibility of direct chemical binding between fexofenadine and EGCG in the gastrointestinal tract, fexofenadine (400 μg/mL, based on the maximum concentration when a 60‐mg dose was dissolved in 150 mL) was incubated in the presence or absence of EGCG (1 mg/mL) in saline at 37°C for 1, 2, and 24 hours. Bortezomib (20 μg/mL) was used as a positive control for the chemical interaction with EGCG. The chromatographic separation of the samples was achieved by UPLC (Waters) system, as stated in detail in Methods S1 .
Pharmacokinetics
The peak plasma concentration (C max), time to C max (T max), area under the concentration‐time curve (AUC0–∞), AUC0–8, AUC0–24, and elimination half‐life (t 1/2), were calculated by noncompartmental analysis using WinNonlin software (version 5.1; Certara, Princeton, NJ). The renal clearance was obtained from the equation CLrenal = A e/AUC0–24, in which A e is the amount of fexofenadine excreted into urine up to 24 hours.
Statistical analysis
In vitro data are expressed as mean ± standard error mean. Clinical pharmacokinetic data are expressed as geometric means and coefficient of variation (geoCV, %) unless otherwise noted. The number of subjects was deemed to be sufficient to detect a potentially clinically meaningful effect size of 35% difference in AUC0–∞ of fexofenadine between 2 phases with a power of 80% (α‐level 5%) based on the previous pharmacokinetic data of fexofenadine in healthy Japanese subjects. 27 Effects of GTE on pharmacokinetics of test drugs were accepted if the 90% confidence interval (CI) of the geometric mean ratios (GMRs) did not fall within the bioequivalence boundary of 0.8–1.25. Correlations were examined using Pearson’s correlation coefficients. In vitro data were analyzed by one‐way analysis of variance and Tukey’s multiple comparison test. Pharmacokinetic parameters were analyzed by a paired t‐test. Statistical analyses were performed using GraphPad Prism software (version 8.4; GraphPad Software, San Diego, CA). Differences were regarded as statistically significant when P values were < 0.05.
RESULTS
All participants completed the study. None of the subjects experienced any adverse events related to the drugs. Three individuals were genotyped as SLCO2B1 c.1457CC (*1/*1) carriers, 5 individuals as SLCO2B1 c.1457CT (*1/*3) carriers, and 2 individuals as SLCO2B1 c.1457TT (*3/*3) carriers.
Fexofenadine pharmacokinetics
Plasma concentration‐time profiles and pharmacokinetic parameters of fexofenadine are shown in Figure 1 and Table 1 . There were no apparent differences in pharmacokinetic parameters of fexofenadine stratified for SLCO2B1 *3 SNV in water (control) phase (data not shown). Fexofenadine C max, AUC0‐8, AUC0‐∞, and A e in GTE phase were significantly decreased by 70% (P < 0.001), 71% (P < 0.001), 70% (P < 0.001), and 67% (P < 0.001), respectively, compared with control phase (Figures 1, 2 1 ). The GMR (GTE/water) for C max and AUC0‐∞ of fexofenadine were 0.296 (90% CI, 0.197–0.396) and 0.296 (90% CI, 0.225–0.366), respectively. Changes in pharmacokinetic parameters for each individual with SLCO2B1 *3 SNV are shown in Figure S1 . The decrease in fexofenadine AUC0‐∞ by GTE was negatively correlated with fexofenadine AUC0‐∞ in control phase (r = −0.9249, P < 0.001) (Figure S2 a). There were no differences in T max and t 1/2 of fexofenadine between control and GTE phases. CL renal of fexofenadine of this study was comparative to that of the previous study. 28 A slight but statistically significant (P = 0.017) increase in CL renal of fexofenadine was observed by co‐administration with GTE (Figure 2 c ).
Figure 1.
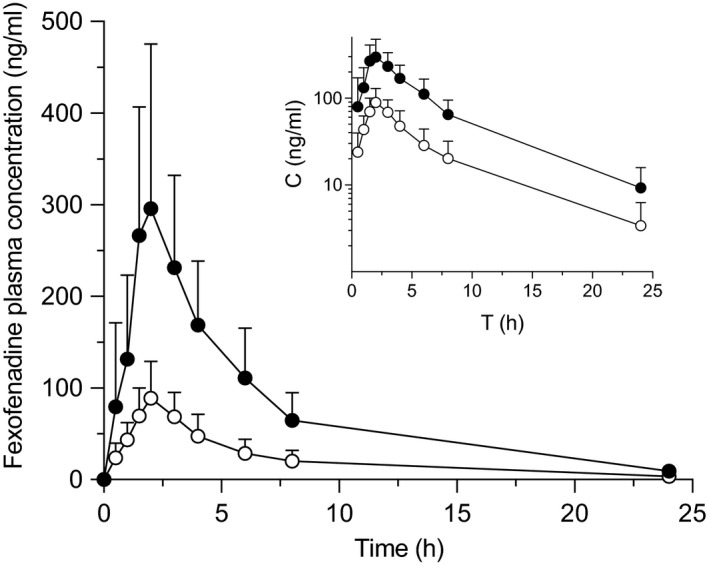
Plasma concentration profile of fexofenadine after oral administration of fexofenadine (60 mg) with 150 mL of an aqueous solution of (−)‐epigallocatechin gallate (EGCG)‐concentrated green tea extract (○), or water (●) in 10 healthy volunteers. Data are expressed as the arithmetic mean ± SD. The inset is the log‐concentration vs. time profile.
Table 1.
Pharmacokinetic parameters of fexofenadine after oral administration with GTE or water in healthy volunteers
| Fexofenadine | Water phase (control) | GTE phase | ||
|---|---|---|---|---|
| Geometric mean | Geo CV(%) | Geometric mean | Geo CV(%) | |
| C max (ng/mL) | 278.7 | 48.3 | 82.6 | 47.1 |
| GMR (90% CI) | 0.296 | (0.197–0.396) | ||
| T max (h) | 2.0 | (1.5–6.0) | 2.0 | (1.5–3.0) |
| AUC0–8 (h ng/mL) | 1131.7 | 47.5 | 323.6 | 45.7 |
| GMR (90% CI) | 0.286 | (0.214–0.357) | ||
| AUC0–∞ (h ng/mL) | 1765.0 | 44.8 | 521.9 | 50.6 |
| GMR (90% CI) | 0.296 | (0.225–0.366) | ||
| t 1/2 (h) | 5.2 | 31.4 | 5.7 | 48.7 |
| GMR (90% CI) | 1.096 | (0.927–1.265) | ||
| A e (mg) | 6.0 | 35.0 | 2.0 | 27.3 |
| GMR (90% CI) | 0.335 | (0.251–0.418) | ||
| CLR (mL/min) | 59.2 | 27.2 | 68.5 | 33.5 |
| GMR (90% CI) | 1.158 | (1.074–1.242) | ||
Fexofenadine (60 mg) was orally administered with water (150 mL), or an aqueous solution of EGCG‐concentrated green tea extract (150 mL) in 10 healthy Japanese volunteers. The T max values are expressed as median (range).
A e, amount excreted unchanged into urine over 24 hours; AUC, area under the plasma concentration‐time curve; CI, confidence interval; CL R, renal clearance; C max, peak plasma concentration; CV, coefficient of variation; GMR, geometric mean ratio; GTE, green tea extract; T max, time to C max; t 1/2, terminal half‐life.
Figure 2.
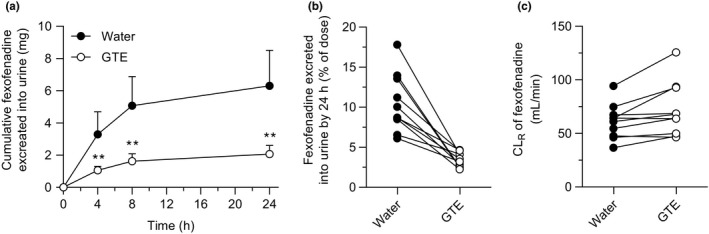
Urinary excretion of fexofenadine after oral administration of fexofenadine (60 mg) with 150 mL of an aqueous solution of (−)‐epigallocatechin gallate (EGCG)‐concentrated green tea extract (○), or water (●) in 10 healthy volunteers. (a) Cumulative urinary excretion (mg) of fexofenadine over 24 hours after administration, (b) % of dose, and (c) renal clearance (CLR) of fexofenadine. Data are expressed as the arithmetic mean ± SD. **, P < 0.01 with respect to water phase. GTE, green tea extract.
Pseudoephedrine pharmacokinetics
Plasma concentration‐time profiles and pharmacokinetic parameters of pseudoephedrine are shown in Figure 3 and Table 2 . No differences were observed in any pharmacokinetic parameters of pseudoephedrine, including AUC, T max, t 1/2, and CL renal between phases, whereas C max was slightly decreased by GTE (P = 0.048; Figure S1 ). Urinary excretion of pseudoephedrine in GTE phase was nearly superimposed on those in water phase (Figure 4 a–c ). The change in pseudoephedrine AUC0‐∞ by GTE was not correlated with pseudoephedrine AUC0‐∞ in control phase (r = −0.3548, P = 0.3144; Figure S2 b).
Figure 3.
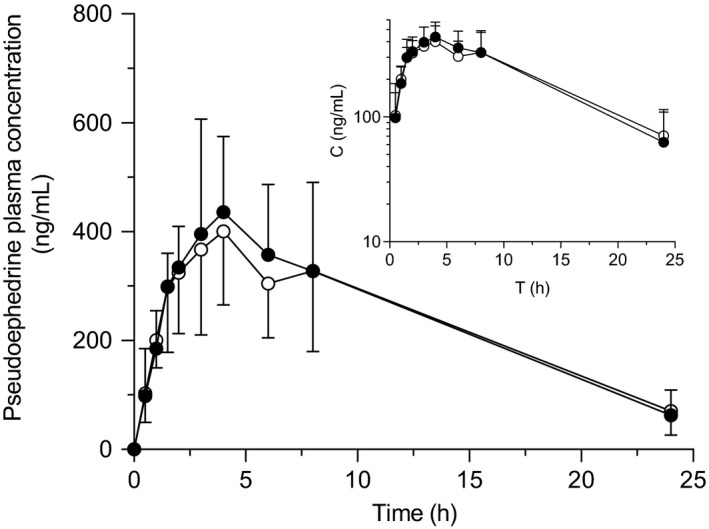
Plasma concentration profile of pseudoephedrine after oral administration of pseudoephedrine (120 mg) with 150 mL of an aqueous solution of (−)‐epigallocatechin gallate (EGCG)‐concentrated green tea extract (○), or water (●) in 10 healthy volunteers. Data are expressed as the arithmetic mean ± SD. The inset is the log‐concentration vs. time profile.
Table 2.
Pharmacokinetic parameters of pseudoephedrine after oral administration with GTE or water in healthy volunteers
| Pseudoephedrine | Water phase (control) | GTE phase | ||
|---|---|---|---|---|
| Geometric mean | Geo CV (%) | Geometric mean | Geo CV (%) | |
| C max (ng/mL) | 474.8 | 34.6 | 407.3 | 27.4 |
| GMR (90% CI) | 0.858 | (0.757–0.958) | ||
| T max (h) | 4.0 | (1.5–6.0) | 3.5 | (1.5–8.0) |
| AUC0–24 (h ng/mL) | 5396.5 | 37.7 | 5309.6 | 36.6 |
| GMR (90% CI) | 0.984 | (0.931–1.037) | ||
| AUC0–∞ (h ng/mL) | 5965.8 | 43.1 | 6107.8 | 42.4 |
| GMR (90% CI) | 1.024 | (0.906–1.142) | ||
| t 1/2 (h) | 6.5 | 31.1 | 7.7 | 31.6 |
| GMR (90% CI) | 1.174 | (0.908–1.439) | ||
| A e (mg) | 68.7 | 21.0 | 63.2 | 20.4 |
| GMR (90% CI) | 0.920 | (0.846–0.994) | ||
| CLR (mL/min) | 212.1 | 53.3 | 198.3 | 50.2 |
| GMR (90% CI) | 0.935 | (0.813–1.058) | ||
Pseudoephedrine (120 mg) was orally administered with water (150 mL), or an aqueous solution of EGCG‐concentrated green tea extract (150 mL) in 10 healthy Japanese volunteers. The T max values are expressed as median (range).
A e, amount excreted unchanged into urine over 24 hours; AUC, area under the plasma concentration‐time curve; CI, confidence interval; CL R, renal clearance; C max, peak plasma concentration; CV, coefficient of variation; GMR, geometric mean ratio; GTE, green tea extract; T max, time to C max; t 1/2, terminal half‐life.
Figure 4.
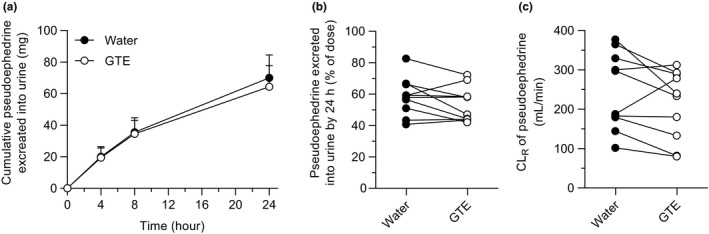
Urinary excretion of pseudoephedrine after oral administration of pseudoephedrine (120 mg) with 150 mL of an aqueous solution of (−)‐epigallocatechin gallate (EGCG)‐concentrated green tea extract (GTE; ○), or water (●) in 10 healthy volunteers. (a) Cumulative urinary excretion (mg) of pseudoephedrine over 24 hours after administration, (b) percent of dose, and (c) renal clearance (CLR) of pseudoephedrine. Data are expressed as the arithmetic mean ± SD.
Inhibition of fexofenadine uptake by EGCG and GTE
Cellular accumulation of fexofenadine (10 μM) in HEK‐OATP1A2 cells was 22.14‐fold higher than that in vector control (PQX) cells (P < 0.01) at pH 7.3. Moreover, OATP1A2‐mediated fexofenadine uptake was nearly completely inhibited in the presence of EGCG (100 μM and 1 mM) or GTE (0.1 and 1 mg/mL; P < 0.01; Figure 5 a ). On the other hand, only slight but significant uptake of fexofenadine (10 μM) mediated by OATP2B1 was observed in HEK‐OATP2B1 cells compared with vector control cells at both pH 6.3 and pH 7.3 (Figure S3 ), which is in accordance with recent papers. 29 , 30 However, due to low uptake ratio for OATP2B1‐mediated fexofenadine transport, inhibition studies at both pH conditions revealed no consistent effect for both EGCG and GTE (data not shown). We used BSP as a typical substrate of OATP2B1, and found that EGCG (100 μM and 1 mM) and GTE (0.1 and 1 mg/mL) significantly reduced OATP2B1‐mediated uptake of BSP at both pH 6.3 and pH 7.3 (P < 0.01; Figure 5 b,c ).
Figure 5.
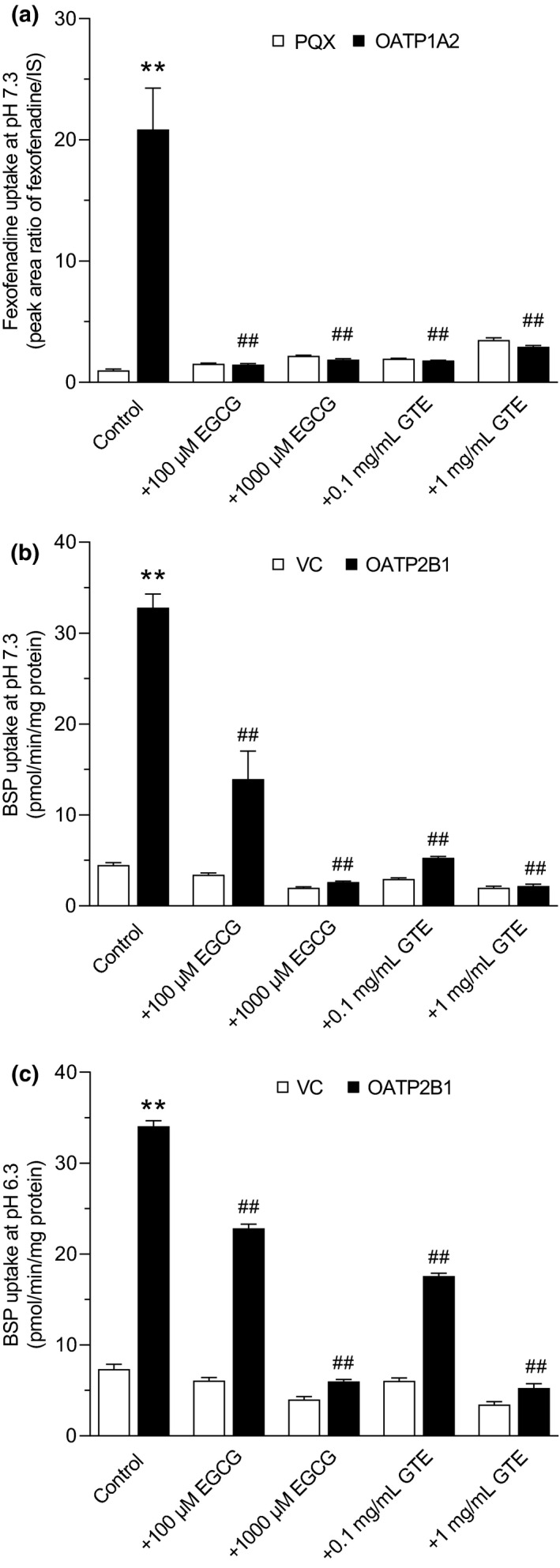
In vitro uptake assays of fexofenadine in HEK‐OATP1A2 cells or sulfobromophthalein (BSP) in HEK‐OATP2B1 cells (black bars), and in the respective HEK control cells (PQX) for OATP1A2 or (VC) for OATP2B1 (white bars). Cellular accumulation of 10 μM fexofenadine at pH 7.3 (a), or 1 μM BSP at pH 7.3 (b) and pH 6.3 (c) after 10 minutes incubation was measured by liquid chromatography–tandem mass spectrometry (LC–MS/MS) (fexofenadine) or liquid scintillation counting (BSP) in the presence or absence of (−)‐epigallocatechin gallate (EGCG at 100 and 1,000 μM) or EGCG‐concentrated green tea extract (GTE at 0.1 and 1 mg/mL) used in the clinical study. Data are presented as mean ± SEM. (n = 6). **P < 0.01, vs. uptake into HEK293 control cells (PQX or VC); ## P < 0.01, vs. uptake into HEK293‐OATP1A2 or HEK‐OATP2B1 cells without inhibitors.
Chemical binding between fexofenadine and EGCG
It has been reported that GT catechins chemically interacted with various kinds of drugs including bortezomib, a proteasome inhibitor (Figure S4 a). 31 , 32 , 33 , 34 Accordingly, we investigated whether EGCG also directly interacts with fexofenadine in vitro. As shown in the representative chromatograms (Figure S4 b,c), the bortezomib peak found in 0 hour was gradually decreased, and 2 new peaks appeared in the presence of EGCG over 24 hours compared with bortezomib alone. Bortezomib concentration was significantly decreased by co‐incubation with EGCG (P < 0.01; Figure S4 d), indicating that EGCG interacted with bortezomib molecule and enhanced its degradation. By contrast, EGCG did not reduce fexofenadine concentration in an aqueous solution through 24 hours at 37°C (Figure S4 e).
DISCUSSION
In this study, we report that a single concomitant ingestion of an aqueous solution (150 mL) of GTE containing about 300 mg of EGCG (~ 4.4 mM), significantly decreases plasma concentrations and urinary excretion of fexofenadine when compared with water (control) in healthy volunteers (Figures 1, 2). In contrast, no substantial differences were observed in the pharmacokinetics of pseudoephedrine between GTE and control phases (Figures 3, 4). Fexofenadine and pseudoephedrine have common properties in terms of high hydrophilicity and negligible oxidative metabolism in the body, whereas the oral bioavailability of fexofenadine (35%) is considerably lower than that of pseudoephedrine (≈100%). 23 , 35 In addition, whereas fexofenadine is mainly eliminated by hepato‐biliary elimination, pseudoephedrine is mostly excreted into urine. Previous clinical studies imply that GT and its main component, EGCG, may reduce the intestinal absorption of hydrophilic drugs with a relatively low bioavailability, including nadolol and lisinopril. 2 , 3 , 4 Therefore, taken together with data showing that GTE did not alter the apparent t 1/2 of fexofenadine, it is suggested that EGCG in the GTE mainly inhibits the intestinal absorption of fexofenadine.
Regarding the molecular mechanisms for reduced exposure of fexofenadine after oral administration, it is well known that the intestinal absorption of fexofenadine is highly dependent on transporter‐mediated uptake and/or efflux, and that inhibition of the relevant transporters by co‐administered drugs or food could result in a significant impact on its pharmacokinetics. 9 OATP1A2 and OATP2B1 are reported to be expressed in enterocytes, 36 , 37 , 38 , 39 and fexofenadine is a substrate of both transporters with a kinetic metabolite value of 6.4 μM for OATP1A2, and kinetic metabolite values for high‐ and low‐affinity binding sites of 0.14 μM and 885 μM, respectively, for OATP2B1. 11 , 40 EGCG is also a substrate of OATP1A2, and competitively inhibits OATP1A2‐mediated nadolol transport with inhibition constant value of ~ 20 μM. 2 , 5 In the present study, we confirmed significant OATP1A2‐mediated uptake of fexofenadine, and which was potently inhibited by GTE used in the clinical study and EGCG (Figure 5 a ). In addition, EGCG inhibits OATP2B1‐mediated uptake of estrone‐3‐sulfate with half‐maximal inhibitory concentration values ranging from 7.1–101 μM. 5 , 6 This was confirmed in BSP transport assay for OATP2B1 (Figure 5 b,c ). On the other hand, only slight uptake of fexofenadine was observed in HEK‐OATP2B1 cells at pH 6.3 and pH 7.3, which is in accordance with recently published data. 29 , 30 , 41 Due to low uptake ratio (HEK‐OATP2B1/HEK‐Co), we found no consistent effects of GTE and EGCG on OATP2B1‐mediated fexofenadine transport. As mentioned above, the subjects orally ingested the GTE aqueous solution containing ~ 4.4 mM of EGCG, and therefore the concentration of EGCG in the gastrointestinal tract may be sufficient to inhibit the uptake transport of fexofenadine by OATP1A2 into enterocytes, which may lead to a reduction in the oral bioavailability. It is noted that intestinal expression of OATP1A2 is still controversial. However, recent highly sensitive proteomic analyses reported the expression of OATP1A2 in human jejunum and ileum samples. 37 , 39 Moreover, Hirvensalo et al. recently demonstrated that intestinal OATP1A2 and P‐glycoprotein play a role in the pharmacokinetics of celiprolol in humans, 42 further supporting the molecular mechanism underlying fexofenadine‐GT interaction through OATP1A2.
A second possible mechanism is a direct chemical interaction between fexofenadine and EGCG. Indeed, various drugs, including aripiprazole, bortezomib, cetirizine, and sunitinib, has been reported to interact with EGCG. 31 , 32 , 33 , 34 In line with a previous study by Golden et al., 31 our data show that EGCG interacted with bortezomib and significantly enhanced its degradation over 24 hours at 37°C (Figure S3 ). In contrast, EGCG did not reduce but rather stabilized fexofenadine in solution presumably by the anti‐oxidative properties, suggesting that EGCG is unlikely to chemically interact with fexofenadine. Another possibility is the osmotic effects of co‐ingested beverages on fexofenadine pharmacokinetics. Fruit juices containing a high amount of nonabsorbed carbohydrates, such as apple juice, show volume‐dependent interaction with fexofenadine. 16 , 27 , 43 In such cases, water volume in intestinal lumen may be increased by an osmotic gradient, 44 and the luminal drug concentration is decreased, which may result in a reduction in intestinal absorption with a tendency of delay in the absorption rate (T max). 27 , 43 In this study, in order to avoid osmotic effects of GT components, we used an aqueous solution of EGCG‐concentrated GTE with water volume of 150 mL. These solutions are reported to be even more hypotonic than GT infusion. 45 Indeed, we found no prolongation in T max of fexofenadine in GTE phase, indicating that GTE used in this study has different interaction mechanisms from high‐volume fruit juices.
In contrast to fexofenadine, the plasma concentration‐time profile and cumulative urinary excretions of pseudoephedrine were similar in both phases, suggesting that EGCG had no impact on pseudoephedrine pharmacokinetics (Figures 3, 4). Due to high solubility and low permeability, pseudoephedrine was provisionally classified as BCS class 3 drug. 21 However, subsequent studies have demonstrated that the permeability of pseudoephedrine is dependent on pH in the gastrointestinal tract. 46 , 47 Pseudoephedrine was shown to exhibit a relatively low permeability at the proximal regions of intestine (pH 6.5), which increases with its transition to distal regions (pH 7.5), possibly accounting for its excellent intestinal absorption. In addition, the molecular size may be another factor contributing to the lack of interaction between GTE and pseudoephedrine. Compared with the molecular weights of nadolol (309.4 g/mol), lisinopril (405.5 g/mol), and fexofenadine (501.7 g/mol), the molecular weight of pseudoephedrine (165.2 g/mol) is much smaller, which offers advantages of (i) higher concentration at the surface of the intestinal epithelium; and (ii) easier passive diffusion through the plasma membrane or the paracellular pathway. 48 Although drug transporters responsible for membrane trafficking of pseudoephedrine in the intestine remains to be investigated, the differences in physicochemical and pharmacokinetic properties may explain the distinct effects of GTE on the intestinal absorption of fexofenadine and pseudoephedrine.
A limitation of this study is that all volunteers were Japanese, so it remains unclear whether ethnic differences effect pharmacokinetic interactions between GT and fexofenadine. In relation to ethnicity, there was no influence of the SLCO2B1*3 SNV on fexofenadine pharmacokinetics, although we confirmed that this polymorphism is common in Japanese population. 18 Fexofenadine disposition is influenced by multiple transporters, such as OATP1Bs (SLCO1B1 and SLCO1B3) and P‐glycoprotein (ABCB1), in addition to OATP1A2 and OATP2B1. 13 , 49 , 50 For example, ABCB1 c.2677G>T/A and SLCO1B1 c.521T>C (OATP1B1*5) polymorphisms might contribute to pharmacokinetic differences of fexofenadine, 40 , 41 however, we did not plan to identify these genotypes in the subjects because of our working hypothesis that GTE could mainly affect intestinal absorption process of test drugs. Finally, we did not differentiate the plasma concentrations and urinary excretions between fexofenadine enantiomers. It would be worth investigating the influence of EGCG on the pharmacokinetics of each stereoisomer of fexofenadine in the future.
In conclusion, we show for the first time that co‐administration of GTE markedly reduces exposure to fexofenadine in humans by inhibiting OATP1A2‐mediated intestinal absorption, whereas the pharmacokinetics of pseudoephedrine are unlikely to be influenced by co‐administration of GTE. Considering that brewed GT contains more catechins and flavonoids in addition to EGCG, we propose that GT should not be taken during anti‐allergic therapy using, at least, fexofenadine.
FUNDING
This study was partly supported by JSPS KAKENHI Grant Number 17K17983 and 17K08949.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
S.M., Y.O., R.T., J.K., H.W., M.F., and K.S. wrote the manuscript. S.M., J.K., H.W., M.F., and K.S. designed the research. S.M., Y.O., R.T., E.H., T.O., H.O., and K.S. performed the research. S.M., Y.O., R.T., J.K., H.W., M.F., and K.S. analyzed the data.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Appendix S1
ACKNOWLEDGMENTS
The authors thank Dr. Kazuo Minamikawa, Dr. Akihiro Inano, Ms. Hazuki Ito, and Ms. Momoka Ito for support on conducting the clinical trial.
References
- 1. Misaka, S. et al. Green tea ingestion greatly reduces plasma concentrations of nadolol in healthy subjects. Clin. Pharmacol. Ther. 95, 432–438 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Abe, O. et al. Role of (−)‐epigallocatechin gallate in the pharmacokinetic interaction between nadolol and green tea in healthy volunteers. Eur. J. Clin. Pharmacol. 74, 775–783 (2018). [DOI] [PubMed] [Google Scholar]
- 3. Misaka, S. et al. Effects of single green tea ingestion on pharmacokinetics of nadolol in healthy volunteers. Br. J. Clin. Pharmacol. 86, 2314–2318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Misaka, S. et al. Impact of green tea catechin ingestion on the pharmacokinetics of lisinopril in healthy volunteers. Clin. Transl. Sci. 14, 476–480 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roth, M. , Timmermann, B.N. & Hagenbuch, B. Interactions of green tea catechins with organic anion‐transporting polypeptides. Drug Metab. Dispos. 39, 920–926 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iijima, R. , Watanabe, T. , Ishiuchi, K. , Matsumoto, T. , Watanabe, J. & Makino, T. Interactions between crude drug extracts used in Japanese traditional Kampo medicines and organic anion‐transporting polypeptide 2B1. J. Ethnopharmacol. 214, 153–159 (2018). [DOI] [PubMed] [Google Scholar]
- 7. Fuchikami, H. , Satoh, H. , Tsujimoto, M. , Ohdo, S. , Ohtani, H. & Sawada, Y. Effects of herbal extracts on the function of human organic anion‐transporting polypeptide OATP‐B. Drug Metab. Dispos. 34, 577–582 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Paśko, P. , Rodacki, T. , Domagała‐Rodacka, R. , Palimonka, K. , Marcinkowska, M. & Owczarek, D. Second generation H1 – antihistamines interaction with food and alcohol – a systematic review. Biomed. Pharmacother. 93, 27–39 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Akamine, Y. & Miura, M. An update on the clinical pharmacokinetics of fexofenadine enantiomers. Expert Opin. Drug Metab. Toxicol. 14, 429–434 (2018). [DOI] [PubMed] [Google Scholar]
- 10. Wu, C.Y. & Benet, L.Z. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm. Res. 22, 11–23 (2005). [DOI] [PubMed] [Google Scholar]
- 11. Cvetkovic, M. , Leake, B. , Fromm, M.F. , Wilkinson, G.R. & Kim, R.B. OATP and P‐glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab. Dispos. 27, 866–871 (1999). [PubMed] [Google Scholar]
- 12. Ma, J.D. , Tsunoda, S.M. , Bertino, J.S. Jr. , Trivedi, M. , Beale, K.K. & Nafziger, A.N. Evaluation of in vivo P‐glycoprotein phenotyping probes: a need for validation. Clin. Pharmacokinet. 49, 223–237 (2010). [DOI] [PubMed] [Google Scholar]
- 13. Kusuhara, H. et al. Effect of coadministration of single and multiple doses of rifampicin on the pharmacokinetics of fexofenadine enantiomers in healthy subjects. Drug Metab. Dispos. 41, 206–213 (2013). [DOI] [PubMed] [Google Scholar]
- 14. Shon, J.H. et al. Effect of itraconazole on the pharmacokinetics and pharmacodynamics of fexofenadine in relation to the MDR1 genetic polymorphism. Clin. Pharmacol. Ther. 78, 191–201 (2005). [DOI] [PubMed] [Google Scholar]
- 15. Hamman, M.A. , Bruce, M.A. , Haehner‐Daniels, B.D. & Hall, S.D. The effect of rifampin administration on the disposition of fexofenadine. Clin. Pharmacol. Ther. 69, 114–121 (2001). [DOI] [PubMed] [Google Scholar]
- 16. Dresser, G.K. et al. Fruit juices inhibit organic anion transporting polypeptide‐mediated drug uptake to decrease the oral availability of fexofenadine. Clin. Pharmacol. Ther. 71, 11–20 (2002). [DOI] [PubMed] [Google Scholar]
- 17. Bailey, D.G. , Dresser, G.K. , Leake, B.F. & Kim, R.B. Naringin is a major and selective clinical inhibitor of organic anion‐transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin. Pharmacol. Ther. 81, 495–502 (2007). [DOI] [PubMed] [Google Scholar]
- 18. Imanaga, J. et al. The effects of the SLCO2B1 c.1457C > T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet. Genomics 21, 84–93 (2011). [DOI] [PubMed] [Google Scholar]
- 19. Mansfield, L.E. Once‐daily immediate‐release fexofenadine and sustained‐release pseudoephedrine combination: a new treatment option for allergic rhinitis. Expert. Opin. Pharmacother. 7, 941–951 (2006). [DOI] [PubMed] [Google Scholar]
- 20. Howard, D.R. , Haribhakti, R. , Kittner, B. & Agrawala, P. Single‐dose and steady‐state bioequivalence of fexofenadine and pseudoephedrine combination tablets compared with individual formulations in healthy adults. Curr. Med. Res. Opin. 21, 769–776 (2005). [DOI] [PubMed] [Google Scholar]
- 21. Takagi, T. , Ramachandran, C. , Bermejo, M. , Yamashita, S. , Yu, L.X. & Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 3, 631–643 (2006). [DOI] [PubMed] [Google Scholar]
- 22. Benet, L.Z. , Broccatelli, F. & Oprea, T.I. BDDCS applied to over 900 drugs. AAPS J. 13, 519–547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanfer, I. , Dowse, R. & Vuma, V. Pharmacokinetics of oral decongestants. Pharmacotherapy 13, 116S–128S (1993). [PubMed] [Google Scholar]
- 24. Wecker, M.T. , Graves, D.A. , Amsel, L.P. , Hinsvark, O.N. & Rotenberg, K.S. Influence of a standard meal on the absorption of controlled‐release pseudoephedrine capsules. J. Pharm. Sci. 76, 29–31 (1987). [DOI] [PubMed] [Google Scholar]
- 25. Li, Q.S. , Wang, Y.Q. , Liang, Y.R. & Lu, J.L. The anti‐allergic potential of tea: a review of its components, mechanisms and risks. Food Funct. 12, 57–69 (2021). [DOI] [PubMed] [Google Scholar]
- 26. Mandery, K. et al. Influence of the flavonoids apigenin, kaempferol, and quercetin on the function of organic anion transporting polypeptides 1A2 and 2B1. Biochem. Pharmacol. 80, 1746–1753 (2010). [DOI] [PubMed] [Google Scholar]
- 27. Luo, J. et al. The pharmacokinetic exposure to fexofenadine is volume‐dependently reduced in healthy subjects following oral administration with apple juice. Clin. Transl. Sci. 9, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Russell, T. , Stoltz, M. & Weir, S. Pharmacokinetics, pharmacodynamics, and tolerance of single‐ and multiple‐dose fexofenadine hydrochloride in healthy male volunteers. Clin. Pharmacol. Ther. 64, 612–621 (1998). [DOI] [PubMed] [Google Scholar]
- 29. Bednarczyk, D. & Sanghvi, M.V. Organic anion transporting polypeptide 2B1 (OATP2B1), an expanded substrate profile, does it align with OATP2B1's hypothesized function? Xenobiotica 50, 1128–1137 (2020). [DOI] [PubMed] [Google Scholar]
- 30. Morita, T. et al. Citrus fruit‐derived flavanone glycoside narirutin is a novel potent inhibitor of organic anion‐transporting polypeptides. J. Agric. Food Chem. 68, 14182–14191 (2020). [DOI] [PubMed] [Google Scholar]
- 31. Golden, E.B. et al. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid‐based proteasome inhibitors. Blood 113, 5927–5937 (2009). [DOI] [PubMed] [Google Scholar]
- 32. Ge, J. et al. Interaction of green tea polyphenol epigallocatechin‐3‐gallate with sunitinib: potential risk of diminished sunitinib bioavailability. J. Mol. Med. 89, 595–602 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Ohata, T. et al. Drug‐tea polyphenol interaction (II) complexation of piperazine derivatives with green tea polyphenol. Thermochim. Acta 653, 1–7 (2017). [Google Scholar]
- 34. Ikeda, H. et al. Drug‐tea polyphenol interaction (III) incompatibility between aripiprazole Oral solution and green tea. Chem. Pharm. Bull. 70, 230–234 (2022). [DOI] [PubMed] [Google Scholar]
- 35. Lappin, G. et al. Pharmacokinetics of fexofenadine: evaluation of a microdose and assessment of absolute oral bioavailability. Eur. J. Pharm. Sci. 40, 125–131 (2010). [DOI] [PubMed] [Google Scholar]
- 36. Glaeser, H. et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin. Pharmacol. Ther. 81, 362–370 (2007). [DOI] [PubMed] [Google Scholar]
- 37. Couto, N. et al. Quantitative proteomics of clinically relevant drug‐metabolizing enzymes and drug transporters and their intercorrelations in the human small intestine. Drug Metab. Dispos. 48, 245–254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Drozdzik, M. et al. Protein abundance of clinically relevant drug transporters in the human liver and intestine: a comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 105, 1204–1212 (2019). [DOI] [PubMed] [Google Scholar]
- 39. Akazawa, T. , Uchida, Y. , Miyauchi, E. , Tachikawa, M. , Ohtsuki, S. & Terasaki, T. High expression of UGT1A1/1A6 in monkey small intestine: comparison of protein expression levels of cytochromes P450, UDP‐glucuronosyltransferases, and transporters in small intestine of Cynomolgus monkey and human. Mol. Pharm. 15, 127–140 (2018). [DOI] [PubMed] [Google Scholar]
- 40. Shirasaka, Y. , Mori, T. , Murata, Y. , Nakanishi, T. & Tamai, I. Substrate‐ and dose‐dependent drug interactions with grapefruit juice caused by multiple binding sites on OATP2B1. Pharm. Res. 31, 2035–2043 (2014). [DOI] [PubMed] [Google Scholar]
- 41. Won, C.S. et al. A modified grapefruit juice eliminates two compound classes as major mediators of the grapefruit juice‐fexofenadine interaction: an in vitro‐in vivo “connect”. J. Clin. Pharmacol. 53, 982–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirvensalo, P. et al. Pharmacogenomics of celiprolol – evidence for a role of P‐glycoprotein and organic anion transporting polypeptide 1A2 in celiprolol pharmacokinetics. Clin. Transl. Sci. 15, 409–421 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dresser, G.K. , Kim, R.B. & Bailey, D.G. Effect of grapefruit juice volume on the reduction of fexofenadine bioavailability: possible role of organic anion transporting polypeptides. Clin. Pharmacol. Ther. 77, 170–177 (2005). [DOI] [PubMed] [Google Scholar]
- 44. Grimm, M. et al. Gastric emptying and small bowel water content after administration of grapefruit juice compared to water and isocaloric solutions of glucose and fructose: a four‐way crossover MRI pilot study in healthy subjects. Mol. Pharm. 15, 548–559 (2018). [DOI] [PubMed] [Google Scholar]
- 45. Adeli, F. et al. Comparative in vitro study of the effectiveness of green tea extract and common storage media on periodontal ligament fibroblast viability. Eur J Dent 10, 408–412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fairstein, M. , Swissa, R. & Dahan, A. Regional‐dependent intestinal permeability and BCS classification: elucidation of pH‐related complexity in rats using pseudoephedrine. AAPS J. 15, 589–597 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wolk, O. et al. Segmental‐dependent intestinal drug permeability: development and model validation of in silico predictions guided by in vivo permeability values. J. Pharm. Sci. 108, 316–325 (2019). [DOI] [PubMed] [Google Scholar]
- 48. Camenisch, G. , Alsenz, J. , van de Waterbeemd, H. & Folkers, G. Estimation of permeability by passive diffusion through Caco‐2 cell monolayers using the drugs' lipophilicity and molecular weight. Eur. J. Pharm. Sci. 6, 317–324 (1998). [PubMed] [Google Scholar]
- 49. Niemi, M. , Kivisto, K.T. , Hofmann, U. , Schwab, M. , Eichelbaum, M. & Fromm, M.F. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1). Br. J. Clin. Pharmacol. 59, 602–604 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vanhove, T. , Bouillon, T. , de Loor, H. , Annaert, P. & Kuypers, D. Fexofenadine, a putative in vivo P‐glycoprotein probe, fails to predict clearance of the substrate tacrolimus in renal recipients. Clin. Pharmacol. Ther. 102, 989–996 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Appendix S1